

Abstract
Glioblastoma multiforme (GBM) is an aggressive brain cancer for which there is no effective treatment. Oncolytic HSV vectors (oHSVs) are attenuated lytic viruses that have shown promise in the treatment of human GBM models in animals, but their efficacy in early phase patient trials has been limited. Instead of attenuating the virus with mutations in virulence genes, we engineered four copies of the recognition sequence for miR-124 into the 3′UTR of the essential ICP4 gene to protect healthy tissue against lytic virus replication; miR-124 is expressed in neurons but not in glioblastoma cells. Following intracranial inoculation into nude mice, the miR-124-sensitive vector failed to replicate or show overt signs of pathogenesis. To address the concern that this safety feature may reduce oncolytic activity, we inserted the miR-124 response elements into an unattenuated, human receptor (EGFR/EGFRvIII)-specific HSV vector. We found that miR-124 sensitivity did not cause a loss of treatment efficiency in an orthotopic model of primary human GBM in nude mice. These results demonstrate that engineered miR-124 responsiveness can eliminate off-target replication by unattenuated oHSV without compromising oncolytic activity, thereby providing increased safety.
Introduction
Glioblastoma multiforme (GBM) is one of the most malignant forms of cancer for which effective treatment remains elusive. Standard medical practices such as surgery and radio- and chemotherapy have shown limited long-term clinical benefit. Oncolytic vectors, including those derived from herpes simplex virus type-1 (oHSV-1), are under development in a number of laboratories as a potential alternative therapeutic strategy.1 Although the application of oncolytic HSV (oHSV) as a therapeutic agent for GBM has proven safe in patient trials with some evidence of efficacy, barriers remain to effective treatment.2,3
The most common method to achieve HSV attenuation has been to functionally delete nonessential genes that circumvent host innate immune responses to infection, provide nucleotide (nt) pools for replication in nondividing cells such as neurons, and prevent cellular apoptosis.2 Virus replication in cancer cells is facilitated by the loss of certain innate immune responses,4 as well as by rapid cell division and inactive apoptotic pathways.2 However, these properties are not uniformly sufficient for vigorous replication of current oHSVs in tumors.
As a first step to improve vector efficacy, we previously developed methods for complete retargeting of HSV in order to redirect infection from the canonical HSV entry receptors to highly expressed tumor cell-surface receptors (e.g., epidermal growth factor receptor (EGFR), and epidermal growth factor receptor variant III (EGFRvIII)).5 Retargeted oHSV showed robust oncolytic activity and high specificity for human GBM cells, resulting in a high level of human tumor destruction in an orthotopic mouse model. Moreover, this treatment vector produced long-term survival of the majority of treated animals without vector-associated toxicity. However, most highly expressed tumor-associated cell surface markers are shared to some degree with normal cell types and thus we sought to increase the safety of a tumor-targeted, unattenuated vector using an independent mechanism to block virus replication in normal brain without reducing replication in the tumor.
Recent studies have taken advantage of differences in the microRNA (miRNA) expression profiles between normal and cancer cells as an alternative approach to tumor targeting.6 At least 30 miRNAs have been identified that are differentially expressed in glioblastoma, neurons, and neural progenitor cells,7,8 suggesting that these differences can be used to limit virus replication in normal brain cells while permitting unimpeded replication in tumor cells. Here, we demonstrate that the incorporation of miR-124 recognition elements into the essential ICP4 gene of essentially wild type virus prevented HSV replication in normal brain tissue where miR-124 is highly expressed. Furthermore, we show that the miR-124 response elements did not reduce the oncolytic activity of an EGFR-retargeted vector. Importantly, since the tumor phenotype depends on the continued absence of miR-124, potential up-regulation of miR-124 as a cellular escape mechanism from lytic viral replication will limit the uncontrolled proliferative capacity of the cell and thereby not compromise vector effectiveness. Vector production is carried out in cells lacking miR-124 and thus there is no selective pressure to produce miR-124-resistant virus mutants during stock preparation. Together, these features provide for vector safety and tumor selectivity and suggest a general strategy for oncolytic vector design suitable for a broad range of tumor types.
Results
Validation of a miR-124 response element
Among multiple miRNAs that are expressed at higher levels in neurons than in GBM cells, miR-124 is the most abundant with minimal expression in GBM.6 We designed a miR-124 response element (T124) consisting of four tandem copies of the reverse complement of mature miR-124 separated by different eight nt spacers. To assess the functionality of this sequence, we inserted it into the 3′UTR of a firefly luciferase (fLuc) expression plasmid and performed cotransfection experiments with a specific (pre-miR-124) or nonspecific (pre-miR-21) precursor miRNA on HEK293AD cells that reportedly express little or no miR-1249; a Renilla luciferase (rLuc) expression plasmid was included for normalization. The results (pfLuc-T124, Figure 1) showed severely reduced fLuc activity at 24 hours in cells cotransfected with pre-miR-124 compared to mock cotransfected cells or cells cotransfected with pre-miR-21. In contrast, little difference in fLuc expression was observed between cells transfected with a control fLuc plasmid containing four copies of the miR-21 sequence in reverse (pfLuc-Ctrl, mock) and cotransfections of pfLuc-Ctrl with either pre-miR-21 or pre-miR-124 (Figure 1). These results demonstrated the functionality of the T124 element as an efficient and specific target for miR-124-mediated restriction of gene expression.
Figure 1.

Effectiveness and specificity of the T124 element. Firefly luciferase (fLuc) expression plasmids containing T124 (pfLuc-T124) or a control sequence (pfLuc-Ctrl) in the 3′UTR were cotransfected with a renilla luciferase (prLuc) internal control plasmid into HEK293AD cells transfected 24 hours earlier with synthetic pre-miR-124 or pre-miR-21. Luciferase activities were measured 48 hours later. The results are shown as the means ± standard deviations from three determinations for fLuc activity normalized to rLuc activity. Statistically significant differences between pairs are indicated by brackets underneath the corresponding P values (unpaired t-test).
