

Abstract
The phosphorylation of histone H3 is known to play a role in regulation of transcription as well as preparation of chromosomes for mitosis. Various signaling cascades induce H3 phosphorylation, particularly at genes activated by these pathways. In this study we show that signaling can also have the opposite effect. Activators of cAMP signaling induce a rapid and potent loss of H3 phosphorylation. This effect is not mediated through a cAMP metabolite since a membrane-permeable form of AMP had no effect on H3 phosphorylation and a phosphodiesterase-resistant cAMP analog efficiently reduced it. cAMP is also the likely regulator of H3 phosphorylation under physiological conditions since only supra-pharmacological doses of cGMP induce the loss of H3 phosphorylation. The loss of phosphorylation is specific for histone H3 since we do not observe drastic losses in total phosphorylation of other histones. In addition, other H3 modifications are unaffected with the exception of lysine 9 methylation, which is elevated. Analysis of cell growth and cell cycle show that cAMP signaling inhibits cell growth and arrests cells at both G1 and G2/M. Similar effects of cAMP signaling on H3 phosphorylation are observed in a variety of mammary adenocarcinoma-derived cell lines. In syngeneic human breast derived cell lines, one diploid and non-transformed, the other derived from a ductal carcinoma, the loss of H3 phosphorylation is significantly more sensitive to cAMP concentration in the transformed cell line.
Keywords: cAMP signaling, cell cycle, chromatin, histone H3, phosphorylation
INTRODUCTION
The nucleosome is the basic structural and functional unit of chromatin and consists of approximately 180 bp of DNA wrapped around an octameric complex of core histones (H2A, H2B, H3, H4) and a linker histone (H1) [1]. The histone tails, which constitute 30% of the nucleosomal mass, extend out of the nucleosome between the gyres of wrapped DNA and are platforms for a multitude of post-translational modifications (phosphorylation, poly-ADP-ribosylation, acetylation, methylation, ubiquitylation and sumoylation) (reviewed in [2,3]). It is postulated that combinations of such modifications generate an epigenetic “histone code” that is read by a variety of protein factors to control basic DNA processes [4,5].
Although its function is not yet completely understood, one of the most-studied of histone modifications is serine/threonine phosphorylation. Experimental evidence supports the notion that this modification is integrally involved in basic chromatin-template processes such as gene expression, DNA repair, and preparation of chromosomes for mitosis [6]. Histone H3 can be phosphorylated at serines 10 and 28 as well as threonines 3 and 11 [7,8]. H3 phosphorylation occurs at low levels during interphase, but is greatly increased during mitosis. An interesting aspect of H3 phosphorylation is its regulation by signal transduction pathways. Phosphorylation of histone H3 at serine 10 has been shown to be increased in mammalian interphase nuclei by the MAP kinase [9], protein kinase A (PKA) [10,11], and NF-κB-cytokine signaling pathways [12,13]. In each case increased H3 phosphorylation was found to occur at particular genes activated by these pathways which suggests a role in regulation of gene expression [11-15]. Kinases shown to play important roles in the regulation of H3 phosphorylation at serines 10 and 28 include MSK1/2 [16,17], IKKα [12,13] and aurora kinases (mitotic phosphorylation) [18].
In the current study we find that activation of cAMP signaling induces a significant downregulation of histone H3 phosphorylation which occurs with rapid kinetics. Histone H3 phosphorylation is specifically targeted since phosphorylation of other histones is relatively unaffected as are most other modifications of the H3 amino terminal tail. We have observed this downregulation of H3 phosphorylation in a variety of mammary adenocarcinoma-derived cell lines. Interestingly, a transformed human breast cancer cell line is significantly more sensitive to this pathway than its matched normal counterpart, indicating that transformed cells may upregulate the components of this pathway.
MATERIALS AND METHODS
Cell Culture and Reagents
Cell line 1470.2 is derived from C127 mouse mammary adenocarcinoma cells and contains stably replicating copies of a bovine papilloma virus (BPV)-MMTV-LTR chloramphenicol acetyltransferase (CAT) fusion. It was maintained in DMEM (Invitrogen) containing 10% fetal bovine serum (Atlanta Biologicals). Human mammary adenocarcinoma-derived cell lines, MCF-7 and T47D, were maintained in the same medium except that insulin (10 μg/ml) was added. Hs 587T and Hs 587Bst cells were grown in DMEM containing 10% FBS; in the case of Hs 587Bst cells the medium was supplemented with 30 ng/ml epidermal growth factor. Exponentially growing cells were seeded at 30-40% confluence in 30, 90 or 150 mm dishes (Falcon) and grown for 14-16 hours prior to treatment with various reagents at concentrations and times indicated in the figure legends. 8-Br-cAMP was purchased from Sigma. Forskolin was purchased from Calbiochem. 8-Br-AMP and Sp-cAMPS were kind gifts from Dr. Hans-Gottfried Geneiser, Biolog Institute, Bremen, Germany.
Histone Extraction
For most immunoblotting experiments cells were rapidly washed 2 times with pre-warmed (37 °C) DMEM after treatment. Cell lysates were generated by the direct addition of 2X reducing sample buffer (60 mM Tris-HCl pH 7, 120 mM DDT, and 0.6 % SDS). For some immunoblotting experiments and reverse-phase high performance liquid chromatography (RP-HPLC) histones were extracted as follows to preserve in vivo histone phosphorylation patterns as much as possible,. After cell washes, cold 0.2 M H2SO4 (1 ml) was spread directly onto cells. After scraping, cellular material was incubated on ice for 1-2 hours. The extracted histones were separated from cell debris by centrifugation at 14,000 rpm for 10 min. at 4 °C. Histones were then precipitated through addition of trichloroacetic acid (TCA) to a final concentration of 20%. This procedure resulted in extraction of the vast majority of histone proteins (data not shown). For analysis of histone H1, core histones were separated from linker histones by 5% perchloric acid precipitation for 1-2 hours on ice. H1 was then precipitated with 20% TCA from the perchloric acid supernatant. TCA precipitates were recovered by centrifugation at 14,000 rpm for 10 min at 4°C and washed once with acidified (0.2% HCl) acetone and twice with acetone alone. Pellets were air dried and stored at -20°C until use.
