

Abstract
The inherited bone marrow failure syndromes are a diverse group of genetic diseases associated with inadequate production of one or more blood cell lineages. Examples include Fanconi anemia, dyskeratosis congenita, Diamond-Blackfan anemia, thrombocytopenia absent radii syndrome, severe congenital neutropenia, and Shwachman-Diamond syndrome. The management of these disorders was once the exclusive domain of pediatric subspecialists, but increasingly physicians who care for adults are being called upon to diagnose or treat these conditions. Through a series of patient vignettes, we highlight the clinical manifestations of inherited bone marrow failure syndromes in adolescents and young adults. The diagnostic and therapeutic challenges posed by these diseases are discussed.
Keywords: anemia, Diamond-Blackfan, aplastic anemia, congenital neutropenia, dyskeratosis congenita, Fanconi anemia, myelodysplastic syndromes, Shwachman-Diamond syndrome, thrombocytopenia absent radii syndrome
1. Overview
The inherited bone marrow failure syndromes (IBMFS) are a group of genetic disorders associated with inadequate production of one or more blood cell lineages (Table 1). The gene mutations responsible for these conditions often impact the development or function of extramedullary tissues, resulting in birth defects or clinical disease in specific organs. These disorders are characterized by a predisposition for malignancies, such as myelodysplastic syndrome (MDS), acute leukemia, and solid tumors.
Table 1. Examples of IBMFS.
| Disease | Hematologic manifestation | Pathway disrupted |
|---|---|---|
| Fanconi anemia | Pancytopenia | DNA repair |
| Dyskeratosis congenita | Pancytopenia | Telomere maintenance |
| Diamond-Blackfan anemia | Red cell aplasia | Ribosome biogenesis |
| Thrombocytopenia absent radii syndrome | Thrombocytopenia | mRNA processing and export |
| Severe congenital neutropenia | Neutropenia | Protein folding/trafficking |
| Shwachman-Diamond syndrome | Pancytopenia | Ribosome maturation |
| Congenital amegakaryocytic thrombocytopenia | Thrombocytopenia | Growth factor receptor signaling |
| Familial platelet disorder with propensity to AML | Thrombocytopenia | Transcriptional regulation |
| MonoMAC and related GATA2 deficiency disorders | Monocytopenia and lymphopenia | Transcriptional regulation |
Traditionally, IBMFS have been classified on the basis of clinical features, but increasingly this classification scheme is being replaced by one based on biochemical pathways (Table 1). Intriguingly, these pathways often entail “housekeeping” functions important for many cell types (e.g., DNA repair, ribosome biogenesis) rather than functions unique to hematopoietic progenitors. Why blood cell production is so profoundly and disproportionately affected by the disruption of certain housekeeping pathways remains an enigma.
The management of IBMFS was once the exclusive realm of pediatric subspecialists. Increasingly, however, physicians who care for adults are being called upon to diagnose or treat these disorders. The prompt recognition of an IBMFS is important, because this ensures optimal therapy while minimizing treatment-related toxicity. Moreover, the timely diagnosis of an IBMFS facilitates genetic counseling and (in some instances) cancer surveillance for the extended family. This article focuses on the manifestations of certain IBMFS in adolescents and young adults. Clinical vignettes are included to highlight the diagnostic and treatment challenges posed by these disorders (note, though inspired by actual cases, aspects of these vignettes have been altered for patient confidentiality).
2. Fanconi Anemia: a DNA repair defect
Fanconi anemia (FA) is a genetically and phenotypically heterogeneous disorder characterized by birth defects, progressive BMF, and a predisposition for cancer (Figure 1A-C). Originally, the diagnosis of FA was based on the presence of both aplastic anemia and certain congenital abnormalities. With the advent of more sensitive and specific diagnostic tests, it has become evident that 25% to 40% of patients with FA lack congenital abnormalities or fail to develop bone marrow failure (BMF) (1, 2). Nevertheless, these individuals remain at risk of other complications, such as leukemia and solid tumors.
Figure 1.
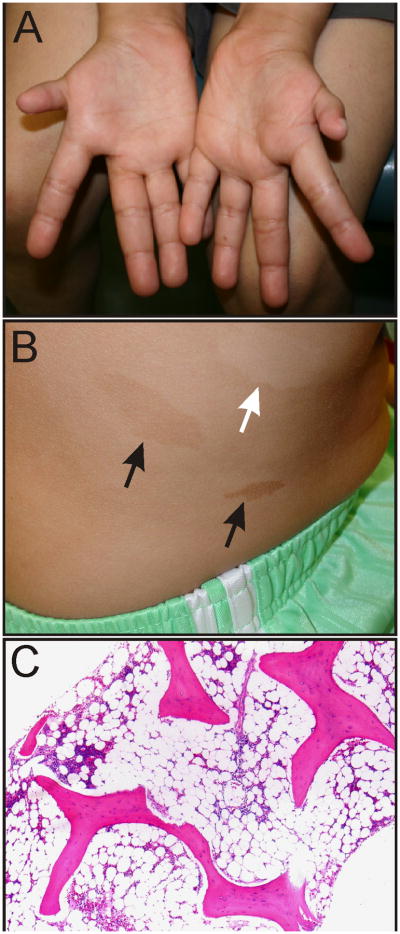
Phenotypic features of FA. (A) Hypoplastic thumbs in an 11-year-old. (B) Hypopigmented (white arrow) and hyperpigmented (black arrows) skin lesions on the trunk of an adolescent. Photograph courtesy of Susan J. Bayliss M.D. (C) Progressive aplastic anemia in a 20-year-old with FA. Most of the marrow space is occupied by fat.
Congenital and developmental defects associated with FA are listed in Table 2. Phenotypic variability makes the diagnosis of FA challenging (1-3). Indeed, patients with FA may have no (or only subtle) dysmorphic features, so physicians must maintain a high index of suspicion when other aspects of the clinical presentation suggest the possibility of FA (Vignette 1). Further obfuscating the diagnosis, pathogenic mutations in FA (and in other IBMFS) may exhibit incomplete penetrance or variable expressivity (i.e., different clinical manifestations), even within a family (Vignette 2).
Table 2. Congenital and developmental anomalies associated with FA (3, 95).
| Anomaly | % Affected |
|---|---|
| Skin (pigmentation changes) | 55-64 |
| Short stature | 51-63 |
| Upper limb | 43-49 |
| Testis | 20-32 |
| Eye | 23-38 |
| Kidney | 21-34 |
| Head (microcephaly and other anomalies) | 26 |
| Developmental disability | 11-16 |
| Birth weight < 2500 g | 11 |
| Ears (hearing loss) | 9-11 |
| Heart and lungs | 6-13 |
| Gastrointestinal tract | 5-14 |
Vignette 1. The physical manifestations of an IBMFs may be subtle. A 25-year-old woman with MDS is invited to “tell her story” to a group of students studying the pathophysiology of leukemia. Her physician, an internist, interviews her in front of the class. The patient, who is of Northern European ancestry, has a tan complexion, small chin, and high-pitched voice. She was well until a few months earlier when she was found to have cytopenias on routine laboratory testing. Bone marrow examination demonstrated MDS with acquired cytogenetic abnormalities, including monosomy 7 and trisomy 1q. An allogeneic bone marrow transplant is planned in the coming months. By happenstance, the interview is overheard by two physician-scientists, who urge that the patient be screened for FA. A diepoxybutane (DEB) chromosome fragility test on peripheral blood leukocytes is abnormal, establishing the diagnosis of FA. Accordingly, her pre-transplant conditioning regimen is modified so as to minimize toxicity from full dose alkylating agents such as cyclophosphamide.
Vignette 2. The phenotype of a given IBMFS may vary even within a single family. A 12-year-old male is diagnosed with severe aplastic anemia. He has no history of birth defects, and his height and weight are average for age. His younger sister is found to be a human leukocyte antigen (HLA) match and is referred to a physician for evaluation as a potential hematopoietic stem cell donor. The sister has short stature, congenital hearing loss, and normal peripheral blood counts. DEB chromosome fragility testing on blood leukocytes establishes the diagnosis of FA in both siblings. The sister is deemed to be an inappropriate source of hematopoietic stem cells, and a search for an unrelated donor is initiated.
BMF is one of the hallmarks of FA, although hematologic manifestations may vary. Thrombocytopenia and macrocytosis usually precede anemia or neutropenia. The median age at which hematologic abnormalities are discovered is 7 years. The BMF of FA is progressive; 50% of individuals presenting with isolated thrombocytopenia progress to pancytopenia within 4 years, and the cumulative incidence of BMF is 50-90% by 40 years of age (4-6). In some patients, hematologic abnormalities never become clinically evident.
