

Abstract
Gene therapy for hematological disorders relies on the genetic modification of CD34+ cells, a heterogeneous cell population containing about 0.01% long-term repopulating cells. Here, we show that the lentiviral vector CD133-LV, which uses a surface marker on human primitive hematopoietic stem cells (HSCs) as entry receptor, transfers genes preferentially into cells with high engraftment capability. Transduction of unstimulated CD34+ cells with CD133-LV resulted in gene marking of cells with competitive proliferative advantage in vitro and in immunodeficient mice. The CD133-LV-transduced population contained significantly more cells with repopulating capacity than cells transduced with vesicular stomatitis virus (VSV)-LV, a lentiviral vector pseudotyped with the vesicular stomatitis virus G protein. Upon transfer of a barcode library, CD133-LV-transduced cells sustained gene marking in vivo for a prolonged period of time with a 6.7-fold higher recovery of barcodes compared to transduced control cells. Moreover, CD133-LV-transduced cells were capable of repopulating secondary recipients. Lastly, we show that this targeting strategy can be used for transfer of a therapeutic gene into CD34+ cells obtained from patients suffering of X-linked chronic granulomatous disease. In conclusion, direct gene transfer into CD133+ cells allows for sustained long-term engraftment of gene corrected cells.
Introduction
A series of phase 1/2 clinical trials have provided convincing evidence that correction of genetic defects by ex vivo gene transfer into hematopoietic CD34+ cells is an alternative therapeutic approach to allogeneic hematopoietic stem cell transplantation (HSCT), in particular for patients lacking a suitable matched donor.1,2,3,4,5 Usually, CD34+ cells from granulocyte colony-stimulating factor (GCSF)-mobilized peripheral blood (mPB) are genetically modified in this approach. This cell population is heterogeneous and contains, in addition to a few cells with long-term repopulating capability (~0.01%),6 a vast excess of multilineage progenitors with short-term engraftment properties as well as more differentiated lineage-restricted progenitors with low or no engraftment capabilities.7,8,9 The relevant target cell for sustained gene correction is the primitive hematopoietic stem cell (HSC) with long-term repopulating and self-renewal capacity (LT-HSC).
A series of elegant studies have characterized LT-HSC based on their multilineage repopulating capacity in nonobese diabetic/severe combined immunodeficient (NOD/SCID) mice.10,11 This SCID-repopulating cell (SRC) is exclusively contained within the CD34+CD38− fraction of human bone marrow, cord blood, and mobilized peripheral blood (mPB) cells.7,12 In addition, expression of CD133 (prominin-1) correlates with the capacity of the cells to engraft long-term, thereby defining CD133 as an additional cell surface marker of LT-HSCs. 13,14 Hematopoietic cells isolated on the basis of CD133 expression possess a higher content of long-term culture initiating cells (LTC-IC) and increased proliferative capacity in vitro than cells isolated on CD34 expression.15,16 Coexpression of CD34 and CD133 is highest in samples from mPB reaching up to 80% compared to CB (50%) and BM (13%), and most of the SRC activity is contained within this cell population.17,18 First clinical trials have shown that cells isolated for CD133+ expression can substitute for standard CD34+ cells in HSC transplantation.19
Thus, one alternative to direct gene transfer to LT-HSCs is to enrich for primitive HSCs based on cell surface marker expression before transduction. Indeed, lentiviral transduction of mPB CD34+CD38−Lin− cells resulted in high gene transfer efficiencies and stable gene marking of LTC-IC and colony-forming cells derived thereof for more than 10 weeks in liquid cultures.20 However, bystander cells are beneficial for accelerated hematopoietic reconstitution after full myeloablative conditioning and thus isolation and transplantation of pure LT-HSCs might be disadvantageous.21 Hence, an ideal approach for gene therapy directs gene transfer to the LT-HSC population present within the heterogeneous pool of CD34+ cells.
The most widely used envelope for pseudotyping lentiviral vectors (LVs) is the vesicular stomatitis virus (VSV) glycoprotein G. The LDL receptor family members were recently identified as entry receptors for VSV-LV particles.22 Therefore, VSV-G pseudotyped vectors have the capacity to transduce a wide range of cell types and have been successfully used for the genetic modification of cells in the context of gene therapy trials (reviewed in ref. 5).
A strictly defined tropism can be achieved by the versatile targeting strategy relying on the two measles virus envelope proteins: the hemagglutinin (H) mediates receptor attachment while the fusion protein (F) is responsible for vector particle cell membrane fusion. Upon blinding the H protein for recognition of its natural receptors23,24 and linking it to a single-chain antibody (scFv) recognizing the cell surface antigen of choice, receptor-targeted vectors highly specific for a variety of cell types have been generated.25,26 Among these, CD133-LV, which displays a scFv derived from the CD133-specific monoclonal antibody 141.7, efficiently targets CD133+ cells in mPB cells.25
Here, we show that CD133-LV preferentially transduces a population of human hematopoietic stem cells with high proliferative potential in vitro and multilineage engraftment in vivo, highlighting the potential of CD133-LVs as an alternative to VSV-pseudotyped lentiviral vectors for gene therapy of hematological disorders.
