

Abstract
Oncolytic viral therapy utilizes a tumor-selective replicating virus which preferentially infects and destroys cancer cells and triggers antitumor immunity. The Western Reserve strain of vaccinia virus (VV) is the most virulent strain of VV in animal models and has been engineered for tumor selectivity through two targeted gene deletions (vvDD). We performed the first-in-human phase 1, intratumoral dose escalation clinical trial of vvDD in 16 patients with advanced solid tumors. In addition to safety, we evaluated signs of vvDD replication and spread to distant tumors, pharmacokinetics and pharmacodynamics, clinical and immune responses to vvDD. Dose escalation proceeded without dose-limiting toxicities to a maximum feasible dose of 3 × 109 pfu. vvDD replication in tumors was reproducible. vvDD genomes and/or infectious particles were recovered from injected (n = 5 patients) and noninjected (n = 2 patients) tumors. At the two highest doses, vvDD genomes were detected acutely in blood in all patients while delayed re-emergence of vvDD genomes in blood was detected in two patients. Fifteen of 16 patients exhibited late symptoms, consistent with ongoing vvDD replication. In summary, intratumoral injection of the oncolytic vaccinia vvDD was well-tolerated in patients and resulted in selective infection of injected and noninjected tumors and antitumor activity.
Introduction
Oncolytic viral therapy has been considered a novel approach to cancer treatment with preclinical investigations using genetically engineered oncolytic viruses being initiated over 23 years ago.1,2,3 The concept of using a tumor-selective replicating virus to infect and spread throughout the tumor microenvironment, destroying cancer cells, and inducing an antitumor immune response in the process is appealing, however, the development of this process has been fraught with obstacles. Early attempts to use replicating adenovirus in patients largely failed due to the inefficiency of replication and poor systemic virus spread. Subsequent efforts with more efficient viral backbones have also failed to demonstrate significant clinical benefits.4 Clinical trials with ONYX-015, an E1B-55kD gene-deleted replication selective adenovirus, however, have provided the “proof of principle” that cancer cells can be infected, and that viral replication and spread to tumors occurs in human beings.5 The most successful oncolytic virus to date, talimogene laherparepvec (T-Vec), an oncolytic granulocyte-macrophage colony-stimulating factor (GM-CSF) producing herpes virus, was recently shown in a randomized phase 3 clinical trial to have a better durable response rate and a trend toward better overall survival compared to GM-CSF for unresectable melanoma.6 There have also been encouraging results with the use of replicating vaccinia virus (VV) (New York City Board of Health Strain) expressing GM-CSF in patients with hepatocellular carcinoma where intralesional Pexa-Vec therapy (Sillajen) resulted in a 15% response rate (mRECIST) and prolongation of survival when high dose was compared to low-dose treatment.7,8,9
VV has many potential advantages over other oncolytic viral backbones. VV is a remarkably efficient, cytoplasmic virus, with DNA synthesis beginning within 2 hours of infection, viral assembly beginning 6 hours after infection10 and 100–200 pfu (2,500–5,000 particles) produced per cell within 20–40 hours.11 VV spreads efficiently using a method of direct cell to cell spread where the virus remains attached to the cell membrane, coated with an extracellular envelope derived from the host cell plasma membrane, thereby avoiding immune recognition; but VV particles can also be released from the cell surface to spread systemically, still protected by an outer host cell derived membrane which can evade immune clearance.12,13,14 VV will infect and replicate in all tumor cells with relatively small variability and a 1.0E2 to 1.0E6-fold improved cytotoxic effect compared to the replicating adenovirus ONYX-015 in NCI 60 cancer cell line panel, suggesting that VV may be effective against all solid tumors.15 VV delivered systemically in a tumor-bearing host, targets the tumor via leaky vasculature and selectively replicates in tumor cells leading to an antitumor response.16,17 Indeed, IV delivery of VV to metastatic tumors has been demonstrated clinically.8 Specific strains of VV have different properties, and the Western Reserve (WR) strain of VV has been demonstrated to be more virulent in animal models, but it has never before been administered to humans.15,18
We have taken the approach of starting with the most virulent, efficient WR strain of VV and engineering genetic mutations for tumor selectivity, such that replication within the tumor will be rapid and potent, while normal cells will be spared and pathogenicity to patients will be minimal. We found that the best tumor selective mutation was the combination of deletional mutations of viral genes encoding vaccinia growth factor (VGF) and thymidine kinase (TK).17 The VGF and TK proteins are essential for viral replication in normal cells, but in tumor cells are compensated for by upregulation of growth factors and nucleotides as part of neoplastic transformation.15 The deletions of the viral genes lead to selective targeting of cancer cells with activation of the transcription factor E2F and the EGFR/Ras pathway, common features of many cancer cells.15,19
Preclinical rodent studies demonstrated the safety of the double deleted (VGF/TK) mutant (vvDD) with very low levels of viral recovery compared to wild type VV in organs harvested in nude mice and nearly no viral recovery in organs harvested from immunocompetent mice.17 We also confirmed the safety of vvDD in nonhuman primates. When compared to wild-type WR strain vaccinia, intradermal injection, intravenous infection, and isolated limb perfusion of vvDD in rhesus macaques caused no clinical signs or symptoms of viremia, no viral recovery from serum saliva, urine, or feces, and no long-term toxicity.18 We also confirmed the antitumor potency of vvDD as a systemic injection in rodents bearing subcutaneous MC38 colon adenocarcinoma with significant antitumor effects in all animals, and one complete response to vvDD therapy.17 The TK or TK/VGF deleted WR strain VV were also demonstrated to be effective antitumor therapy in additional models of carcinoma including rodent studies of both human and murine ovarian carcinomatosis, in a solid tumor model (when combined with hyperthermia), and in a model of murine liver metastases.20,21,22
Given its excellent safety profile in nonhuman primates and its impressive effects in preclinical in vitro and rodent models, it was important to define vvDD safety, ability to spread selectively and antitumor activity in cancer patients. We report here a phase 1 study of first-in-human WR strain VV mutant vvDD (JX-929) as a direct injection into tumors. This phase 1 study had classic toxicity endpoints, but we also examined vvDD pharmacokinetics and pharmacodynamics, systemic vvDD spread to noninjected tumors, antitumor activity, and antitumor immune responses.
Results
Treatment and safety
Patient population. A total of 17 patients were treated with intratumoral injections of vvDD. Patient characteristics are described in Table 1. Twenty-one patients were screened and 16 patients were treated on the phase 1 protocol. A 17th male patient was treated as a compassionate exemption due to steroid therapy for arthritis. This patient is not included in the analysis and results related to this patient are presented separately. Four male and 12 females were treated with a median age of 53.6 years. Tumor histology included: 10 colorectal cancer, 4 breast cancer, and 2 pancreas cancer. All patients had advanced bulky tumors that progressed through standard systemic chemotherapy with 100% having prior surgery and a median of 3 systemic chemotherapy regimens. Patients had an ECOG performance status of 0 or 1. Fifteen of the 16 had known prior exposure to VV due to vaccinia vaccination, and baseline neutralizing antibodies to vaccinia were detected in serum from all evaluated patients (12 of 17).
Table 1. Baseline characteristics.