FA is a bona fide cancer predisposition syndrome. The risk of cancer in individuals with FA is ∼50 times that of the general population, and the risk of leukemia is about ∼500 times that of the general population (6-8). By the age of 40 years, the cumulative incidences of MDS, acute myelogenous leukemia (AML), and solid tumors are 30%, 10%, and 30%, respectively (5, 6). Patients with mild hematologic abnormalities are at the highest risk for cancer because of survival bias (i.e., premature death from BMF precludes the opportunity to develop cancer). The most frequent solid tumors, affecting 5-11% of individuals with FA, are squamous cell carcinomas of the head and neck, esophagus, vulva, and anus (5-8). Hepatocellular adenomas and carcinoma have been reported in ∼3%.
The fundamental biochemical defect in FA is impaired DNA repair. At least 16 different DNA repair genes (FANCA, FANCB, FANCC, etc.) have been linked to this disorder. Inheritance of FA is autosomal recessive or, in the case of one gene (FANCB), X-linked. The proteins encoded by FANC genes form multimeric complexes involved in various DNA repair pathways, including nucleotide excision repair and homologous recombination (9). These multimeric complexes are critical for the repair of DNA interstrand crosslinks (10, 11).
Cells from FA patients exhibit a low tolerance for DNA-damage, both in vitro and in vivo. Accordingly, chemotherapy and radiation therapy must be administered at reduced dosages or avoided altogether in patients with FA (Vignette 3). The most widely used diagnostic tests for FA are based on hypersensitivity to the DNA-crosslinking agents DEB or mitomycin C (12). DEB- or mitomycin C-treated lymphocytes from individuals with FA demonstrate a significant increase in chromosomal breakage over normal controls. These tests are highly sensitive and (with rare exceptions) specific for FA (12); however, lymphocyte mosaicism caused by spontaneous gene reversion may confound the interpretation of these assays (13). Chromosome fragility testing of peripheral blood lymphocytes in somatic mosaics may demonstrate only a few cells with chromosomal breaks, so the average number of breaks per cell may fall within the normal range. The molecular mechanisms underlying gene reversion include intragenic recombination (in compound heterozygotes) and mitotic gene conversion (nonreciprocal exchange of genetic information during heteroduplex formation between nonsister chromatids) (14). Gene reversion confers a proliferative advantage to stem cells, leading to expansion of the revertant clone and progressive replacement of defective hematopoietic cells in bone marrow (15). Spontaneous gene reversion can be viewed as a natural form of gene therapy. Gene reversion has been documented in hematopoietic cells but not in fibroblasts from patients with FA (16). Therefore, chromosome fragility testing of fibroblasts should be considered in patients with clinical or genetic suspicion of FA but a negative DEB or mitomycin C test in peripheral blood lymphocytes (Vignette 3). Mosaicism caused by gene reversion contributes to phenotypic variability.
Vignette 3. Certain cancers are distinctively characteristic of IBMFS. An otherwise healthy 23-year-old man develops a tongue ulcer, and biopsy demonstrates squamous cell carcinoma. Human papilloma virus and p16 testing on the biopsy specimen are negative, and there is no history of carcinogen exposure. He has no obvious congenital anomalies, and his peripheral blood counts and family history are unremarkable. Partial glossectomy and neck dissection are performed. Recognizing the increased incidence of tongue cancer in young adults with FA, the astute otorhinolaryngologist orders a DEB chromosome fragility test on blood cells. The test is borderline abnormal. Subsequent DEB testing on skin fibroblasts is overtly abnormal, establishing the diagnosis of FA with somatic cell mosaicism. In light of this diagnosis, adjuvant radiation and chemotherapy are deemed to be contraindicated.
Patients with FA and only mild hematologic manifestations can be managed by monitoring of peripheral blood counts and periodic bone marrow examination for evidence of MDS. Contingency plans should be made for hematopoietic stem cell transplantation (HSCT), because hematologic complications may develop rapidly. Given the inherent sensitivity of patients with FA to radiation and chemotherapy, reduced intensity conditioning regimens are employed for HSCT (17). Androgen therapy can be used to improve bone marrow function in patients with FA (response rates of 50-80%) (18), although this therapy may be associated with significant side effects, including increased risk of hepatocellular carcinoma.
Adolescent and young adult patients with FA warrant close surveillance for development of malignancies. For example, the high risk of early vulvar cancer warrants initiation of regular gynecologic examinations during adolescence. Likewise, surveillance for head and neck squamous cell carcinoma should be performed on a semiannual basis. The Fanconi Anemia Research Foundation offers a monograph with surveillance and treatment guidelines (http://www.fanconi.org/images/uploads/other/FAHandbook3.pdf).
3. Dyskeratosis congenita: a telomere maintenance disorder
Dyskeratosis congenita (DC) is a phenotypically heterogeneous disorder caused by defects in telomere maintenance. Telomeres are DNA/protein structures that serve to protect chromosome ends from degradation (19). Human telomere DNA consists of thousands of repeats of TTAGGG, which are synthesized by the enzyme telomerase. This enzyme contains two core components: a reverse transcriptase (TERT) and an RNA molecule (TERC) that acts as a template for the synthesis of telomere repeats. Most somatic cells lack telomerase, so telomeres shorten with each cell division owing to the inability of DNA polymerase to copy to the very end of a DNA strand (20, 21). When telomeres become critically short, cell cycle arrest or cell death ensues. Stem cells contain telomerase, which ensures that telomere length is maintained over the course of repeated cell divisions.
Originally, DC was recognized by the classic diagnostic triad of reticular skin pigmentation, nail dystrophy, and mucosal leukoplakia (Figure 2A,B) (22). Other clinical manifestations of DC, including BMF, are summarized in Table 3. The cumulative incidence of BMF in patients with DC is approximately 50% by 50 years of age (6). In the prototypical patient with DC, mucocutaneous features appear in childhood, BMF develops in adolescence, and death occurs as a young adult. This inexorably deteriorating clinical course reflects progressive stem cell dysfunction in tissues with high cell turnover.
Figure 2.

Phenotypic features of DC. (A) Nail dystrophy (arrow) in an adolescent. (B) Oral leukoplakia in an adult. (C) Skin changes in an adult. Hypopigmented lesions and the typical reticular rash of DC are evident on the upper chest (black arrow). Hypopigmentation is more pronounced in the sun-damaged skin of the neck (white arrow). (D,E) Pulmonary fibrosis on chest radiograph and CT scan of an adult. Photographs courtesy of Susan J. Bayliss, M.D. and William H. McAlister, M.D.
Table 3. Prevalence of clinical features in patients with classic DC (3, 22, 95).
| Clinical feature | % Affected |
|---|---|
| Diagnostic mucocutaneous features | |
| • Skin pigmentation chagnes | 88-89 |
| • Nail dystrophy | 73-88 |
| • Leukoplakia | 64-78 |
| Eye abnormalities, excessive tearing | 31-38 |
| Pulmonary fibrosis | 20 |
| Learning difficulties or developmental delay | 14-25 |
| Teeth abnormalities | 17-19 |
| Hair loss or early graying | 16-18 |
| Hyperhidrosis | 10-15 |
| Short stature | 14 |
| Gastrointestinal abnormalities (liver fibrosis, esophageal strictures, ulcers, malabsorption) | 14 |
| Malignancy | 10-12 |
| Microcephaly | 8 |
| Urinary tract abnormalities (urethral stricture, phimosis) | 7 |
| Cerebellar hypoplasia, ataxia | 7 |
| Gonadal abnormalities (hypogonadism, cryptorchidism) | 1-5 |
The identification of 9 of the genes responsible for DC (DKC1, TERC, TERT, TINF2, NOP10, NHP2, WRAP53, CTC1, RTEL1) and the realization that these genes converge on the common pathway of telomere maintenance have changed the perception of DC. Now it is thought that individuals with the classic constellation of mucocutaneous features and BMF represent only a fraction of the patients with DC (20-22). MDS, MDS/AML, or an extramedullary complication may be the initial manifestation of DC in adolescents or young adults (Vignette 4). Pulmonary fibrosis due to disruption of lung stem cells is another common manifestation of DC (Figure 2C,D). Liver cirrhosis is another frequent complication.