Replication sensitivity of T124-modified HSV to miR-124 expression
We used double Red recombination in Escherichia coli10 to introduce a series of modifications into KOS-37 BAC, a full-length genomic clone of the KOS strain of HSV-1 on a bacterial artificial chromosome (BAC).11 The product, KGBAC (Figure 2a), is deleted for the internal repeat (joint) region containing one copy each of the diploid genes ICP0, ICP34.5, LAT, and ICP4 along with the promoter for the ICP47 gene. This deletion facilitates manipulation of the remaining copies of the four deleted genes, provides abundant space for the potential incorporation of transgenes that enhance the oncolytic activity of the virus, and increases tumor specificity by reducing expression of the neurovirulence factor ICP34.512; elimination of ICP47 expression benefits immune recognition of infected cancer cells by virus-specific T cells.4 KGBAC also contains the green fluorescent protein open reading frame fused to the glycoprotein C open reading frame via a 2A peptide sequence13,14 to allow monitoring of late (postreplication) viral gene expression. Lastly, KGBAC contains a pair of mutations in the glycoprotein B gene shown by us to enhance HSV entry through noncanonical receptors.15,16 We recombined the T124 sequence into the 3′UTR of the remaining ICP4 gene of KGBAC to generate KG4:T124BAC (Figure 2a). Both BAC constructs were converted to virus particles with simultaneous removal of the BAC sequences located between loxP sites by transfection of U2OS-Cre cells. Following plaque purification, KG and KG4:T124 virus stocks were prepared and titered on U2OS cells.
Figure 2.
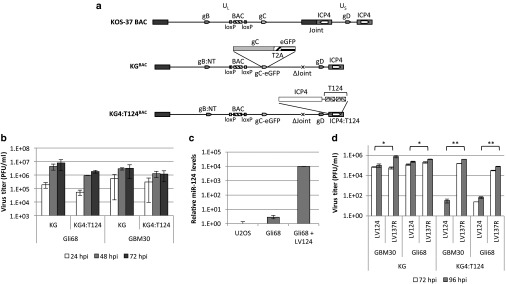
Virus replication in glioma cells. (a) Vector diagrams. The parental KOS-37 BAC contains loxP-flanked BAC, chloramphenicol-resistance and lacZ sequences (“BAC”) between the viral UL37 and UL38 genes (Gierasch et al., 2006). Modifications to generate KGBAC and KG4:T124BAC are illustrated, as follows: gB:NT, virus entry-enhancing double mutation in the gB gene; gC-eGFP, fusion of the complete gC ORF to GFP via a 2A peptide sequence; ΔJoint, deletion of the complete internal repeat region, including one copy of the ICP4 gene; ICP4:T124, insertion of T124 in the 3′UTR of the remaining ICP4 gene; UL, unique long segment of the viral genome; US, unique short segment. (b) Effect of T124 on virus replication in patient-derived glioma cells in culture. Gli68 and GBM30 cells were infected with KG or KG4:T124 viruses in triplicate at an MOI of 0.01. At the indicated time, points post infection, cell lysates, and supernatants were collected and titered on U2OS cells. While 48 hpi, we observed that the kinetic of replication of KG4:T124 was statistically different from KG (Gli68 = 0.0437; GBM30 = 0.0278), at 72 hpi, we did not observed any difference (Gli68 = 0.1434; GBM30 = 0.2859), suggesting that overall these vectors have a comparable efficiency of replication. Values are means ± standard deviation. (c) MiR-124 expression in LV124-infected Gli68 cells. Cells were infected at 5 cfu/cell, selected the following day for 3 days in puromycin-containing media, and harvested for total RNA extraction. Control RNAs were from uninfected Gli68 and U2OS cells. miR-124 levels were determined in triplicate by qRT-PCR and normalized to RNU43 levels. Shown is the fold increase ± standard deviation relative to U2OS cells. P < 0.05 for all pairs (unpaired t-test). (d) KG and KG4:T124 virus replication in miR-124-transduced and control GBM30 and Gli68 cells. Cells were infected with LV124 or LV137R at 5 cfu/cell, selected with puromycin for 3 days, and superinfected at MOIs of 0.01 with KG or KG4:T124. Infectious HSV in combined cell lysates and supernatants collected 72 and 96 hours later was titered on U2OS cells. Results are the mean values ± standard deviation from triplicate HSV infections. Brackets indicate significantly different pairs with the corresponding P values shown (unpaired t-test).
We first determined whether inclusion of the four tandem miR-124 target sites in the 3′UTR of ICP4 affected virus replication in human GBM cells in culture. The results (Figure 2b) showed that KG4:T124 replicated with similar kinetics as KG in spheroids of two primary glioblastoma lines, Gli68 and GBM30, and the yields of the two viruses were not statistically different at 72 hpi (Gli68 = 0.1434, GBM30 = 0.2859). We then determined whether replication and virus yield were sensitive to transduction of these lines with a human miR-124 expressing lentivirus (LV124). Figure 2c shows the relative levels of miR-124 in U2OS, Gli68, and Gli68-LV124 cells measured by quantitative real-time PCR (qPCR) on reverse transcribed small RNAs and standardized to endogenous RNU43 levels. KG grew equally well and to similar titers on Gli68-LV124 and Gli68 cells transduced with a lentiviral construct expressing the reverse complement of human miR-137 (LV137R; Figure 2d). In contrast, KG4:T124 grew poorly on the former compared to the latter, and similar results were obtained with LV124- versus LV137R-transduced GBM30 cells (Figure 2d). In combination, these observations strongly indicated that (i) the T124 element in the ICP4 gene was effective as a means to limit HSV replication in a miR-124-dependent manner and (ii) the levels of endogenous miR-124 in the two GBM lines were low enough to minimize this effect. In addition, the qRT-PCR data confirmed the suitability of U2OS cells for unimpaired growth and titration of KG4:T124 compared to KG.
