

Abstract
Despite the proven safety of oncolytic viruses (OV) in clinical trials for glioblastoma (GBM), their efficacy has been hindered by suboptimal spreading within the tumor. We show that hyaluronan or hyaluronic acid (HA), an important component of extracellular matrix (ECM), is highly expressed in a majority of tumor xenografts established from patient-derived GBM lines that present both invasive and nodular phenotypes. Intratumoral injection of a conditionally replicating adenovirus expressing soluble hyaluronidase (ICOVIR17) into nodular GBM, mediated HA degradation and enhanced viral spread, resulting in a significant antitumor effect and mice survival. In an effort to translate OV-based therapeutics into clinical settings, we encapsulated human adipose-derived mesenchymal stem cells (MSC) loaded with ICOVIR17 in biocompatible synthetic extracellular matrix (sECM) and tested their efficacy in a clinically relevant mouse model of GBM resection. Compared with direct injection of ICOVIR17, sECM-MSC loaded with ICOVIR17 resulted in a significant decrease in tumor regrowth and increased mice survival. This is the first report of its kind revealing the expression of HA in GBM and the role of OV-mediated HA targeting in clinically relevant mouse model of GBM resection and thus has clinical implications.
Introduction
Glioblastoma (GBM) is the most common and aggressive primary brain tumor in adults, with median survival of 14.6 months despite aggressive multimodality treatments that include surgical resection, radiotherapy, and chemotherapy.1 The failure of these current therapies can be explained by the resistance and disseminated nature of these tumors. Therefore, the development of novel therapies is urgently needed in order to increase patients' survival.
Oncolytic virotherapy for cancer is a novel approach in which viruses are modified to preferentially replicate in tumor cells and selectively destroy them. Among different oncolytic virus (OV) types, such as Herpes Simplex Virus, Myxoma virus, or Vaccinia virus, oncolytic adenovirus is a promising therapy for different tumor types including GBMs.2,3 During the past decade, a number of engineered oncolytic adenoviruses have been used in early phase 1 and phase 2 clinical trials in GBM patients showing signs of antitumor activity and safety.3,4,5,6 However, the clinical response rates have been suboptimal mainly due to the inefficient viral spread in the tumor mass.4 This may be partially explained by the fact that human GBM tumors, like other types of tumors, contain multiple barriers to viral spread that represent hurdles for successful viral-mediated tumor eradication. The presence of high amounts of extracellular matrix (ECM) and high interstitial fluid pressure in the tumor interstitium are the main source of physical resistance to therapeutic agents. Hyaluronan or hyaluronic acid (HA), one of the most important structural elements of ECM, is involved in tumor development, proliferation, invasion, and therapeutic resistance to chemo- and radiotherapy through the binding to CD44 receptor.7,8 Also, the expression of HA presents a physical barrier that limits viral spreading within the tumor mass.9 The degradation of HA has been shown to induce antitumor effect in breast cancer and also permit anticancer agents to reach malignant cells.10 Several studies have suggested that GBMs also express high amounts of HA, and this expression may be associated with poor prognosis in GBM patients.7,8 Therefore, degradation of HA could pave a way to improve therapeutic efficacy of antitumor agents in GBMs. In this study, we assessed the levels of HA expressed by established and patient-derived GBM cancer stem cell (CSC) lines11,12 and the intracranial xenografts derived from these GBM lines. CSCs are defined as tumor-initiating cancer cell subpopulations that possess stem cell–like properties and resist chemotherapy and radiotherapy, causing tumor relapse. Recent publications have shown that oncolytic viruses can target unique CSC signaling pathways and are promising therapeutic agents against CSCs.13 We also tested the ability of ICOVIR17, an oncolytic adenovirus expressing a soluble form of PH20 hyaluronidase,9 to degrade HA and efficiently spread in the tumor mass.
The standard procedure in oncolytic adenovirus clinical trials for GBM involves direct injections of purified virus into brain parenchyma adjacent to the tumor resection cavity after tumor debulking.4,14 This surgical intervention induces secondary bleeding and influx of cerebrospinal fluid into the resection cavity that may result in rinsing out and dispersing injected virus and contribute to inefficient delivery of oncolytic viruses in clinical trials.15,16 Stem cells (SC) have been explored as vehicles to deliver antitumor agents including oncolytic viruses to brain tumors since they have been shown to migrate preferentially toward tumor cells. SC can improve the therapeutic efficacy of viral therapy by enhancing the targeting specificity of oncolytic adenovirus to the localized tumor environment, increasing the viral payload and protecting the virus from attack by the immune system.17
Biodegradable synthetic ECM (sECM) have been used in various rodent models owing to their ability to provide a physiologic environment that promotes SC survival while permitting easy in vivo transplantation and cell retention.18,19 Three-dimensional ECM collagen-based (3-DECM) purified from tissue-engineered skin cultures has also shown the potential to increase transplantation efficiency of SC by reducing metabolic stress and providing mechanical support, especially in the surgical resection cavity after brain tumor removal.20 We have recently shown that the use of sECM, based on two biocompatible liquid components: thiol-modified hyaluronan and a thiol-reactive crosslinker, polyethylene glycol diacrylate (PEGDA)21,22 that solidify when they are mixed, provides biocompatibility and customizability to encapsulate therapeutic SC and also significantly improves the survival and therapeutic efficacy of SC in the tumor resection cavity in mouse model of GBM resection.23,24 In this study, we loaded human mesenchymal stem cells (MSC) with ICOVIR17 and explored the dynamics of ICOVIR17-loaded MSC in real time in vitro and in vivo, in malignant GBM mice models. We then assessed the therapeutic efficacy of sECM encapsulated MSC-ICOVIR17 compared with direct ICOVIR17 injection in our mouse resection model which more accurately reflects the current clinical setting which involves GBM tumor resection as standard treatment.
Results
Expression of HA in established and patient-derived GBMs
HA is highly expressed in many cancers such as lung, breast, colon, kidney, and prostate and is associated with poor prognosis.7 To determine if HA expression levels are also increased in GBMs, three established and eight patient-derived (three nodular and five invasive) GBM lines were intracranially implanted in mice, and tumor formation and HA production in GBM xenografts was evaluated by hematoxylin and eosin and histochemical staining, respectively.11 For each xenografts, an adjacent section pretreated with soluble hyaluronidase served as negative staining control (Figure 1a). We showed that U87 and U373vIII GBM lines express high levels of HA, while Gli36vIII expresses almost no detectable level of HA (Figure 1b,e). Interestingly, most of the patient-derived GBMs, either nodular (Figure 1c,e) or invasive (Figure 1d,e), express high amounts of HA. Compared to the HA near-negative Gli36vIII tumor model, HA expression was significantly increased in all the other GBM xenografts. U87 (established GBM) and GBM4 (patient-derived GBM), which produce high amounts of HA in vitro (Supplementary Figure S1) and in vivo (Figure 1b,c), were selected for subsequent in vivo studies. As expected, the high expression of HA was associated with GBM tumor mass and was not detected either in the normal brain (Supplementary Figure S2a) or tumor blood vessels (Supplementary Figure S2b). These results determine that HA is highly expressed in xenografts established from most of the patient-derived GBMs, irrespective of tumor invasiveness.
Figure 1.
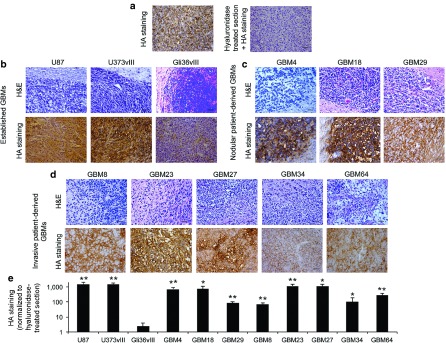
Expression of hyaluronic acid (HA) in established and patient-derived GBM xenografts. (a–d) GBM xenografts were generated by intracranial implantation of different established or patient-derived GBM lines in mice. (a) Evaluation of HA levels in established U87 by histochemical analysis using the biotinylated hyaluronan-binding protein (HABP-b) in untreated (left) and pretreated tumor section with soluble hyaluronidase (right). (b–e) Evaluation of tumor formation (hematoxylin and eosin staining) and HA levels (histochemical staining) from brain sections within (b) different established, (c) nodular patient-derived, (d) and invasive patient-derived GBM xenografts. (e) Plot showing HA staining quantification normalized to the staining measured in hyaluronidase-treated tumor sections. Bar = 50 µm. Mean + SE (n = 3) is plotted. **P < 0.01; *P < 0.05 versus Gli36vIII HA-negative tumor model (t-test, two-sided).