Vignette 4. An acquired, non-neoplastic disorder affecting an extramedullary tissue may be the first manifestation of an IBMFS. An 18-year-old man develops recurrent urethral strictures necessitating surgical intervention. A routine preoperative CBC demonstrates thrombocytopenia, triggering a consultation from a hematologist, who notes the presence of nail dystrophy. The family history is not informative. Bone marrow biopsy shows hypocellularity. Telomere length analysis demonstrates marked shortening in peripheral blood leukocytes, consistent with the diagnosis of DC. No pathogenic mutation is detected in any of the known DC genes. The patient subsequently develops avascular necrosis of the hip, another known complication of DC.
Like FA, DC is a cancer predisposition syndrome, and malignancy is the third leading cause of death (after BMF and pulmonary fibrosis) in these patients. The cumulative incidence of malignancy is estimated to be 20-30% by the age of 50 years (6). Malignancies usually develop in the third decade of life and are therefore diagnosed more often in individuals with milder forms of the disease. The most common solid tumors in patients with DC are squamous cell carcinomas of the head and neck, gastrointestinal tract, and female genital tract (23, 24). By 50 years of age, the cumulative incidences of MDS and AML are about 30% and 10%, respectively (6). Although DC rarely causes MDS in children, it should be considered in the differential diagnosis in young adults with MDS or MDS/AML.
The presence of short telomeres in circulating blood cells is a hallmark of patients with BMF and DC (25, 26). Peripheral blood leukocyte telomere length in individuals with BMF secondary to DC is far below that of age-matched healthy controls. In the setting of BMF, measurement of telomere length is a sensitive but nonspecific screening method for DC. Telomere shortening is not pathognomonic of DC, as approximately 30% of patients with BMF due to other causes have peripheral blood leukocyte telomere lengths ≤ 1st percentile (26). Therefore, clinicians should not make the diagnosis of DC solely on the basis of BMF and shortened telomeres. Caution should be exercised in interpreting the results of telomere length measurements in individuals without BMF, because in the absence of marrow failure telomere length measurements may not reliably identify mutation carriers (26, 27).
DC exhibits different modes of inheritance (20-22). The most common form is X-linked, due to mutations in the DKC1 gene encoding dyskerin, a protein involved in telomere maintenance and ribosome biogenesis. Autosomal dominant forms of DC are caused by mutations in the TERC or TERT genes. These autosomal dominant forms of DC exhibit genetic anticipation, a phenomenon in which an inherited disease manifests at increasingly younger ages or with increased severity in each succeeding generation (owing the inheritance of progressively shorter telomeres in successive generations) (20-22). In adults the most common manifestions of TERC or TERT mutations are cytopenias and pulmonary fibrosis (20-22). Thus, individuals with pulmonary fibrosis and a macrocytic anemia or aplastic anemia in the family should be examined for TERC or TERT mutations (28). Another gene linked to autosomal dominant DC is TINF2, which encodes a component of the shelterin complex that protects telomeres. TINF2 mutations usually manifest in early childhood (29, 30). Because patients with TINF2 mutations seldom have offspring, most TINF2 mutations are sporadic (29). Autosomal recessive forms of DC are caused by biallelic mutations in other genes involved in telomere maintenance, including CTC1, RTEL1, NOP10, NHP2, and WRAP53. In approximately half of patients with the classic constellation of findings (BMF, mucocutaneous features, and short telomeres), none of the known DC genes is mutated, suggesting that additional causal genes will be discovered in the coming years.
Treatment of patients with DC is similar to that of patients with FA. Patients with DC and only mild hematologic manifestations can be managed by monitoring of peripheral blood counts and serial bone marrow examinations for evidence of MDS. Contingency plans for a reduced intensity HSCT should be in place, as hematologic complications may develop rapidly. HSCT-related morbidity and mortality is greater for DC than other IBMFS. Pulmonary and vascular complications are the major cause of HSCT-associated mortality in patients with DC (31-33). In patients without a suitable donor, androgen therapy can be used to improve bone marrow function. Given the risk of pulmonary fibrosis, patients with DC should refrain from smoking, avoid drugs with known pulmonary toxicity, and have their lungs shielded during radiation therapy. Lung transplantation is an option for some patients with DC-associated pulmonary fibrosis (34). As with FA, surveillance for head and neck squamous cell carcinoma should be performed on a regular basis.
4. Diamond-Blackfan anemia: a defect in ribosomal biogenesis
Diamond-Blackfan anemia (DBA) is a genetically and phenotypically heterogeneous condition that is associated with reduced or absent erythroid precursors in bone marrow. Macrocytic anemia and reticulocytopenia are the other major diagnostic criteria for DBA. In some individuals with DBA the anemia is transient. In rare cases DBA progresses to aplastic anemia (35). Elevated levels of erythrocyte adenosine deaminase (eADA), a critical enzyme in the purine salvage pathway, are present in 80% to 89% of patients with DBA (36). The diagnosis of DBA is usually made before 1 year of age, but occasionally the condition is diagnosed in older children or adults. In ∼50% of cases, DBA is associated with congenital anomalies or growth retardation (Table 4), but this estimate may be inaccurate because case definitions often stipulate diagnosis at < 1 year of age. Among the more common birth defects are thumb anomalies, midline facial defects, and congenital heart disease (Figure 3A,B). In some individuals with DBA the clinical manifestations are subtle (Vignette 5).
Table 4. Congenital and developmental anomalies associated with DBA (35, 96-98).
| System | Range of anomalies | % Affected |
|---|---|---|
| Skeletal | Short stature | 30-33 |
| Head, face, palate | Hypertelorism, cleft palate, high arched palate, microcephaly, micrognathia, microtia, low set ears, low hairline, epicantus, ptosis, flat broad nasal bridge | 21-36 |
| Upper limb | Triphalangeal, duplex or bifid, hypoplastic thumb, syndactyly, absent radial artery | 9-21 |
| Renal, urogenital | Absent kidney, horseshoe kidney, hypospadias | 7-19 |
| Cardiopulmonary | Ventricular or atrial septal defects, coarctation of the aorta, complex cardiac anomalies | 7-15 |
| Eyes | Congenital glaucoma, strabismus, congenital cataract | 12 |
| Neck | short neck, webbed neck | |
| Brain | Learning difficulties | 7 |
Figure 3.

Phenotypic features of DBA. (A) Abnormal thumb in an adult with an RPL11 mutation. (B) Muscular ventricular septal defect. Color indicates blood flow across the defect. Abbreviations: LV, left ventricle; RV, right ventricle. Echocardiogram courtesy of Patrick Jay, M.D., Ph.D.
Vignette 5. Macrocytosis is an often overlooked clue to the diagnosis of an IBMFS and may be the sole clinical manifestation. An ostensibly healthy woman of normal stature gives birth to 2 children with DBA. The older child is transfusion-dependent, while the younger is glucocorticoid-responsive. Genetic testing demonstrates a heterozygous nonsense mutation in RPS19 in the woman and her affected children. The woman's peripheral blood counts are normal, but her erythrocytes are macrocytic (MCV = 101 fL).
Like patients with FA and DC, patients with DBA are predisposed to malignancy, including MDS, AML, and solid tumors (37). The median age at diagnosis of malignancy is 41 years. The relative risk of all cancers (excluding MDS) in DBA is 5.4-fold with a cumulative incidence of 22% by 46 years of age (37). It should be noted, however, that the accumulated cancer data in the DBA Registry is limited and biased towards younger patients. Consequently, there is insufficient data to define appropriate cancer surveillance recommendations for adult DBA patients.
DBA is caused by mutations in genes encoding either the small 40 S (e.g., RPS19, RPS17) or large 60 S (e.g., RPL5, RPL11) ribosomal subunits. About 25% of mutations occur in RPS19 (38). The ribosomal gene mutations in DBA are always heterozygous and include missense, nonsense, frameshift, and splice site mutations as well as large deletions; consequently, haploinsufficiency is thought to be responsible for disease (39). Although ribosomal proteins do not participate directly in the protein synthesis activity of the ribosome, they are important for ribosome biogenesis, so DBA is a disorder of ribosome biogenesis or ribosomopathy. Genetic testing can currently provide a definite diagnosis in 50% to 70% of cases (38).