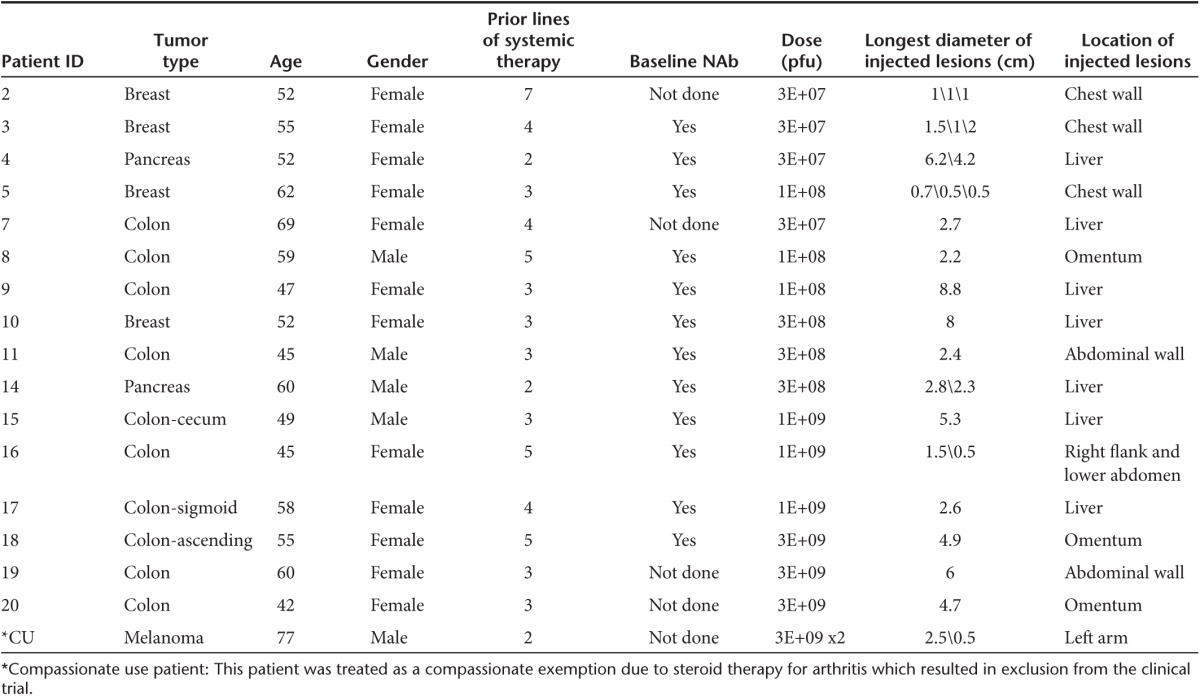
Treatment characteristics. Patients were treated in one of five dose cohorts and received intratumoral injections of vvDD at doses ranging from 3 × 107 pfu to 3 × 109 pfu (Table 1). Three patients received injections into superficial, visible, cutaneous tumors and disease was monitored with photographs (patient #2, 3, and 5), while all other patients received ultrasound guided injections into deep lesions and disease was assessed by CT scans. Up to three sites could be injected per patient, dividing the total dose accordingly, but 59% of patients (n = 10) had only a single site injected. Four individual, equally spaced needle tracts could be used per lesion. vvDD-CDSR was suspended in 0.9% of phosphate-buffered saline in a volume equivalent to 25% of the estimated tumor volume. A standard 21 gauge needle was used for vvDD injection in six patients, whereas the multipronged Quadrafuse needle was used in 11 patients with larger lesions. The size of injected lesions ranged from 0.5 to 8.8 cm in longest diameter (median 2.4 cm) (Table 1). The most common site of injection was liver metastases in nine patients and superficial skin or palpable subcutaneous lesions in five patients.
Safety. Dose escalation proceeded without dose-limiting toxicities. Therefore, 3 × 109 pfu was considered the maximum feasible dose, given the achievable vvDD concentration with current purification methods. One treatment-related serious adverse event (SAE) was reported on study. Seven non-treatment-related SAEs were reported. SAEs were not dose-related (Table 2).
Table 2. Adverse events.
The only severe adverse event possibly related to vvDD included one patient with grade 3 pain (breast cancer patient #3, 3 × 107 pfu dose). Patient #3 had pain in a rib requiring hospital admission 7 days after injection of cutaneous breast cancer metastases, with no radiologic evidence of pulmonary problems (pneumonia, pneumonitis, embolism, effusion) and no evidence on plain film or CT scan of tumor or infection in the rib. The timing of the pain correlated with the peak inflammatory response and clearing of the replicating vvDD in preclinical animal studies. The patient was treated with narcotic pain medication and the pain resolved in 48 hours.
SAEs deemed unrelated to vvDD by an independent safety review board, included one patient (pancreatic cancer patient #4; 3 × 107 pfu) with an upper gastrointestinal bleed from an ulcerated pancreatic cancer metastasis in the stomach 21 days after vvDD injection (noninjected lesion). To evaluate gastrointestinal bleeding, this patient underwent upper endoscopy and tissue was sent for both PCR and plaque assay, neither of which revealed the presence of vvDD. This patient also experienced dehydration, pneumonia, and lower extremity edema classified as SAEs. This patient went on to die of disease progression 36 days after vvDD injection and postmortem examination failed to reveal any residual vvDD in any tissues by plaque forming assays or quantitative polymerase chain reaction (qPCR). Other unrelated Grade 3 or 4 adverse events included pancreatitis, abdominal pain, ascites and elevated aspartate aminotransferase all related to progression of disease outside the injected lesion.
Adverse events are summarized by grade and dose level in Table 2. At doses of 3 × 108 pfu and higher, all patients except one experienced fever and/or chills within 24 hours of injection. No hypotension occurred. Notable Grade 1 or 2 side effects related to treatment included in one patient a glove/stocking distribution of a macular, erythematous rash 15 days after vvDD injection into cutaneous breast cancer metastases. The rash was self-limiting, and a biopsy revealed nonspecific vasculitis with no vvDD antigens by immunohistochemistry (using a polyclonal antivaccinia antibody). Tissue was not obtained for DNA analysis.
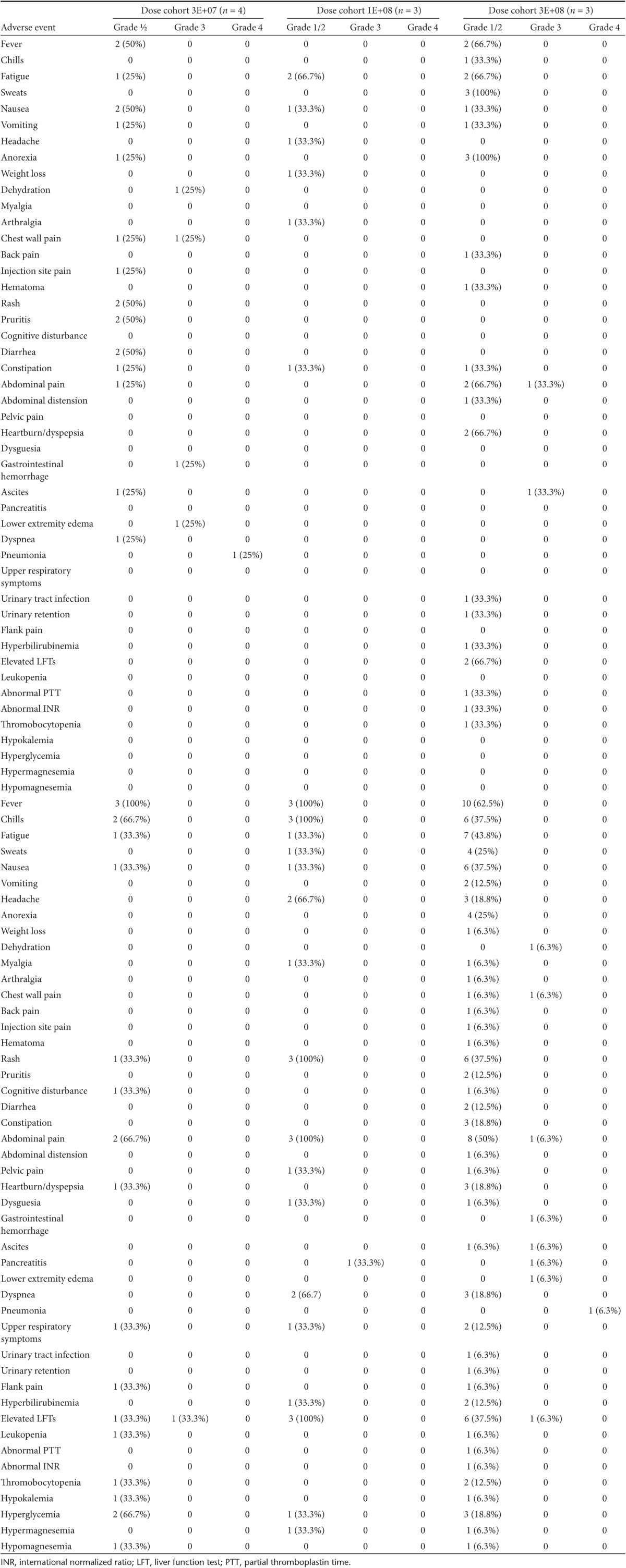
Delayed systemic symptoms. Fifteen of 17 patients had late symptoms (day 5 to 15) consistent with the body's inflammatory response to the virus (Table 3). In most cases, the patients felt well for many days, then developed fever, malaise and/or pain consistent with the expected peak in vvDD replication and the immune response against vvDD. In nonhuman primate studies as well as murine studies, we found vvDD recovery peaked at day 4, then was cleared by the immune system by day 8. This course is typical for patients receiving live vaccinia skin scarification for vaccinia vaccination. Patients' symptoms in this trial correlated with a similar timeline.