KG4:T124 does not replicate in mouse brain or cause disease
Having shown that exogenous miR-124 expression in primary glioma cells in culture is highly effective in preventing KG4:T124 vector growth, we next tested whether the endogenous levels of miR-124 in mouse brain were sufficient to prevent vector replication and the typical neuropathogenesis associated with wild-type virus; we note that mature human and mouse miR-124 are identical in sequence.17 We used nude mice for these experiments to limit the effect of the host antiviral response and thereby facilitate the identification of direct effects of the T124 insertion in the virus. BALB/cnu/nu mice were chosen because these animals are highly sensitive to HSV replication and pathogenesis18,19,20 and have been used previously for tumor treatment efficacy experiments with human tumor cells.5,12,21,22 We compared the KG control vector and the miR-124-sensitive test vector KG4:T124 for their ability both to replicate in nude mouse brain and cause a lethal infection following intracranial inoculation of equal genome copy (gc) numbers (4.8 × 109 gc) into the right hemisphere. The results showed that injection of the control vector resulted in rapid animal death within 5 days (Figure 3a,c) with a twofold increase in total gc number present within the infected brains (Figure 3b). In contrast, there was no observable change in the health of the KG4:T124 injected mice over the 33-day observation period, as exemplified by their normal weight gain until sacrifice (Figure 3a), and the viral gc content declined steadily over this time period to ~0.4% of input (Figure 3b). The difference in survival between the animals inoculated with control or test vector (Figure 3c) was highly significant (P = 0.0058, log-rank test), indicating that four copies of the miR-124 recognition sequence inserted into the 3′UTR of the ICP4 gene were capable of blocking lethal vector replication in the brains of highly HSV-sensitive nude mice. Thus, these sequences alone were sufficient to prevent vector toxicity in the brain.
Figure 3.

KG4:T124 virus replication and toxicity in nude mouse brains. 4.8 × 109 genome copies of KG or KG4:T124 were intracranially injected into four BALB/c nude mice each (n = 4/group). (a) Animal weights over time post vector injection. Left, KG-injected animals; X, animal death. Right, KG4:T124-injected mice; filled circles, animal sacrifice. (b) Total viral genome copies over time in mouse brains following vector injection. Brains from single KG4:T124-injected mice sacrificed on days 5, 14, 21, and 33 post vector injection and the last surviving animal from the KG-injected group euthanized on day 5 with severe symptoms of disease were collected, DNA was isolated, and the total numbers of viral vector genomes per brain were determined by qPCR. (c) Kaplan–Meier survival plot of the animals in this experiment. Arrows indicate the days of sacrifice of single animals from the KG4:T124-injected group. P = 0.0058, log-rank test.
In a parallel experiment, using the similar conditions, we inoculated the animals with 4.36 × 109 gc of either KG4:T-124 or KG. As a negative control four animals received phosphate buffered saline (PBS). All the animals injected with KG virus died by day 7. In contrast, all mice injected with the retargeted virus remained alive and symptom-free throughout the 30-day observation period. We analyzed brain sections of injected mice for the presence of virus by immunostaining for the viral HSV protein. The representative images (see Supplementary Figure S1) illustrate the abundant presence of virus in the brain of a mouse injected with KG (a–f) that died at 5 dpi, while no evidence of virus replication was detected in brain sections from a mouse that had been sacrificed on day 30 (symptom-free) after injection of the same amount of miR-124 controlled virus (g–l) or from the one injected with PBS (m–o).
To confirm the suggestion from these results that loss or mutational inactivation of the miR-124 target sites during virus stock preparation was rare at best, DNA was isolated from the KG4:T124 viral stock and subjected to PCR through the T124 insertion site in the ICP4 3′UTR. Analysis of the products by gel electrophoresis and DNA sequencing showed no abnormal PCR product sizes or evidence of nt variability (data not shown). Likewise, PCR and sequence analyses of total brain DNA isolated at 3 hours or 21 days post intracranial inoculation of normal BALB/c mice with KG4:T124 virus (1.5 × 1010 gc) showed no abnormalities through the T124 region (data not shown). These results allayed concerns about potential selection of miR-124-insensitive variants during KG4:T124 virus growth or in vivo.
The miR-124 response elements do not impair EGFR-targeted oncolytic HSV activity
We next sought to ascertain whether the protective miR-124 recognition elements adversely affected the viral tumor-killing activity in a nude-mouse model of human GBM. Since KG was highly toxic when inoculated into the brains of these animals (Figure 3c), the use of this virus as a treatment control in survival experiments of tumor-bearing mice could result in animal death due to the virus rather than the tumor and thus was not attractive. Instead, we introduced the four copies of the miR-124 binding site into a fully EGFR-retargeted derivative of KG based on our published observations that fully EGFR-retargeted wild-type HSV-1 KOS is nontoxic for nude mouse brain but is effective in the treatment of orthotopic human GBM in nude mice.5 Thus, comparison of EGFR-retargeted versions of KG and KG4:T124, referred to as KGE and KGE-4:T124, respectively, should identify any limiting effects of the miR-124 sites on viral oncolytic activity. We used patient-derived, sphere-forming GBM30 cells to establish aggressive intracranial tumors in nude mice.5 Animals were observed daily and euthanized when showing signs of morbidity. Similar to our published results, mice injected with PBS 5 days after tumor-cell inoculation at the same stereotactic coordinates died within weeks of tumor-cell implantation (median 21.5 days; Figure 4a,b). In contrast, tumor treatments using either the EGFR-retargeted control virus, KGE, or the T124-containing retargeted vector, KGE-4:T124, protected half of the animals for the duration of the experiment (90 days) and the median survival times for these two groups were comparable (79.5 and 85.5 days, respectively; P = 0.83, log-rank test). These results indicated that the miR-124 sites in the ICP4 gene of KGE-4:T124 did not impair GBM30 tumor treatment efficacy.
Figure 4.

EGFR-retargeted miR-124-sensitive HSV vector treatment of a nude mouse model of human glioblastoma. Triturated GBM30 cells were implanted intracranially and 5 days later, PBS or 1.8 × 108 gc of KGE or KGE-4:T124 virus were injected at the same coordinates. (a) Kaplan–Meier survival plot. Log-rank statistics: KGE versus PBS, P = 0.0188; KGE-4:T124 versus PBS, P = 0.0009; KGE versus KGE-4:T124, P = 0.8327. (b) Animal weights over time post tumor-cell implantation. X, animal death or euthanasia.