Results
Targeted gene transfer into human CD133hi/CD34hi/CD38− cells
To identify the target cell population of CD133-LV, we first analyzed mPB CD34+ cells for coexpression of CD133 and CD38 (Figure 1a). Next, we sorted CD34+ cells into discrete populations of cells expressing high or low CD34 or CD38, while for CD133+ an additional population with intermediate mean fluorescence intensity was isolated (Figure 1b). These cell populations were individually transduced with CD133-LV or lentiviral vector pseudotyped with the VSV-G envelope protein (VSV-LV). Transductions were performed at low multiplicity of infection and in the absence of cytokine prestimulation, as the CD133 marker is rapidly downregulated upon cultivation of freshly isolated CD34+ cells in the presence of cytokines (Supplementary Figure S1). Under these conditions, transduction efficiencies were rather low (~10%), but high enough to identify the transduced cell types. CD133-LV preferentially transduced CD133hi, CD34hi, and CD38lo but also CD133mid, CD34lo, CD38hi, and CD133lo cells, albeit at a markedly reduced efficiency (Figure 1c). In contrast, this more mature cell population was preferentially transduced by VSV-LV, while the CD133hi, CD38lo, and CD34hi populations were barely transduced under the conditions used in these experiments. From these observations, we conclude that CD133-LV transduces preferentially CD133bright cells, which in addition are positive for CD34 and negative for CD38, a human hematopoietic cell population with phenotypic characteristics attributed to stem cells.
Figure 1.

Targeted gene transfer into human CD133hi/CD34hi/CD38low cells. (a) Correlation of CD133 expression with established hematopoietic stem cell (HSC) markers in freshly isolated mPB CD34+ cells (purity >95%). (b) Sorting of mPB CD34+ cells into discrete subpopulations according to the expression intensities of CD34, CD38, and CD133. For CD34 and CD38, the highest and lowest 15% of cells were isolated via FACS; for CD133 expression, cells were subdivided into three intervals (lowest, mid, highest), each 30% of the total population. (c) Sorted cells obtained from b were cultured at identical conditions and transduced with equal amounts of transducing units of VSV- or CD133-LV to estimate the relative transduction efficiency on different hematopoietic stem-progenitor cells (HSPCs) 3 days post-transduction (n = 3, error bars = SD).
CD133-LV transduces a cell population with high proliferative potential
Freshly isolated mPB CD34+ cells were transduced overnight with CD133-LV and VSV-LV, expressing tuBFP and eGFP, respectively (Figure 2a). Transduction efficiencies ranged from 2% up to 20% for CD133-LV(tuBFP) and 5–40% for VSV-LV(GFP). To avoid skewing of the results due to pseudotransduction, events with low fluorescence intensity were excluded at early time points by stringent gating. Transduced cells were kept in culture for 17 days under cytokine stimulation to assess their proliferative potential. While CD133-LV-transduced cells continuously expanded during culture, the proportion of VSV-LV transduced cells declined gradually from initially 17–10% (Figure 2b,c). Thus, CD133-LV transduces a cell population with extensive proliferative capacity, a hallmark of hematopoietic stem and primitive progenitor cells.7 This effect was also observed when cells were prestimulated for 12, 24, or 36 hours prior to transduction. Although overall transduction efficiencies for CD133-LV decreased with extended prestimulation time, the overall dynamics of transduced cell fractions remained surprisingly consistent (Supplementary Figure S2). In contrast, transduction of CD34+ cells with VSV-LVs resulted in increased transduction efficiencies with increased prestimulation time. However, gene marking was not sustained over time. These observations confirm preferential transduction by CD133-LV of cells with high proliferative potential.
Figure 2.

In vitro competitive proliferation kinetics of mPB CD34+ cells transduced with VSV-LV and CD133-LV. (a) Lentiviral vectors encoding green or blue fluorescent proteins were packaged using VSV-G or CD133-targeting envelopes, respectively. Subsequently freshly isolated mPB CD34+ cells were transduced using both vectors and cultivated for up to 17 days in vitro in cytokine-supplemented media. (b) The fraction of transduced cells was analyzed in regular intervals by flow cytometry. (c) The average fold change of percentage transduced cells from six independent experiments is shown. Error bars: SD, P ≤ 0.001 at days 7 and 14.
In order to maintain the HSC phenotype and to support optimal transduction conditions for CD133-LV, subsequent experiments were performed in the absence of prestimulation.
Preferential competitive engraftment of CD133-LV-transduced human cells in NSG mice
As human HSCs are functionally best defined by their property to engraft into NSG animals, we performed a competitive repopulation assay in vivo with CD133-LV and VSV-LV transduced cells. Mobilized PB CD34+ cells were transduced with color-coded lentiviral vectors, mixed at a cell ratio of 1:1 and transplanted at a total cell dose of 5 × 105 or 1 × 106 cells into NSG animals. Initial transduction efficiencies ranged from 2 to 20% for CD133-LV and VSV-LV. Bone marrow, spleen, and thymus of transplanted animals were analyzed for human engraftment (huCD45+) in the myeloid and lymphoid compartment at weeks 8, 12, and 16 post-transplantation according to the gating strategy shown in Figure 3a. We found high and sustained engraftment levels in transplanted animals, likely resulting from the short one day ex vivo culture used in our experimental procedure (Supplementary Figure S3a). Analysis of bone marrow, spleen, and thymus of transplanted animals at week 16 post-transplantation revealed that the most prominent gene-marked cell population engrafted in bone marrow and spleen were derived from CD133-LV transduced cells, while no significant difference between groups was observed in the thymus (Figure 3b). These findings were also reflected by the main hematopoietic cell subsets present in the different organs. We found a clear dominance of CD133-LV-transduced myeloid and B cells in bone marrow and spleen, while the contribution of either CD133-LV- or VSV-LV-transduced cells to the CD3 T-cell compartment in spleen and thymus was more balanced (Figure 3c and Supplementary Figure S3b). This likely resulted from different repopulation kinetics of T cells compared to myeloid and B cells. The superior competitive repopulation potential of CD133-LV-transduced cells was stable throughout week 8, 12, and 16 post-transplantation, indicating that this effect is established within the first 8 weeks after transplantation and remains constant thereafter (Supplementary Figure S3c).