Glucocorticoid administration improves erythropoiesis in a majority of patients with DBA (60% to 70% achieve transfusion independence in response to glucocorticoids) (40-42). The mechanistic basis for this effect is unclear, although studies suggest that glucocorticoids expand burst-forming unit-erythroid (BFU-E) progenitors (43, 44). Red cell transfusion remains the mainstay of therapy for glucocorticoid-resistant DBA. Recently, the amino acid leucine has been reported to be beneficial for the treatment of some patients with DBA (45). In addition to serving as a substrate for protein synthesis, leucine regulates protein metabolism through activation of the mTOR pathway (46). Studies of mouse and zebrafish models of DBA have shown that leucine treatment lessens the degree of anemia (47, 48).
HSCT is curative in patients with DBA and may be considered in transfusion-dependent patients, particularly those with an HLA-matched sibling donor or those progressing to aplastic anemia. Data from the DBA registry show survival rates of 70% in individuals ≥ 9 years of age who receive transplants from sibling donors (42).
The decision to perform HSCT in patients with transfusion-dependent DBA is confounded by the possibility of spontaneous hematologic remission. Appoximately 25% of patients experience this phenomenon, which is defined as a stable, physiologically acceptable hemoglobin level maintained for at least 6 months in the absence of therapy (49). In the majority of cases hematologic remission occurs within the first or second decade of life in patients who were glucocorticoid-responsive. Hematologic relapses may occur, especially during pregnancy or when estrogen-containing oral contraceptives are used (50). Gene reversion in hematopoietic stem/progenitor cells may account for the normalization of erythropoiesis in some patients (51).
5q− syndrome, a condition that typically affects older adults, shares several features with DBA, including macrocytic anemia with a paucity of erythroid precursors (52). Acquired deletions of RPS14 (in combination with neighboring genes) have been implicated as causative in 5q− syndrome, implying that 5q- syndrome, like DBA, is at least in part a ribosomopathy. Adolescent and young adult patients with atypical presentations of DBA (macrocytic hypoplastic anemia with normal eADA activity, no congenital anomalies, age > 1 year at presentation) have been shown to harbor small, acquired, microdeletions encompassing RPS14 and adjacent genes (52).
5. Thrombocytopenia with absent radii (TAR): a defect in mRNA processing and export
The hallmarks of this IBMFS are bilateral defects in development of the radii (Figure 4A) and severe thrombocytopenia at birth. Bone marrow aspiration shows a decrease in megakaryocytes, although this is not required to make the diagnosis (53). During infancy, patients often require platelet transfusions to maintain a platelet count above 10,000/μL. After the first year of life, platelet transfusion requirements usually diminish. Extraskeletal manifestations observed in individuals with TAR syndrome include short stature, facial dysmorphism, cardiac defects, and genitourinary malformations. Orthopedic interventions dominate the treatment of TAR syndrome later in life. The development of aplastic anemia has not been observed, but AML and ALL have been reported, albeit rarely, in patients with TAR syndrome (54). Vignette 6 describes a patient with TAR syndrome who developed MDS.
Figure 4.

Phenotypic features of TAR syndrome. (A) Absence of the radius. (B) Late-onset MDS in an adolescent with TAR syndrome (see Vignette 6). Radiograph courtesy of William H. McAlister, M.D.
Vignette 6. IBMFS are associated with an increased risk of late-onset MDS. A 14-year-old female with TAR syndrome, who has been transfusion-independent since infancy, develops severe macrocytic anemia and worsening thrombocytopenia. Bone marrow aspiration/biopsy demonstrates MDS (refractory anemia with excess blasts type 2; Figure 4B) with acquired cytogenetic abnormalities including ins(3;3)(q26;q21q26). She receives treatment with decitabine and then undergoes an unrelated donor HSCT.
TAR syndrome is caused by compound inheritance of mutations in the RBM8A gene, encoding Y14, an RNA-binding protein. Y14 is a component of the exon junction complex that is involved in a variety of cellular functions, including nuclear export of transcripts, nonsense mediated decay, and translational enhancement (55). Most individuals with TAR harbor one null RBM8A allele, caused by an interstitial deletion of chromosome 1q21.1 (56), and one hypomorphic RBM8A allele, caused by a polymorphism in either the 5′-untranslated region or first intron (55). These polymorphisms reduce expression of the hypomorphic allele in a cell type- and developmental stage-specific manner.
6. Severe congenital neutropenia: a defect in protein folding/trafficking
Severe congenital neutropenia (SCN) is a genetically heterogeneous group of disorders characterized by severe neutropenia at birth, with neutrophil counts generally < 200 cells/μL. Bone marrow evaluation demonstrates an arrest at the promyelocyte/myelocyte stage of neutrophil maturation. Bacterial infections, such as omphalitis, skin abscesses, deep tissue abscesses, oral ulcers, and pneumonia develop in 90% of patients with SCN by the age of 6 months.
Historically, patients with SCN had a poor prognosis and generally died in the first or second decade of life due to overwhelming infection. The advent of G-CSF therapy changed the natural history of this disease by reducing in the incidence and severity of bacterial infections (57). Despite G-CSF therapy, mortality due to infection remains a major concern in SCN, with a 12% cumulative incidence of death due to sepsis by 15 years of age.
Patients with SCN also have a markedly increased risk for MDS or AML with an estimated hazard rate of 2% per year (58-60). A consistent feature of myeloid transformation in SCN is a high rate of chromosome 7 abnormalities. In one series, nearly 60% of AML/MDS samples displayed complete or partial loss of chromosome 7. Patients with SCN who require very high doses of G-CSF to achieve an adequate neutrophil count are at increased risk for transformation to MDS/AML (60), although this observation has not been shown to be a result of G-CSF promoting a malignant clone. SCN exhibits multiple patterns of inheritance, including autosomal recessive, autosomal dominant, X-linked, and sporadic forms. Accordingly, mutations of several different genes have been shown to cause SCN, including ELANE, HAX1, G6PC3, GFI1, CSF3R, and the X-linked gene WAS. Mutations in ELANE, encoding neutrophil elastase, account for 60% of the cases (61-63). While it is unclear how ELANE mutations cause neutropenia, nearly all of the reported mutations are missense substitutions, small insertions or deletions preserving translational reading frame, or chain-terminating mutations near the C-terminus of the protein. This mutational spectrum suggests that the pathogenic mechanism involves synthesis of an abnormal protein rather than haploinsufficiency. One model posits that protein misfolding activates the unfolded protein response, which induces apoptosis of neutrophil precursors (64-66). Aberrant intracellular trafficking of a mutant protein may also contribute to pathogenesis.
Allogeneic HSCT is the only curative therapy for SCN. In the absence of MDS or MDS/AML, the overall survival for patients with SCN who undergo HSCT from an HLA-matched sib is 89% (67). Widely accepted indications for HSCT in patients with SCN include inadequate neutrophil response to G-CSF and the development of MDS or AML. The more vexing question is whether to perform HSCT in patients who appear well on G-CSF therapy (Vignette 7).
Vignette 7. The looming risk of AML may impact clinical decision making in patients with IBMFS. A 13-year-old girl with SCN due to an ELANE missense mutation is referred for consideration of HSCT. She was diagnosed at 3 months of age after presenting with omphalitis and carbuncles. She has received daily injections of G-CSF since that time. Annual bone marrow examinations have shown no overt morphological or cytogenetic evidence of MDS, but her requirements for G-CSF have increased significantly over the past year (currently 12 μg/kg/day to maintain the neutrophil count between 500/μL and 1000/μL). Her healthy brother is not an HLA match. In light of the declining neutrophil response to G-CSF and the high risk of progression to MDS/AML, her physicians recommend a matched unrelated donor HSCT using a non-myeloablative conditioning regimen.
7. Shwachman-Diamond Syndrome: a defect in ribosome maturation
Shwachman-Diamond syndrome (SDS) is characterized by exocrine pancreatic insufficiency, marrow dysfunction (including increased risk for MDS/AML), and skeletal abnormalities (Table 5) (68-70). Most cases of SDS are diagnosed in early childhood, although some patients, especially those with mild pancreatic insufficiency, are diagnosed as adults. Approximately 90% of patients with SDS have a biallelic mutations in the Shwachman-Bodian-Diamond syndrome (SBDS) gene located on chromosome 7(q11) (69, 71-74).