Gel Electrophoresis, Western Transfer, and Immunoblotting
Cellular extracts were resolved by electrophoresis in 15 % sodium dodecylsulfate (SDS)-polyacrylamide gels. Proteins were visualized by staining of gels with Coomassie Blue (GelCode Blue Stain Reagent, Pierce) or staining of membranes with Ponceau S after Western transfer. When 32P-labeled histones were resolved by polyacrylamide gels, proteins were stained as above; the gel was dried, and exposed to film (Kodak) with an intensifying screen at −80°C for 3-5 days.
Western transfer of proteins onto either PDVF or nitrocellulose membranes was carried out for 1 hour at 400 mA in 1x Tris-Glycine buffer containing 0.02% SDS. Immunoblotting was performed with antibodies against specific H3 modifications (Upstate Biotechnology) according to manufacturer’s instructions. After exposure of peroxidase-conjugated secondary antibodies (Jackson Immunoresearch), bound antibodies were detected using a chemiluminescent assay (Pierce). Chemiluminescent signals were detected by exposure to film or by use of a chemiluminescence imager (Alpha Innotech Corp.). Quantitation of signals was carried out with Fluorchem software (Alpha Innotech Corp.). When necessary, membranes were stripped either using Restore buffer (Pierce) or repeated washes with a solution containing 9 M Urea/10% acetic acid.
Metabolic Labeling of Histones and Reverse Phase HPLC
For analysis of core histones, exponentially growing 1470.2 cells in 150 mm dishes were incubated for 18-20 hr in 30 ml complete medium containing 20 μCi/ml 32P-orthophosphoric acid (PerkinElmer). Cells were then treated with 8-Br-cAMP for two hours. For analysis of H1, the cells were grown in 6 well plates and labeled via 32P-orthophosphoric acid as described above.
For RP-HPLC acid-extracted core histones were resuspended in water (200 μl) and incubated overnight at 4°C. Trifluoroacetic acid (TFA) at 0.05% (Fisher) and 2% acetonitrile (HPLC grade, Fisher) were added and the material was clarified by centrifugation at 14,000 rpm for 10 min. The supernatant was injected, at a rate of 0.4 ml/min., into a silica C2/C8 reverse-phase column (Sephasil RPC2/C8, 4.6/250, Amersham Bioscience) connected to a Pharmacia SMART System. The column had been previously equilibrated in a 2% acetonitrile/0.05% TFA solution. The concentration of acetonitrile was raised to 30% within 2 min. A shallow gradient in which acetonitrile concentration was raised in increments of 0.24% per minute was run for 140 min. 0.4 ml fractions were collected, dried under vacuum, and resuspended in 50 μl deionized water. Typically 5-10 μl from each sample was used for SDS-PAGE and immunoblots.
Cell Growth and Cell Cycle Analysis
For monitoring cell growth, 1470.2 cells were seeded into 150 mm dishes at a density of 1×106 cells per dish, treated as described, and harvested by trypsinization at various times for counting. Cell cycle analysis was carried as follows. First, harvested cells were washed, resuspended in ice-cold PBS, and fixed in 64% ethanol overnight at 4°C. At least 1×106 cells from each sample were washed and treated with RNase A in PBS. Cellular DNA was labeled by overnight exposure to propidium iodide (40 μg/ml). Cellular DNA content was analyzed using a FACS Calibur (BD Biosciences). Modeling of the DNA content data to determine cell cycle distribution was carried out using Modfit software BD (Verity Software House, Inc.).
RESULTS
Activators of cAMP signaling induce loss of H3 phosphorylation
In the process of using chromatin immunoprecipitation assays to examine gene-specific changes in histone H3 modifications [19], we noticed that activation of cAMP signaling in mouse mammary adenocarcinoma-derived cell lines resulted in a decreased level of bulk histone H3 phosphorylation (data not shown). To characterize this phenomenon further, we carried out a time course of treatment with either 8-Br-cAMP or forskolin (adenylate cyclase activator) and subjected cellular extracts to Western blot analysis using antibodies against H3 phosphorylated at either Ser10 or Ser28. Both drugs, 8-Br-cAMP (Fig. 1A) and forskolin (Fig. 1B), induce a rapid decline in H3 phosphorylation at Ser10 and Ser28, which is clearly visible after 5-10 minutes and persists for at least two hours. An inactive analog of forskolin, 1,9-dideoxyforskolin, did not change H3 phosphorylation (data not shown), strongly implicating the cAMP-generating activity of adenylate cyclase in this process. To ensure that the loss of H3 phosphorylation could be induced by physiological stimulators of cAMP synthesis, we treated cells with either epinephrine or isoproterenol, agonists of the β-adrenergic receptor (β-AR), and monitored phosphorylation of serines 10 and 28 by immunoblot as shown in Fig 1C. Both ligands induced rapid and potent loss of H3 phosphorylation with the same kinetics and to the same extent as treatment with 8-Br-cAMP (Fig. 1E). The effects of these ligands are mediated through the β-AR since the β-AR antagonist, propranolol, completely blocked their ability to reduce H3 phosphorylation (shown in Fig. 1D for epinephrine). Remarkably, 80-90% of H3 serine phosphorylation was lost in 60 min. of treatment with inducers of cAMP signaling (Fig. 1E), indicating that this effect is mediated directly through the action of a signaling cascade and does not require changes in gene expression.