Figure 3.
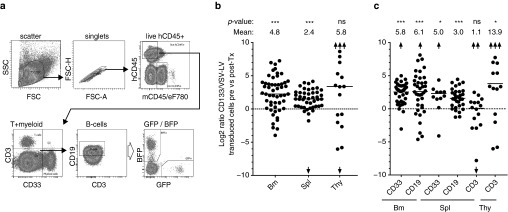
Competitive repopulation assays of gene modified hCD34+ cells in NSG mice. (a) Representative gating scheme used for the identification of transduced cells within human derived myeloid, B- and T-cell populations. (b) Representation of all transplanted animals as described in Supplementary Information on a log2 scale. Total hCD45+ cells from the bone marrow (BM, n = 53), spleen (n = 45), or thymus (n = 19) are shown. A value of zero indicates a constant ratio of CD133-LV versus VSV-LV gene modified cells, above zero indicates superior performance of CD133-LV transduced cells. (c) Same analysis as in b, but subdivided into myeloid, B- and T-cells to reveal a possible lineage preference of cells transduced with either VSV- or CD133-LVs. eF780: Viability dye, *, **, and *** correspond to P ≤ 0.05, 0.01, or 0.001 respectively. Fold change is given as linear value. Arrows represent out of scale data points.
The observed differences in the repopulation capacity of CD133-LV- and VSV-LV-transduced cells could be simply explained from toxic effects of the VSV-LV vector preparation, resulting in the ablation of engrafting cells in the VSV-LV-transduced population. Therefore, we cultured hCD34+ cells with increasing amounts of either CD133-LV or VSV-LV. The percentage of 7AAD+/AnnexinV+ cells was measured 2 days after transduction. No significant differences in cell toxicity were observed between both vector preparations. At the dilutions used for transduction of hCD34+ cells (1% for VSV-LV and 10% for CD133-LV), the CD133-LV supernatant was more toxic than the VSV-LV vector preparation (Supplementary Figure S4).
Next, we performed secondary transplantation to confirm the stem cell character of transduced cells. In analogy to the previous experiment, primary animals were transplanted with a 1:1 mixture of CD133- and VSV-LV-transduced cells together with a 15-fold excess of unmodified cells. Animals were analyzed for myeloid and lymphoid engraftment in the bone marrow at week 12 post-transplantation (Figure 4a). In agreement with the results shown in Figure 3b,c, the myeloid and B-cell compartment contained substantially more CD133-LV-transduced cells than VSV-LV-transduced cells, while contribution to the CD3+ population was similar (Figure 4a). Human CD34+ cells were isolated from the bone marrow of primary transplanted animals and retransplanted into secondary NSG mice. At this time point, gene marked hCD34+ cells were predominantly derived from CD133-LV-transduced cells (Figure 4b). Secondary animals were analyzed 12 weeks after transplantation for myeloid and lymphoid engraftment in bone marrow and spleen. While most of the animals (63–88%) engrafted with CD133-LV-derived cells, only a few of the secondary animals showed engraftment of VSV-LV-transduced cells (13% in the bone marrow, 0% in the spleen) (Figure 4b). Thus, this result confirms that CD133-LV-derived vectors transduced a cell population with long-term multilineage repopulation capabilities in primary and secondary animals under competitive conditions.
Figure 4.

Competitive serial transplantation assay. (a) Primary mice were transplanted with indicated numbers of gene modified cells along with a 15-fold excess of unmodified cells. See legend to Figure 3 for further information. (b) Twelve weeks later, hCD34+ cells were isolated from the bone marrow of 12 mice and transferred into 8 secondary recipient mice for further 12 weeks. The table indicates the frequency of mice in which VSV-LV or CD133-LV gene modified cells were identified in the bone marrow or spleen. Tx, transplantation.
Higher frequency of repopulating HSCs with multilineage contribution within the CD133-LV-transduced cell population
The SRC frequency within the transduced cell fraction was determined in competitive repopulation experiments using limiting dilutions of CD133-LV- and VSV-LV-transduced cells. NSG mice were transplanted with a total of 1 × 106 cells or serial dilutions thereof containing a 1:1 cell ratio of CD133-LV- and VSV-LV-transduced hCD34+ cells. After correction for transduction efficiency (20% for VSV-LV and 5% for CD133-LV), the high-dose cohort was transplanted with 2.4 × 104 CD133-LV and 9.9 × 104 VSV-LV-transduced cells together with 8.8 × 105 nontransduced competitor cells. We analyzed the mice 16 weeks after transplantation for contribution of gene marked cells to the total CD45+ cell population (cutoff: 0.1%). From this analysis, the frequency of SRCs in the CD133-LV-transduced population was 1 in 12,547 (95% confidence interval (CI): 7,055–22,316), while this number was 1 in 55,136 for VSV-LV-transduced cells (95% CI: 30,805–98,683) (Figure 5a and Supplementary Table S1).
Figure 5.