Discussion
Our goal was to engineer an oHSV that expresses the full complement of viral functions but can only infect cells expressing a GBM-associated receptor and replicate with high efficiency only in the tumor and not in normal brain cells. Tumor-selective infection and lytic virus growth relied on a combination of complete viral entry retargeting5 and cellular miRNA-mediated restriction of virus replication in normal brain tissue. Recent studies have taken advantage of differences in the miRNA expression profiles between normal and target cells to achieve cell-specific replication of several viruses, including amplicon-based HSV vector and adenoviral oncolytic virus.23,24,25,26,27 This combination of transductional and posttranscriptional tumor targeting promises to provide a very safe and effective oHSV since lytic infection requires two separate characteristics of the target cell that are important for maintenance of the tumor phenotype, the targeted receptor and a tumor-specific miRNA expression profile. This general strategy is broadly applicable using targeting and miRNA-response elements tailored to different cancers; its application can be optimized for personalized therapy by taking into account potential differences in specific antigen and miRNA expression between individual tumors of the same type.
In GBM, altered gene expression includes substantial down-regulation of multiple miRNAs compared to normal brain tissue,28,29,30 presenting several possible miRNAs that may be used to preferentially attenuate engineered virus replication in normal brain. Because miR-124 is recognized as a potent inducer of neuronal differentiation,31 it is among the most highly down-regulated miRNAs in GBM6 and inhibits STAT3 signal in glioma cancer stem cells,32 we focused on this miRNA as a means to block oHSV replication in normal brain tissue. Repeat recognition sites for miR-124 (T124) were introduced into the 3′UTR of the viral ICP4 gene whose product is absolutely required for launching the HSV lytic cycle. We found that in glioma cells, the T124+ virus could replicate essentially as robustly as the control virus lacking T124, whereas lentiviral expression of miR-124 selectively blocked its replication. Furthermore, the T124 element was sufficient to completely protect nude mice from very high intracranial vector dosing (4.8 x 109 particles) while the control vector killed all animals within 5 days. Determination of total viral gc numbers in the brains of these animals showed no evidence of T124+ vector replication but rather a gradual decrease in viral genome content over time. Furthermore, while the animals injected with the control vector (KG) showed the abundant presence of viral antigens, no evidence of virus replication was detected in brain sections from symptom-free mice injected with miR-124 controlled virus and sacrificed on day 30 dpi. The T124 sequence was stable as assessed by size and sequence analysis of the ICP4 3′UTR amplified on purified DNA from virus stocks and infected animals, consistent with the lack of overt neuropathogenesis in tumor-free animals or long-term survivors from our tumor treatment experiment. Finally, we used a retargeted virus that fails to infect mouse cells to demonstrate that the T124 element did not reduce the oncolytic efficacy of this virus in a human GBM model in nude mice.
The combination of virus targeting to tumor receptors and miRNA-mediated blocking of virus replication in normal cells enhances the target specificity of the lytic virus by blocking productive infection of normal cells that may share the targeted receptor with the tumor (e.g., EGFR). While our results show that the insertion of four copies of the target sequence for miR-124 into the 3′UTR of the ICP4 gene completely blocks very high dose viral neuropathogenesis in nude mice, not all brain cells express miR-124. For example, neural progenitor cells (NPCs) located in the hippocampus and subventricular zone are not expected to be protected by the miR-124 target sequences since these cells have an miRNA expression profile that is similar to that of GBM cells, including minimal expression of miR-124.33 However, several miRNAs are expressed at up to 100-fold higher levels in NPCs than in gliomas,33,34,35,36 suggesting the possibility of using target sites for additional miRNAs engineered into the same or other essential genes of the same virus to block replication in a wider range of brain cells without compromising tumor specific virus replication.
Although our study suggests that the combination of virus targeting to a tumor antigen and miRNA-restricted replication in normal tissue is an attractive strategy for effective and highly specific tumor virotherapy, it is likely that individual tumors will differ in their response to the treatment due to variability in tumor antigen levels and perhaps miRNA content. For example, there are significant differences between tumors classified as GBM, and even within the molecularly defined GBM subtypes, heterogeneity in gene expression profiles remains.37 Thus, a single retargeted virus will not be effective against all GBM or all GBM of the same subtype. In addition, it may be anticipated that resistant cell populations can emerge in largely oHSV-sensitive tumors as a consequence of preexisting or treatment-induced cell-to-cell variability within the tumor. Developments over the past several years suggest that the small population of self-renewing, chemo- and radio-resistant cancer stem cells identified in many different tumor types are the most relevant targets for therapy.38 Although comparison of individual cancer stem cells from a given tumor is problematic, it is likely that their variability within a tumor is limited relative to that of the complete tumor-cell population. Reports in the literature describe different glioma stem cell markers39 and retargeted oncolytic viruses can be used to distinguish the significance of each of these for human GBM establishment and maintenance in nude mice. We anticipate that tumors showing partial responses to individual retargeted vectors may be more effectively treated with combinations of vectors retargeted to different glioma stem cell candidate markers. Since each of these vectors may also target certain normal cells, similar to our EGFR-retargeted viruses, miRNA-mediated blockage of virus replication in these normal cells will be of increasing importance. In addition, it may be possible to gain further specificity using cell type- or developmental stage-specific promoters to control the expression of key viral replication functions, as pioneered in the oHSV field by Kambara and colleagues.40 While these features may provide highly active and specific oncolytic vector cocktails, it is noteworthy that vectors such as KGE-4:T124 have ample space to accommodate transgenes that may enhance therapeutic efficacy such as genes encoding immune modulators, inhibitors of tumor cell migration, or proteolytic enzymes that degrade the tumor extracellular matrix and thereby facilitate intratumoral virus spread.