Table 3. Reproducible evidence of tumor selective vvDD infection, replication, and systemic spread.
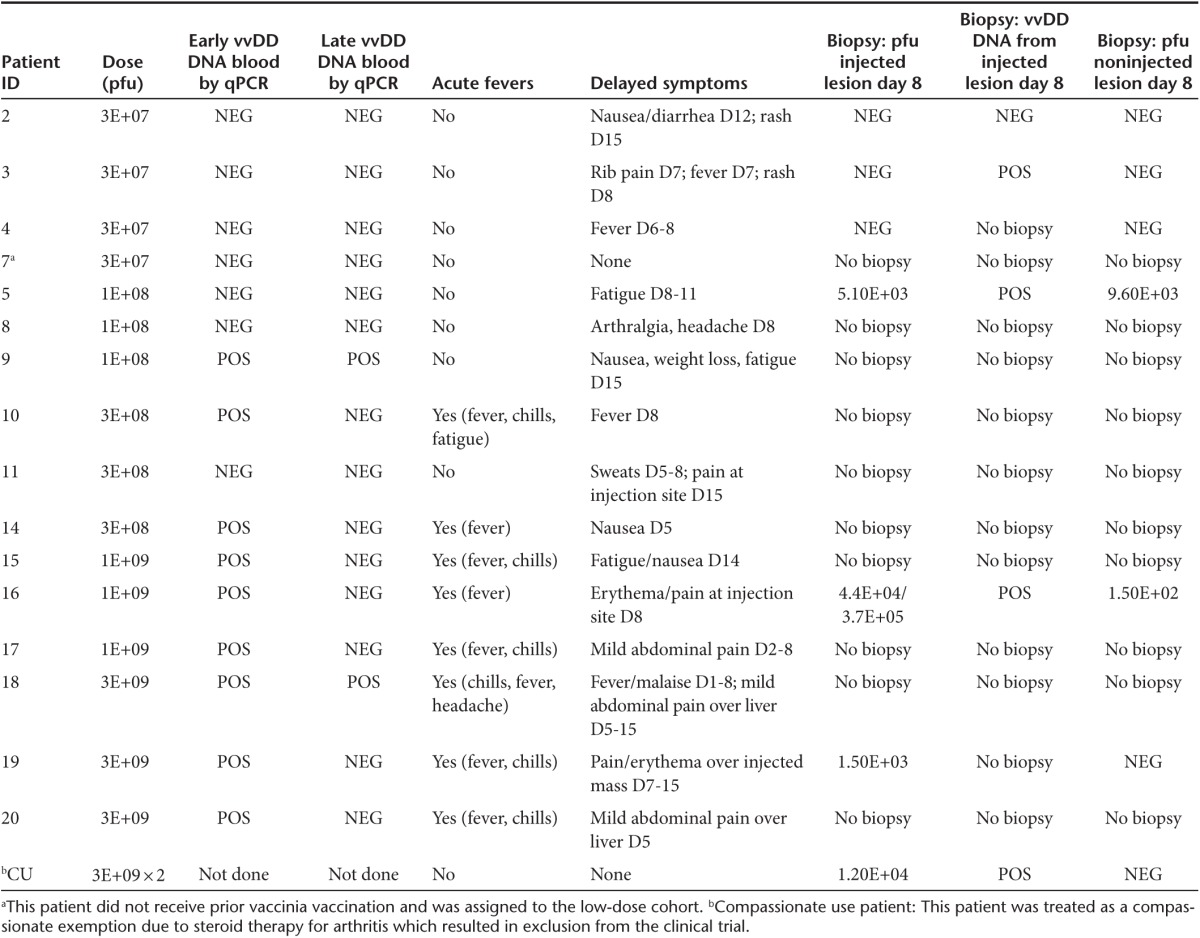
The detection of viral replication in blood by qPCR is a specific, but not sensitive marker for the presence in viral genomes in blood, in part due to the short half-life (~70 minutes) of vaccinia virus.8 Additionally, qPCR testing for viral genomes was performed at predetermined time points, and not necessarily when patients reported specific side effects, such as fever. In patient #18, viral genomes were detected at day 3 and day 5 with symptoms of fever and malaise occurring days 1–8 and pain over the injection site beginning on day 5. However, in other patients, the detection of viral genomes did not correlate contemporaneously with a rise in viral genomes however given vaccinia virus's short half-life in the blood and predetermined time points for testing, it is possible that some late symptoms may represent ongoing viral replication.
Laboratory findings. Laboratory analysis revealed no grade 3 or 4 toxicities related to treatment. Mild liver function test abnormalities were common, however many patients presented with a mild increase in transaminase values prior to vvDD administration. Ten patients had a mild elevation in aspartate aminotransferase after vvDD injection, but it did not correspond with a consistent pattern related to the time from vvDD administration. Of these 10 patients, aspartate aminotransferase levels in 5 returned to below baseline by last follow-up. The remaining 5 patients had persistent mild elevation in aspartate aminotransferase above their own baseline (maximum 160 IU/l above baseline). Eleven patients demonstrated elevation in alkaline phosphatase (ALP) with a trend toward increasing elevation as time from treatment progressed with a median peak value at day 28 post-treatment (Figure 1a). Of the 11 patients who demonstrated an elevated ALP above their baseline measurement, 4 eventually returned to below baseline by day 22. The ALP in the remaining 6 patients remained elevated above their own baseline at the time of last follow-up. Nine of the 11 patients with elevated ALP underwent vvDD administration into intrahepatic lesions. Four patients experienced an elevated total bilirubin with no evidence of baseline hyperbilirubinemia, each at single time points with rapid resolution. The highest total bilirubin noted was 4.1 mg/dl (normal high 1.3 mg/dl). Two patients demonstrated leukopenia at a single time point, but there were no instances of neutropenia. Most patients (14) experienced a relative lymphopenia compared to baseline with a median nadir at day 3 after vvDD administration. Nearly all patients (11) recovered their lymphocyte count to above baseline at a median of 8 days after initial treatment with vvDD (Figure 1b). The remaining three patients were not lymphopenic at last follow-up but did not recover their lymphocyte count to above their baseline measurement. Thirteen patients had an elevation in lactate dehydrogenase (LDH). In seven of these patients, a high LDH was present at baseline, however in all but one, LDH further increased after VV administration. In all patients, there was a trend toward increasing LDH compared to baseline with a median peak value at day 22 after VV administration (Figure 1c). LDH is known to be released by oncolytic virus infected and dying tumor cells, so it may indicate tumor necrosis.23 LDH returned to below baseline levels in three patients within 4 weeks, but high LDH did not correlate with the recovery of viral DNA or viral particles from noninjected tumors.
Figure 1.
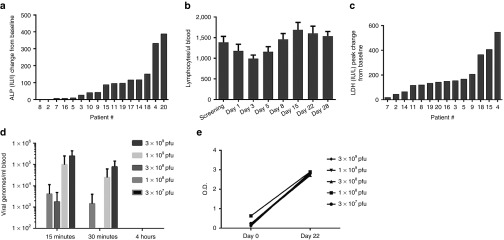
Toxicity of vvDD administration and acute pharmacokinetics. Blood samples were collected at various time points before and after virus administration. The enzyme activities in the serum and lymphocyte numbers in peripheral blood mononuclear cells were quantified. (a) Peak alkaline phosphatase levels (IU/l) relative to the baseline values before virus administration. (b) Median absolute lymphocyte count over time. (c) Lactate dehydrogenase peak levels (IU/l) relative to the baseline. (d) Recovery of serum vvDD DNA by qPCR at 15 minutes, 30 minutes, and 4 hours postinjection. The level of detection was 666 copies/ml and the level of quantification was 3333 copies/ml. At the 30-minute time point, viral DNA was detectable in patients treated at dose levels of 1.00E+08 and 3.00E+08 but below the level of quantification (at least 666 copies/ml but less than 3,333 copies/ml). This was also true at the 4-hour time point for patients treated at the two highest dose levels (1.00E+09 and 3.00E+09). The values displayed graphically represent serum vvDD DNA in copies/ml only in cases in which the detected level was greater than 3,333 copies/ml (the level of quantification). (e) Antibody response to viral administration demonstrating baseline and day 22 post-vvDD administration levels of anti-VV antibodies as measured by enzyme-linked immunosorbent assay in 12 of 17 patients. A 1.0 OD value represents a >50-fold induction of antibody.