Preferential transduction of primitive hematopoietic stem cells (HSCs) by CD133-LV. (a) A limiting dilution competitive repopulation experiment was performed by cotransplantion of serial dilutions of limited numbers of CD133-LV- and VSV-LV-transduced cells. A total of 27 mice were transplanted in four dose groups. The frequency of repopulating HSCs in the total transduced graft was calculated using ELDA software based on a threshold of 0.1% of total CD45+ cells (mouse or human). (b) CD34+ cells from mobilized peripheral blood were transduced with barcoded VSV- or CD133-LV vectors. After 16 weeks, CD45-positive bone marrow cells were used to measure the clonal repertoire after high-throughput sequencing. The total number of different clones recovered in vivo is shown in relation to the number of initially injected barcoded clones and expressed as the percentage recovered clones. (c) XCGD derived BM CD34+ cells were transduced with CD133-LV expressing the gp91-transgene under control of a strong viral promoter. Cells were transplanted into NSG mice along with a control mouse which received identically treated mPB CD34 cells from a healthy donor. Sixteen weeks after transplantation, bone marrow cells were stained for hCD45 and gp91 expression (n = 2).
We also analyzed the frequency of engrafting cells within the CD133-LV- and VSV-LV-transduced population using DNA barcoded CD133-LV and VSV-LV vector libraries as unique identifier for individual transduced cells. Deep sequencing was used to determine the identity of bar-codes in isolated hCD45+ cells 16 weeks after transplantation. The sum of barcodes determined in all mice in the experimental group totaled 338 and 1,218 in the CD133-LV- and VSV-LV-transplanted groups, respectively (nine mice per group). After correcting for the total input of gene marked cells per animal (845 and 20,685 for CD133-LV- and VSV-LV-transduced cells, respectively), we found in average a 6.7-fold difference in the recovery of DNA barcodes for CD133-LV- versus VSV-LV-transduced cells (Figure 5b). Taken together, our data suggest that the CD133-LV targets gene transfer to a more primitive HSC fraction with increased long-term engraftment potential than vectors pseudotyped with VSV-G.
Lastly, we asked whether the CD133-targeting strategy would be applicable for the transfer of therapeutic genes into primitive repopulating cells. For this we used mPB CD34+ cells derived from a patient suffering from the X-linked form of chronic granulomatous disease (X-CGD), a primary immunodeficiency characterized by a defective elimination of microbes by phagocytic cells. In this case, frozen X-CGD CD34+ cells were used and allowed to recover for 24 hours prior to transduction. The proportion of CD34+ CD38− cells, which contains most of the CD133+ cells (see Figure 1a), after thawing and culture was 5%. Cells were transduced with CD133-LV expressing gp91phox from an internal viral promoter. One million cells per mouse were infused and the bone marrow was harvested 10 weeks after transplantation and analyzed for gp91phox expression in the human CD45 cell fraction. As shown in Figure 5c, a clear gp91phox signal was observed in the human cell fraction, representing 14–18% of the gp91phox expression recovered from the BM of animals transplanted with CD34+ cells derived from healthy donors.
Discussion
Lentiviral vectors equipped with the VSV-G envelope protein have a proven record of being beneficial for patients suffering from genetic diseases that can be treated by HSC-based gene therapy.1,2,3 While this clearly demonstrates that VSV-LV transduces long-term repopulating HSCs, its broad tropism supports gene transfer into all cell types present within the CD34+ cell population making a high vector dose necessary to achieve sufficient transduction of the minor fraction of LT-HSC present within the CD34+ cell population.20,27,28,29 VSV-G protein recognizes the ubiquitously expressed LDL receptor (LDL-R) family as cellular entry port, explaining its broad cell tropism.22 The LDL-R is poorly expressed in quiescent T, B, and hematopoietic stem cells but increases after cytokine stimulation, making prestimulation of these cells prior to VSV-LV-mediated gene transfer necessary in order to achieve high transduction efficiencies.30,31,32 However, quiescent HSCs have a higher long-term engraftment capacity than cytokine-stimulated HSCs.33,34,35 The activation of CD34+ cells during ex vivo culture therefore reduces the proportion of target cells with long-term repopulating capacity further.
Here, we provide evidence that these limitations can be overcome by targeting LVs to CD133+ cells. CD133-targeted gene transfer into human hematopoietic long-term repopulating cells improves engraftment and multilineage repopulation while at the same time reduces vector dose, thereby potentially lowering the risk of insertional mutagenesis. Previous work aiming at improved rates of genetic modification of engrafting HSCs included optimization of ex vivo culture and transduction conditions as well as the use of envelope proteins derived from diverse viruses to pseudotype lentiviral vectors. For example, lentiviral vectors pseudotyped with a chimeric envelope containing the extracellular and transmembrane domains of the feline leukemia virus RD114 (RD114tr) or the prototype foamy virus envelope (PFVenv) have been reported to transduce HSC with high efficiency and to be less toxic than VSV-G.36,37,38 Nonetheless, these pseudotypes still possess a broad tropism and require an excess of vector particles. Preferential genetic modification of human CD34+ cells has also been achieved using envelopes containing membrane bound “early acting” cytokines, which have the capacity to activate quiescent HSC thereby facilitating gene transfer into these cells at low multiplicity of infection and even allow for in vivo gene transfer.39,40 However, neither for this vector type nor for the pseudotypes mentioned above, a side by side comparison with state of the art vector technology for the transduction of LT-HSCs has been provided. Considering the high variability in mouse repopulation assays only a competitive approach, in which both vector types are applied to the same donor cells, cultivated under the same conditions, and, more importantly, implanted into the same individual mouse, will allow reliable conclusions.