Antitumor effect of ICOVIR17 in an established HA-expressing GBM model
Degradation of HA has been shown to improve antitumor therapies.10 To investigate the effect of HA degradation in GBMs, we used ICOVIR17, a conditionally replicating adenovirus armed with a soluble PH20 hyaluronidase. A significant reduction in tumor volume was seen in mice bearing intracranial U87 GBM tumors engineered to express Fluc mCherry (FmC) after intratumoral treatment with purified ICOVIR15 (P < 0.001) or ICOVIR17 (P < 0.001) compared with phosphate-buffered saline (PBS) treatment (Figure 2a). Furthermore, survival studies showed a significant difference in median survival between ICOVIR15-treated (30 days) and ICOVIR17-treated animals (58 days; P = 0.033; Figure 2b). Immunofluorescent detection of viral capsid proteins on brain sections of treated mice revealed the presence of wider areas of ICOVIR17 replication in the tumor tissue compared to ICOVIR15-treated mice (Figure 2c,d). The better distribution of the oncolytic virus ICOVIR17 was associated with a decrease in HA levels in the tumor (Figure 2e,f). These results demonstrate that intratumoral injection of ICOVIR17 targets HA, improves viral spreading in the tumor mass, and results in a significant reduction of GBM tumor growth.
Figure 2.
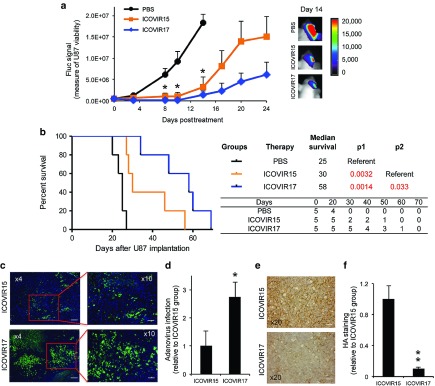
Anti-tumor effect of ICOVIR17 in established HA-expressing GBMs. (a–d) Mice bearing established intracranial U87-FmC tumors were treated with intratumoral injection of PBS, purified ICOVIR15, or purified ICOVIR17. (a) Plot showing changes in Fluc activity as a measure of tumor growth monitored over time. A representative bioluminescence image from each group at day 14 after treatment is shown. (b) Kaplan–Meier survival curves of treated mice. Overall survival time was compared between groups. p1 represents P value when PBS-treated group is used as reference and p2 represents P value when ICOVIR15 group serves as a reference. The number of mice at risk is shown below the graph. (c–f) Analysis of HA expression and adenovirus distribution in U87 tumors in the different treatment groups at day 24 posttreatment. (c) Adenovirus immunodetection and (e) HA staining was performed on brain sections from ICOVIR15 or ICOVIR17 treated groups. (c) Representative images from adenovirus staining are shown. (d) Plot showing quantification of adenovirus staining. (e) Representative images from HA staining are shown. (f) Plot showing quantification of HA levels. In all panels, Bars, +SE (n = 5) and *P < 0.05 ICOVIR15 and ICOVIR17 versus PBS control; P = 0.15 ICOVIR17 versus ICOVIR15 at day 24 (t-test, two-sided). Bar = 200 µm in c left, 100 µm in c right, and 50 µm in e.
ICOVIR17 production and therapeutic effect of MSC loaded with ICOVIR17 in established and patient-derived GBM cells
To determine whether human adipose derived MSC (MSC) have the capability of delivering ICOVIR17 to GBMs, we first studied ICOVIR17 production in MSC in vitro. Upon infection of MSC, both ICOVIR15 and ICOVIR17 showed similar replication kinetics, reaching the peak of viral production at day 7, with the ICOVIR15 yields slightly higher than those of ICOVIR17 (Figure 3a). Expression of hyaluronidase in MSC infected with ICOVIR17 at multiplicity of infections = 7 and 70 (plaque forming unit (PFU)/cell) was detected at days 3 and 5 postinfection (Figure 3b). To assess ICOVIR17 production in MSC in vivo, U87-FmC established tumors were treated intratumorally with MSC-GFP-ICOVIR17, and viral production and spread was monitored over time. Adenovirus staining in tumor sections revealed an increase in virus-positive areas within the tumor mass over time (Figure 3c–e). To monitor MSC and GBM viability in vitro and in vivo, we created MSC expressing a bimodal imaging marker GFP Rluc and human GBM lines expressing FmC by transducing MSC or GBM cells with LV-GFP-Rluc or LV-FmC, respectively (Figure 3f). MSC-GFP-Rluc infected with ICOVIR17 or ICOVIR15 were then cocultured with U87-FmC. A decrease in MSC cell viability was seen over time (Figure 3g) followed by a reduction in GBM viability (Figure 3h), suggesting the infection and subsequent killing of GBM cells by viral progeny released from MSC. Similar experiments performed with other types of human stem cells, such as bone marrow MSC and umbilical cord blood cells, cocultured with U87 (Supplementary Figure S3a) or LN229 (Supplementary Figure S3b) GBM cells, showed comparable results. Cocultures of different proportions of MSC-ICOVIR17 with a broad spectrum of established (Figure 3i and Supplementary Figure S4a) and patient-derived (Figure 3j and Supplementary Figure S4b) GBMs showed that 2.5% of MSC-ICOVIR17 or MSC-ICOVIR15 was sufficient to drastically reduce GBM viability in vitro. Importantly, MSC-ICOVIR17 and MSC-ICOVIR15 showed similar antitumor effects, suggesting that the incorporation of the hyaluronidase gene in ICOVIR17 does not reduce its oncolytic potency in vitro. These results reveal that MSC-ICOVIR17 effectively produces ICOVIR17 in vitro and in vivo, which results in a decrease in tumor viability in established and patient-derived GBMs in vitro.
Figure 3.
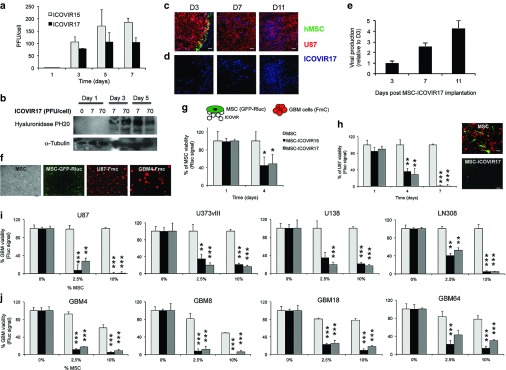
ICOVIR17 production and therapeutic effect of MSC loaded with ICOVIR17 in established and patient-derived GBM cells. (a) Human adipose stem cells (MSC) were infected with ICOVIR15 or ICOVIR17, and cells were collected at different time points. Plot showing virus production. (b) Western blotting analysis shows increased expression over time of hyaluronidase PH20 in MSC infected with ICOVIR17 at different multiplicity of infections (7 or 70 PFU/cell). (c,d) Mice bearing established intracranial U87-FmC tumors were treated with MSC expressing GFP loaded with ICOVIR17. Representative images of brain sections showing MSC in green, (c) U87 in red and (d) adenovirus staining in blue over time are shown. (e) Plot showing quantification of adenovirus staining areas over time. Bars, +SE (n = 2). (f) MSC and GBMs were engineered to express GFP-Rluc or Fluc-mCherry fusion markers, respectively. Photomicrograph showing MSC, modified MSC-GFP-Rluc, established GBM cell line U87-FmC, and patient-derived cell line GBM4-Fmc. Original magnifications: ×4. (g,h) MSC-GFP-Rluc were infected with ICOVIR15 or ICOVIR17 and cocultured with U87-FmC in a ratio of 1:10. Plots showing changes in (g) Rluc activity as a measure of MSC viability and (h) Fluc activity as a measure of tumor viability monitored over time. Photomicrographs showing cocultures of MSC-GFP-Rluc or MSC-GFP-Rluc-ICOVIR17 with U87-FmC at day 7. (i,j) A panel of (i) established and (j) patient-derived GBM lines expressing FmC were cocultured with increasing proportions of MSC, MSC-ICOVIR15, or MSC-ICOVIR17. Plots showing changes in Fluc signal as a measure of tumor viability. In all panels, Bars, +SD (n = 3). ***P < 0.001; **P < 0.01; *P < 0.05 versus MSC control (t-test, two-sided). Bar = 100 µm in c,d, 200 µm in f, and 100 µm in h.