In summary, the KGE-4:T124 vector described in this report represents a novel type of oHSV that contains the complete complement of virus replicative functions, but derives tumor specificity from a combination of viral envelope retargeting to tumor-associated receptors and replication sensitivity to miRNAs that are expressed in normal tissue but not in the tumor. This combination of control systems can be applied to other tumor types but has not been previously described in oncolytic vectors. Key advantages of our strategy are (i) that the vector does not contain any defective genes, allowing maximal virus replication in tumors to provide optimal oncolytic virotherapy, and (ii) that vector replication requires both the expression of important tumor-associated cell-surface markers and a tumor-specific profile of miRNA expression that differs substantially from that of normal tissue. The most compelling argument for our strategy is that miRNAs chosen to control vector replication in normal brain cannot be up-regulated in glioblastoma without compromising the tumor phenotype7,30,41; loss of the targeted receptor such as the tumor-specific EGFRvIII variant recognized by our vector may have a similar effect. Thus, while in most cancer therapies the tumor develops the ability to escape treatment, this outcome is less likely with tumor antigen-targeted, miRNA-regulated viruses. Together, these arguments support the expectation that our approach will provide highly selective, safe, and effective oHSV systems for the treatment of GBM and other cancers. Ultimately, the combination vector must be tested in patient trials to confirm the safety and utility of this oncolytic vector strategy.
Materials and Methods
Cell culture. U2OS, HEK293T, and HEK293AD cells were from ATCC (Manassas, VA) and were grown in a 5% CO2 incubator at 37 °C in ATCC-recommended medium supplemented with 5–10% (v/v) fetal bovine serum (Sigma, St. Louis, MO). A U2OS cell line stably expressing Cre recombinase (U2OS-Cre) was generated by retroviral transduction (Y.M. and J.C.G., unpublished results). GBM30 and Gli68 patient-derived primary glioma spheroid lines, generously provided by E.A. Chiocca (Harvard Medical School, MA), were grown in Neurobasal medium (Gibco/Invitrogen/Life Technologies, Carlsbad, CA) plus 2% (v/v) B27 w/o vitamin A, 2 mg/mL amphotericin B (Lonza, Walkersville, MD), 100 μg/mL gentamycin (Lonza), 2 mmol/l l-glutamine (Cellgro, Manassas, VA), plus 10 ng/mL recombinant human epidermal growth factor and 10 ng/mL recombinant human basic fibroblast growth factor (both from Shenandoah Biotechnology, Warwick, PA).
Plasmids. pfLuc-T124 contains four tandem repeats of the reverse complement of the hsa-miR-124 sequence separated by 8 nt, while pfLuc-Ctrl contains four tandem repeats of the hsa-miR-21 reverse sequence separated by 8 nt. Both plasmids were constructed by insertion of annealed complementary oligonucleotides into the 3′UTR of the luciferase gene in pMIR-REPORT™ (miRNA Expression Reporter Vector System; Ambion, Austin, TX). Oligonucleotides were T124-F, T124-R, TconF, and TconR (Table 1). Annealed oligonucleotides were digested with SpeI and SacI, and ligated to SpeI-SacI-digested pMIR-REPORT™.
Table 1. Primers and probe used in this study.

HSV genome engineering. KOS-37 BAC,11 containing the complete strain KOS HSV-1 genome on a bacterial artificial chromosome (BAC), was kindly provided by David Leib (Dartmouth Medical School, NH). The HSV unique short region in this BAC is in the reverse orientation relative to the published sequence (positions 132,275–145,608) of HSV-1 KOS42 (GenBank Accession number JQ673480). Modifications detailed further below were introduced by double Red recombination, essentially as described by Tischer et al.10 Plasmids pEPkan-S and pBAD-I-sceI10 were generously provided by Nikolaus Osterrieder (Free University of Berlin, Germany). Changes were verified by PCR analysis, FIGE analysis of restriction enzyme digests, and local DNA sequencing.
Vectors used in this study were sequentially derived as follows. KGBAC was derived from KOS-37 BAC by deletion of the complete HSV internal repeat region or “joint” (IRL, IRS), fusion of the green fluorescent protein open reading frame to the glycoprotein C open reading frame via the Thosea asigna virus 2A (T2A) translation termination/reinitiation sequence,13,43 and introduction of two missense mutations in the glycoprotein B coding sequence (gB:N/T;15 KG4:T124BAC was created from KGBAC by insertion of the T124 element from pfLuc-T124 into the 3′UTR of the ICP4 gene. The retargeted vector KGEBAC was derived from KGBAC by replacement of the amino-terminal region of the glycoprotein D (gD) gene with the corresponding region of gD-scEGFR containing the sequence for a human EGFR-specific single chain antibody between gD positions 1 and 25 and a missense mutation at codon 38.5 KGE-4:T124BAC combines the modifications from KG4:T124BAC and KGEBAC.
Virus growth and purification. BAC DNAs were converted to infectious virus by transfection of U2OS-Cre cells using Lipofectamine™ LTX Reagent (Invitrogen); Cre recombinase expressed in these cells allowed the removal of the virus growth-inhibitory BAC elements and adjacent lacZ gene located in KOS-37 BAC and derivatives between loxP recombination signals.11 Single plaques were isolated by limiting dilution and tested for elimination of the lacZ gene by X-gal staining.44 Colorless plaques were subjected to two additional rounds of limiting dilution and accurate removal of the BAC/lacZ region was confirmed by local DNA sequencing of purified virion DNA. Biological titers of virus stocks (PFU/mL) were established on U2OS cells; physical titers in genome copies (gc)/mL were determined by qPCR for the viral gD gene, as described below.
Luciferase assay. HEK293AD were transfected with the renilla luciferase expression plasmid prLuc together with combinations of different fLuc expression plasmids and pre-miR™ miRNA Precursors (Ambion) using Lipofectamine 2000 (Invitrogen). The next day, cells were lysed and the firefly-to-renilla luciferase expression ratios were determined using a Berthold LB-953 AutoLumat luminometer (Berthold Technologies USA, Oak Ridge, TN).