We have used the CD133 antigen to target gene transfer preferentially to a subset of CD34+ cells. Compared to CD34+ cells, CD133+ cells have a higher proliferative capacity in vitro, threefold higher LTC-IC and higher G0-content and most of the SRC activity.16,41,42 Although there is a substantial overlap between the expression of CD34 and CD133, CD133+/CD34− cells can generate CD133+/CD34+ cells, suggesting that the CD133+ cells comprise a more primitive cell type than the CD34+ fraction.43
Consistent with this, we found a 3.5-fold higher proliferation of CD133-LV- compared to VSV-LV-transduced cells during 17 days of culture (Figure 2). This difference was even more pronounced when cells were transplanted into NSG mice. Then, CD133-LV was 4.8-fold more effective in the transduction of cell with competitive repopulation capacity (Figures 3b and 5a). Moreover, CD133-LV-derived cells engrafted secondary recipients with high efficiency, further supporting the primitive nature of CD133-LV-transduced cells (Figure 4). It is worth mentioning that also VSV-LV is capable of transducing serially transplantable HSCs when used at higher multiplicity of infection and under conditions optimized for VSV-mediated gene transfer, which routinely includes cytokine prestimulation. Lastly, the superior recovery rate of barcoded CD133-LV sequences over VSV-LV sequences from animals 16 weeks after transplantation (6.7-fold difference) provides clear evidence for the prolonged persistence of CD133-LV marked cells in vivo (Figure 5b).
The transduction conditions applied in our experiments disfavored gene transfer by VSV-LV, as no cytokine stimulation and low multiplicity of infections have been used.28,30 These conditions ensured the presence of as many as possible CD133+ cells after isolation, since CD133 expression is rapidly downregulated after cytokine stimulation and hematopoietic differentiation (Supplementary Figure S1 and refs. 13,14). However, we also confirmed superior transduction of cells with high proliferative capacity in ex vivo culture by CD133-LV when prestimulation of up to 36 hours was used (Supplementary Figure S2).
Our data suggest, that directly isolating CD133+ cells and transducing with VSV-LV may represent a valid alternative procedure, although CD133-LV mediated gene transfer can be easily implemented into the well-established clinical protocols based on CD34 enrichment. Furthermore, there is a considerable heterogeneity within the CD133-positive fraction of HSCs both in terms of functionality and CD133 expression levels. We have previously demonstrated that entry targeted LVs preferentially transduce cells that express the highest levels of the targeted cell surface receptor.25,44 This holds true also for CD133-LV (Figure 1). Thus, even when transducing CD133-purified cells, CD133-LV will be advantageous as it preferentially modifies those cells expressing high levels of CD133, which are particularly enriched for CD38-negative cells and correlate with high expression of CD34 (Figure 1). The more committed CD34low/CD38high/CD133low progenitor pool is also transduced by CD133-LV, albeit to a lower degree, which might facilitate fast hematopoietic recovery upon conditioning and transplantation of gene modified cells.
Under the conditions used in our experiments about 10% of the CD34highCD133high cells were transduced. This is at least partly due to the moderate titers of CD133-LV vector supernatants, which were 1–2 log below the titers routinely obtained with VSV-LV.25 Higher transduction rates may also be prevented by CD133-containing membrane vesicles which become released during differentiation of HSCs and potentially sequester CD133-LV particles.45 Removing these vesicles may thus further enhance the transduction of repopulating cells in future. Nevertheless, the moderate transduction efficiencies achieved so far were compensated by the high proliferative potential of the transduced CD133+ cells and by their capacity to engraft long-term. Indeed, most of the animals transplanted with CD133-LV-modified cells engrafted. Thus, it appears that a few gene modified CD133+ cells may be sufficient to regenerate all hematopoietic lineages with gene modified cells after transplantation into myeloablated recipients.
Besides HSCs, CD133 is also present in endothelial and neural progenitor cells and other progenitor cells derived from somatic tissues like prostate, liver, skin, and retina46,47 and in a variety of cancer stem cells, including acute and chronic myeloid and lymphoblastic leukemia, glioblastoma, and colon cancer.48 Thus, CD133-LV may have a wider application beyond gene transfer into HSCs. Even an in vivo application of CD133-LV is conceivable in the near future. Although a humoral immune response may preclude the use of wild-type measles envelope in vivo as most of the human population is vaccinated against measles, immune escape mutants exists and can be used to pseudotype gene transfer vectors.49,50,51 Thus, further improvements of the CD133 targeting strategy are feasible and may generate an extremely useful tool to direct gene transfer to normal and malignant stem and progenitor cells.
Materials and Methods
Healthy donor human CD34+ cells from mobilized peripheral blood were isolated from mononuclear cell concentrates after written informed consent was obtained from donors according to the declaration of Helsinki. All procedures involving human samples were approved by the Institutional Ethics Committee. CD34+ cells were isolated using the CD34-microbead kit from Miltenyi, Bergisch-Gladbach, Germany according to the manufacturer's instructions. After isolation, cells were cultured at a density of 106 cells/ml in SpemSpan SFEM supplemented with human stem cell factor (50 ng/ml), human thrombopoietin (10 ng/ml), hIGFBP2 (100 ng/ml), and hFLT3L (50 ng/ml) and immediately transduced. Vector doses were adjusted to achieve similar transduction efficiencies in both VSV- and CD133-LV groups. After transduction cells were kept in culture for 17 days at a density of 0.2 to 1 × 106 cells/ml.
Animal experiments were performed in accordance with the German animal welfare legislation and were approved by the local authorities. Details on vector production, transplantations, flow cytometry, and sequence analysis of barcoded libraries are given in Supplementary Data.