Pharmacokinetic and pharmacodynamic endpoints
Acute pharmacokinetics. For pharmacokinetic (PK) analyses, qPCR was used to measure the vvDD genome concentration in serum 15 minutes, 30 minutes and 4 hours after injection. vvDD injections into tumors resulted in vvDD diffusion into the blood acutely, thus resulting in systemic distribution. vvDD DNA was more likely to be recovered in the serum within hours of injection at higher doses: 0/4 of patients at the 3 × 107 pfu dose, 1/3 of patients at the 1 × 108 pfu dose, 2/3 of patients at 3 × 108 pfu dose, and all patients at 1 × 109 pfu, and 3 × 109 pfu doses (Figure 1d; Table 3).
Maximum genome concentrations were observed 15 minutes postinjection (Figure 1d) and peak genome concentrations were dose-related. Concentrations decreased about 65% within 15 minutes.
Neutralizing Ab kinetics over time. Baseline anti-VV antibodies were detectable in all evaluable patients (five patients had insufficient samples) (Table 1), and a marked induction of anti-VV antibodies (neutralizing Ab) was detected in all patients evaluated in response to vvDD treatment, as analyzed by enzyme-linked immunosorbent assay (Figure 1e), and by neutralization assays (data not shown). The antibody concentration was greatly induced at 22 days after vvDD administration. The difference of 1.0 OD value represents the difference of at least 50-fold antibody induction. When averaged by dose cohort, there was between a 2.4 and 2.8 increase in OD value when anti-VV antibodies were measured at day 22 compared to baseline. Therefore, as expected, vvDD administration resulted in an induction of anti-VV antibodies.
vvDD shedding and recovery. Shedding was analyzed by qPCR and plaque assay (measuring infectious particles) from the urine and saliva on days 1, 3, 8, 15, and 22 following vvDD injection at all doses. No vvDD DNA or infectious particles were detected in urine or saliva at any time point.
Evidence for vvDD replication
Visible signs of replication. All patients with superficial cutaneous lesions receiving vvDD injection demonstrated classic vaccinia necrosum (Figure 2a,b) consistent with vvDD replication as described previously with vaccinia vaccination in the setting of immune compromised host,24 and in nonhuman primate studies.18
Figure 2.

Tumor selective infection, systemic spread, and clinical activity of vvDD in patients. (a) Evolution of response in patient #5 with breast cancer metastatic to the skin. vvDD replication leads to an ulcerated region encompassing only the tumor, which is resolving and scabbed over by day 28. (b) Another example of antitumor activity in patient #3 with breast cancer metastases to skin. A noninjected lesion 12 cm from the injected lesion became pustular and virus was recovered by plaque assay(*) on day 8. A similar lesion became pustular and resolved by day 28. (c) Patient #16 with metastatic colon cancer to the subcutaneous tissue and skin demonstrates significant erythema on day 8 at the two injected sites, consistent with active vvDD replication, and vvDD was recovered from the biopsy by plaque assay(*). A noninjected cutaneous lesion on the shoulder also became pustular and vvDD was recovered(*) on day 8. (D) In patient # 8, an omental mass of metastatic colon cancer was injected and a CT scan at day 30 revealed a markedly necrotic tumor and surrounding inflammation.
vvDD recovery from blood and biopsy samples. After initial systemic vvDD exposure postinjection, delayed re-emergence of vvDD genomes was detected in two patients. vvDD re-emergence in blood is consistent with replication at the tumor site and leakage of newly produced virus into the systemic circulation. vvDD DNA was detected in blood in two patients on day 5 following injection (1 × 108 and 3 × 109 pfu dose levels) (Table 3). Superficial tumors were biopsied in seven cases (patient's decision) on day 8, and replicating vvDD (by plaque assay measuring infectious particles) was recovered in all cases except the three patients at the lowest dose level (Table 3). When evaluated by qPCR, vvDD genomes were recovered in four out of five evaluated samples in injected lesions on day 8 post-treatment (Table 3). The patient without detectable vvDD by qPCR was treated at the lowest dose level.
vvDD spread to noninjected tumors. Patients #3 and #5 had superficial breast cancer metastases injected with 3 × 107 pfu vvDD and developed classic vaccinia necrosum lesions encompassing the injected tumors (Figure 2a,b). In patient #3, a pustule formed at the site of a tumor nodule 12 cm away from the injected lesion on day 8 (Figure 2b). Replicating vvDD was recovered by plaque assay from both the injected lesion and the noninjected lesion 12 cm away on the skin (Figure 2b). One colorectal cancer patient (patient #6) at the 1 × 109 pfu dose level had replicating vvDD by plaque assay recovered from two injected lesions and one noninjected lesion on day 8. This patient had a left lower quadrant abdominal wall mass and a right subcutaneous flank mass injected, but developed an inflamed right axillary lymph node (which was positive on biopsy for tumor) and an inflamed nodule on the right shoulder (Figure 2c). Replicating vvDD was recovered from both of the injected lesions and the right shoulder on day 8. This was evidence of systemic vvDD spread and secondary infection of new tumors.
Antitumor activity
Antitumor effects were seen in two of three patients (the nonresponder was treated at the lowest dose level) following injection of cutaneous lesions, including two patients with metastatic breast cancer to the skin. The lesions typically became erythematous by day 2, ulcerated by day 8, and scabbed over by day 28 (Figure 2a,b)—all consistent with the expected replicating virus effect and reminiscent of the evolution of live vaccinia vaccination lesions from Dryvax. One patient with an omental mass from metastatic colon cancer injected with 1 × 108 pfu vvDD, demonstrated an inflamed, necrotic response by CT scan on day 30 (Figure 2d).
In patient #3, two small nodules of breast cancer metastases were noted a distance of greater than 10 cm from the injected tumor, and both became pustular by day 8 after injection. One was excised as discussed above, demonstrating replicating vvDD, and the other was observed and resolved by 14 days post-treatment, representing effective treatment of a distant lesion by replicating vvDD having spread from the injected lesion (Figure 2b). Unfortunately, numerous other tumor nodules in the same patient did not become infected or respond.
Most patients were not evaluable by RECIST criteria, a more traditional measure of response, because all of their tumors were not measured over time.
Tumor specificity
There was no evidence for vvDD replication in normal tissues. When infection could be observed on cutaneous tumors, vvDD led to ulceration and necrosis of the tumor only, and at no time was normal tissue affected, demonstrating the remarkable vvDD selectivity for tumor tissue.
Evidence for immune cell activation
We analyzed peripheral blood mononuclear cells (PBMCs) collected at multiple time points by Phosflow analysis, which measures immune cell stimulation by monitoring phosphorylation of enzymes involved in proliferative signaling pathways. pERK, pS6, and Ki67 were upregulated in CD4 and CD8 T cells after vvDD treatment. Comparing the two highest dose levels, there exists a dose response, where the 3 × 109 pfu dose leads to increased pERK, PS6, and Ki67 in CD4 and CD8 cells compared to the 1 × 109 pfu dose (Figure 3a). Finally, serum was assayed for 13 chemokines and cytokines pre- and post-vvDD injection using a Luminex platform, and no significant patterns were observed.
Figure 3.

Immunologic effects of vvDD. (a) By phosflow analysis, pERK, pS6, and Ki67 were upregulated in circulating CD4 and CD8 T cells after vvDD injection in a dose-dependent manner. (b) Comparison of injected and noninjected tumor biopsies in compassionate use patient with melanoma on day 8 by qPCR for inflammatory markers. All proinflammatory markers are upregulated in the injected tumor, but the anti-inflammatory marker CCL22 is unchanged.