Figure 1. cAMP signaling induces rapid loss of histone H3 phosphorylation at serines 10 and 28.
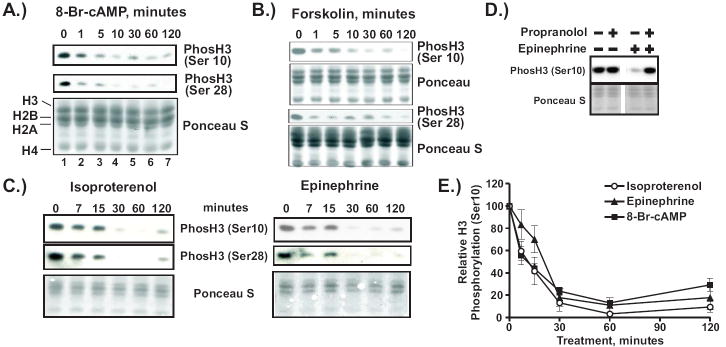
A-C.) 1470.2 cells were treated with 1 mM 8-Br-cAMP (A), 10 μM forskolin (B), 20 μM isoproterenol (C, left panel) or 20 μM epinephrine (C, right panel) for the times shown. Cellular proteins were extracted as described in Materials and Methods. Equal volumes of each extract were resolved by SDS-PAGE and transferred onto nitrocellulose membranes. To visualize transferred proteins, membranes were stained with Ponceau S. Immunoblotting was carried out with antibodies against H3 phosphorylated at either serine 10 or 28. D.) 1470.2 cells were treated with either epinephrine (1 μM, 60 minutes) or propranolol (20 μM, 90 minutes) or both. Immunoblotting of cell lysates was carried out with antibody against histone H3 phosphorylated at serine 10. E.) Statistical analysis of data from at least 6 independent experiments using the anti-phosH3 (ser10) antibody. In each experiment the signal intensity from untreated cells (0 minutes) was set to 100 and all other intensity values were expressed relative to that. Error bars represent SEM.
Effects of other nucleotides on H3 phosphorylation
In the cell, cAMP is broken down to AMP by phosphodiesterases. Elevated levels of cellular AMP are known to activate signaling cascades initiated in part by allosteric binding to AMP-activated protein kinase (AMPK) [20]. We therefore measured the ability of membrane-permeable AMP (8-Br-AMP) to induce loss of H3 phosphorylation at various doses and found that, unlike 8-Br-cAMP, it failed to do so (Fig. 2A, upper panels). We also tested a cAMP analog unable to be metabolized by phosphodiesterases. Sp-cAMPS efficiently induced loss of H3 phosphorylation (Fig. 2A, lower panel). Taken together, these data indicate that cAMP metabolites do not mediate this effect.
Figure 2. Downregulation of H3 phosphorylation is not mediated through degradation products of cAMP or by cGMP.
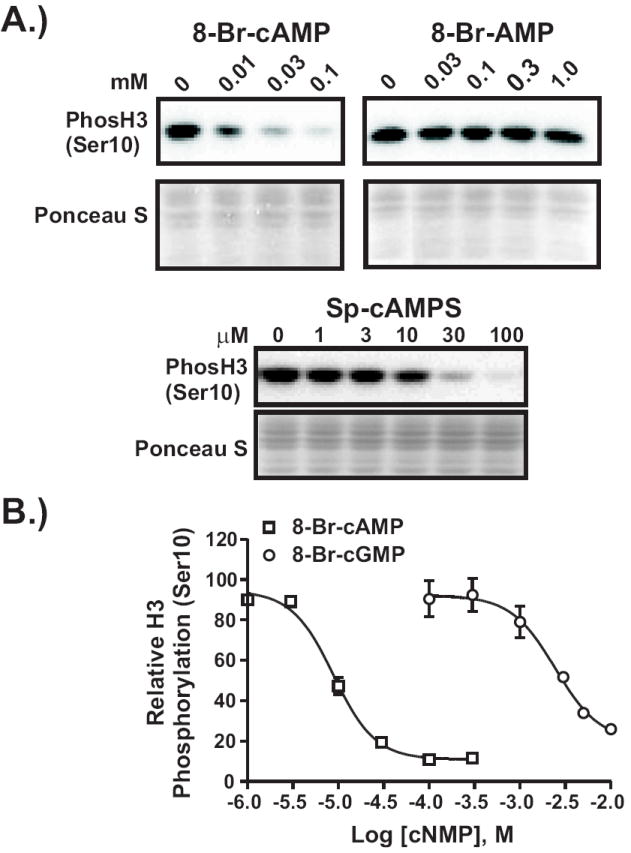
A.) 1470.2 cells were treated with various doses of 8-Br-cAMP, 8-Br-AMP, or Sp-cAMPS for 1 h. Lysates were processed as described above. B.) Cells were treated with varying doses of either 8-Br-cAMP or 8-Br-cGMP for 1 h. Lysates were assayed for H3 phosphorylation as described above. Dose response curves were generated as described in the legend to Fig. 8A. The graph represents the results from 2-3 independent experiments. Error bars represent SEM.
Known cyclic nucleotide binding proteins are physiologically regulated by either cAMP or cGMP, or both. To test the cyclic nucleotide specificity of the binding protein which mediates loss of H3 phosphorylation, we carried out a dose response experiment using either 8-Br-cAMP or 8-Br-cGMP. As shown in Fig. 2B, 8-Br-cAMP is 2.5 orders of magnitude more effective than 8-Br-cGMP at inducing loss of H3 phosphorylation. Thus, cAMP is the likely physiological activator of this signaling pathway.