SUPPLEMENTARY MATERIAL Supplementary Material and Methods Supplementary References Figure S1. Cell surface marker loss during prolonged culture of CD34+ cells. Figure S2. Impact of prestimulation on transduction rate and population dynamics over time. Figure S3. Competitive repopulation experiments in NSG mice. Figure S4. Toxicity of CD133-LV and VSV-LV. Table S1. Limiting dilution competitive repopulation assay.
Acknowledgments
This work was supported by a grant from the European Union (FP7 integrated project CELL-PID HEALTH-2010–261387 to MG), the LOEWE Center for Cell and Gene Therapy Frankfurt funded by the Hessische Ministerium für Wissenschaft und Kunst (HMWK; funding reference number: III L 4- 518/17.004 (2010)) to C.B., S.K., B.G., C.J.B., and M.G. and by the Deutsche Forschungsgemeinschaft (DFG) Graduate Program GK1172-Biologicals to C.B. and K.B.K. The Georg-Speyer-Haus is supported by the Bundesministerium für Gesundheit and the Hessisches Ministerium für Wissenschaft und Kunst. C.J.B. and S.K. are listed as inventors on patents about the retargeting of lentiviral vectors that have been out licensed. All other authors declare that they have no conflict of interest.
Supplementary Material
References
- Aiuti A, Biasco L, Scaramuzza S, Ferrua F, Cicalese MP, Baricordi C, et al. Lentiviral hematopoietic stem cell gene therapy in patients with Wiskott-Aldrich syndrome. Science. 2013;341:1233151. doi: 10.1126/science.1233151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cartier N, Hacein-Bey-Abina S, Bartholomae CC, Veres G, Schmidt M, Kutschera I, et al. Hematopoietic stem cell gene therapy with a lentiviral vector in X-linked adrenoleukodystrophy. Science (New York, N.Y.) 2009;326:818–823. doi: 10.1126/science.1171242. [DOI] [PubMed] [Google Scholar]
- Biffi A, Montini E, Lorioli L, Cesani M, Fumagalli F, Plati T, et al. Lentiviral hematopoietic stem cell gene therapy benefits metachromatic leukodystrophy. Science. 2013;341:1233158. doi: 10.1126/science.1233158. [DOI] [PubMed] [Google Scholar]
- Kaufmann KB, Büning H, Galy A, Schambach A, Grez M. Gene therapy on the move. EMBO Mol Med. 2013;5:1642–1661. doi: 10.1002/emmm.201202287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seymour LW, Thrasher AJ. Gene therapy matures in the clinic. Nat Biotechnol. 2012;30:588–593. doi: 10.1038/nbt.2290. [DOI] [PubMed] [Google Scholar]
- Kim S, Kim N, Presson AP, Metzger ME, Bonifacino AC, Sehl M, et al. Dynamics of HSPC repopulation in nonhuman primates revealed by a decade-long clonal-tracking study. Cell Stem Cell. 2014;14:473–485. doi: 10.1016/j.stem.2013.12.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhatia M, Wang JC, Kapp U, Bonnet D, Dick JE. Purification of primitive human hematopoietic cells capable of repopulating immune-deficient mice. Proc Natl Acad Sci USA. 1997;94:5320–5325. doi: 10.1073/pnas.94.10.5320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guenechea G, Gan OI, Dorrell C, Dick JE. Distinct classes of human stem cells that differ in proliferative and self-renewal potential. Nat Immunol. 2001;2:75–82. doi: 10.1038/83199. [DOI] [PubMed] [Google Scholar]
- McKenzie JL, Gan OI, Doedens M, Wang JC, Dick JE. Individual stem cells with highly variable proliferation and self-renewal properties comprise the human hematopoietic stem cell compartment. Nat Immunol. 2006;7:1225–1233. doi: 10.1038/ni1393. [DOI] [PubMed] [Google Scholar]
- Lapidot T, Pflumio F, Doedens M, Murdoch B, Williams DE, Dick JE. Cytokine stimulation of multilineage hematopoiesis from immature human cells engrafted in SCID mice. Science. 1992;255:1137–1141. doi: 10.1126/science.1372131. [DOI] [PubMed] [Google Scholar]
- Vormoor J, Lapidot T, Pflumio F, Risdon G, Patterson B, Broxmeyer HE, et al. Immature human cord blood progenitors engraft and proliferate to high levels in severe combined immunodeficient mice. Blood. 1994;83:2489–2497. [PubMed] [Google Scholar]
- Notta F, Doulatov S, Laurenti E, Poeppl A, Jurisica I, Dick JE. Isolation of single human hematopoietic stem cells capable of long-term multilineage engraftment. Science. 2011;333:218–221. doi: 10.1126/science.1201219. [DOI] [PubMed] [Google Scholar]
- Yin AH, Miraglia S, Zanjani ED, Almeida-Porada G, Ogawa M, Leary AG, et al. AC133, a novel marker for human hematopoietic stem and progenitor cells. Blood. 1997;90:5002–5012. [PubMed] [Google Scholar]
- Miraglia S, Godfrey W, Yin AH, Atkins K, Warnke R, Holden JT, et al. A novel five-transmembrane hematopoietic stem cell antigen: isolation, characterization, and molecular cloning. Blood. 1997;90:5013–5021. [PubMed] [Google Scholar]