Compassionate use patient: clinical response, tumor specificity, and evidence of immune cell activation
Patient # 21, with metastatic melanoma received a clinical benefit from vvDD injection. The patient had a 19 × 11 cm tumor on the left arm causing significant dysfunction of the extremity and it was deemed unresectable without an amputation. An adjacent 2.5 cm tumor and 0.5 cm tumor on the left arm were both injected with 1.5 × 109 pfu vvDD and complete resolution of those tumors occurred (Figure 4a). The large noninjected tumor on the same arm had a significant reduction in size as well. Six weeks after the initial injection, a second injection of vvDD was performed into this large lesion (previously uninjected), and the lesion continued to shrink (Figure 4b). Finally, the tumor was able to be surgically resected with negative margins, and covered with a skin graft. Treatment results in this patient verified tumor specificity of vvDD as even a 1mm strip of skin between two responding lesions was spared in the patient with melanoma injected with 3 × 109 pfu (Figure 4a). The patient had unresectable lung metastases that did not respond and he succumbed to metastatic disease 11.3 months after treatment. We examined the tumor biopsies for infiltrating immune cell markers, cytokines, and chemokines by qPCR. While small biopsy samples limited these results overall, we were able to compare an injected and two noninjected lesions in this patient. The results by real-time PCR revealed elevations of proinflammatory chemokines (type-1 chemokines; known attractants for CTLs, NK, and Th1 cells) such as CCL5, CXCL9, CXCL10 and evidence of CD4 and CD8 T cell and NK cell infiltration in the vvDD-injected lesion (Figure 3b). In contrast, the Treg-attracting chemokine CCL22 was not elevated. At the time of resection (~3 months after the first vaccinia injection), pathologic examination revealed karyorrhectic as well as coagulative necrosis and fibrosis, consistent with regression (Figure 4c). No significant active immune response was identified, as the lymphocytic infiltrate was sparse. The specimen was not collected appropriately for detection of vaccinia viral DNA.
Figure 4.

Antitumor activity in melanoma (compassionate use patient). (a) Evolution of tumor response demonstrating ulceration caused by vvDD replication specific for the injected tumors, with intervening skin unaffected. The tumor scabs over by day 22 and completely heals by day 75. (b) A larger adjacent uninjected lesion also responded. The larger lesion was reinjected on day 42, and surgically resected 5 months after the beginning of treatment. (c) A representative hematoxalyin-eosin stained slide from tissue obtained at the time of resection (5 months after the first vaccinia injection) demonstrating karyorrhectic as well as coagulative necrosis and visible tumor nest surrounded by hyalinized fibrous tissue.
Discussion
This is a report of the first-in-human trial of vvDD, a WR oncolytic vaccinia virus with a combination of deletional mutations of genes encoding VGF and TK. This also represents the first clinical evaluation of an oncolytic VV not expressing GM-CSF. vvDD was administered by local injection into tumors to assess its direct oncolytic activity at the site of injection and to study the ability of vvDD to spread to noninjected tumors throughout the body in the setting of preformed antiviral immunity. vvDD was well-tolerated. No dose-limiting toxicities were observed, and an maximum feasible dose of 3 × 109 pfu was defined. Furthermore, we did not find any evidence of vvDD replication in normal tissue (based on the lack of observed toxicities and lack of infection of normal skin). We also did not find any evidence of vvDD shedding in saliva or urine at multiple time points after injection, suggesting that vvDD is not easily spread to human contacts. We had concern that toxicity might be more significant in patients who had not received prior vaccinia vaccination. In this study only one patient had not been vaccinated, and at the lowest dose level had no evidence of side effects whatsoever. In other patients, the duration since vaccination varied, however this did not correlate with toxicity, systemic vvDD spread, or antitumor activity in this phase 1 study.
Here we report evidence of vvDD replication in both injected and noninjected tumors. Tumor-selective replication was measured by visual inspection of cutaneous tumors, pfu or DNA recovery from biopsied tumor sites, delayed re-emergence of vvDD genomes in blood as well as fever and malaise occurring 1–2 weeks post-vvDD administration (consistent with timing of peak vvDD replication). In all biopsied patients receiving a dose of 1 × 108 pfu or higher, we were able to demonstrate evidence of vvDD replication in the injected lesions, and in two cases, evidence of local or systemic vvDD spread to noninjected tumors. An additional two patients had delayed re-emergence of vvDD genomes in blood. The injected, purified form of vvDD is predominately the intracellular mature virus form, which lacks the outer membrane that is partly derived from the plasma membrane. After in vivo infection, subsequent spread is secondary to newly produced vvDD particles which are released from the cell membrane of the host, the EEV form, which is relatively resistant to neutralization and complement destruction.12,13 Either this released progeny is responsible for the distant spread, or vvDD that escaped into the circulation at the time of initial injection was able to seed other systemic sites. One patient who had evidence of systemic vvDD spread, also had vvDD recovery from the circulation at early time points, suggesting that vvDD did escape into the circulation at the time of injection, and might lead to systemic uptake and replication. Future studies of intravenous vvDD delivery may differentiate the source of distant infection.
Unfortunately, true clinical benefit was not achieved in the patients treated on this phase 1 study, other than the melanoma patient who was able to have surgical resection after shrinkage of his extremity tumor. Also, none of the deep tumors injected had radiographic responses to treatment. Evidence of antitumor activity, however, was noted on this study, with all of the injected skin lesions treated at 1 × 108 pfu or greater undergoing ulceration and destruction similar to the dermonecrosis known to be induced by VV (note the effect was not painful and was limited to the tumor). Surrounding normal skin was never affected. As we extended the trial to include noncutaneous lesions, one 3 cm deep tumor (colorectal cancer mass in omentum) injected by ultrasound guidance also had a dramatic necrotic response. We were able to demonstrate evidence of response in multiple histologies, including breast cancer, colorectal cancer, and melanoma. For those deep tumors that did not respond, it is impossible to differentiate the reason for nonresponse, including the possibility of resistant cells (although we have not observed tumor cell resistance in vitro), inaccurate injection (ultrasound guided injection being much more challenging than injection of cutaneous tumors), or true response that could not be imaged (inflammation secondary to vvDD infection may falsely image as increased growth by CT scans). All patients enrolled on this phase 1 study had advanced tumors that were refractory to all available therapies prior to study entry. Therefore, we did not expect to observe elimination of tumor throughout the body and long term disease free survival in these patients. Indeed, all patients exhibited progression of bulky noninjected lesions, preventing long-term follow-up.
We cannot differentiate the direct replicative oncolysis of tumor from an immune response to the tumor. The responding cutaneous lesions were consistent with vaccinia dermonecrosis, indicating replicative virus tissue destruction. Unlike the only other clinical studies with replicating VV expressing GM-CSF (Pexa-Vec), vvDD did not express any immunologic adjuvant. In addition, our assays did not demonstrate a consistent systemic inflammatory chemokine milieu, nor a definitive immune response. In 1 patient, we demonstrated evidence of induction of circulating T-cell reactivity to CEA antigen (data not shown), suggesting an antitumor immune response was generated, but no systemic effect was evident. While investigators have described the importance of the immune response for oncolytic viral effect,25 we and others have found with VV that the antitumor effect is enhanced in the immunocompromised host or in conjunction with immunosuppression, consistent with the importance of the viral replication cytotoxicity and the negative effect of premature immune clearance of the virus.26,27 Previously, others have found that it is the case for oncolytic adenovirus and herpes simplex virus.28,29,30 Nevertheless, ultimately the immunologic effect of the viral tumor destruction may be the most efficient means of achieving systemic tumor clearance. We and others have examined ways of enhancing this effect through cytokine/chemokine expression, and immunologic adjuvant treatments. The fact that some tumor specific antigen immune reactivity is induced, suggests that this can be optimized with additional adjuvants which create an immunologically favorable tumor microenvironment.