HPLC Analysis of Histone Phosphorylation in Metabolically-Labeled Cells
The histone H3 tail is highly dense in amino acid residues which can be modified. It has been shown that antibody recognition of a specific modification in the H3 tail can be disrupted by the presence of a nearby modification [14]. To ensure that the loss of H3 phosphorylation observed by immunoblot was not due to cAMP-induced epitope masking, we examined the phosphorylation status of H3 radioactively-labeled through incubation of cultured 1470.2 cells with 32P-orthophosphate followed by reverse phase HPLC. Core histones were isolated by direct acid extraction from untreated cells and cells treated with 8-Br-cAMP for 2 h. The H3 elution profiles from a Sephasil RP-C2/8 column are shown in Fig. 3A,B, upper panels. Consistent with previous reports [21,22] histone H3 eluted in two distinct fractions, termed H3f and H3s. The autoradiograms (Fig. 3A,B – lower panels) for H3 extracted from untreated or 8-Br-cAMP treated cells shows 32P incorporation into both H3 species. However, there is a drastic reduction in phosphate incorporation observed in 8-Br-cAMP-treated cells. The radioactive bands were excised and subjected to scintillation counting to obtain the data shown in Fig. 3C. Relative to H3 derived from untreated cells, levels of phosphate incorporation in H3 derived from cAMP-treated cells were reduced 75-85%, which is in good agreement with the results from immunoblotting (Fig. 1B). Using the antibody specific for H3 phosphorylated at serine 10, we confirmed that the peaks recovered from RP-HPLC were H3 proteins (Fig. 3D). Antibody-reactive species corresponded to the bands observed in Coomassie-stained gels and were greatly reduced in intensity after 8-Br-cAMP treatment. Thus, cAMP signaling reduces total H3 phosphorylation and the loss of H3 phosphorylation we observed by western blotting was not due to masking of phosphoepitopes.
Figure 3. HPLC analysis of histone H3 phosphorylation in metabolically-labeled cells.

A, B.) 1470.2 cells exposed to 32P-orthophosphoric acid were treated with or without 8-Br-cAMP (1 mM) for 2 h. Acid-extracted cellular proteins were resolved by reverse-phase HPLC. Equal aliquots of histone-containing fractions were separated on 15% SDS-polyacrylamide gels and stained with Coomassie Blue (upper panels). The gels were dried and exposed to X-ray film for autoradiography (lower panels). Histone H3 from untreated (A) and 8-Br-cAMP-treated (B) cells eluted in two fractions, termed fast (H3f) and slow (H3s). C.) Radiolabeled bands were excised from the gels shown in A and B and subjected to scintillation counting. The results are expressed graphically for both fast (H3f) and slow (H3s) fractions of H3. D.) Histones from cells treated with or without 8-Br-cAMP (1mM, 2 hrs) were resolved by HPLC. Fractions containing histone H3 were separated by SDS-PAGE. Gels were either stained with Coomassie Blue or subjected to Western blotting with antibodies against histone H3 phosphorylated at serine 10.
To determine whether phosphorylation of other histones was also affected by cAMP signaling, we measured total phosphorylation of histones H2A, H2B and H4 by the same method, as shown in Fig. 4A,B. As expected [22,23], H2B had the shortest retention time in the RP-C8 matrix (Fig. 4A,B, upper panels, fractions 91-95 and 93-96 respectively) and the autoradiogram shows no detectable phosphate incorporation in either untreated or cAMP-treated cells (Fig. 4A,B, lower panels). The identity of the phosphorylated species (marked with an asterisk) in the fractions containing H2B is unknown but has a significantly faster electrophoretic mobility than H2B. H2A eluted from the resin in at least two different fractions (Fig. 4A,B, upper panels, fractions 97-108 and 99-109, respectively). These fractions, termed H2A fast (H2Af) and H2A slow (H2As), have been observed previously and suggested to be variants with differing levels of hydrophobicity [22]. Both H2A species are significantly more phosphorylated than the other core histones (Fig. 4A,B, lower panels). Histone H4 shows a relatively low level of phosphorylation (Fig. 4A,B, fractions 98-102 and 99-103, respectively). Quantitation of radioactive species in two independent experiments did not reveal dramatic cAMP-induced changes in overall phosphate incorporation for the H2A fractions or H4; the magnitude of these changes was less than 30% (Fig. 4C).
Figure 4. HPLC analysis of histone phosphorylation (H2B, H2A, H4, and H1) in metabolically-labeled cells.

A, B.) 1470.2 cells were treated and processed as described in the legend to Fig. 2. Acid extracted histones H2B, H2A, and H4 from either untreated cells (A) or 8-Br-cAMP-treated cells (B) were resolved by reverse phase HPLC. Upper panels in (A) and (B) show Coomassie-stained histones while the lower panels are the corresponding autoradiograms. Retention times (in minutes) are indicated at the bottom of each panel. (C) Radiolabeled bands were excised from the autoradiograms shown in A and B and subjected to scintillation counting. The results are expressed graphically for H4 as well as both fast (H2Af) and slow (H2As) fractions of H2A. D.) Histone H1 was extracted with perchloric acid from metabolically-labeled 1470.2 cells treated for various times with 8-Br-cAMP (1 mM) and separated by SDS-PAGE. Gels were stained with Coomassie Blue (upper panel), dried, and exposed to film. The autoradiogram is shown in the lower panel.