Antitumor effect of MSC-ICOVIR17 in established and patient-derived HA-expressing GBMs in vivo
To explore the therapeutic efficacy of MSC-ICOVIR17 in vivo, GBM bearing mice were treated with MSC-ICOVIR17 or controls, MSC or PBS. MSC-ICOVIR17 resulted in a drastic tumor volume reduction in U87-FmC model (MSC-ICOVIR17 versus PBS = 97% and P = 0.0015; MSC-ICOVIR17 versus MSC = 95% and P < 0.001; Figure 4a) and in GBM4-FmC model (MSC-ICOVIR17 versus PBS = 68% and P = 0.04; Figure 4b). Survival analysis also showed a significant extension in median survival time by MSC-ICOVIR17 in U87-FmC (PBS = 24 days versus MSC-ICOVIR17 = 77 days, P = 0.0018; MSC = 26 days versus MSC-ICOVIR17, P = 0.0018; Figure 4c) and in GBM4-FmC models (PBS = 53 days versus MSC-ICOVIR17 = 62 days, P = 0.017; MSC = 52 days versus MSC-ICOVIR17, P = 0.0064; Figure 4d). Immunohistological analysis showed wide areas of ICOVIR17 replication in the tumor tissue (Figure 4e,h) that were accompanied by a decrease in HA levels compared to the PBS control group (Figure 4f,g,i, and j).
Figure 4.
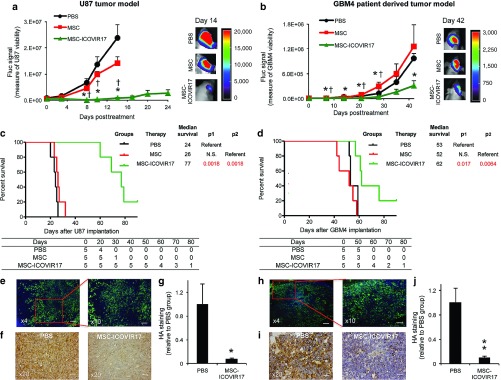
Antitumor effect of MSC-ICOVIR17 in established and patient-derived HA-expressing GBMs in vivo. (a–j) Mice bearing intracranial U87-FmC (a,c,e–g) or patient-derived GBM4 (b,d,h–j) tumors were treated with PBS, MSC, or MSC-ICOVIR17. (a,b) Plot showing changes in Fluc activity as a measure of tumor growth monitored over time. Representative bioluminescence images from each group at day 14 (U87) or day 42 (GBM4) after treatment are shown. (c,d) Kaplan–Meier survival curves of treated mice. Overall survival time was compared between groups. p1 represents P value when PBS-treated group is used as reference, and p2 represents P value when MSC group serves as a reference. The number of mice at risk is shown below the graph. (e–j) Analysis of adenovirus distribution and HA expression on brain sections at day 24 (U87 model) or day 42 (GBM4 model) posttreatment. (e,h) Representative images of adenovirus immunodetection in MSC-ICOVIR17 group are shown. (f,i) Representative images of HA staining from PBS and MSC-ICOVIR17-treated mice. (g,j) Plot showing quantification of HA levels. In all panels, Bars, +SE (n = 5) and **P < 0.01; *P < 0.05 versus PBS control, †P < 0.05 versus MSC control (t-test, two-sided). Bar = 200 µm in e left and h left, 100 µm in e right and h right, and 50 µm in f and i.
We next sought to determine how the MSC-ICOVIR17 administration routes, intratumoral versus systemic, affect the biodistribution of ICOVIR17 in mice with intracerebral U87-FmC tumors. After intratumoral treatment of MSC-ICOVIR17 or ICOVIR17, viral shedding to circulation was at very low levels (<2 × 104 PFU/ml at 5 hours and <1 × 104 PFU/ml at 96 hours; Supplementary Figure S5a), and no viral infection was observed in the liver (Supplementary Figure S5b), while tumor infection was confirmed in the tumor (Supplementary Figure S5c). By contrast, we observed high blood viral titers after intravenous administration of ICOVIR17 (Supplementary Figure S5a) and viral infection in the liver (Supplementary Figure S5b). However, tumor sections revealed no viral replication in mice treated with intravenous MSC-ICOVIR17 or ICOVIR17 (Supplementary Figure S5c) in line with our published study23 that showed that systemic delivery of MSC does not reach intracranial tumors. These results demonstrate that intratumoral administration of MSC-ICOVIR17 results in strong antitumor effects and extensive prolongation of survival in mice bearing established and patient-derived HA-expressing GBMs.
Therapeutic effect of sECM-encapsulated MSC-ICOVIR17 in a clinically relevant mouse model of GBM resection
Our group recently developed a clinically relevant mouse model of GBM resection and showed that sECM encapsulation of therapeutic MSC infected with oncolytic herpes simplex virus (oHSV) allows the retention of the therapeutic MSC after tumor resection, providing higher therapeutic efficacy.23 Based on these studies and taking into account that sECM is based on thiol-modified HA, we first assessed the ability of sECM encapsulated MSC-ICOVIR17 to release the virus and kill GBM cells. In vitro, sECM-encapsulated MSC-ICOVIR17 reduced viability of surrounding U87-FmC (Figure 5a), other established (Supplementary Figure S6a) and patient-derived (Supplementary Figure S6b) GBM cells. To determine the impact of the hyaluronidase expressed by ICOVIR17 on virus release from reconstituted sECM to the media, we coinfected MSC with the nonreplicating adenovirus expressing GFP-Fluc (AdTL) and ICOVIR15 or ICOVIR17, encapsulated in sECM and cocultured with U87-FmC target cells. Fluorescence images showed that AdTL was released more efficiently from sECM encapsulated MSC-AdTL-ICOVIR17 (Supplementary Figure S7a). Exogenously added soluble hyaluronidase also promoted AdTL release from encapsulated sECM-MSC-AdTL-ICOVIR15 and U87-FmC infection in a dose-dependent manner (Supplementary Figure S7b). In addition, adenovirus immunodetection on brain sections of mice bearing U87 tumors treated with sECM-MSC-ICOVIR17 showed better viral spread on day 7 posttreatment compared with mice treated with sECM-MSC-ICOVIR15 (Figure 5b,c). These results suggest that hyaluronidase expressed by ICOVIR17 could accelerate the degradation of reconstituted HA-based sECM and the release of viral progeny from sECM capsules in vitro and in vivo.
Figure 5.