In summary, we have multiple key findings in this study. First we have shown that vvDD (WR strain) exhibits exquisite tumor specificity in human cancer patients, with vvDD replication demonstrated in injected and non-injected tumors. Second, vvDD is well-tolerated, having reached a maximum feasible dose of 3 × 109 pfu. Finally, we have evidence of tumor response in multiple histologies without infection of normal tissue or systemic toxicity. One patient had a true clinical benefit where the tumor response converted an unresectable dominant arm melanoma into a resectable lesion which was completely removed with negative margins. As this was a first-in-man study of oncolytic vaccinia virus, the Food and Drug Administration required an intratumoral trial before proceeding to an intravenous trial. However, our next trial will examine the intravenous delivery of vvDD to determine the ability to achieve systemic infection of metastatic tumors as seen in the animal models. It may be that elimination of the intracellular mature virus form of vvDD by circulating antibodies and complement will prevent efficient systemic infection of metastatic tumors, but prior clinical studies with Pexa-Vec have suggested the feasibility of intravenous delivery.8 Multiple dosing regimens may also lead to improved antitumor effects, and should be considered in subsequent studies. Preclinical studies with immune stimulating transgenes and deletions leading to improved spreading of the virus have demonstrated promise, and these constructs may lead to more clinical activity in patients. We would also like to combine VV infection with transient immune suppression to enhance viral delivery and replication for destruction of larger tumors and more efficient vvDD systemic spread, as seen in the preclinical studies.26,31
Materials and Methods
Clinical trial
Patients. Over 26 months, 20 patients were enrolled in this phase 1 dose escalation trial. Four patients failed final screening; so, 16 patients were treated. One patient was treated identically to the previous patients at the highest dose level (3 × 109 pfu) as a compassionate exemption because he did not meet the inclusion criteria. He was on prednisone for severe arthritis and could not tolerate the 4 weeks off prednisone required by the protocol. The prednisone was discontinued 1 week prior to treatment. In order to enter the trial, patients had to be greater than 18 years of age with histologically-confirmed cancer that had progressed despite standard therapy and was not surgically curable. Patients had to have a Karnofsky performance status of ≥ 70 with an anticipated survival of at least 16 weeks. Patients were required to have adequate bone marrow function: WBC > 3,500, absolute neutrophil count > 1,500 cells/mm3, CD4 T cell count >350 per µl blood, hemoglobin > 10 g/dl, and platelet count > 150,000 cells/mm3. Patients had to have a serum creatinine level ≤ 1.2 × upper limit of normal and an international normalized ratio < 1.1 × upper limit of normal. Patients were not required to have evidence of neutralizing antibodies to vaccinia virus for inclusion in the study. Patients were excluded if they were pregnant or nursing an infant, had an active viral infection (including HIV, Hepatitis B and C), or used systemic corticosteroid or other immunosuppressive medication within 4 weeks of the study treatment. Any significant immunodeficiency (e.g., due to underlying illness and/ or medication) in subject or household contacts was also an exclusion criteria. Patients with a history of eczema requiring systemic therapy were also excluded due to the risk of systemic vaccinia and overwhelming skin infection. Also excluded were patients with unstable cardiac disease, patients whose target tumor(s) were adherent to a major vascular structure (e.g., carotid artery), subjects who received radiation, chemotherapy or other potentially immunosuppressive therapy within 4 weeks prior to study screening; patients with clinically significant and/or rapidly accumulating ascites, pericardial and/or pleural effusions, patients who experienced a severe systemic reaction or side-effect as a result of a previous smallpox vaccination.
The Declaration of Helsinki protocols were followed and all patients gave written informed consent. The study protocol and the consent forms were approved by the Recombinant Advisory Committee, the US Food and Drug Administration, the Institutional Review Board, the Institutional Protocol Review Committee, and the Institutional Biosafety Committee. An independent data-safety monitoring board reviewed all dose-escalation decisions and major safety assessments.
Viral production. vvDD-CDSR, also called JX-929 or vvDD, was constructed using homologous recombination of the cytosine deaminase and somatostatin receptor genes into the TK-locus of VSC20 (a VGF deleted WR strain of VV), which has been used in preclinical studies.20,32 While the potential for imaging with octreotide scans existed, it was not included in this trial due to a limited budget. Likewise, the enzyme/prodrug treatment with 5-fluorocytosine (5-FC) was possible but not undertaken in this phase 1 trial, where the determination of safety and efficacy of the viral backbone alone was the objective. The VGF gene had been previously deleted by inserting β-galactosidase gene into the VGF gene loci by homologous recombination. We subsequently selected (color selection after x-gal staining of plaques) for a spontaneous mutant of β-galactosidase, such that the β-galactosidase protein was not expressed. The TK and VGF inserts in the final product were both completely sequenced. Sequencing confirmed a deletional mutation of the β-galactosidase gene.
Trial material was generated according to Good Manufacturing Practice guidelines in Vero cells (American Type Culture Collection, Manassas, VA) and purified through sucrose-gradient centrifugation, by contract with Novovax (Rockville, MD). The genome-to-pfu ratio was about 50:1. vvDD-CDSR was formulated in phosphate-buffered saline with 10% glycerol, 138 mmol/l sodium chloride at pH 7.4. Final product quality control tests included assays for sterility, endotoxin, and potency. The clinical trial material was also tested for TK- and VGF- deletion. vvDD-CDSR was diluted in 0.9% bicarbonate buffered saline in a volume equivalent to 25% of the estimated total volume of target tumors.
Treatment. This was a phase 1, open-label, dose-escalation trial using a single dose group sequential dose-escalating design. Subjects were stratified into two groups, depending on a history of vaccination for smallpox (but not upon measured antibody titres). Only one patient (patient #1) was not known to be previously vaccinated, and was treated at the lowest dose level of 3 × 107 pfu. All other patients were previously vaccinated against smallpox and were treated with a standard dose escalation format, starting at 3 × 107 pfu, then 1 × 108 pfu, 3 × 108 pfu, 1 × 109 pfu, and ending at 3 × 109 pfu (the maximum feasible dose given the current concentration limit of VV purification). Three patients were treated at each dose level, and 4 weeks were allowed prior to dose escalation. The maximum tolerated dose was defined as the dose level at which < 2 out of six patients had a dose limiting toxicity.
Subjects were admitted to the Clinical and Translational Research Center Montefiore University Hospital for at least 24 hours on injection days. Subjects were given a physical exam and vital signs were monitored for 24 hours postinjection. For cutaneous tumor injection, the total dose could be divided between 1–3 lesions, using four equally spaced needle tracts per tumor radiating out from a central puncture site (extending to within 2–3 millimeters of edge of tumor). Deep tumors were injected using ultrasound guidance and a straight needle or a multipronged QuadraFuse needle (Rex Medical, Conshohocken, PA) with a variable tine deployment of 1, 2, 3, 4, and 5 cm diameters. The injections were performed while retracting the needle and the needle was rotated once to maximize vvDD distribution throughout the tumor. The strictly defined injection protocol allows for consistency of treatment, but for deep injections, the radiologic guided injections are operator dependent and may be subject to variability. A nonocclusive dressing was kept on the skin overlying the tumor injection site at all times during the study.
Pharmacokinetic blood draws were obtained at 15 minutes, 30 minutes, 60 minutes, 3–4 hours, and 4–6 hours postinjection. Patients with superficial tumors could undergo a punch biopsy of the injected tumor and a control noninjected tumor on day 8 following injection (patient's choice). Patients with deep tumors were not requested to undergo biopsies. Blood, saliva, and urine samples were collected on days 1, 4, 8, 14, 21, and 28 following vvDD injection. Tumor response was assessed by direct measurements of superficial skin tumors at each follow-up time. Deep lesions were assessed by contrast enhanced computed tomography scan (unless contraindicated), comparing pretreatment scans and day 28 scans. If there was no evidence of progression, a repeat scan was obtained at day 90, and every three months thereafter.