The linker histone H1 is also known to be phosphorylated, especially during mitosis [24,25]. Thus, we investigated the possibility that H1 phosphorylation could be altered upon cAMP treatment. Histone H1 was extracted from cells with perchloric acid after incubation with 32P-orthophosphate and separated by SDS-PAGE. As shown in Fig. 4D, the total level of H1 (Coomassie-stained gel) and the level of phosphate incorporation (autoradiogram) remains unchanged in cells incubated with cAMP for up to 120 minutes. Thus, cAMP signaling specifically targets H3 phosphorylation and does not alter total phosphorylation of other histones in a rapid and dramatic fashion.
Analysis of Acetylation and Methylation of Histone H3
In addition to multiple phosphorylation sites, the histone H3 tail also contains multiple sites for acetylation and methylation, some of which are in very close proximity to the phosphorylated serine residues. To determine whether the cAMP-induced changes in H3 modification are specific to phosphorylation, we assayed for changes in the levels of other H3 modifications by Western blot analysis using a panel of modification-specific antibodies. As shown in Fig. 5, we observed no consistent or significant changes in acetylation of lysines 9, 14, or 27 or in methylation of lysines 4 and 36. However, levels of methylated lysine 9 increase about 2-fold at 30 minutes of cAMP treatment.
Figure 5. Analysis of H3 acetylation and methylation in response to cAMP signaling.

1470.2 cells were treated for various times with 1 mM 8-Br-cAMP and processed as described in the legend to Fig. 2. Western blotting was carried out with the antibodies indicated.
cAMP Signaling Inhibits Cell Growth and Induces Cell Cycle Arrest
Previous studies have shown that cAMP signaling can either stimulate or inhibit cell cycle progression, depending on the cell type [26,27]. Since H3 phosphorylation is subject to cell cycle regulation, we investigated the possibility that cAMP signaling affects cell cycle progression in our mammary adenocarcinoma-derived cell line. Analyses of cell growth are shown in Fig. 6A,B. Cells grown for 5 days without treatment are completely confluent while cells grown in the presence of 8-Br-cAMP appear to have undergone very little, if any, proliferation (Fig 6A). In addition, the treated cells have an altered morphology compared to cells on Day 0, suggesting that cAMP signaling might induce the cells to stop dividing and differentiate. To quantitate the effects of cAMP signaling on cell growth, we generated growth curves in the presence and absence of 8-Br-cAMP (Fig. 6B). It is clear that cAMP signaling rapidly stops cell proliferation; after 48 h treatment, the treated cells have not even doubled in number. Cell cycle analysis was carried out to determine at which point in the cell cycle cAMP signaling arrests progression (Fig. 6C). The untreated cells had a cell cycle distribution typical of asynchronously cycling cells. In contrast, the cells treated with 8-Br-cAMP for 48 h were enriched in the G0/G1 and G2/M phases of the cell cycle, while S phase cells were depleted, indicating a block in both G1 and G2/M. None of our growth analyses indicated that cAMP signaling caused any cell toxicity (data not shown).
Figure 6. cAMP signaling inhibits cell growth and cell cycle progression.

A.) Cell line 1470.2 was plated at low density and either untreated or treated with 0.125 mM 8-Br-cAMP for 5 days. Photographs were taken on day 0 and day 5. B.) Cells were either untreated or treated with 0.125 mM 8-Br-cAMP for up to 48 hrs. Cells were harvested and counted at the times indicated. The results of 4 independent experiments are shown graphically as fold change in cell number relative to time zero. C.) Cells were untreated or treated for 48 hours with 0.125 mM 8-Br-cAMP. Cells were fixed and processed for cell cycle analysis. Representative histograms showing cellular DNA content are shown on the right. Average percentages of cells in the various phases of the cell cycle are shown graphically on the left for untreated and 8-Br-cAMP-treated cells and represent statistical analysis of results from 5 independent experiments.
cAMP signaling decreases H3 phosphorylation in several mammary-derived cell lines
Cell line 1470.2 was generated from a mouse mammary adenocarcinoma-derived cell line, C127i. We examined other mammary-derived cell lines to determine if cAMP regulates H3 phosphorylation in a similar manner as shown in Fig. 7. As observed in 1470.2 cells, 8-Br-cAMP causes a significant loss of H3 phosphorylation at serine 10 in both T47D and MCF7 cells, although the kinetics of the change are slightly slower. We also measured H3 phosphorylation in syngeneic human mammary-derived cell lines, Hs 587Bst and Hs 587T, isolated from non-malignant and malignant breast tissue in a single patient. The former is diploid and considered to model normal, untransformed human myoepithelial breast cells. The latter was derived from a ductal breast carcinoma and is aneuploid and transformed. As shown in Fig. 7C,D, 8-Br-cAMP treatment rapidly induces loss of H3 phosphorylation in both cell lines, but the tumor-derived cell line, Hs 587T, is much more sensitive, requiring a tenth as much 8-Br-cAMP to efficiently reduce it. Taken together the results suggests that cAMP signaling regulates H3 phosphorylation by a similar mechanism in a variety of mammary-derived cell lines and that transformed cells may be more sensitive to this form of regulation than non-transformed cells.
Figure 7. Regulation of H3 phosphorylation by cAMP signaling in mammary-derived cell lines.

A, B.) T47D (A) or MCF-7 cells (B) were treated with 8-Br-cAMP for the times indicated. Cellular proteins were extracted and subjected to Western blotting with an antibody against histone H3 phopshorylated at serine 10. C, D.) Human normal breast-derived Hs 587Bst (C) and human breast carcinoma-derived Hs 587T (D) cells were treated for up to 2 h with the concentrations of 8-Br-cAMP shown. Cellular proteins were extracted and subjected to Western blotting with an antibody against histone H3 phopshorylated at serine 10.