Therapeutic effect of sECM-encapsulated MSC-ICOVIR17 in a clinically relevant mouse model of GBM resection. (a) sECM encapsulated MSC, MSC-ICOVIR15, or MSC-ICOVIR17 were cocultured with U87-FmC. Plots showing tumor cell viability at day 7. (b,c) Mice bearing established intracranial U87-FmC tumors were treated intratumorally with sECM-MSC-ICOVIR15 or sECM-MSC-ICOVIR17, sacrificed at day 7 posttreatment, and adenovirus immunodetection was performed on brain sections from treated mice. (b) Representative images from adenovirus staining from each treated group are shown. (c) Plot showing quantification of adenovirus staining. (d–h). Mice bearing established intracranial U87-FmC tumors were resected and treated with intracavitary injections of PBS, sECM-encapsulated MSC, ICOVIR17, or sECM-encapsulated MSC-ICOVIR17. (d) Plot showing changes in Fluc activity as a measure of tumor growth monitored over time. Representative bioluminescence images from each group at day 11 after treatment are shown. (e) Kaplan–Meier survival curves of treated mice. Overall survival time was compared between groups. p1 represents P value when PBS-treated group is used as reference, p2 represents P value when sECM-MSC group serves as a reference, and p3 when ICOVIR17 is used as reference. The number of mice at risk is shown below the graph. (f–h) Analysis of adenovirus distribution and HA expression in U87 resected tumors at day 15 posttreatment. (f) Representative images showing adenovirus staining from ICOVIR17 or sECM-MSC-ICOVIR17 group. (g) Representative images showing HA staining from PBS and sECM-MSC-ICOVIR17 groups. (h) Plot showing quantification of HA levels. In vitro panel, Bars, +SD (n = 3). In vivo panels, Bars, +SE (n = 4–6) and ***P < 0.001 versus sECM-MSC control, *P < 0.05 versus PBS control, †P < 0.05 versus ICOVIR17 control (t-test, two-sided). Bar = 200 µm in b left, 100 µm in b right, 200 µm in f left, 100 µm in f right, and 50 µm in g.
Next, we performed a partial tumor resection in U87-FmC tumor bearing mice (Supplementary Figure S8a,b) and compared the efficacy of sECM-encapsulated MSC-ICOVIR17 with that of direct injection of purified ICOVIR17. Serial bioluminescence imaging of tumor volume showed that the treatment with sECM-MSC-ICOVIR17 potently suppressed regrowth of residual GBM tumors compared to controls (tumor volume reduction at day 11 posttreatment: sECM MSC-ICOVIR17 versus ICOVIR17 = 67%; sECM MSC-ICOVIR17 versus sECM MSC = 65%; sECM MSC-ICOVIR17 versus PBS = 71% and P = 0.048; Figure 5d). sECM MSC-ICOVIR17-mediated reduction of tumor burden resulted in significant increase in median survival times compared to other groups (sECM-MSC-ICOVIR17 = 45.5 days versus PBS = 34.5 days, P = 0.036; sECM-MSC-ICOVIR17 versus ICOVIR17 = 29 days, P = 0.015; Figure 5e). Immunohistological analysis on brain sections revealed the presence of adenovirus replication in the tumors in sECM-MSC-ICOVIR17 group but not in ICOVIR17 group (Figure 5f), which was associated with a decrease in HA levels in the tumor (Figure 5g,h). These results demonstrate that sECM-encapsulated MSC-ICOVIR17 have therapeutic effects in a mouse model of GBM resection.
Discussion
In this study, we show that GBMs express high levels of HA and demonstrate the strong therapeutic effect of ICOVIR17, a conditionally replicating adenovirus armed with soluble hyaluronidase, in high HA-expressing GBM models. In an effort to effectively apply ICOVIR17 in clinically relevant mouse models of GBM resection, we used encapsulated MSC loaded with ICOVIR17 and showed that this therapeutic strategy degraded HA and enhanced intratumoral viral distribution in resected GBM models, resulting in a significant tumor regrowth control and extension in mice survival.
ECM components, especially HA and collagen, are secreted by tumor cells and form barriers precluding intratumoral distribution of nanosized particles, including oncolytic viruses. This effect could be partly responsible for the suboptimal response rates observed in clinical trials testing oncolytic adenovirus.25 Also, a recent publication from Pedron et al.26 demonstrated that HA induces significant alterations of GBM malignant markers. Degradation of ECM has been proposed as a promising mechanism to increase small particle diffusion in the tumor mass resulting in an improved antitumor therapy for malignant central nervous system tumors, such as GBMs, astrocytomas, or diffuse pontine gliomas.27 In fact, predigestion of ECM with protease,28 collagenases,29 hyaluronidase,30 or matrix metalloproteinases31 results in degradation of ECM and enhancement of oncolytic virus distribution in the tumor mass. A recent study has shown that antiangiogenic agents, such as bevacizumab, increase MMP-2 expression and decrease the amount of collagen IV, enhancing viral distribution in GBMs.32 Although tumor cells expressing hyaluronidase were shown to be more invasive in the presence of HA than hyaluronidase nonexpressing cells,33 we did not observe any increase in invasiveness after ICOVIR17 therapy. This may be because in our therapeutic strategy, the expression of hyaluronidase is restricted to tumor cells that have been already infected with ICOVIR17 and therefore will eventually die within days.
Although it is well known that solid malignancies such as lung, breast, colon, kidney, prostate and non-Hodgkin's lymphoma express high amounts of HA in the tumor mass,34 the levels of HA expression in a broad spectrum of GBM tumors have not been studied yet. In this study, we show that HA is highly expressed in a majority of established and both nodular and invasive patient-derived GBMs. Our in vivo studies showed that ICOVIR17 was able to target HA, efficiently spread throughout the tumor mass, and improve antitumor efficacy as well as mice survival compared to its counterpart ICOVIR15. Difference in efficacy between ICOVIR15 and ICOVIR17 was observed in latest stages of tumor growth (Figure 2a,b). This may be associated with the abundance of HA in mature GBM since HA production in the tumor microenvironment is a dynamic process that escalates during tumor growth.35 Several studies have suggested that the interaction between HA and CD44 receptor plays an important role in GBM cell motility and invasion, inducing the expression of osteopontin, a highly phosphorylated glycoprotein of the ECM, via the phosphatidyliniositol 3-kinase/AKT pathway.36 In our GBM models we did not observe any correlation between HA levels and GBM invasiveness. It will be important to perform more studies to determine if the use of ICOVIR17 may reduce GBM invasion as well as tumor growth.
The sub-optimal efficacy of oncolytic adenovirus in GBM patients could be attributed to the inefficient delivery methods used in clinical trials.4,5,6 The clinical settings in GBM trials involve tumor debulking that induces secondary bleeding and influx of cerebrospinal fluid into the resection cavity, rinsing out and dispersing the virus that has been injected into brain parenchyma adjacent to the resection cavity. In the last decade, stem cells (SC) have been used to deliver anticancer agents to GBMs because of their tumor tropism.37 Our group and others have previously shown that NSC and MSC can effectively deliver oncolytic virus to both established and patient-derived GBMs in vivo resulting in significant therapeutic efficacy.17,24,38,39 In this study we used adipose derived MSC to deliver oncolytic adenovirus because they exhibit similar tumor tropism to NSC in vivo,40 and can be easily isolated from patients. In addition, they are highly immunosuppressive, which allows them to protect the viral load from neutralizing antibodies against adenovirus.41 Further, the therapeutic efficacy of MSC-ICOVIR17 that we observed in GBM models are in line with the recently published study by Ahmed et al.39 showing that the efficacy of oncolytic based therapies was much stronger in established than in patient-derived GBM models in vivo. One of the reasons for this may be the higher growth rate of established GBMs compared with patient-derived GBMs that is associated with better replication of oncolytic virus. In addition, in our case, GBM4 is a highly angiogenic tumor model that may hinder viral spread in vivo.12
Previously, we employed biodegradable sECM, that is based on two components: Hystem and Extralink,21,22 and showed that sECM protects therapeutic SC from clearance with the cerebrospinal fluid after GBM resection and allows them to secrete therapeutic agents in the resection cavity.22 We observed that hyaluronidase expressed by ICOVIR17 accelerates the degradation of sECM and allows the release of viral progeny from the sECM capsule compared to ICOVIR15, an oncolytic virus that does not bear hyaluronidase. Furthermore, we showed that sECM-MSC-ICOVIR17, but not intracavital injection of purified ICOVIR17, exhibits significant therapeutic effect in resected GBM models. We speculate that the limited effect mediated by purified ICOVIR17 in this GBM resection model is mainly because the virus was washed out by the cerebrospinal fluid after the injection in the resection cavity, as has been shown in clinical trials with oncolytic virus.15,16 These results along with our recently published data about MSC loaded with oHSV23 establish that MSC are useful to deliver oncolytic virus and provide therapeutic efficacy in resected GBMs. To improve this therapeutic approach, several studies have shown that oncolytic virus has a synergistic effect with chemotherapy, such as temozolomide (TMZ)42 or TGFβ inhibitors (unpublished data). Since HA degradation could sensitize solid tumors to chemotherapeutic agents and small molecule inhibitors in patients,43 the combination of TMZ or TGFβ inhibitors with sECM-MSC-ICOVIR17 might enhance efficacy in resected GBMs.