Lentiviral expression of miRNAs. Genomic DNA from U-87 human glioblastoma cells was used as template for PCR amplification of the human pri-miR-124 sequence from the hsa-miR-124-3 gene using High Fidelity Accuprime GC-rich DNA Polymerase (Invitrogen) and the miR-124 primer pair listed in Table 1. The 320-bp product was digested with BamHI and NheI, cloned between the corresponding sites in the intron of miRNASelect pEP-miR vector (Cell Biolabs, San Diego, CA), and sequence confirmed. The promoter-intron-pri-miR-124 region was subsequently transferred into pCDH-CMV-MCS-EF1-Puro (System Biosciences, Mountain View, CA) by replacement of the resident EF1 promoter to generate lentiviral expression plasmid pCDH-miR-124. The same procedures were used to construct the control lentiviral plasmid (pCDH-miR-137R) containing the pri-miR-137 sequence in the reverse orientation; the PCR primers used for pri-miR-137 cloning are listed in Table 1. Lentiviruses LV124 and LV137R were produced by cotransfection of pCDH-miR-124 or pCDH-miR-137R, respectively, with packaging plasmids pLP1, pLP2, pLP-VSVG (Invitrogen) into HEK293T cells. Supernatants were harvested 72 hours later, passed through a 0.45 µm filter (Millipore, Billerica, MA), and concentrated by centrifugation for 16 hours at 4 °C and 6,800 × g. Pellets were resuspended in DMEM and titered as puromycin-resistant colony-forming units (cfu) per mL on HEK293T cells.
2 × 105 triturated Gli68 or GBM30 cells were infected in suspension with either LV124 or LV137R at 5 cfu/cell in the presence of 8 µg/mL polybrene for 90 minutes and plated. The cells were fed the following day with fresh media containing 30 µg/mL puromycin and superinfected 72 hours later with either KG or KG4:T124 virus at an MOI of 0.01 pfu/cell. At 72 and 96 hours post HSV infection, infectious virus particles were collected from cells and supernatants and titered on U2OS cells. RNA was isolated from parallel cultures of LV124-infected Gli68 cells after 72 hours of puromycin selection for determination of miR-124 levels by qRT-PCR, as described below.
RNA isolation and reverse transcription (RT)-qPCR. Total RNA was extracted from U2OS, Gli68, and LV124-infected Gli68 cells using TRIzol Reagent (Invitrogen) according to the manufacturer's instructions. RNA samples were treated with DNase I (Invitrogen), quantified using a NanoDrop 2000c spectrophotometer (Thermo-Fisher, Pittsburgh, PA) and visualized on a MOPS-formaldehyde gel for quality assurance. Mature hsa-miR-124 levels were determined relative to RNU43 according to the TaqMan Small RNA Assays Protocol (Applied Biosystems/Life Technologies, Carlsbad, CA). TaqMan primers and probes were from Applied Biosystems. All TaqMan PCR reactions were performed in triplicate.
Animals. 3–4 week-old BALB/c athymic nu/nu mice were purchased from Charles River Laboratory (Wilmington, MA) and housed in a BSL2 facility. All animal procedures were performed in accordance with the requirements and recommendations in the Guide for the Care and the Use of Laboratory Animals (Institute for Laboratory Animal Research, 1985) as approved by the University of Pittsburgh Institutional Animal Care and Use Committee.
Intracranial toxicity. Intracranial virus inoculations were performed a described.5 For the qPCR: Mice received 4.8 × 109 gc of KG or KG4:T124 virus (n = 4/group). The animals were monitored daily for signs of morbidity and were weighed every other day. All mice of the KG group died by day 5 and one mouse of the other group was sacrificed the same day. Remaining animals of the KG4:T124 group were sacrificed on days 14, 21, and 33. Whole brains were collected from euthanized mice for total DNA extraction and qPCR for viral genomes, as described below.
For the IHC: Mice received 4.36 × 109 gc of KG or KG4:T124 virus (n = 4/group) or PBS. The animals were monitored daily for signs of morbidity and were weighed every other day. All mice of the KG group died by day 7 while the animals injected with KG4:T124 were human sacrificed 30 dpi. Ten micrometers cryosections were stained with anti-HSV antibody (1:500 Abcam ab9533) and secondary antibody (goat anti-rabbit 1:200).
qPCR for viral genomes. DNA was extracted from mouse brains or virus stocks using the DNeasy Blood & Tissue kit (Qiagen, Valencia, CA) according to the manufacturer's procedure. A standard curve for qPCR was generated on DNA from a pENTR1A (Invitrogen) plasmid containing the complete HSV-1 (strain KOS) gD coding sequence (pE-gD18) using the protocol described in the Applied Biosystems StepOne™ and StepOnePlus™ Real-Time PCR Systems manual. Primers and probe sequences are listed in Table 1.
Tumor model and treatment. Intracranial implantation of human GBM30 cells into nude mice was performed as described.5 At 5 days, viruses (1.8 × 108 gc of KGE or KGE-4:T124, n = 8/group) or PBS (n = 2) were inoculated at the same coordinates, as also described.5 Animal health and well-being were monitored as described above under “Intracranial toxicity.” Animals were euthanized when showing signs of morbidity.
Statistical analysis. Unpaired t-test with Welch's correction was performed using GraphPad Prism version 6.01 for Windows (GraphPad Software, La Jolla, CA; www.graphpad.com). Animal survival data were charted as Kaplan–Meier plots and compared by Mantel–Cox log-rank test using the same software.
SUPPLEMENTARY MATERIAL Figure S1. Evaluation of KG4:T124 virus replication and toxicity in nude mouse brains.
Acknowledgments
This work was supported by grants from the National Institutes of Health (CA119298, NS40923, NS064988-02 and CA163205-01A1) to J.C.G. B.R. was supported in part by an NIH T32 postdoctoral training grant in Molecular Microbial Persistence and Pathogenesis. We thank Nikolaus Osterrieder for the generous gift of pEPkan-S and pBAD-I-sceI plasmids; Chiocca for the GBM30 and Gli68 cell lines; Leib for KOS-HSV BAC. We also thank Mingdi Zhang for excellent technical assistance in all in vivo studies and Shaohua Huang for preparation of oHSV virus stocks.