Table 5. Congenital and developmental anomalies associated with SDS (70, 99, 100).
| Feature | % Affected |
|---|---|
| Hematologic | |
| • Neutropenia | 77-100 |
| • Severe neutropenia (< 500/μL) | 23-67 |
| • Anemia | 17-66 |
| • Thrombocytopenia | 24-70 |
| Gastrointestinal | |
| • Exocrine pancreatic insufficiency | 85-100 |
| • Elevated transaminases | 45-100 |
| Skeletal abnormalites | |
| • Metaphyseal dysostosis | 41-100 |
| • Rib cage abnormalities | 12-92 |
| Short stature | 52-95 |
Intermittent or chronic neutropenia (of varying severity) is evident in the majority of individuals with SDS. Anemia, typically with mild macrocytosis, is present in ∼50% of cases (72). Similarly, thrombocytopenia is present in ∼50% of cases. Bone marrow examination usually shows hypoplasia (75). Patients with SDS may experience transient or protracted exacerbations of cytopenia, including progression to severe aplastic anemia (74, 76). Such exacerbations may occur in adults who have outgrown the care of pediatric subspecialists.
Another noteworthy hematologic feature of SDS is the propensity to develop clonal cytogenetic abnormalities, such as i(7)(q10) and del(20)(q11), in the absence of MDS or AML. In one prospective study of patients with SDS followed for a maximum of 5 years, acquired cytogenetic abnormalities were observed in 29% of individuals (77). Such hematopoietic clones may persist for years and are not necessarily harbingers of progressive marrow dysfunction (Vignette 8). Indeed, neither i(7)(q10) nor del(20)(q11) is associated with an increased risk of transformation to MDS or MDS/AML (78, 79).
Vignette 8. An acquired chromosomal abnormality in hematopoietic progenitors does not portend a negative clinical outcome in all IBMFS. A young girl is diagnosed with SDS on the basis of exocrine pancreatic insufficiency, short stature, and metaphyseal dysostosis. Subsequent genetic testing demonstrates bilallelic mutations in SBDS. Over the ensuing years her peripheral blood cell counts remain relatively stable with a neutrophil count of ∼1,200/μL, Hb of ∼11 g/dL, and a platelet count of ∼80,000/μL. At age 15 years, routine bone marrow testing demonstrates an acquired cytogenetic abnormality [46,XX,i(7)(q10)] but no histological evidence of myelodysplasia. Subsequent bone marrow evaluations document persistence of this benign hematopoietic clone for over the next decade. At 26 years of age she gives birth to a healthy child.
The reported rate of transformation to MDS/AML for patients with SDS ranges from 0% to 36% (6, 59, 74). Part of this variability may reflect differences in study populations, because median patient age was greater in the studies with the higher reported rates of transformation. Published reports of solid tumors in patients with SDS are rare.
Exocrine pancreatic deficiency can be confirmed by demonstration of low serum isoamylase, which decreased in both children and adults with SDS (80). Imaging of the pancreas may reveal characteristic fatty infiltration (Figure 5A). For unclear reasons, pancreatic insufficiency improves with age in many patients; by 4 years of age, ∼50% of patients no longer require pancreatic enzyme supplements based on evidence of fat absorption (81).
Figure 5.

Phenotypic features of SDS. (A) MRI showing fatty infiltration of the pancreas (white arrow). (B) Metaphyseal dysostosis (black arrows). Images courtesy of William H. McAlister, M.D.
Skeletal manifestations of SDS include metaphyseal dysostosis (Figure 5B), rib cage dysplasia, and osteoporosis (82, 83). Spinal compression fractures are common among adults with SDS (75). Even with adequate pancreatic enzyme replacement, most patients with SDS remain below or at the 3rd percentile for height (84).
The majority of SBDS mutations cause a dramatic reduction in SBDS protein production (71, 85, 86). Patients carrying homozygous null alleles for SBDS have not been reported, suggesting that the gene product is essential for life. SBDS encodes a protein involved in ribosomal maturation and implicated in additional functions, such as cell proliferation, mitosis, and maintence of the stromal microenvironment (68, 87). Some of the acquired chromosomal abnormalities associated with SDS appear to mitigate the effects of intracellular SBDS protein depletion. For example, duplication of the SBDS locus, as occurs in i(7)(q10), is thought to increase the level of SBDS protein in progenitor cells (88). Similarly, del(20)(q11) results in loss of the EIF6 gene whose protein product competes with SBDS (89). Thus, the appearance of benign chromosomal abnormalities in SDS is thought to reflect a clonal proliferation advantage.
Allogeneic HSCT, the only cure for the hematologic manifestations of SDS, is generally reserved for patients with severe cytopenias or MDS/AML. HSCT in patients with SDS is associated with increased regimen-related toxicity, notably of the heart, lung and liver (90). In the absence of MDS/AML, the overall survivial for patients with SDS who undergo allogeneic HSCT is ∼65% (91). This survival rate may improve in the coming years with the more widespread use of non-myeloablative conditioning regimens that minimize organ toxicity.
The management of individuals with SDS, including adolescents and adults, was summarized recently in a consensus guideline (75).
8. Other IBMFS
It should be noted that this review article is not all-inclusive. There are other IBMFS that can impact the health of adolescents and adults, including the following: 1) congenital amegakaryocytic thrombocytopenia, caused by bialleic mutations in the MPL gene encoding the receptor for thrombopoietin (92), 2) familial platelet disorder with propensity to AML (FDP/AML), due to monoallelic mutations in RUNX1 (93), and 3) GATA2 haploinsufficiency disorders [monocytopenia and mycobacterial infections (MonoMAC); dendritic cell, monocyte, B, and natural killer (NK) lymphoid deficiency; familial MDS/AML; and Emberger syndrome (primary lymphedema with MDS)] (94). The principles discussed in this review are applicable to these other syndromes.
9. Conclusions
As the cases in this article illustrate, IBMFS are phenotypically heterogeneous disorders associated with congenital anomalies and a predisposition to malignancy. The phenotypic variablility of any given IBMFS may be striking, even within a single family. A number of factors contribute to the incomplete penetrance and variable expressivity of these disorders, including modifier genes and spontaneous gene conversion leading to somatic cell mosaicism. In some patients with IBMFS, the hematologic and physical abnormalities are subtle or nonexistent, and the diagnosis is made later in life as a result of other complications such as MDS/AML, a characteristic solid tumor, or a non-malignant condition affecting an extramedullary tissue (e.g., pulmonary fibrosis, urethral stricture, avascular necrosis). Accordingly, physicians who care for adolescents and young adults must consider the diagnosis of an IBMFS in patients who manifest these features.
As rapid genetic diagnostic tools are developed, the true prevalence of IBMFS among adolescents and adults will be elucidated. With such tools, it seems plausible that patients with atypical presentations of known BMF syndromes and novel BMF disorders will be diagnosed more often in adolescents and adults than in young children.
Key Messages.
Inherited bone marrow failure syndromes are genetically and phenotypically heterogeneous disorders associated with inadequate blood cell production, congenital anomalies, and a predisposition to malignancy.
In some patients with inherited bone marrow failure syndromes, the hematologic and physical abnormalities are subtle or nonexistent, and the diagnosis is made later in life as a result of other complications, such as cancer or pulmonary fibrosis, or through genotyping of a whole family.
Acknowledgments
We thank Joshua Rubin, Monica Hulbert, Shalini Shenoy, and Markku Heikinehimo for their comments on this manuscript. We thank William McAlister, Susan Bayliss, and Patrick Jay for providing image. M.B., P.M., and D.W. are supported by NIH grant CA105312. M.B. has additional support from Buck Family Endowed Chair in Hematology at the Children's Hospital of Philadelphia and by NIH grant DK084188.
Footnotes
Disclosure of Interests: The authors have nothing to disclose.