In this study, we used an established (U87) and a nodular patient-derived GBM cell line (GBM4) for in vivo studies. However, future work should explore the use of MSC-ICOVIR17 in invasive patient-derived tumors that mirror the human disease and provide the challenge of tumor invasion.11 Although MSC are known to be nonimmunogenic following transplantation in patients,44 in an effort to translate this therapeutic approach to the clinics, it would be ideal to use patient's own MSC or reprogrammed induced pluripotent cells loaded with ICOVIR17.45 Another important area of research is how to turn immune and inflammatory response elicited by oncolytic adenovirus infection into beneficial antitumor responses such as tumor-specific cytotoxic lymphocytes. The combination of MSC loaded with ICOVIR17 with immune checkpoint inhibitors, such as PD-1/PDL-1 antibodies, is an intriguing approach toward completely eliminating GBM tumors.
In summary, this study reveals that GBMs express high levels of HA and that HA-targeting ICOVIR17 improves viral spreading and therapeutic efficacy in GBM. We also demonstrate that MSC loaded with ICOVIR17 exerts potent antitumor effect in established and patient-derived GBMs. Furthermore, we show that sECM-MSC-ICOVIR17 but not ICOVIR17 alone exhibits therapeutic effect in resected GBMs, suggesting that the use of MSC and sECM encapsulation is an effective strategy to deliver ICOVIR17 to resected GBM. In conclusion, our data support that sECM-MSC-ICOVIR17 is a promising candidate to be tested in phase 1 clinical trials in resected GBMs.
Materials and Methods
Parental cell lines. Adipose-derived MSC were obtained from Celleng-tech (www.celleng-tech.com) and grown in Dulbecco's modified Eagle's medium (DMEM) with low glucose (Invitrogen/GIBCO, Grand Island, NY) with 20% fetal bovine serum (FBS) and 1% penicillin/streptomycin 100 U/ml penicillin and 100μg/ml streptomycin (P/S). HEK293 and human GBM cells, U87-MG, LN308, U138, and LN229, were obtained from American Type Culture Collection (ATCC, Manassas, VA). Gli36vIII and U373vIII were generated by infecting Gli36 or U373 with lentivirus expressing EGFRvIII, a constitutively active variant of EGFR. All GBMs were cultured in DMEM supplemented with 10% FBS and P/S. All patient-derived CSC lines (GBM4, GBM8, GBM18, GBM23, GBM27, GBM34, and GBM64) were obtained from surgical specimens at Massachusetts General Hospital and cultured in EF medium composed of Neurobasal medium (Invitrogen) supplemented with 3 mmol/l l-glutamine (Mediatech, Manassas, VA), 1× B27 supplement (Life Technologies, Carlsbad, CA), 0.5× N2 supplement (Life Technologies), 2 μg/ml heparin (Sigma Aldrich, St Louis, MO), 20 ng/ml recombinant human EGF (R & D systems, Minneapolis, MN), 20 ng/ml recombinant human FGF2 (Peprotech, Rocky Hill, NJ), and 1% penicillin/streptomycin (Invitrogen) as described.11
Recombinant oncolytic adenovirus and viral growth assay. ICOVIR15 and ICOVIR17 have been described previously.9 ICOVIR15 is a conditionally replicating adenovirus in which expression of the adenovirus E1A-Δ24 gene is regulated by a modified endogenous E1A promoter, which contains eight E2F-1–binding sites and one Sp1-binding site.46 ICOVIR17 is an armed ICOVIR15 engineered with PH20 hyaluronidase gene under the major late promoter downstream of the fiber gene.9 AdTL is a nonreplicating adenovirus expressing a bimodal imaging marker GFP-Fluc. All adenoviruses were amplified in A549 cells and purified on CsCl gradients according to standard techniques.
HA quantification in vitro. To determine the HA content in the supernatants of various cell lines, cell cultures were grown to 60% confluence in DMEM containing 5% FBS. Medium was removed, and cells were incubated in serum-free medium. After 24 hours, medium was collected and digested overnight with 0.1 mg/ml of pronase. HA content in samples was measured using an enzyme-linked immunosorbent assay as previously described.9
In vitro production and coculture experiments. MSC were plated in 12-well plates (1 × 104 cells/well), infected with ICOVIR15 or ICOVIR17 for 4 hours at 70 PFU/cell, washed twice with PBS, and incubated with fresh medium. At the indicated time points, cell extracts (CE) were harvested and subjected to three rounds of freeze–thaw lysis. Viral titers of CE were determined in triplicate according to an antihexon staining–based method in HEK293 cells.47
To assess the viability of MSC infected with ICOVIR17 and the antitumor effect in cocultures with GBMs over time, MSC-expressing Rluc GFP (500 cells/well) uninfected or infected with ICOVIR15 or ICOVIR17 as previously described were cocultured with U87 expressing FmC (5 × 103 cells/well). Viability of MSC (Rluc signal) was measured at days 1 and 4. On the other hand, tumor viability (Fluc signal) was monitored at days 1, 4, and 7 as described previously.48
To determine the therapeutic effect of MSC-ICOVIR17 in established and patient-derived GBMs, GBM cells expressing FmC (5 × 103 cells/well) were cocultured with different proportions of MSC, MSC-ICOVIR15, or MSC-ICOVIR17. The viability of GBM cells was assessed after 7 days by measuring the Fluc activity of GBM cells. All experiments were performed in triplicates.
In vitro encapsulation experiments. To encapsulate MSC, sECM based on two biocompatible components: Hystem (thiol-modified hyaluronan, a major constituent of native ECM) and Extralink (thiol-reactive crosslinker, polyethylene glycol diacrylate; PEGDA) (Glycosan Hystem-C; Biotime, San Francisco, CA)21,22 was reconstituted according to the manufacturer's protocol. MSC cells (5 × 103 cells), MSC infected with ICOVIR15 or ICOVIR17 for 4 hours at 70 PFU/cell (5 × 103 cells) were resuspended in Hystem (3 µl), and Extralink (1.5 µl) was added. sECM encapsulated MSC, MSC-ICOVIR15, or MSC-ICOVIR17 were plated in 96 wells, and GBM-Fluc cells (2.5 × 103 cells) were added to the plate, and Fluc signal was measured at day 8. All experiments were performed in triplicates.
To study the effect of hyaluronidase on oncolytic virus release from reconstituted sECM, MSC were infected with the nonreplicating adenovirus AdTL or coinfected with AdTL and ICOVIR15 or ICOVIR17. MSC (5 × 103) were then encapsulated in sECM as described above and plated in 96 wells. Next, U87-FmC cells were added, and colocalization of GFP (AdTL) and mCherry (U87 cells) was determined on day 4.
To determine the effect of exogenous hyaluronidase on virus release from reconstituted sECM, MSC loaded with AdTL and ICOVIR15 were encapsulated in sECM and cocultured with U87-FmC. Next day, serial dilutions (Units/ml) of bovine testes hyaluronidase (Sigma) were added to cultures, and colocalization of GFP and mCherry was determined on day 4.
Western blotting analysis. MSC infected with ICOVIR17 for 4 hours at 7 or 70 PFU/cell (1 × 105 cells) were lysed at different time points (days 1, 3, and 5) with NP40 buffer supplemented with protease (Roche, San Francisco, CA) and phosphatase inhibitors (Sigma). Twenty micrograms of harvested proteins from each lysate were denatured and resolved on 10% SDS–PAGE, immunoblotted with antibodies against hyaluronidase PH20 (Abcam, Cambridge, MA; 1:500 dilution) or α-tubulin (Sigma; 1:5000 dilution), and detected by chemiluminescence after incubation with horseradish peroxidase–conjugated secondary antibodies.