Supplementary Material
Evaluation of KG4:T124 virus replication and toxicity in nude mouse brains.
References
- Parker JN, Bauer DF, Cody JJ, Markert JM. Oncolytic viral therapy of malignant glioma. Neurotherapeutics. 2009;6:558–569. doi: 10.1016/j.nurt.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grandi P, Peruzzi P, Reinhart B, Cohen JB, Chiocca EA, Glorioso JC. Design and application of oncolytic HSV vectors for glioblastoma therapy. Expert Rev Neurother. 2009;9:505–517. doi: 10.1586/ern.09.9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert JM, Medlock MD, Rabkin SD, Gillespie GY, Todo T, Hunter WD, et al. Conditionally replicating herpes simplex virus mutant, G207 for the treatment of malignant glioma: results of a phase I trial. Gene Ther. 2000;7:867–874. doi: 10.1038/sj.gt.3301205. [DOI] [PubMed] [Google Scholar]
- Todo T. Oncolytic virus therapy using genetically engineered herpes simplex viruses. Front Biosci. 2008;13:2060–2064. doi: 10.2741/2823. [DOI] [PubMed] [Google Scholar]
- Uchida H, Marzulli M, Nakano K, Goins WF, Chan J, Hong CS, et al. Effective treatment of an orthotopic xenograft model of human glioblastoma using an EGFR-retargeted oncolytic herpes simplex virus. Mol Ther. 2013;21:561–569. doi: 10.1038/mt.2012.211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaur A, Jewell DA, Liang Y, Ridzon D, Moore JH, Chen C, et al. Characterization of microRNA expression levels and their biological correlates in human cancer cell lines. Cancer Res. 2007;67:2456–2468. doi: 10.1158/0008-5472.CAN-06-2698. [DOI] [PubMed] [Google Scholar]
- Karsy M, Arslan E, Moy F. Current Progress on understanding microRNAs in glioblastoma multiforme. Genes Cancer. 2012;3:3–15. doi: 10.1177/1947601912448068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riddick G, Fine HA. Integration and analysis of genome-scale data from gliomas. Nat Rev Neurol. 2011;7:439–450. doi: 10.1038/nrneurol.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- Tischer BK, von Einem J, Kaufer B, Osterrieder N. Two-step red-mediated recombination for versatile high-efficiency markerless DNA manipulation in Escherichia coli. Biotechniques. 2006;40:191–197. doi: 10.2144/000112096. [DOI] [PubMed] [Google Scholar]
- Gierasch WW, Zimmerman DL, Ward SL, Vanheyningen TK, Romine JD, Leib DA. Construction and characterization of bacterial artificial chromosomes containing HSV-1 strains 17 and KOS. J Virol Methods. 2006;135:197–206. doi: 10.1016/j.jviromet.2006.03.014. [DOI] [PubMed] [Google Scholar]
- Bennett JJ, Delman KA, Burt BM, Mariotti A, Malhotra S, Zager J, et al. Comparison of safety, delivery, and efficacy of two oncolytic herpes viruses (G207 and NV1020) for peritoneal cancer. Cancer Gene Ther. 2002;9:935–945. doi: 10.1038/sj.cgt.7700510. [DOI] [PubMed] [Google Scholar]
- Szymczak AL, Vignali DA. Development of 2A peptide-based strategies in the design of multicistronic vectors. Expert Opin Biol Ther. 2005;5:627–638. doi: 10.1517/14712598.5.5.627. [DOI] [PubMed] [Google Scholar]
- Doronina VA, Wu C, de Felipe P, Sachs MS, Ryan MD, Brown JD. Site-specific release of nascent chains from ribosomes at a sense codon. Mol Cell Biol. 2008;28:4227–4239. doi: 10.1128/MCB.00421-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Chan J, Goins WF, Grandi P, Kumagai I, Cohen JB, et al. A double mutation in glycoprotein gB compensates for ineffective gD-dependent initiation of herpes simplex virus type 1 infection. J Virol. 2010;84:12200–12209. doi: 10.1128/JVI.01633-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchida H, Chan J, Shrivastava I, Reinhart B, Grandi P, Glorioso JC, et al. Novel mutations in gB and gH circumvent the requirement for known gD receptors in herpes simplex virus 1 entry and cell-to-cell spread. J Virol. 2013;87:1430–1442. doi: 10.1128/JVI.02804-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao X, Pfaff SL, Gage FH. A functional study of miR-124 in the developing neural tube. Genes Dev. 2007;21:531–536. doi: 10.1101/gad.1519207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujioka N, Akazawa R, Ohashi K, Fujii M, Ikeda M, Kurimoto M. Interleukin-18 protects mice against acute herpes simplex virus type 1 infection. J Virol. 1999;73:2401–2409. doi: 10.1128/jvi.73.3.2401-2409.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manickan E, Rouse RJ, Yu Z, Wire WS, Rouse BT. Genetic immunization against herpes simplex virus. Protection is mediated by CD4+ T lymphocytes. J Immunol. 1995;155:259–265. [PubMed] [Google Scholar]
- Sethi KK, Omata Y, Schneweis KE. Protection of mice from fatal herpes simplex virus type 1 infection by adoptive transfer of cloned virus-specific and H-2-restricted cytotoxic T lymphocytes. J Gen Virol. 1983;64 Pt 2:443–447. doi: 10.1099/0022-1317-64-2-443. [DOI] [PubMed] [Google Scholar]
- Currier MA, Gillespie RA, Sawtell NM, Mahller YY, Stroup G, Collins MH, et al. Efficacy and safety of the oncolytic herpes simplex virus rRp450 alone and combined with cyclophosphamide. Mol Ther. 2008;16:879–885. doi: 10.1038/mt.2008.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong CS, Fellows W, Niranjan A, Alber S, Watkins S, Cohen JB, et al. Ectopic matrix metalloproteinase-9 expression in human brain tumor cells enhances oncolytic HSV vector infection. Gene Ther. 2010;17:1200–1205. doi: 10.1038/gt.2010.66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee CY, Rennie PS, Jia WW. MicroRNA regulation of oncolytic herpes simplex virus-1 for selective killing of prostate cancer cells. Clin Cancer Res. 2009;15:5126–5135. doi: 10.1158/1078-0432.CCR-09-0051. [DOI] [PubMed] [Google Scholar]