References
- 1.Giampietro PF, Verlander PC, Davis JG, Auerbach AD. Diagnosis of Fanconi anemia in patients without congenital malformations: an international Fanconi Anemia Registry Study. Am J Med Genet. 1997 Jan 10;68(1):58–61. [PubMed] [Google Scholar]
- 2.Yoshimi A, Niemeyer C, Baumann I, Schwarz-Furlan S, Schindler D, Ebell W, et al. High incidence of Fanconi anaemia in patients with a morphological picture consistent with refractory cytopenia of childhood. Br J Haematol. 2013 Jan;160(1):109–11. doi: 10.1111/bjh.12083. [DOI] [PubMed] [Google Scholar]
- 3.Shimamura A, Alter BP. Pathophysiology and management of inherited bone marrow failure syndromes. Blood Rev. 2010 May;24(3):101–22. doi: 10.1016/j.blre.2010.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Butturini A, Gale RP, Verlander PC, Adler-Brecher B, Gillio AP, Auerbach AD. Hematologic abnormalities in Fanconi anemia: an International Fanconi Anemia Registry study. Blood. 1994 Sep 1;84(5):1650–5. [PubMed] [Google Scholar]
- 5.Kutler DI, Singh B, Satagopan J, Batish SD, Berwick M, Giampietro PF, et al. A 20-year perspective on the International Fanconi Anemia Registry (IFAR) Blood. 2003 Feb 15;101(4):1249–56. doi: 10.1182/blood-2002-07-2170. [DOI] [PubMed] [Google Scholar]
- 6.Alter BP, Giri N, Savage SA, Peters JA, Loud JT, Leathwood L, et al. Malignancies and survival patterns in the National Cancer Institute inherited bone marrow failure syndromes cohort study. Br J Haematol. 2010 Jul;150(2):179–88. doi: 10.1111/j.1365-2141.2010.08212.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Alter BP. Cancer in Fanconi anemia, 1927-2001. Cancer. 2003 Jan 15;97(2):425–40. doi: 10.1002/cncr.11046. [DOI] [PubMed] [Google Scholar]
- 8.Rosenberg PS, Greene MH, Alter BP. Cancer incidence in persons with Fanconi anemia. Blood. 2003 Feb 1;101(3):822–6. doi: 10.1182/blood-2002-05-1498. [DOI] [PubMed] [Google Scholar]
- 9.Sengerova B, Wang AT, McHugh PJ. Orchestrating the nucleases involved in DNA interstrand cross-link (ICL) repair. Cell Cycle. 2011 Dec 1;10(23):3999–4008. doi: 10.4161/cc.10.23.18385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer. 2011 Jul;11(7):467–80. doi: 10.1038/nrc3088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Crossan GP, Patel KJ. The Fanconi anaemia pathway orchestrates incisions at sites of crosslinked DNA. J Pathol. 2012 Jan;226(2):326–37. doi: 10.1002/path.3002. [DOI] [PubMed] [Google Scholar]
- 12.Oostra AB, Nieuwint AW, Joenje H, de Winter JP. Diagnosis of fanconi anemia: chromosomal breakage analysis. Anemia. 2012;2012:238731. doi: 10.1155/2012/238731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Joenje H, Arwert F, Kwee ML, Madan K, Hoehn H. Confounding factors in the diagnosis of Fanconi anaemia. Am J Med Genet. 1998 Oct 12;79(5):403–5. doi: 10.1002/(sici)1096-8628(19981012)79:5<403::aid-ajmg16>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- 14.Gross M, Hanenberg H, Lobitz S, Friedl R, Herterich S, Dietrich R, et al. Reverse mosaicism in Fanconi anemia: natural gene therapy via molecular self-correction. Cytogenet Genome Res. 2002;98(2-3):126–35. doi: 10.1159/000069805. [DOI] [PubMed] [Google Scholar]
- 15.Hirschhorn R. In vivo reversion to normal of inherited mutations in humans. J Med Genet. 2003 Oct;40(10):721–8. doi: 10.1136/jmg.40.10.721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Waisfisz Q, Morgan NV, Savino M, de Winter JP, van Berkel CG, Hoatlin ME, et al. Spontaneous functional correction of homozygous fanconi anaemia alleles reveals novel mechanistic basis for reverse mosaicism. Nat Genet. 1999 Aug;22(4):379–83. doi: 10.1038/11956. [DOI] [PubMed] [Google Scholar]
- 17.Pasquini R, Carreras J, Pasquini MC, Camitta BM, Fasth AL, Hale GA, et al. HLA-matched sibling hematopoietic stem cell transplantation for fanconi anemia: comparison of irradiation and nonirradiation containing conditioning regimens. Biol Blood Marrow Transplant. 2008 Oct;14(10):1141–7. doi: 10.1016/j.bbmt.2008.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rose SR, Kim MO, Korbee L, Wilson KA, Ris MD, Eyal O, et al. Oxandrolone for the treatment of bone marrow failure in Fanconi anemia. Pediatr Blood Cancer. 2014 Jan;61(1):11–9. doi: 10.1002/pbc.24617. [DOI] [PubMed] [Google Scholar]
- 19.Palm W, de Lange T. How shelterin protects mammalian telomeres. Annu Rev Genet. 2008;42:301–34. doi: 10.1146/annurev.genet.41.110306.130350. [DOI] [PubMed] [Google Scholar]
- 20.Bessler M, Wilson DB, Mason PJ. Dyskeratosis congenita and telomerase. Curr Opin Pediatr. 2004 Feb;16(1):23–8. doi: 10.1097/00008480-200402000-00006. [DOI] [PubMed] [Google Scholar]
- 21.Bessler M, Du HY, Gu B, Mason PJ. Dysfunctional telomeres and dyskeratosis congenita. Haematologica. 2007 Aug;92(8):1009–12. doi: 10.3324/haematol.11221. [DOI] [PubMed] [Google Scholar]
- 22.Vulliamy T, Dokal I. Dyskeratosis congenita. Semin Hematol. 2006;43(3):157–66. doi: 10.1053/j.seminhematol.2006.04.001. [DOI] [PubMed] [Google Scholar]
- 23.Handley TP, Ogden GR. Dyskeratosis congenita: oral hyperkeratosis in association with lichenoid reaction. J Oral Pathol Med. 2006 Sep;35(8):508–12. doi: 10.1111/j.1600-0714.2006.00434.x. [DOI] [PubMed] [Google Scholar]
- 24.Atkinson JC, Harvey KE, Domingo DL, Trujillo MI, Guadagnini JP, Gollins S, et al. Oral and dental phenotype of dyskeratosis congenita. Oral Dis. 2008 Jul;14(5):419–27. doi: 10.1111/j.1601-0825.2007.01394.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Alter BP, Baerlocher GM, Savage SA, Chanock SJ, Weksler BB, Willner JP, et al. Very short telomere length by flow fluorescence in situ hybridization identifies patients with dyskeratosis congenita. Blood. 2007 Sep 1;110(5):1439–47. doi: 10.1182/blood-2007-02-075598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du HY, Pumbo E, Ivanovich J, An P, Maziarz RT, Reiss UM, et al. TERC and TERT gene mutations in patients with bone marrow failure and the significance of telomere length measurements. Blood. 2009 Jan 8;113(2):309–16. doi: 10.1182/blood-2008-07-166421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Walne AJ, Bhagat T, Kirwan M, Gitiaux C, Desguerre I, Leonard N, et al. Mutations in the telomere capping complex in bone marrow failure and related syndromes. Haematologica. 2013 Mar;98(3):334–8. doi: 10.3324/haematol.2012.071068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Parry EM, Alder JK, Qi X, Chen JJ, Armanios M. Syndrome complex of bone marrow failure and pulmonary fibrosis predicts germline defects in telomerase. Blood. 2011 May 26;117(21):5607–11. doi: 10.1182/blood-2010-11-322149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Walne AJ, Vulliamy T, Beswick R, Kirwan M, Dokal I. TINF2 mutations result in very short telomeres: analysis of a large cohort of patients with dyskeratosis congenita and related bone marrow failure syndromes. Blood. 2008 Nov 1;112(9):3594–600. doi: 10.1182/blood-2008-05-153445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Savage SA, Giri N, Baerlocher GM, Orr N, Lansdorp PM, Alter BP. TINF2, a component of the shelterin telomere protection complex, is mutated in dyskeratosis congenita. Am J Hum Genet. 2008 Feb;82(2):501–9. doi: 10.1016/j.ajhg.2007.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Rocha V, Devergie A, Socie G, Ribaud P, Esperou H, Parquet N, et al. Unusual complications after bone marrow transplantation for dyskeratosis congenita. Br J Haematol. 1998 Oct;103(1):243–8. doi: 10.1046/j.1365-2141.1998.00949.x. [DOI] [PubMed] [Google Scholar]