DISCUSSION
Previous studies have shown that histone H3 phosphorylation can be increased by various signaling pathways through the activation of kinases [9]. We have now demonstrated that it can also be specifically and potently decreased in response to both pharmacologic and physiologic inducers of cAMP signaling. By immunoblotting we found that cAMP signaling rapidly induced an 80-90% loss of H3 phosphorylation at serines 10 and 28 in the amino terminal tail region. This result was confirmed through HPLC analysis of metabolically-labeled histones, indicating that the loss of H3 phosphorylation demonstrated through the use of phospho-specific antibodies was not due to masking of epitopes caused by changes in modification of nearby residues. In addition, the HPLC results showed that cAMP signaling targets histone H3 specifically because we did not observe large changes in total phosphorylation of other histones. However, we cannot rule out the possibility of site-specific changes in phosphorylation of other histones, especially in the case of H2A, which is heavily phosphorylated.
The cAMP-initiated signal did not induce changes of similar magnitude in four other H3 modifications, including acetylation and methylation that occur at lysine residues adjacent to serines 10 and 28. This lends further support to the idea that changes in nearby modifications do not contribute to the loss of H3 phosphorylation. However, we did observe a modest increase in H3 lysine 9 methylation by 30 minutes of cAMP treatment. Since this change is delayed relative to the rapid kinetics of the change in H3 phosphorylation, it is more likely to be a consequence of the loss of H3 phosphorylation than a cause. This may be due to an increase in good substrates for SUV39H1, a histone methyltransferase which specifically methylates lysine 9 in the histone H3 tail. This enzyme has been shown to be inhibited from methylating lysine 9 when serine 10 is phosphorylated in vitro [28]. Thus, a global loss of serine 10 phosphorylation would be predicted to lead to an increase in lysine 9 methylation in areas of chromatin where SUV39H1 is localized. In support, inhibition of mitotic H3 phosphorylation at serine 10 by knockdown of Aurora B kinase expression has been shown to result in increased association of SUV39H1 with mitotic chromosomes [29]. This study further supports a reciprocal relationship between serine 10 phosphorylation and lysine 9 methylation in vivo.
Once generated as a result of adenylate cyclase activity, cAMP is rapidly degraded through the action of phosphodiesterases. The product of this reaction is AMP, which also has second messenger function through binding and activation of AMP-activated kinase (AMPK) [30]. Treatment of cells with 8-Br-AMP however, does not induce loss of H3 phosphorylation. In addition, a non-hydrolyzable cAMP analog, Sp-cAMPS, downregulates H3 phosphorylation very efficiently. Thus, this pathway is mediated through the action of a cyclic nucleotide binding protein. Most of these proteins bind either cAMP or cGMP with high affinity with the exception of some of the cyclic nucleotide gated ion channels, which bind both [31]. Comparing the dose response curves of 8-Br-cAMP and 8-Br-cGMP allow us to conclude that the loss of H3 phosphorylation is mediated physiologically through a cAMP-binding protein. Further study of this phenomenon has shown that PKA expression is required for regulation of H3 phosphorylation (Rodriguez et al., in preparation).
Our observation of rapid, cAMP-mediated loss of H3 phosphorylation stands in contrast to that of Salvador and colleagues who reported that cAMP signaling rapidly and transiently induces H3 phosphorylation in primary cultures of ovarian granulosa cells and at cAMP target promoters [11]. Clearly, the response of a given cell to cAMP signaling depends on the array of cyclic nucleotide binding proteins and downstream effectors expressed. In addition, it is unclear whether the granulosa cells were actively dividing. cAMP signaling has been shown to delay G2 progression in several mammalian cell lines [32-34]. Since mitotic H3 phosphorylation is dramatically increased in late G2/early M phase, a delay in G2 progression may have the effect of decreasing overall H3 phosphorylation. In fact, we show that cAMP signaling inhibits cell growth in our mammary-adenocarcinoma cell line through an arrest to cell cycle progression Fig. 6). Cells treated with 8-Br-cAMP were arrested at both G1 and G2/M. Global levels of H3 phosphorylation are relatively low at G1, but are extremely high during mitosis. A block to G1 and G2 progression would very likely result in decreased global levels of H3 phosphorylation due to a reduction in the number of mitotic cells rather than a general dephosphorylation of H3 at cAMP target genes. This is supported by our previous study in which we showed that rapid cAMP-induced repression of the mouse mammary tumor virus promoter in 1470.2 cells was associated with loss of H3 phosphorylation from a very localized region of the promoter; other areas of the promoter were unaffected [19]. The rapidity of the effect on global H3 phosphorylation indicates that it is controlled through a direct kinase cascade. This cascade may target the kinases/phosphatases which regulate H3 phosphorylation and/or the proteins which directly regulate cell cycle progression. The mechanism is currently under investigation.
A large variety of hormones use cAMP as a second messenger to elicit specific responses in different cell types. To determine the generality of cAMP-induced downregulation of H3 phosphorylation in mammary-derived cell lines, we tested several lines in addition to the 1470.2 cells. Three human breast cancer cell lines, T47D, MCF-7, and Hs 587T, responded to activation of cAMP signaling similarly, showing significant and rapid loss of H3 phosphorylation. Interestingly, a pair of cell lines derived from a human breast tumor and normal surrounding tissue, Hs 587T and Hs 587Bst respectively, both downregulated H3 phosphorylation in response to cAMP signaling. However, we observed that 10 times more 8-Br-cAMP was required to achieve this downregulation in the non-transformed Hs 587Bst cells, indicating that the transformed cells were highly sensitive to cAMP regulation of H3 phosphorylation. These results suggest that components of the pathway which mediates the loss of H3 phosphorylation may be upregulated in some breast cancers thus increasing the sensitivity of the response to cAMP generation. In fact, the cAMP-binding regulatory subunit I (RI) has been shown to be upregulated to varying extents in human breast cancer samples relative to surrounding breast tissue [35]. This has led to the development of antisense oligonucleotides targeting RI as therapeutic agents to treat a variety of cancers. They are particularly effective when used in combination with other agents which inhibit other key signaling pathways such as those induced by epidermal growth factor or estrogen [35,36]. The results of our study suggest that cAMP signaling may provide targets for development of therapeutics which act on growth regulatory pathways in breast cancer. Activation of the pathway may inhibit growth of tumors. Alternatively, inhibitors of the pathway may work as abrogators of the G2 checkpoint, which G1-checkpoint deficient tumor cells rely on to repair damage caused by chemotherapeutic agents [37].