- Kelly EJ, Nace R, Barber GN, Russell SJ. Attenuation of vesicular stomatitis virus encephalitis through microRNA targeting. J Virol. 2010;84:1550–1562. doi: 10.1128/JVI.01788-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cawood R, Chen HH, Carroll F, Bazan-Peregrino M, van Rooijen N, Seymour LW. Use of tissue-specific microRNA to control pathology of wild-type adenovirus without attenuation of its ability to kill cancer cells. PLoS Pathog. 2009;5:e1000440. doi: 10.1371/journal.ppat.1000440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ylösmäki E, Hakkarainen T, Hemminki A, Visakorpi T, Andino R, Saksela K. Generation of a conditionally replicating adenovirus based on targeted destruction of E1A mRNA by a cell type-specific microRNA. J Virol. 2008;82:11009–11015. doi: 10.1128/JVI.01608-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barnes D, Kunitomi M, Vignuzzi M, Saksela K, Andino R. Harnessing endogenous miRNAs to control virus tissue tropism as a strategy for developing attenuated virus vaccines. Cell Host Microbe. 2008;4:239–248. doi: 10.1016/j.chom.2008.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Chao T, Li R, Liu W, Chen Y, Yan X, et al. MicroRNA-128 inhibits glioma cells proliferation by targeting transcription factor E2F3a. J Mol Med (Berl) 2009;87:43–51. doi: 10.1007/s00109-008-0403-6. [DOI] [PubMed] [Google Scholar]
- Shi L, Cheng Z, Zhang J, Li R, Zhao P, Fu Z, et al. hsa-mir-181a and hsa-mir-181b function as tumor suppressors in human glioma cells. Brain Res. 2008;1236:185–193. doi: 10.1016/j.brainres.2008.07.085. [DOI] [PubMed] [Google Scholar]
- Silber J, Lim DA, Petritsch C, Persson AI, Maunakea AK, Yu M, et al. miR-124 and miR-137 inhibit proliferation of glioblastoma multiforme cells and induce differentiation of brain tumor stem cells. BMC Med. 2008;6:14. doi: 10.1186/1741-7015-6-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maiorano NA, Mallamaci A. The pro-differentiating role of miR-124: indicating the road to become a neuron. RNA Biol. 2010;7:528–533. doi: 10.4161/rna.7.5.12262. [DOI] [PubMed] [Google Scholar]
- Wei J, Wang F, Kong LY, Xu S, Doucette T, Ferguson SD, et al. miR-124 inhibits STAT3 signaling to enhance T cell-mediated immune clearance of glioma. Cancer Res. 2013;73:3913–3926. doi: 10.1158/0008-5472.CAN-12-4318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lavon I, Zrihan D, Granit A, Einstein O, Fainstein N, Cohen MA, et al. Gliomas display a microRNA expression profile reminiscent of neural precursor cells. Neuro Oncol. 2010;12:422–433. doi: 10.1093/neuonc/nop061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karpowicz P, Willaime-Morawek S, Balenci L, DeVeale B, Inoue T, van der Kooy D. E-cadherin regulates neural stem cell self-renewal. J Neurosci. 2009;29:3885–3896. doi: 10.1523/JNEUROSCI.0037-09.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Katoh Y, Katoh M. Hedgehog signaling, epithelial-to-mesenchymal transition and miRNA (review) Int J Mol Med. 2008;22:271–275. [PubMed] [Google Scholar]
- Ocaña OH, Nieto MA. A new regulatory loop in cancer-cell invasion. EMBO Rep. 2008;9:521–522. doi: 10.1038/embor.2008.84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verhaak RG, Hoadley KA, Purdom E, Wang V, Qi Y, Wilkerson MD, et al. Cancer Genome Atlas Research Network Integrated genomic analysis identifies clinically relevant subtypes of glioblastoma characterized by abnormalities in PDGFRA, IDH1, EGFR, and NF1. Cancer Cell. 2010;17:98–110. doi: 10.1016/j.ccr.2009.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nduom EK, Hadjipanayis CG, Van Meir EG. Glioblastoma cancer stem-like cells: implications for pathogenesis and treatment. Cancer J. 2012;18:100–106. doi: 10.1097/PPO.0b013e3182452e0d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He J, Liu Y, Lubman DM. Targeting glioblastoma stem cells: cell surface markers. Curr Med Chem. 2012;19:6050–6055. [PubMed] [Google Scholar]
- Kambara H, Okano H, Chiocca EA, Saeki Y. An oncolytic HSV-1 mutant expressing ICP34.5 under control of a nestin promoter increases survival of animals even when symptomatic from a brain tumor. Cancer Res. 2005;65:2832–2839. doi: 10.1158/0008-5472.CAN-04-3227. [DOI] [PubMed] [Google Scholar]
- Xia H, Cheung WK, Ng SS, Jiang X, Jiang S, Sze J, et al. Loss of brain-enriched miR-124 microRNA enhances stem-like traits and invasiveness of glioma cells. J Biol Chem. 2012;287:9962–9971. doi: 10.1074/jbc.M111.332627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald SJ, Mostafa HH, Morrison LA, Davido DJ. Genome sequence of herpes simplex virus 1 strain KOS. J Virol. 2012;86:6371–6372. doi: 10.1128/JVI.00646-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaji K, Norrby K, Paca A, Mileikovsky M, Mohseni P, Woltjen K. Virus-free induction of pluripotency and subsequent excision of reprogramming factors. Nature. 2009;458:771–775. doi: 10.1038/nature07864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krisky DM, Marconi PC, Oligino T, Rouse RJ, Fink DJ, Glorioso JC. Rapid method for construction of recombinant HSV gene transfer vectors. Gene Ther. 1997;4:1120–1125. doi: 10.1038/sj.gt.3300497. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Evaluation of KG4:T124 virus replication and toxicity in nude mouse brains.