- 32.Langston AA, Sanders JE, Deeg HJ, Crawford SW, Anasetti C, Sullivan KM, et al. Allogeneic marrow transplantation for aplastic anaemia associated with dyskeratosis congenita. Br J Haematol. 1996 Mar;92(3):758–65. doi: 10.1046/j.1365-2141.1996.424984.x. [DOI] [PubMed] [Google Scholar]
- 33.Amarasinghe K, Dalley C, Dokal I, Laurie A, Gupta V, Marsh J. Late death after unrelated-BMT for dyskeratosis congenita following conditioning with alemtuzumab, fludarabine and melphalan. Bone Marrow Transplant. 2007 Nov;40(9):913–4. doi: 10.1038/sj.bmt.1705839. [DOI] [PubMed] [Google Scholar]
- 34.Giri N, Lee R, Faro A, Huddleston CB, White FV, Alter BP, et al. Lung transplantation for pulmonary fibrosis in dyskeratosis congenita: Case Report and systematic literature review. BMC Blood Disord. 2011;11:3. doi: 10.1186/1471-2326-11-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lipton JM, Atsidaftos E, Zyskind I, Vlachos A. Improving clinical care and elucidating the pathophysiology of Diamond Blackfan anemia: an update from the Diamond Blackfan Anemia Registry. Pediatr Blood Cancer. 2006 May 1;46(5):558–64. doi: 10.1002/pbc.20642. [DOI] [PubMed] [Google Scholar]
- 36.Fargo JH, Kratz CP, Giri N, Savage SA, Wong C, Backer K, et al. Erythrocyte adenosine deaminase: diagnostic value for Diamond-Blackfan anaemia. Br J Haematol. 2012 Dec 17; doi: 10.1111/bjh.12167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vlachos A, Rosenberg PS, Atsidaftos E, Alter BP, Lipton JM. Incidence of neoplasia in Diamond Blackfan anemia: a report from the Diamond Blackfan Anemia Registry. Blood. 2012 Apr 19;119(16):3815–9. doi: 10.1182/blood-2011-08-375972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Vlachos A, Dahl N, Dianzani I, Lipton JM. Clinical utility gene card for: Diamond - Blackfan anemia - update 2013. Eur J Hum Genet. 2013 Oct;21(10) doi: 10.1038/ejhg.2013.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gazda HT, Zhong R, Long L, Niewiadomska E, Lipton JM, Ploszynska A, et al. RNA and protein evidence for haplo-insufficiency in Diamond-Blackfan anaemia patients with RPS19 mutations. Br J Haematol. 2004 Oct;127(1):105–13. doi: 10.1111/j.1365-2141.2004.05152.x. [DOI] [PubMed] [Google Scholar]
- 40.Vlachos A, Ball S, Dahl N, Alter BP, Sheth S, Ramenghi U, et al. Diagnosing and treating Diamond Blackfan anaemia: results of an international clinical consensus conference. Br J Haematol. 2008 Sep;142(6):859–76. doi: 10.1111/j.1365-2141.2008.07269.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Vlachos A, Muir E. How I treat Diamond-Blackfan anemia. Blood. 2010 Nov 11;116(19):3715–23. doi: 10.1182/blood-2010-02-251090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Lipton JM. Diamond blackfan anemia: New paradigms for a “not so pure” inherited red cell aplasia. Semin Hematol. 2006 Jul;43(3):167–77. doi: 10.1053/j.seminhematol.2006.04.002. [DOI] [PubMed] [Google Scholar]
- 43.Ohene-Abuakwa Y, Orfali KA, Marius C, Ball SE. Two-phase culture in Diamond Blackfan anemia: localization of erythroid defect. Blood. 2005 Jan 15;105(2):838–46. doi: 10.1182/blood-2004-03-1016. [DOI] [PubMed] [Google Scholar]
- 44.Zhang L, Prak L, Rayon-Estrada V, Thiru P, Flygare J, Lim B, et al. ZFP36L2 is required for self-renewal of early burst-forming unit erythroid progenitors. Nature. 2013 Jul 4;499(7456):92–6. doi: 10.1038/nature12215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Pospisilova D, Cmejlova J, Hak J, Adam T, Cmejla R. Successful treatment of a Diamond-Blackfan anemia patient with amino acid leucine. Haematologica. 2007 May;92(5):e66–7. doi: 10.3324/haematol.11498. [DOI] [PubMed] [Google Scholar]
- 46.Boultwood J, Yip BH, Vuppusetty C, Pellagatti A, Wainscoat JS. Activation of the mTOR pathway by the amino acid (L)-leucine in the 5q- syndrome and other ribosomopathies. Adv Biol Regul. 2013 Jan;53(1):8–17. doi: 10.1016/j.jbior.2012.09.002. [DOI] [PubMed] [Google Scholar]
- 47.Payne EM, Virgilio M, Narla A, Sun H, Levine M, Paw BH, et al. L-Leucine improves the anemia and developmental defects associated with Diamond-Blackfan anemia and del(5q) MDS by activating the mTOR pathway. Blood. 2012 Sep 13;120(11):2214–24. doi: 10.1182/blood-2011-10-382986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Jaako P, Debnath S, Olsson K, Bryder D, Flygare J, Karlsson S. Dietary L-leucine improves the anemia in a mouse model for Diamond-Blackfan anemia. Blood. 2012 Sep 13;120(11):2225–8. doi: 10.1182/blood-2012-05-431437. [DOI] [PubMed] [Google Scholar]
- 49.Vlachos A, Klein GW, Lipton JM. The Diamond Blackfan Anemia Registry: tool for investigating the epidemiology and biology of Diamond-Blackfan anemia. J Pediatr Hematol Oncol. 2001 Aug-Sep;23(6):377–82. doi: 10.1097/00043426-200108000-00015. [DOI] [PubMed] [Google Scholar]
- 50.Faivre L, Meerpohl J, Da Costa L, Marie I, Nouvel C, Gnekow A, et al. High-risk pregnancies in Diamond-Blackfan anemia: a survey of 64 pregnancies from the French and German registries. Haematologica. 2006 Apr;91(4):530–3. [PubMed] [Google Scholar]
- 51.Farrar JE, Vlachos A, Atsidaftos E, Carlson-Donohoe H, Markello TC, Arceci RJ, et al. Ribosomal protein gene deletions in Diamond-Blackfan anemia. Blood. 2011 Dec 22;118(26):6943–51. doi: 10.1182/blood-2011-08-375170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Vlachos A, Farrar JE, Atsidaftos E, Muir E, Narla A, Markello TC, et al. Diminutive somatic deletions in the 5q region lead to a phenotype atypical of classical 5q- syndrome. Blood. 2013 Oct 3;122(14):2487–90. doi: 10.1182/blood-2013-06-509935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Letestu R, Vitrat N, Masse A, Le Couedic JP, Lazar V, Rameau P, et al. Existence of a differentiation blockage at the stage of a megakaryocyte precursor in the thrombocytopenia and absent radii (TAR) syndrome. Blood. 2000 Mar 1;95(5):1633–41. [PubMed] [Google Scholar]
- 54.Fadoo Z, Naqvi SM. Acute myeloid leukemia in a patient with thrombocytopenia with absent radii syndrome. J Pediatr Hematol Oncol. 2002 Feb;24(2):134–5. doi: 10.1097/00043426-200202000-00015. [DOI] [PubMed] [Google Scholar]
- 55.Albers CA, Paul DS, Schulze H, Freson K, Stephens JC, Smethurst PA, et al. Compound inheritance of a low-frequency regulatory SNP and a rare null mutation in exon-junction complex subunit RBM8A causes TAR syndrome. Nat Genet. 2012 Apr;44(4):435–9. S1–2. doi: 10.1038/ng.1083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klopocki E, Schulze H, Strauss G, Ott CE, Hall J, Trotier F, et al. Complex inheritance pattern resembling autosomal recessive inheritance involving a microdeletion in thrombocytopenia-absent radius syndrome. Am J Hum Genet. 2007 Feb;80(2):232–40. doi: 10.1086/510919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dale DC, Bonilla MA, Davis MW, Nakanishi AM, Hammond WP, Kurtzberg J, et al. A randomized controlled phase III trial of recombinant human granulocyte colony-stimulating factor (filgrastim) for treatment of severe chronic neutropenia. Blood. 1993;81(10):2496–502. [PMC free article] [PubMed] [Google Scholar]
- 58.Carlsson G, Fasth A, Berglof E, Lagerstedt-Robinson K, Nordenskjold M, Palmblad J, et al. Incidence of severe congenital neutropenia in Sweden and risk of evolution to myelodysplastic syndrome/leukaemia. Br J Haematol. 2012 Aug;158(3):363–9. doi: 10.1111/j.1365-2141.2012.09171.x. [DOI] [PubMed] [Google Scholar]