In vivo experiments in intact GBMs. To assess the therapeutic effect of oncolytic virus or virus-loaded MSC in established GBMs, U87-Fmc cells (1.5 × 105 cells/mouse) were stereotactically implanted (from bregma, AP: −2 mm, ML: 1.5 mm V (from dura): 2 mm) into right frontal lobe of adult nude mice brains (n = 30) as described.49 Tumor bearing mice (day 4 postimplantation) were injected with ICOVIR15 (1.4 × 107 PFU/mouse; n = 5), ICOVIR17 (1.4 × 107 PFU/mouse, n = 5), PBS (n = 10), MSC (2 × 105 cells/mouse, n = 5), or MSC-ICOVIR17 (2 × 105 cells/mouse preinfected with 70 PFU/cell; n = 5) intratumorally at the same coordinate as the tumor cell implantation. Mice were followed for changes in tumor volumes by Fluc bioluminescence imaging as described previously49 as well as for survival and sacrificed when neurological symptoms became apparent. To obtain brain sections for immunohistochemical analysis, we performed the same experiment as above, but mice were sacrificed at day 24 posttreatment. To test ICOVIR17 production after intratumoral implantation of MSC-ICOVIR17, U87-FmC tumor bearing mice (2 × 105 cells/mouse) were treated at day 7 postimplantation with MSC-GFP loaded with ICOVIR17 (2 × 105 cells preinfected with 70 PFU/cell, n = 6) as previously described and sacrificed at days 3, 7, and 11 (n = 2).
To determine the viral spread of ICOVIR17 from sECM encapsulated MSC-ICOVIR17, mice bearing established intracranial U87-FmC tumors (day 7 postimplantation; 2.5 × 105 cells/mouse) were treated intratumorally with sECM-MSC-ICOVIR17 (2 × 105 cells preinfected with 70 PFU/cell, n = 3) or sECM-MSC-ICOVIR15 (2 × 105 cells preinfected with 70 PFU/cell, n = 3) and sacrificed at day 7 posttreatment.
To study the therapeutic effect of MSC-ICOVIR17 in patient-derived GBMs, GBM4-Fmc cells (3 × 105 cells/mouse) were stereotactically implanted into the brains of nude mice (n = 15) as described above. Tumor-bearing mice (day 12 postimplantation) were injected with PBS (n = 5), MSC (n = 5), or MSC-ICOVIR17 (2 × 105 cells/mouse preinfected with 70 PFU/cell, n = 5) intratumorally. Mice were followed up for changes in tumor volumes by bioluminescence imaging and for survival. To obtain brain sections for immunohistochemical analysis, we performed the same experiment as above, and mice were sacrificed at day 42 posttreatment.
In vivo experiments in resected GBMs. To assess the efficacy of MSC-ICOVIR17 in a mouse model of tumor resection, a cranial window was created over the original implantation site for tumor debulking using a SZX10 stereo microscope system (Olympus, Waltham, MA) for fluorescence-guided surgery. Two weeks later, U87-FmC (1.5 × 105 cells/mouse) were stereotactically implanted (right striatum, 2.5-mm lateral from bregma and 0.5-mm deep) into the brains of 18 mice, and tumor debulking was performed 7 days postimplantation as previously described.22,50 For MSC encapsulation, MSC-ICOVIR17 (2 × 105 cells preinfected with 70 PFU/cell) were resuspended in Hystem (5 µl), and the matrix cross-linker (2.5 µl) was added. sECM encapsulated MSC-ICOVIR17 (2 × 105 cells/mouse, n = 6), sECM-MSC (2 × 105 cells/mouse, n = 4), ICOVIR17 (1.4 × 107 PFU/mouse, n = 4), or PBS (n = 4) were injected into the resection cavity. Mice were serially imaged for Fluc activity over time and followed up for survival. For immunohistochemical analysis, we performed the same experiment as above, and mice were sacrificed at day 15 posttreatment. All in vivo procedures were approved by the Subcommittee on Research Animal Care at Massachusetts General Hospital.
Tissue processing and immunohistochemistry. Mice were perfused by pumping ice-cold 4% paraformaldehyde directly into the heart, and the brains were fixed in 4% paraformaldehyde, and frozen sections (10 µm) were obtained for immunohistochemistry. To detect adenoviral hexon proteins in the brain sections, 10-μm-thick OCT-embedded brain sections were blocked with normal goat serum for 30 minutes and incubated with an antiadenovirus type 5 antibody (ab6982; Abcam; 1:100 dilution) for 3 hours at room temperature. Sections were washed twice with PBS, incubated at room temperature for 2 hours in Alexa Fluor 488 or 647 goat antirabbit secondary antibody diluted in blocking solution (Invitrogen; 1:300 dilution), mounted with DAPI media, and visualized on a confocal microscope (LSM Pascal; Zeiss, Thornwood, NY). To detect adenoviral infection in liver sections, we used the same procedure as above except that we used an AdV 5 E1A antibody (sc-58657; Santa Cruz, Dallas, TX; 1:100 dilution). To quantify adenovirus titers in mice blood, serum was collected from treated mice. Viral titers of serum were determined in triplicate according to an antihexon staining-based method in HEK293 cells.47 Quantification of adenovirus staining was performed using Fiji software (http://www.softpedia.com/get/Science-CAD/Fiji.shtml).
Hematoxylin and eosin and histochemical staining of HA. For hematoxylin and eosin staining, 10-μm-thick OCT-embedded brain sections were incubated with hematoxylin and eosin Y (1% alcohol), dehydrated with 95 and 100% EtOH, and mounted in xylene-based media. For HA staining, paraffin-embedded blocks (n = 3/GBM line) were cut into 10-μm-thick sections. Sections were deparaffinized, and endogenous peroxidase activity was blocked by incubation for 30 minutes in 0.3% H2O2 in methanol. After rehydration, sections were blocked for 30 minutes with 10% normal goat serum diluted in PBS. Then, the slides were incubated with 5 μg/ml of a biotinylated HA-binding protein (HABP-b; Seikagaku, Tokyo, Japan) overnight at 4 °C. The specificity of HA staining was tested by pretreating an adjacent section with 20 U/ml of bovine testes hyaluronidase (Sigma) at 37 °C for 1 hour, prior to the addition of the HABP-b. After incubation with HABP-b, the slides were washed in PBS and treated with avidin–biotin–peroxidase kit (ABC KIT PK-4000; Vectastain, Burlingame, CA). After washings, sections were developed with DAB (Dako Laboratories, Glostrup, Denmark) and counterstained with diluted hematoxylin. Quantification of HA staining was performed using Fiji software.
Statistical analysis. Comparison of data in cell viability and tumor growth was performed using a two-sided t-test (unpaired). Mice survival analyses were performed by Kaplan–Meier curves, and their comparison was evaluated by a two-sided long-rank (Mantel-Cox) test. P values less than 0.05 were considered statistically significant. Data were expressed as mean + SDs in in vitro studies and SEs in in vivo studies. Prism (GraphPad Software San Diego, CA) and Fiji (ImageJ's Jenkins server, Bethesda, MD) software packages were used for analysis and quantification of HA.
SUPPLEMENTARY MATERIAL Figure S1. Expression of hyaluronic acid (HA) in established U87 and patient-derived GBM4 tumor cells in vitro. Figure S2. Expression of HA in peritumoral normal brain. Figure S3. Anti-tumor efficacy of different types of stem cells loaded with ICOVIR17 in established GBMs in vitro. Figure S4. Anti-tumor effect of MSC-ICOVIR17 co-cultured with established or patient-derived GBM cells. Figure S5. Biodistribution of ICOVIR17 after systemic of intratumor administration of MSC-ICOVIR17. Figure S6. Anti-tumor effect of sECM-encapsulated MSC-ICOVIR17 co-cultured with established or patient-derived GBM cells. Figure S7. Effect of hyaluronidase on virus release from sECM in vitro. Figure S8. Mouse model of GBM resection.