- Freund D, Oswald J, Feldmann S, Ehninger G, Corbeil D, Bornhäuser M. Comparative analysis of proliferative potential and clonogenicity of MACS-immunomagnetic isolated CD34+ and CD133+ blood stem cells derived from a single donor. Cell Prolif. 2006;39:325–332. doi: 10.1111/j.1365-2184.2006.00386.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Summers YJ, Heyworth CM, de Wynter EA, Hart CA, Chang J, Testa NG. AC133+ G0 cells from cord blood show a high incidence of long-term culture-initiating cells and a capacity for more than 100 million-fold amplification of colony-forming cells in vitro. Stem Cells. 2004;22:704–715. doi: 10.1634/stemcells.22-5-704. [DOI] [PubMed] [Google Scholar]
- Gordon PR, Leimig T, Babarin-Dorner A, Houston J, Holladay M, Mueller I, et al. Large-scale isolation of CD133+ progenitor cells from G-CSF mobilized peripheral blood stem cells. Bone Marrow Transplant. 2003;31:17–22. doi: 10.1038/sj.bmt.1703792. [DOI] [PubMed] [Google Scholar]
- Tura O, Barclay GR, Roddie H, Davies J, Turner ML. Absence of a relationship between immunophenotypic and colony enumeration analysis of endothelial progenitor cells in clinical haematopoietic cell sources. J Transl Med. 2007;5:37. doi: 10.1186/1479-5876-5-37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lang P, Bader P, Schumm M, Feuchtinger T, Einsele H, Führer M, et al. Transplantation of a combination of CD133+ and CD34+ selected progenitor cells from alternative donors. Br J Haematol. 2004;124:72–79. doi: 10.1046/j.1365-2141.2003.04747.x. [DOI] [PubMed] [Google Scholar]
- Géronimi F, Richard E, Redonnet-Vernhet I, Lamrissi-Garcia I, Lalanne M, Ged C, et al. Highly efficient lentiviral gene transfer in CD34+ and CD34+/38-/lin- cells from mobilized peripheral blood after cytokine prestimulation. Stem Cells. 2003;21:472–480. doi: 10.1634/stemcells.21-4-472. [DOI] [PubMed] [Google Scholar]
- Catlin SN, Busque L, Gale RE, Guttorp P, Abkowitz JL. The replication rate of human hematopoietic stem cells in vivo. Blood. 2011;117:4460–4466. doi: 10.1182/blood-2010-08-303537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finkelshtein D, Werman A, Novick D, Barak S, Rubinstein M. LDL receptor and its family members serve as the cellular receptors for vesicular stomatitis virus. Proc Natl Acad Sci USA. 2013;110:7306–7311. doi: 10.1073/pnas.1214441110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yanagi Y, Takeda M, Ohno S, Hashiguchi T. Measles virus receptors. Curr Top Microbiol Immunol. 2009;329:13–30. doi: 10.1007/978-3-540-70523-9_2. [DOI] [PubMed] [Google Scholar]
- Mühlebach MD, Mateo M, Sinn PL, Prüfer S, Uhlig KM, Leonard VH, et al. Adherens junction protein nectin-4 is the epithelial receptor for measles virus. Nature. 2011;480:530–533. doi: 10.1038/nature10639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anliker B, Abel T, Kneissl S, Hlavaty J, Caputi A, Brynza J, et al. Specific gene transfer to neurons, endothelial cells and hematopoietic progenitors with lentiviral vectors. Nat Methods. 2010;7:929–935. doi: 10.1038/nmeth.1514. [DOI] [PubMed] [Google Scholar]
- Buchholz CJ, Mühlebach MD, Cichutek K. Lentiviral vectors with measles virus glycoproteins - dream team for gene transfer. Trends Biotechnol. 2009;27:259–265. doi: 10.1016/j.tibtech.2009.02.002. [DOI] [PubMed] [Google Scholar]
- Ailles L, Schmidt M, Santoni de Sio FR, Glimm H, Cavalieri S, Bruno S, et al. Molecular evidence of lentiviral vector-mediated gene transfer into human self-renewing, multi-potent, long-term NOD/SCID repopulating hematopoietic cells. Mol Ther. 2002;6:615–626. [PubMed] [Google Scholar]
- Amsellem S, Ravet E, Fichelson S, Pflumio F, Dubart-Kupperschmitt A. Maximal lentivirus-mediated gene transfer and sustained transgene expression in human hematopoietic primitive cells and their progeny. Mol Ther. 2002;6:673–677. [PubMed] [Google Scholar]
- Miyoshi H, Smith KA, Mosier DE, Verma IM, Torbett BE. Transduction of human CD34+ cells that mediate long-term engraftment of NOD/SCID mice by HIV vectors. Science. 1999;283:682–686. doi: 10.1126/science.283.5402.682. [DOI] [PubMed] [Google Scholar]
- Sutton RE, Reitsma MJ, Uchida N, Brown PO. Transduction of human progenitor hematopoietic stem cells by human immunodeficiency virus type 1-based vectors is cell cycle dependent. J Virol. 1999;73:3649–3660. doi: 10.1128/jvi.73.5.3649-3660.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zielske SP, Gerson SL. Cytokines, including stem cell factor alone, enhance lentiviral transduction in nondividing human LTCIC and NOD/SCID repopulating cells. Mol Ther. 2003;7:325–333. doi: 10.1016/s1525-0016(03)00005-4. [DOI] [PubMed] [Google Scholar]
- Amirache F, Lévy C, Costa C, Mangeot PE, Torbett BE, Wang CX, et al. Mystery solved: VSV-G-LVs do not allow efficient gene transfer into unstimulated T cells, B cells, and HSCs because they lack the LDL receptor. Blood. 2014;123:1422–1424. doi: 10.1182/blood-2013-11-540641. [DOI] [PubMed] [Google Scholar]
- Glimm H, Oh IH, Eaves CJ. Human hematopoietic stem cells stimulated to proliferate in vitro lose engraftment potential during their S/G(2)/M transit and do not reenter G(0) Blood. 2000;96:4185–4193. [PubMed] [Google Scholar]