Acknowledgments
We are grateful to the members of the Laboratory of Receptor Biology and Gene Expression (NCI) for helpful input and discussion. This work was supported by an Intramural Research Award to CLS and grants from the Emmerling Fund of the Pittsburgh Foundation and the Rett Syndrome Research Fund to SHL and JZ.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.van Holde KE. Chromatin. Springer-Verlag; Heidelberg: 1988. [Google Scholar]
- 2.Berger SL. Histone modifications in transcriptional regulation. Curr Opin Genet Dev. 2002;12:142–148. doi: 10.1016/s0959-437x(02)00279-4. [DOI] [PubMed] [Google Scholar]
- 3.Nathan D, Sterner DE, Berger SL. Histone modifications: Now summoning sumoylation. Proc Natl Acad Sci U S A. 2003;100:13118–13120. doi: 10.1073/pnas.2436173100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Turner BM. Decoding the nucleosome. Cell. 1993;75:5–8. [PubMed] [Google Scholar]
- 5.Strahl BD, Allis CD. The language of covalent histone modifications. Nature. 2000;403:41–45. doi: 10.1038/47412. [DOI] [PubMed] [Google Scholar]
- 6.Nowak SJ, Corces VG. Phosphorylation of histone H3: a balancing act between chromosome condensation and transcriptional activation. Trends Genet. 2004;20:214–220. doi: 10.1016/j.tig.2004.02.007. [DOI] [PubMed] [Google Scholar]
- 7.Shoemaker CB, Chalkley R. H3-specific nucleohistone kinase of bovine thymus chromatin. Purification, characterization, and specificity for threonine residue 3. J Biol Chem. 1980;255:11048–11055. [PubMed] [Google Scholar]
- 8.Preuss U, Landsberg G, Scheidtmann KH. Novel mitosis-specific phosphorylation of histone H3 at Thr11 mediated by Dlk/ZIP kinase. Nucleic Acids Res. 2003;31:878–885. doi: 10.1093/nar/gkg176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mahadevan LC, Willis AC, Barratt MJ. Rapid histone H3 phosphorylation in response to growth factors, phorbol esters, okadaic acid, and protein synthesis inhibitors. Cell. 1991;65:775–783. doi: 10.1016/0092-8674(91)90385-c. [DOI] [PubMed] [Google Scholar]
- 10.DeManno DA, Cottom JE, Kline MP, Peters CA, Maizels ET, Hunzicker-Dunn M. Follicle-stimulating hormone promotes histone H3 phosphorylation on serine-10. Mol Endocrinol. 1999;13:91–105. doi: 10.1210/mend.13.1.0222. [DOI] [PubMed] [Google Scholar]
- 11.Salvador LM, Park Y, Cottom J, Maizels ET, Jones JC, Schillace RV, Carr DW, Cheung P, Allis CD, Jameson JL, Hunzicker-Dunn M. Follicle-stimulating hormone stimulates protein kinase A-mediated histone H3 phosphorylation and acetylation leading to select gene activation in ovarian granulosa cells. J Biol Chem. 2001;276:40146–40155. doi: 10.1074/jbc.M106710200. [DOI] [PubMed] [Google Scholar]
- 12.Anest V, Hanson JL, Cogswell PC, Steinbrecher KA, Strahl BD, Baldwin AS. A nucleosomal function for IkappaB kinase-alpha in NF-kappaB-dependent gene expression. Nature. 2003;423:659–663. doi: 10.1038/nature01648. [DOI] [PubMed] [Google Scholar]
- 13.Yamamoto Y, Verma UN, Prajapati S, Kwak YT, Gaynor RB. Histone H3 phosphorylation by IKK-alpha is critical for cytokine-induced gene expression. Nature. 2003;423:655–659. doi: 10.1038/nature01576. [DOI] [PubMed] [Google Scholar]
- 14.Clayton AL, Rose S, Barratt MJ, Mahadevan LC. Phosphoacetylation of histone H3 on c-fos- and c-jun-associated nucleosomes upon gene activation. EMBO J. 2000;19:3714–3726. doi: 10.1093/emboj/19.14.3714. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Cheung P, Tanner KG, Cheung WL, Sassone-Corsi P, Denu JM, Allis CD. Synergistic coupling of histone H3 phosphorylation and acetylation in response to epidermal growth factor stimulation. Mol Cell. 2000;5:905–915. doi: 10.1016/s1097-2765(00)80256-7. [DOI] [PubMed] [Google Scholar]
- 16.Soloaga A, Thomson S, Wiggin GR, Rampersaud N, Dyson MH, Hazzalin CA, Mahadevan LC, Arthur JS. MSK2 and MSK1 mediate the mitogen- and stress-induced phosphorylation of histone H3 and HMG-14. EMBO J. 2003;22:2788–2797. doi: 10.1093/emboj/cdg273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Thomson S, Clayton AL, Hazzalin CA, Rose S, Barratt MJ, Mahadevan LC. The nucleosomal response associated with immediate-early gene induction is mediated via alternative MAP kinase cascades: MSK1 as a potential histone H3/HMG-14 kinase. EMBO J. 1999;18:4779–4793. doi: 10.1093/emboj/18.17.4779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pascreau G, Arlot-Bonnemains Y, Prigent C. Phosphorylation of histone and histone-like proteins by aurora kinases during mitosis. Prog Cell Cycle Res. 2003;5:369–374. [PubMed] [Google Scholar]