Laboratory analysis
vvDD titers. The tissues were homogenized using a FastPrep Cell Disrupter (Model FP120; Qbiogene, Carlsbad, CA) to release virions, and the infectious viruses in the resulting cell lysates were used to infect CV-1 cells (American Type Culture Collection) and titres determined by standard plaque assays.17,31
qPCR analysis for vvDD genomes. Quantitative PCR (Q-PCR) was used to measure vvDD genomes in blood serially because of its reproducibility and ability to detect and quantitate genome copies, irrespective of antibody or complement neutralization (or both). vvDD DNA was purified from samples by use of QIAamp DNA Blood Mini Kit (Qiagen GmbH, Hilden, Germany). Q-PCR was performed as described elsewhere.7,8,33 The lower limit of vvDD detection was 666 copies per milliliters of plasma and the limit of vvDD quantitation (i.e., measurable copy numbers) was 3,333 copies per milliliters of plasma. To detect shedding of infectious vvDD into the environment, a plaque-forming assay was used. Infectious shedding of viral units would have public health consequences. Urine and saliva samples were centrifuged at 80 g, resuspended in 10 mmol/l Tris (pH 8.0), and plaque assays were performed. The detection limit was 20 pfu/ml.
Neutralizing antibody titers to vaccinia virus. This procedure is based on the ability of neutralizing antibodies in patient serum samples to reduce the cytopathic effect caused by live VV as described by Breitbach et al.8 Serum samples obtained at baseline (day 0) and on day 22 were heat-inactivated, serially diluted in 96-well format (dilution factor 10–20,480) and incubated with VV for 2 hours before transfer of the mixture onto monolayers of A2780 cells. Cell viability was measured 3 days after inoculation by a colorimetric assay based on live-cell-mediated reduction of tetrazolium salt to formazan (Cell Counting Kit-8, Donjindo Laboratories, Kumamoto, Japan). The neutralizing antibody titer was defined as the reciprocal of the highest dilution of serum that results in ≥50% cell viability.
The analysis of the total antibody against VV in the serum samples was performed using an enzyme-linked immunosorbent assay.
TaqMan qPCR analysis of mRNA levels in tumor tissues and PBMCs. PBMCs or biopsied tissues were placed in Lysing Matrix E Tubes (MP Biomedicals, Santa Ana, CA) containing RLT buffer from RNeasy kit (Qiagen GmbH), and agitated using a FP120 homogenizer (MP Biomedicals). Debris-free supernatant from the lysis matrix tubes were transferred into new tubes. The total RNA was extracted using the RNeasy kit. Usually, one microgram of total RNA was used for cDNA synthesis, and 25–50 ng of cDNA was used for TaqMan PCR analysis for levels of individual mRNAs on the StepOnePlus Real-Time PCR system (Applied Biosystems, Life Technologies, Grand Island, NY), as we have described previously.34 The TaqMan Gene Expression Assays for individual genes including primers were obtained from Applied Biosystems.
Detection of cytokine-producing immune cells by ELISPOT assay. Ag-specific interferon (IFN)-γ-producing cells were detected by an ELISPOT assay. PBMCs from human patients were used directly in ELISPOT assay or were cultured in Iscove's Modified Dulbecco's Medium supplemented with 5% AB human serum in the presence of human IL-2 (20 U/ml) (Chiron, Emeryville, CA) or both IL-2 and one of the antigen proteins (CEA; MyBiosource, San Diego, CA) or survivin (Abcam, Cambridge, MA; 10 µg/ml) for a total of 11 days. For the ELISPOT assay, cells were plated at 1e5 cells per well in triplicates in Millipore (Billerica, MA) Multiscreen 96-well plate coated with anti IFN-γ 1-D1K mAb (Mabtech, Cincinnati, OH) and stimulated for 48 hours with either CEA or survivin proteins (10 µg/ml). Spots were detected with anti IFN-γ 7-B6-1 biotin mAb (Mabtech), Vectastain ABC kit and AEC peroxidase substrate (both from Vector Laboratories, Burlingame, CA). Spots were enumerated by using ImmunoSpot analyzer (Cellular Technologies, Shaker Heights, OH) and the frequency of responding cells was determined. Wells without Ag were included as controls and did not yield cytokine-producing spots.
Phosflow analysis of PBMC. Fixed and permeabilized cells were prepared for flow cytometric analysis as previously described.35 The following Abs were used (purchased from BD Biosciences, San Jose, CA unless otherwise noted): Ki67 (B56), CD4 (RM4-5), CD8, phospho-ERK1/2 (Thr202/Tyr204), phospho-p70 S6 Kinase (Ser371) (Cell Signaling Technology, Danvers, MA). Note that all Ab concentrations were titrated for optimal staining in a 100-ul staining volume with no more than 4 × 106 methanol-permeabilized cells per sample.
Statistical analysis. The study sample size was set to assess safety issues. The primary objectives were to study the safety and maximum tolerated dose of vvDD. Secondary objectives included pharmacokinetics, replication and shedding (urine, throat swabs), immune responses (neutralizing antibodies, cytokines, chemokines, and antitumor immune recognition) and tumor responses. The likelihood of dose escalation, given varying true dose-limiting toxicities in the treated population, was calculated as is routine in phase 1 dose-escalation trials. Expected sample size was 15–21 patients.
Acknowledgments
Bernard Moss, MD, PhD (Chief, Laboratory of Viral Diseases, National Institute of Health) provided advice in designing tumor selective mutations in the WR strain of vaccinia virus. Ravi R. Muthuswamy, PhD (University of Pittsburgh, Department of Surgery) contributed to the analysis of data by performing TaqMan qPCR analysis of mRNA levels in tumor tissues and PBMCs. Roshni Ravindranathan (University of Pittsburgh, Department of Surgery) contributed to analysis of clinical samples by performing plaque assays for viral recovery. Conflicts of interest include a consultancy to Jennerex Biotherapeutics by D.L.B. from 2001–2013. He also holds a patent for Combined growth factor-deleted and thymidine kindase-deleted vaccinia virus vector (#US 20030031681 A1). S.H.T. was a consultant to Jennerex Biotherapeutics from 2006–2012. M.D., T.H.H., A.M., D.H.K. are Sillajen (formerly Jennerex) consultants. C.J.B. is a Sillajen employee. J.C.B. was a cofounder of Jennerex Biotherapeutics. This project was supported by award numbers P01CA132714, R01CA155925, and T32CA113263 from the National Cancer Institute. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Cancer Institute or the National Institutes of Health. This work was also partially funded by generous support from Valerie Koch and the New Era Cap Company.
References
- Martuza RL, Malick A, Markert JM, Ruffner KL, Coen DM. Experimental therapy of human glioma by means of a genetically engineered virus mutant. Science. 1991;252:854–856. doi: 10.1126/science.1851332. [DOI] [PubMed] [Google Scholar]
- Russell SJ, Peng KW, Bell JC. Oncolytic virotherapy. Nat Biotechnol. 2012;30:658–670. doi: 10.1038/nbt.2287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Thorne SH, Bartlett DL. Oncolytic virotherapy: molecular targets in tumor-selective replication and carrier cell-mediated delivery of oncolytic viruses. Biochim Biophys Acta. 2008;1785:217–231. doi: 10.1016/j.bbcan.2008.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirn D. Clinical research results with dl1520 (Onyx-015), a replication-selective adenovirus for the treatment of cancer: what have we learned. Gene Ther. 2001;8:89–98. doi: 10.1038/sj.gt.3301377. [DOI] [PubMed] [Google Scholar]
- Nemunaitis J, Cunningham C, Buchanan A, Blackburn A, Edelman G, Maples P, et al. Intravenous infusion of a replication-selective adenovirus (ONYX-015) in cancer patients: safety, feasibility and biological activity. Gene Ther. 2001;8:746–759. doi: 10.1038/sj.gt.3301424. [DOI] [PubMed] [Google Scholar]
- Andtbacka RHI, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, et al. 2013OPTiM: a randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment of unresected stage IIIB/C and IV melanoma J Clin OncolASCO Annual Meeting Abstracts. Vol. 31, No. 18_suppl (June 20 supplement), 2013: LBA9008.