- Kittler EL, Peters SO, Crittenden RB, Debatis ME, Ramshaw HS, Stewart FM, et al. Cytokine-facilitated transduction leads to low-level engraftment in nonablated hosts. Blood. 1997;90:865–872. [PubMed] [Google Scholar]
- Gothot A, van der Loo JC, Clapp DW, Srour EF. Cell cycle-related changes in repopulating capacity of human mobilized peripheral blood CD34(+) cells in non-obese diabetic/severe combined immune-deficient mice. Blood. 1998;92:2641–2649. [PubMed] [Google Scholar]
- Bell AJ, Jr, Fegen D, Ward M, Bank A. RD114 envelope proteins provide an effective and versatile approach to pseudotype lentiviral vectors. Exp Biol Med (Maywood) 2010;235:1269–1276. doi: 10.1258/ebm.2010.010053. [DOI] [PubMed] [Google Scholar]
- Hanawa H, Kelly PF, Nathwani AC, Persons DA, Vandergriff JA, Hargrove P, et al. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transduce primitive hematopoietic cells from human blood. Mol Ther. 2002;5:242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- Lindemann D, Rethwilm A. Foamy virus biology and its application for vector development. Viruses. 2011;3:561–585. doi: 10.3390/v3050561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhoeyen E, Wiznerowicz M, Olivier D, Izac B, Trono D, Dubart-Kupperschmitt A, et al. Novel lentiviral vectors displaying “early-acting cytokines” selectively promote survival and transduction of NOD/SCID repopulating human hematopoietic stem cells. Blood. 2005;106:3386–3395. doi: 10.1182/blood-2004-12-4736. [DOI] [PubMed] [Google Scholar]
- Frecha C, Costa C, Nègre D, Amirache F, Trono D, Rio P, et al. A novel lentiviral vector targets gene transfer into human hematopoietic stem cells in marrow from patients with bone marrow failure syndrome and in vivo in humanized mice. Blood. 2012;119:1139–1150. doi: 10.1182/blood-2011-04-346619. [DOI] [PubMed] [Google Scholar]
- Wagner W, Ansorge A, Wirkner U, Eckstein V, Schwager C, Blake J, et al. Molecular evidence for stem cell function of the slow-dividing fraction among human hematopoietic progenitor cells by genome-wide analysis. Blood. 2004;104:675–686. doi: 10.1182/blood-2003-10-3423. [DOI] [PubMed] [Google Scholar]
- Hess DA, Wirthlin L, Craft TP, Herrbrich PE, Hohm SA, Lahey R, et al. Selection based on CD133 and high aldehyde dehydrogenase activity isolates long-term reconstituting human hematopoietic stem cells. Blood. 2006;107:2162–2169. doi: 10.1182/blood-2005-06-2284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallacher L, Murdoch B, Wu DM, Karanu FN, Keeney M, Bhatia M. Isolation and characterization of human CD34(-)Lin(-) and CD34(+)Lin(-) hematopoietic stem cells using cell surface markers AC133 and CD7. Blood. 2000;95:2813–2820. [PubMed] [Google Scholar]
- Münch RC, Mühlebach MD, Schaser T, Kneissl S, Jost C, Plückthun A, et al. DARPins: an efficient targeting domain for lentiviral vectors. Mol Ther. 2011;19:686–693. doi: 10.1038/mt.2010.298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bauer N, Wilsch-Bräuninger M, Karbanová J, Fonseca AV, Strauss D, Freund D, et al. Haematopoietic stem cell differentiation promotes the release of prominin-1/CD133-containing membrane vesicles–a role of the endocytic-exocytic pathway. EMBO Mol Med. 2011;3:398–409. doi: 10.1002/emmm.201100147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meregalli M, Farini A, Belicchi M, Torrente Y. CD133(+) cells isolated from various sources and their role in future clinical perspectives. Expert Opin Biol Ther. 2010;10:1521–1528. doi: 10.1517/14712598.2010.528386. [DOI] [PubMed] [Google Scholar]
- Tárnok A, Ulrich H, Bocsi J. Phenotypes of stem cells from diverse origin. Cytometry A. 2010;77:6–10. doi: 10.1002/cyto.a.20844. [DOI] [PubMed] [Google Scholar]
- Grosse-Gehling P, Fargeas CA, Dittfeld C, Garbe Y, Alison MR, Corbeil D, et al. CD133 as a biomarker for putative cancer stem cells in solid tumours: limitations, problems and challenges. J Pathol. 2013;229:355–378. doi: 10.1002/path.4086. [DOI] [PubMed] [Google Scholar]
- Kneissl S, Abel T, Rasbach A, Brynza J, Schneider-Schaulies J, Buchholz CJ. Measles virus glycoprotein-based lentiviral targeting vectors that avoid neutralizing antibodies. PLoS ONE. 2012;7:e46667. doi: 10.1371/journal.pone.0046667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kweder H, Ainouze M, Cosby SL, Muller CP, Lévy C, Verhoeyen E, et al. Mutations in the H, F, or M Proteins Can Facilitate Resistance of Measles Virus to Neutralizing Human Anti-MV Sera. Adv Virol. 2014;2014:205617. doi: 10.1155/2014/205617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lévy C, Amirache F, Costa C, Frecha C, Muller CP, Kweder H, et al. Lentiviral vectors displaying modified measles virus gp overcome pre-existing immunity in in vivo-like transduction of human T and B cells. Mol Ther. 2012;20:1699–1712. doi: 10.1038/mt.2012.96. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.