- 19.Mulholland NM, Snyder SK, Kolla SS, Smith CL. Chromatin-dependent regulation of the MMTV promoter by cAMP signaling is mediated through distinct pathways. Exp Cell Res. 2003;287:361–373. doi: 10.1016/s0014-4827(03)00153-8. [DOI] [PubMed] [Google Scholar]
- 20.Hardie DG, Scott JW, Pan DA, Hudson ER. Management of cellular energy by the AMP-activated protein kinase system. FEBS Lett. 2003;546:113–120. doi: 10.1016/s0014-5793(03)00560-x. [DOI] [PubMed] [Google Scholar]
- 21.Hake SB, Garcia BA, Kauer M, Baker SP, Shabanowitz J, Hunt DF, Allis CD. Serine 31 phosphorylation of histone variant H3.3 is specific to regions bordering centromeres in metaphase chromosomes. Proc Natl Acad Sci U S A. 2005;102:6344–6349. doi: 10.1073/pnas.0502413102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lindner H, Helliger W, Puschendorf B. Histone separation by high-performance liquid chromatography on C4 reverse-phase columns. Anal Biochem. 1986;158:424–430. doi: 10.1016/0003-2697(86)90570-1. [DOI] [PubMed] [Google Scholar]
- 23.Shechter D, Dormann HL, Allis CD, Hake SB. Extraction, purification and analysis of histones. Nat Protoc. 2007;2:1445–1457. doi: 10.1038/nprot.2007.202. [DOI] [PubMed] [Google Scholar]
- 24.Bradbury EM, Inglis RJ, Matthews HR. Control of cell division by very lysine rich histone (F1) phosphorylation. Nature. 1974;247:257–261. doi: 10.1038/247257a0. [DOI] [PubMed] [Google Scholar]
- 25.Lu MJ, Dadd CA, Mizzen CA, Perry CA, McLachlan DR, Annunziato AT, Allis CD. Generation and characterization of novel antibodies highly selective for phosphorylated linker histone H1 in Tetrahymena and HeLa cells. Chromosoma. 1994;103:111–121. doi: 10.1007/BF00352320. [DOI] [PubMed] [Google Scholar]
- 26.Stork PJ, Schmitt JM. Crosstalk between cAMP and MAP kinase signaling in the regulation of cell proliferation. Trends Cell Biol. 2002;12:258–266. doi: 10.1016/s0962-8924(02)02294-8. [DOI] [PubMed] [Google Scholar]
- 27.Richards JS. New signaling pathways for hormones and cyclic adenosine 3’,5’-monophosphate action in endocrine cells. Mol Endocrinol. 2001;15:209–218. doi: 10.1210/mend.15.2.0606. [DOI] [PubMed] [Google Scholar]
- 28.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD, Jenuwein T. Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature. 2000;406:593–599. doi: 10.1038/35020506. [DOI] [PubMed] [Google Scholar]
- 29.Terada Y. Aurora-B/AIM-1 regulates the dynamic behavior of HP1alpha at the G2-M transition. Mol Biol Cell. 2006;17:3232–3241. doi: 10.1091/mbc.E05-09-0906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Long YC, Zierath JR. AMP-activated protein kinase signaling in metabolic regulation. J Clin Invest. 2006;116:1776–1783. doi: 10.1172/JCI29044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kaupp UB, Seifert R. Cyclic nucleotide-gated ion channels. Physiol Rev. 2002;82:769–824. doi: 10.1152/physrev.00008.2002. [DOI] [PubMed] [Google Scholar]
- 32.Stambrook PJ, Velez C. Reversible arrest of Chinese hamster V79 cells in G2 by dibutytyl AMP. Exp Cell Res. 1976;99:57–62. doi: 10.1016/0014-4827(76)90679-0. [DOI] [PubMed] [Google Scholar]
- 33.Blomhoff HK, Blomhoff R, Stokke T, deLange DC, Brevik K, Smeland EB, Funderud S, Godal T. cAMP-mediated growth inhibition of a B-lymphoid precursor cell line Reh is associated with an early transient delay in G2/M, followed by an accumulation of cells in G1. J Cell Physiol. 1988;137:583–587. doi: 10.1002/jcp.1041370327. [DOI] [PubMed] [Google Scholar]
- 34.Kurokawa K, Kato J. Cyclic AMP delays G2 progression and prevents efficient accumulation of cyclin B1 proteins in mouse macrophage cells. Cell Struct Funct. 1998;23:357–365. doi: 10.1247/csf.23.357. [DOI] [PubMed] [Google Scholar]
- 35.Miller WR. Regulatory subunits of PKA and breast cancer. Ann N Y Acad Sci. 2002;968:37–48. doi: 10.1111/j.1749-6632.2002.tb04325.x. [DOI] [PubMed] [Google Scholar]
- 36.Tortora G, Ciardiello F. Antisense targeting protein kinase A type I as a drug for integrated strategies of cancer therapy. Ann N Y Acad Sci. 2003;1002:236–243. doi: 10.1196/annals.1281.026. [DOI] [PubMed] [Google Scholar]
- 37.Kawabe T. G2 checkpoint abrogators as anticancer drugs. Mol Cancer Ther. 2004;3:513–519. [PubMed] [Google Scholar]