- Park BH, Hwang T, Liu TC, Sze DY, Kim JS, Kwon HC, et al. Use of a targeted oncolytic poxvirus, JX-594, in patients with refractory primary or metastatic liver cancer: a phase I trial. Lancet Oncol. 2008;9:533–542. doi: 10.1016/S1470-2045(08)70107-4. [DOI] [PubMed] [Google Scholar]
- Breitbach CJ, Burke J, Jonker D, Stephenson J, Haas AR, Chow LQ, et al. Intravenous delivery of a multi-mechanistic cancer-targeted oncolytic poxvirus in humans. Nature. 2011;477:99–102. doi: 10.1038/nature10358. [DOI] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tolonen N, Doglio L, Schleich S, Krijnse Locker J. Vaccinia virus DNA replication occurs in endoplasmic reticulum-enclosed cytoplasmic mini-nuclei. Mol Biol Cell. 2001;12:2031–2046. doi: 10.1091/mbc.12.7.2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moss B, Earl PL. Overview of the vaccinia virus expression system. Curr Protoc Mol Biol. 2002;Chapter 16:Unit16.15. doi: 10.1002/0471142727.mb1615s60. [DOI] [PubMed] [Google Scholar]
- Ichihashi Y. Extracellular enveloped vaccinia virus escapes neutralization. Virology. 1996;217:478–485. doi: 10.1006/viro.1996.0142. [DOI] [PubMed] [Google Scholar]
- Vanderplasschen A, Mathew E, Hollinshead M, Sim RB, Smith GL. Extracellular enveloped vaccinia virus is resistant to complement because of incorporation of host complement control proteins into its envelope. Proc Natl Acad Sci U S A. 1998;95:7544–7549. doi: 10.1073/pnas.95.13.7544. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roberts KL, Smith GL. Vaccinia virus morphogenesis and dissemination. Trends Microbiol. 2008;16:472–479. doi: 10.1016/j.tim.2008.07.009. [DOI] [PubMed] [Google Scholar]
- Thorne SH, Hwang TH, O'Gorman WE, Bartlett DL, Sei S, Kanji F, et al. Rational strain selection and engineering creates a broad-spectrum, systemically effective oncolytic poxvirus, JX-963. J Clin Invest. 2007;117:3350–3358. doi: 10.1172/JCI32727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Puhlmann M, Brown CK, Gnant M, Huang J, Libutti SK, Alexander HR, et al. Vaccinia as a vector for tumor-directed gene therapy: biodistribution of a thymidine kinase-deleted mutant. Cancer Gene Ther. 2000;7:66–73. doi: 10.1038/sj.cgt.7700075. [DOI] [PubMed] [Google Scholar]
- McCart JA, Ward JM, Lee J, Hu Y, Alexander HR, Libutti SK, et al. Systemic cancer therapy with a tumor-selective vaccinia virus mutant lacking thymidine kinase and vaccinia growth factor genes. Cancer Res. 2001;61:8751–8757. [PubMed] [Google Scholar]
- Naik AM, Chalikonda S, McCart JA, Xu H, Guo ZS, Langham G, et al. Intravenous and isolated limb perfusion delivery of wild type and a tumor-selective replicating mutant vaccinia virus in nonhuman primates. Hum Gene Ther. 2006;17:31–45. doi: 10.1089/hum.2006.17.31. [DOI] [PubMed] [Google Scholar]
- Parato KA, Breitbach CJ, Le Boeuf F, Wang J, Storbeck C, Ilkow C, et al. The oncolytic poxvirus JX-594 selectively replicates in and destroys cancer cells driven by genetic pathways commonly activated in cancers. Mol Ther. 2012;20:749–758. doi: 10.1038/mt.2011.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chalikonda S, Kivlen MH, O'Malley ME, Eric Dong XD, McCart JA, Gorry MC, et al. Oncolytic virotherapy for ovarian carcinomatosis using a replication-selective vaccinia virus armed with a yeast cytosine deaminase gene. Cancer Gene Ther. 2008;15:115–125. doi: 10.1038/sj.cgt.7701110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang E, Chalikonda S, Friedl J, Xu H, Phan GQ, Marincola FM, et al. Targeting vaccinia to solid tumors with local hyperthermia. Hum Gene Ther. 2005;16:435–444. doi: 10.1089/hum.2005.16.435. [DOI] [PubMed] [Google Scholar]
- Gnant MF, Puhlmann M, Bartlett DL, Alexander HR., Jr Regional versus systemic delivery of recombinant vaccinia virus as suicide gene therapy for murine liver metastases. Ann Surg. 1999;230:352–60; discussion 360. doi: 10.1097/00000658-199909000-00008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanderson CM, Parkinson JE, Hollinshead M, Smith GL. Overexpression of the vaccinia virus A38L integral membrane protein promotes Ca2+ influx into infected cells. J Virol. 1996;70:905–914. doi: 10.1128/jvi.70.2.905-914.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aragón TJ, Ulrich S, Fernyak S, Rutherford GW. Risks of serious complications and death from smallpox vaccination: a systematic review of the United States experience, 1963-1968. BMC Public Health. 2003;3:26. doi: 10.1186/1471-2458-3-26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartlett DL, Liu Z, Sathaiah M, Ravindranathan R, Guo Z, He Y, et al. Oncolytic viruses as therapeutic cancer vaccines. Mol Cancer. 2013;12:103. doi: 10.1186/1476-4598-12-103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo ZS, Parimi V, O'Malley ME, Thirunavukarasu P, Sathaiah M, Austin F, et al. The combination of immunosuppression and carrier cells significantly enhances the efficacy of oncolytic poxvirus in the pre-immunized host. Gene Ther. 2010;17:1465–1475. doi: 10.1038/gt.2010.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weibel S, Raab V, Yu YA, Worschech A, Wang E, Marincola FM, et al. Viral-mediated oncolysis is the most critical factor in the late-phase of the tumor regression process upon vaccinia virus infection. BMC Cancer. 2011;11:68. doi: 10.1186/1471-2407-11-68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeda K, Ichikawa T, Wakimoto H, Silver JS, Deisboeck TS, Finkelstein D, et al. Oncolytic virus therapy of multiple tumors in the brain requires suppression of innate and elicited antiviral responses. Nat Med. 1999;5:881–887. doi: 10.1038/11320. [DOI] [PubMed] [Google Scholar]
- Thomas MA, Spencer JF, Toth K, Sagartz JE, Phillips NJ, Wold WS. Immunosuppression enhances oncolytic adenovirus replication and antitumor efficacy in the Syrian hamster model. Mol Ther. 2008;16:1665–1673. doi: 10.1038/mt.2008.162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hasegawa N, Abei M, Yokoyama KK, Fukuda K, Seo E, Kawashima R, et al. Cyclophosphamide enhances antitumor efficacy of oncolytic adenovirus expressing uracil phosphoribosyltransferase (UPRT) in immunocompetent Syrian hamsters. Int J Cancer. 2013;133:1479–1488. doi: 10.1002/ijc.28132. [DOI] [PubMed] [Google Scholar]
- Thirunavukarasu P, Sathaiah M, Gorry MC, O'Malley ME, Ravindranathan R, Austin F, et al. A rationally designed A34R mutant oncolytic poxvirus: improved efficacy in peritoneal carcinomatosis. Mol Ther. 2013;21:1024–1033. doi: 10.1038/mt.2013.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCart JA, Mehta N, Scollard D, Reilly RM, Carrasquillo JA, Tang N, et al. Oncolytic vaccinia virus expressing the human somatostatin receptor SSTR2: molecular imaging after systemic delivery using 111In-pentetreotide. Mol Ther. 2004;10:553–561. doi: 10.1016/j.ymthe.2004.06.158. [DOI] [PubMed] [Google Scholar]
- Kulesh DA, Baker RO, Loveless BM, Norwood D, Zwiers SH, Mucker E, et al. Smallpox and pan-orthopox virus detection by real-time 3'-minor groove binder TaqMan assays on the roche LightCycler and the Cepheid smart Cycler platforms. J Clin Microbiol. 2004;42:601–609. doi: 10.1128/JCM.42.2.601-609.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muthuswamy R, Berk E, Junecko BF, Zeh HJ, Zureikat AH, Normolle D, et al. NF-κB hyperactivation in tumor tissues allows tumor-selective reprogramming of the chemokine microenvironment to enhance the recruitment of cytolytic T effector cells. Cancer Res. 2012;72:3735–3743. doi: 10.1158/0008-5472.CAN-11-4136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schulz KR, Danna EA, Krutzik PO, Nolan GP. Single-cell phospho-protein analysis by flow cytometry. Curr Protoc Immunol. 2007;Chapter 8:Unit 8.17. doi: 10.1002/0471142735.im0817s78. [DOI] [PubMed] [Google Scholar]