Acknowledgments
We thank Juan J. Rojas and Sonia Guedan for engineering ICOVIR15 and ICOVIR17, respectively, and Rafael Moreno, Cristina Puig, Alba Rodriguez, and Raúl Gil for their help and advice in adenovirus techniques. We also acknowledge Manel Cascallo and VCN Biosciences for kindly allowing us to use ICOVIR17. This work was supported by Ministerio de Educación of Spain (J.M.-Q.), RO1 CA138922 (K.S.), RO1 NS071197 (K.S.), and James McDonnell Foundation (K.S.).
The authors indicate no potential conflicts of interest.
J.M-Q. participated in collection and assembly of data, data analysis and interpretation, and manuscript writing; D.H. participated in collection and assembly of data, data analysis, and manuscript writing; H.W. assisted in collection and assembly of data and manuscript writing; R.A. provided reagents and helped with manuscript writing; K.S. performed conception and design, data analysis and interpretation, financial support, and manuscript writing.
Supplementary Material
References
- Stupp R, Mason WP, van den Bent MJ, Weller M, Fisher B, Taphoorn MJ, et al. European Organisation for Research and Treatment of Cancer Brain Tumor and Radiotherapy Groups; National Cancer Institute of Canada Clinical Trials Group Radiotherapy plus concomitant and adjuvant temozolomide for glioblastoma. N Engl J Med. 2005;352:987–996. doi: 10.1056/NEJMoa043330. [DOI] [PubMed] [Google Scholar]
- Alonso MM, Cascallo M, Gomez-Manzano C, Jiang H, Bekele BN, Perez-Gimenez A, et al. ICOVIR-5 shows E2F1 addiction and potent antiglioma effect in vivo. Cancer Res. 2007;67:8255–8263. doi: 10.1158/0008-5472.CAN-06-4675. [DOI] [PubMed] [Google Scholar]
- Parker JN, Bauer DF, Cody JJ, Markert JM. Oncolytic viral therapy of malignant glioma. Neurotherapeutics. 2009;6:558–569. doi: 10.1016/j.nurt.2009.04.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiocca EA, Abbed KM, Tatter S, Louis DN, Hochberg FH, Barker F, et al. A phase I open-label, dose-escalation, multi-institutional trial of injection with an E1B-Attenuated adenovirus, ONYX-015, into the peritumoral region of recurrent malignant gliomas, in the adjuvant setting. Mol Ther. 2004;10:958–966. doi: 10.1016/j.ymthe.2004.07.021. [DOI] [PubMed] [Google Scholar]
- Chiocca EA, Smith KM, McKinney B, Palmer CA, Rosenfeld S, Lillehei K, et al. A phase I trial of Ad.hIFN-beta gene therapy for glioma. Mol Ther. 2008;16:618–626. doi: 10.1038/sj.mt.6300396. [DOI] [PubMed] [Google Scholar]
- Lang FF, Bruner JM, Fuller GN, Aldape K, Prados MD, Chang S, et al. Phase I trial of adenovirus-mediated p53 gene therapy for recurrent glioma: biological and clinical results. J Clin Oncol. 2003;21:2508–2518. doi: 10.1200/JCO.2003.21.13.2508. [DOI] [PubMed] [Google Scholar]
- Sironen RK, Tammi M, Tammi R, Auvinen PK, Anttila M, Kosma VM. Hyaluronan in human malignancies. Exp Cell Res. 2011;317:383–391. doi: 10.1016/j.yexcr.2010.11.017. [DOI] [PubMed] [Google Scholar]
- Park JB, Kwak HJ, Lee SH. Role of hyaluronan in glioma invasion. Cell Adh Migr. 2008;2:202–207. doi: 10.4161/cam.2.3.6320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guedan S, Rojas JJ, Gros A, Mercade E, Cascallo M, Alemany R. Hyaluronidase expression by an oncolytic adenovirus enhances its intratumoral spread and suppresses tumor growth. Mol Ther. 2010;18:1275–1283. doi: 10.1038/mt.2010.79. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shuster S, Frost GI, Csoka AB, Formby B, Stern R. Hyaluronidase reduces human breast cancer xenografts in SCID mice. Int J Cancer. 2002;102:192–197. doi: 10.1002/ijc.10668. [DOI] [PubMed] [Google Scholar]
- Wakimoto H, Mohapatra G, Kanai R, Curry WT, Jr, Yip S, Nitta M, et al. Maintenance of primary tumor phenotype and genotype in glioblastoma stem cells. Neuro Oncol. 2012;14:132–144. doi: 10.1093/neuonc/nor195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wakimoto H, Kesari S, Farrell CJ, Curry WT, Jr, Zaupa C, Aghi M, et al. Human glioblastoma-derived cancer stem cells: establishment of invasive glioma models and treatment with oncolytic herpes simplex virus vectors. Cancer Res. 2009;69:3472–3481. doi: 10.1158/0008-5472.CAN-08-3886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith TT, Roth JC, Friedman GK, Gillespie GY. Oncolytic viral therapy: targeting cancer stem cells. Oncolytic Virother. 2014;2014:21–33. doi: 10.2147/OV.S52749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markert JM, Liechty PG, Wang W, Gaston S, Braz E, Karrasch M, et al. Phase Ib trial of mutant herpes simplex virus G207 inoculated pre-and post-tumor resection for recurrent GBM. Mol Ther. 2009;17:199–207. doi: 10.1038/mt.2008.228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mohyeldin A, Chiocca EA. Gene and viral therapy for glioblastoma: a review of clinical trials and future directions. Cancer J. 2012;18:82–88. doi: 10.1097/PPO.0b013e3182458b13. [DOI] [PubMed] [Google Scholar]
- Kaur B, Chiocca EA, Cripe TP. Oncolytic HSV-1 virotherapy: clinical experience and opportunities for progress. Curr Pharm Biotechnol. 2012;13:1842–1851. doi: 10.2174/138920112800958814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AU, Thaci B, Alexiades NG, Han Y, Qian S, Liu F, et al. Neural stem cell-based cell carriers enhance therapeutic efficacy of an oncolytic adenovirus in an orthotopic mouse model of human glioblastoma. Mol Ther. 2011;19:1714–1726. doi: 10.1038/mt.2011.100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma W, Fitzgerald W, Liu QY, O'Shaughnessy TJ, Maric D, Lin HJ, et al. CNS stem and progenitor cell differentiation into functional neuronal circuits in three-dimensional collagen gels. Exp Neurol. 2004;190:276–288. doi: 10.1016/j.expneurol.2003.10.016. [DOI] [PubMed] [Google Scholar]
- Potter W, Kalil RE, Kao WJ. Biomimetic material systems for neural progenitor cell-based therapy. Front Biosci. 2008;13:806–821. doi: 10.2741/2721. [DOI] [PubMed] [Google Scholar]
- Hansen K, Müller FJ, Messing M, Zeigler F, Loring JF, Lamszus K, et al. A 3-dimensional extracellular matrix as a delivery system for the transplantation of glioma-targeting neural stem/progenitor cells. Neuro Oncol. 2010;12:645–654. doi: 10.1093/neuonc/noq002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich GD. Hyaluronic acid-based clinical biomaterials derived for cell and molecule delivery in regenerative medicine. J Control Release. 2011;155:193–199. doi: 10.1016/j.jconrel.2011.04.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauer TM, Figueiredo JL, Hingtgen S, Shah K. Encapsulated therapeutic stem cells implanted in the tumor resection cavity induce cell death in gliomas. Nat Neurosci. 2012;15:197–204. doi: 10.1038/nn.3019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duebgen M, Martinez-Quintanilla J, Tamura K, Hingtgen S, Redjal N, Wakimoto H, et al. Stem cells loaded with multimechanistic oncolytic herpes simplex virus variants for brain tumor therapy. J Natl Cancer Inst. 2014;106:dju090. doi: 10.1093/jnci/dju090. [DOI] [PubMed] [Google Scholar]
- Stuckey DW, Hingtgen SD, Karakas N, Rich BE, Shah K.2014Engineering toxin-resistant therapeutic stem cells to treat brain tumors Stem Cells(epub ahead of print) [DOI] [PMC free article] [PubMed]
- Pluen A, Boucher Y, Ramanujan S, McKee TD, Gohongi T, di Tomaso E, et al. Role of tumor-host interactions in interstitial diffusion of macromolecules: cranial vs. subcutaneous tumors. Proc Natl Acad Sci USA. 2001;98:4628–4633. doi: 10.1073/pnas.081626898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pedron S, Becka E, Harley BA. Regulation of glioma cell phenotype in 3D matrices by hyaluronic acid. Biomaterials. 2013;34:7408–7417. doi: 10.1016/j.biomaterials.2013.06.024. [DOI] [PubMed] [Google Scholar]
- Maria BL, Gupta N, Gilg AG, Abdel-Wahab M, Leonard AP, Slomiany M, et al. Targeting hyaluronan interactions in spinal cord astrocytomas and diffuse pontine gliomas. J Child Neurol. 2008;23:1214–1220. doi: 10.1177/0883073808321771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuriyama N, Kuriyama H, Julin CM, Lamborn K, Israel MA. Pretreatment with protease is a useful experimental strategy for enhancing adenovirus-mediated cancer gene therapy. Hum Gene Ther. 2000;11:2219–2230. doi: 10.1089/104303400750035744. [DOI] [PubMed] [Google Scholar]
- McKee TD, Grandi P, Mok W, Alexandrakis G, Insin N, Zimmer JP, et al. Degradation of fibrillar collagen in a human melanoma xenograft improves the efficacy of an oncolytic herpes simplex virus vector. Cancer Res. 2006;66:2509–2513. doi: 10.1158/0008-5472.CAN-05-2242. [DOI] [PubMed] [Google Scholar]
- Ganesh S, Gonzalez-Edick M, Gibbons D, Van Roey M, Jooss K. Intratumoral coadministration of hyaluronidase enzyme and oncolytic adenoviruses enhances virus potency in metastatic tumor models. Clin Cancer Res. 2008;14:3933–3941. doi: 10.1158/1078-0432.CCR-07-4732. [DOI] [PubMed] [Google Scholar]
- Mok W, Boucher Y, Jain RK. Matrix metalloproteinases-1 and -8 improve the distribution and efficacy of an oncolytic virus. Cancer Res. 2007;67:10664–10668. doi: 10.1158/0008-5472.CAN-07-3107. [DOI] [PubMed] [Google Scholar]
- Thaci B, Ulasov IV, Ahmed AU, Ferguson SD, Han Y, Lesniak MS. Anti-angiogenic therapy increases intratumoral adenovirus distribution by inducing collagen degradation. Gene Ther. 2013;20:318–327. doi: 10.1038/gt.2012.42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin SG, Jeong YI, Jung S, Ryu HH, Jin YH, Kim IY. The effect of hyaluronic Acid on the invasiveness of malignant glioma cells: comparison of invasion potential at hyaluronic Acid hydrogel and matrigel. J Korean Neurosurg Soc. 2009;46:472–478. doi: 10.3340/jkns.2009.46.5.472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boregowda RK, Appaiah HN, Siddaiah M, Kumarswamy SB, Sunila S, Thimmaiah KN, et al. Expression of hyaluronan in human tumor progression. J Carcinog. 2006;5:2. doi: 10.1186/1477-3163-5-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toole BP. Hyaluronan: from extracellular glue to pericellular cue. Nat Rev Cancer. 2004;4:528–539. doi: 10.1038/nrc1391. [DOI] [PubMed] [Google Scholar]
- Kim MS, Park MJ, Moon EJ, Kim SJ, Lee CH, Yoo H, et al. Hyaluronic acid induces osteopontin via the phosphatidylinositol 3-kinase/Akt pathway to enhance the motility of human glioma cells. Cancer Res. 2005;65:686–691. [PubMed] [Google Scholar]
- Shah K, Bureau E, Kim DE, Yang K, Tang Y, Weissleder R, et al. Glioma therapy and real-time imaging of neural precursor cell migration and tumor regression. Ann Neurol. 2005;57:34–41. doi: 10.1002/ana.20306. [DOI] [PubMed] [Google Scholar]
- Kranzler J, Tyler MA, Sonabend AM, Ulasov IV, Lesniak MS. Stem cells as delivery vehicles for oncolytic adenoviral virotherapy. Curr Gene Ther. 2009;9:389–395. doi: 10.2174/156652309789753347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahmed AU, Thaci B, Tobias AL, Auffinger B, Zhang L, Cheng Y, et al. A preclinical evaluation of neural stem cell-based cell carrier for targeted antiglioma oncolytic virotherapy. J Natl Cancer Inst. 2013;105:968–977. doi: 10.1093/jnci/djt141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee DH, Ahn Y, Kim SU, Wang KC, Cho BK, Phi JH, et al. Targeting rat brainstem glioma using human neural stem cells and human mesenchymal stem cells. Clin Cancer Res. 2009;15:4925–4934. doi: 10.1158/1078-0432.CCR-08-3076. [DOI] [PubMed] [Google Scholar]
- Abumaree M, Al Jumah M, Pace RA, Kalionis B. Immunosuppressive properties of mesenchymal stem cells. Stem Cell Rev. 2012;8:375–392. doi: 10.1007/s12015-011-9312-0. [DOI] [PubMed] [Google Scholar]
- Kanai R, Rabkin SD, Yip S, Sgubin D, Zaupa CM, Hirose Y, et al. Oncolytic virus-mediated manipulation of DNA damage responses: synergy with chemotherapy in killing glioblastoma stem cells. J Natl Cancer Inst. 2012;104:42–55. doi: 10.1093/jnci/djr509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baumgartner G. The impact of extracellular matrix on chemoresistance of solid tumors–experimental and clinical results of hyaluronidase as additive to cytostatic chemotherapy. Cancer Lett. 1998;131:1–2. [PubMed] [Google Scholar]
- Shah K. Mesenchymal stem cells engineered for cancer therapy. Adv Drug Deliv Rev. 2012;64:739–748. doi: 10.1016/j.addr.2011.06.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Somoza RA, Rubio FJ. Cell therapy using induced pluripotent stem cells or somatic stem cells: this is the question. Curr Stem Cell Res Ther. 2012;7:191–196. doi: 10.2174/157488812799859865. [DOI] [PubMed] [Google Scholar]
- Rojas JJ, Guedan S, Searle PF, Martinez-Quintanilla J, Gil-Hoyos R, Alcayaga-Miranda F, et al. Minimal RB-responsive E1A promoter modification to attain potency, selectivity, and transgene-arming capacity in oncolytic adenoviruses. Mol Ther. 2010;18:1960–1971. doi: 10.1038/mt.2010.173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cascallo M, Gros A, Bayo N, Serrano T, Capella G, Alemany R. Deletion of VAI and VAII RNA genes in the design of oncolytic adenoviruses. Hum Gene Ther. 2006;17:929–940. doi: 10.1089/hum.2006.17.929. [DOI] [PubMed] [Google Scholar]
- Martinez-Quintanilla J, Bhere D, Heidari P, He D, Mahmood U, Shah K. Therapeutic efficacy and fate of bimodal engineered stem cells in malignant brain tumors. Stem Cells. 2013;31:1706–1714. doi: 10.1002/stem.1355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sasportas LS, Kasmieh R, Wakimoto H, Hingtgen S, van de Water JA, Mohapatra G, et al. Assessment of therapeutic efficacy and fate of engineered human mesenchymal stem cells for cancer therapy. Proc Natl Acad Sci USA. 2009;106:4822–4827. doi: 10.1073/pnas.0806647106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hingtgen S, Figueiredo JL, Farrar C, Duebgen M, Martinez-Quintanilla J, Bhere D, et al. Real-time multi-modality imaging of glioblastoma tumor resection and recurrence. J Neurooncol. 2013;111:153–161. doi: 10.1007/s11060-012-1008-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.