- 59.Rosenberg PS, Alter BP, Bolyard AA, Bonilla MA, Boxer LA, Cham B, et al. The incidence of leukemia and mortality from sepsis in patients with severe congenital neutropenia receiving long-term G-CSF therapy. Blood. 2006 Feb 23; doi: 10.1182/blood-2005-11-4370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rosenberg PS, Zeidler C, Bolyard AA, Alter BP, Bonilla MA, Boxer LA, et al. Stable long-term risk of leukaemia in patients with severe congenital neutropenia maintained on G-CSF therapy. Br J Haematol. 2010 Jul;150(2):196–9. doi: 10.1111/j.1365-2141.2010.08216.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Bellanne-Chantelot C, Clauin S, Leblanc T, Cassinat B, Rodrigues-Lima F, Beaufils S, et al. Mutations in the ELA2 gene correlate with more severe expression of neutropenia: A study of 81 patients from the French Neutropenia Register. Blood. 2004 Feb 12; doi: 10.1182/blood-2003-10-3518. [DOI] [PubMed] [Google Scholar]
- 62.Dale DC, Person RE, Bolyard AA, Aprikyan AG, Bos C, Bonilla MA, et al. Mutations in the gene encoding neutrophil elastase in congenital and cyclic neutropenia. Blood. 2000 Oct 1;96(7):2317–22. [PubMed] [Google Scholar]
- 63.Xia J, Bolyard AA, Rodger E, Stein S, Aprikyan AA, Dale DC, et al. Prevalence of mutations in ELANE, GFI1, HAX1, SBDS, WAS and G6PC3 in patients with severe congenital neutropenia. Br J Haematol. 2009 Nov;147(4):535–42. doi: 10.1111/j.1365-2141.2009.07888.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Grenda DS, Murakami M, Ghatak J, Xia J, Boxer LA, Dale D, et al. Mutations of the ELA2 gene found in patients with severe congenital neutropenia induce the unfolded protein response and cellular apoptosis. Blood. 2007 doi: 10.1182/blood-2006-11-057299. blood-2006-11-057299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Kollner I, Sodeik B, Schreek S, Heyn H, von Neuhoff N, Germeshausen M, et al. Mutations in neutrophil elastase causing congenital neutropenia lead to cytoplasmic protein accumulation and induction of the unfolded protein response. Blood. 2006 Jul 15;108(2):493–500. doi: 10.1182/blood-2005-11-4689. [DOI] [PubMed] [Google Scholar]
- 66.Nanua S, Murakami M, Xia J, Grenda DS, Woloszynek J, Strand M, et al. Activation of the unfolded protein response is associated with impaired granulopoiesis in transgenic mice expressing mutant Elane. Blood. 2011 Mar 31;117(13):3539–47. doi: 10.1182/blood-2010-10-311704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Connelly JA, Choi SW, Levine JE. Hematopoietic stem cell transplantation for severe congenital neutropenia. Curr Opin Hematol. 2012 Jan;19(1):44–51. doi: 10.1097/MOH.0b013e32834da96e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Myers KC, Davies SM, Shimamura A. Clinical and molecular pathophysiology of Shwachman-Diamond syndrome: an update. Hematol Oncol Clin North Am. 2013 Feb;27(1):117–28, ix. doi: 10.1016/j.hoc.2012.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kuijpers TW, Alders M, Tool AT, Mellink C, Roos D, Hennekam RC. Hematologic abnormalities in Shwachman Diamond syndrome: lack of genotype-phenotype relationship. Blood. 2005 Jul 1;106(1):356–61. doi: 10.1182/blood-2004-11-4371. [DOI] [PubMed] [Google Scholar]
- 70.Smith OP, Hann IM, Chessells JM, Reeves BR, Milla P. Haematological abnormalities in Shwachman-Diamond syndrome. Br J Haematol. 1996 Aug;94(2):279–84. doi: 10.1046/j.1365-2141.1996.d01-1788.x. [DOI] [PubMed] [Google Scholar]
- 71.Boocock GR, Morrison JA, Popovic M, Richards N, Ellis L, Durie PR, et al. Mutations in SBDS are associated with Shwachman-Diamond syndrome. Nat Genet. 2003 Jan;33(1):97–101. doi: 10.1038/ng1062. [DOI] [PubMed] [Google Scholar]
- 72.Hashmi SK, Allen C, Klaassen R, Fernandez CV, Yanofsky R, Shereck E, et al. Comparative analysis of Shwachman-Diamond syndrome to other inherited bone marrow failure syndromes and genotype-phenotype correlation. Clin Genet. 2011 May;79(5):448–58. doi: 10.1111/j.1399-0004.2010.01468.x. [DOI] [PubMed] [Google Scholar]
- 73.Nakashima E, Mabuchi A, Makita Y, Masuno M, Ohashi H, Nishimura G, et al. Novel SBDS mutations caused by gene conversion in Japanese patients with Shwachman-Diamond syndrome. Hum Genet. 2004 Mar;114(4):345–8. doi: 10.1007/s00439-004-1081-2. Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 74.Donadieu J, Fenneteau O, Beaupain B, Beaufils S, Bellanger F, Mahlaoui N, et al. Classification of and risk factors for hematologic complications in a French national cohort of 102 patients with Shwachman-Diamond syndrome. Haematologica. 2012 Sep;97(9):1312–9. doi: 10.3324/haematol.2011.057489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dror Y, Donadieu J, Koglmeier J, Dodge J, Toiviainen-Salo S, Makitie O, et al. Draft consensus guidelines for diagnosis and treatment of Shwachman-Diamond syndrome. Ann N Y Acad Sci. 2011 Dec;1242:40–55. doi: 10.1111/j.1749-6632.2011.06349.x. [DOI] [PubMed] [Google Scholar]
- 76.Woods WG, Krivit W, Lubin BH, Ramsay NK. Aplastic anemia associated with the Shwachman syndrome. In vivo and in vitro observations. Am J Pediatr Hematol Oncol. 1981 Winter;3(4):347–51. [PubMed] [Google Scholar]
- 77.Dror Y, Durie P, Ginzberg H, Herman R, Banerjee A, Champagne M, et al. Clonal evolution in marrows of patients with Shwachman-Diamond syndrome: a prospective 5-year follow-up study. Exp Hematol. 2002 Jul;30(7):659–69. doi: 10.1016/s0301-472x(02)00815-9. [DOI] [PubMed] [Google Scholar]
- 78.Cunningham J, Sales M, Pearce A, Howard J, Stallings R, Telford N, et al. Does isochromosome 7q mandate bone marrow transplant in children with Shwachman-Diamond syndrome? Br J Haematol. 2002 Dec;119(4):1062–9. doi: 10.1046/j.1365-2141.2002.03940.x. [DOI] [PubMed] [Google Scholar]
- 79.Maserati E, Pressato B, Valli R, Minelli A, Sainati L, Patitucci F, et al. The route to development of myelodysplastic syndrome/acute myeloid leukaemia in Shwachman-Diamond syndrome: the role of ageing, karyotype instability, and acquired chromosome anomalies. Br J Haematol. 2009 Apr;145(2):190–7. doi: 10.1111/j.1365-2141.2009.07611.x. [DOI] [PubMed] [Google Scholar]
- 80.Ip WF, Dupuis A, Ellis L, Beharry S, Morrison J, Stormon MO, et al. Serum pancreatic enzymes define the pancreatic phenotype in patients with Shwachman-Diamond syndrome. J Pediatr. 2002 Aug;141(2):259–65. doi: 10.1067/mpd.2002.125849. [DOI] [PubMed] [Google Scholar]
- 81.Mack DR, Forstner GG, Wilschanski M, Freedman MH, Durie PR. Shwachman syndrome: exocrine pancreatic dysfunction and variable phenotypic expression. Gastroenterology. 1996 Dec;111(6):1593–602. doi: 10.1016/s0016-5085(96)70022-7. [DOI] [PubMed] [Google Scholar]
- 82.Makitie O, Ellis L, Durie PR, Morrison JA, Sochett EB, Rommens JM, et al. Skeletal phenotype in patients with Shwachman-Diamond syndrome and mutations in SBDS. Clin Genet. 2004 Feb;65(2):101–12. doi: 10.1111/j.0009-9163.2004.00198.x. [DOI] [PubMed] [Google Scholar]
- 83.Leung R, Cuddy K, Wang Y, Rommens J, Glogauer M. Sbds is required for Rac2-mediated monocyte migration and signaling downstream of RANK during osteoclastogenesis. Blood. 2011 Feb 10;117(6):2044–53. doi: 10.1182/blood-2010-05-282574. In Vitro Research Support, Non-U.S. Gov't. [DOI] [PubMed] [Google Scholar]
- 84.Ginzberg H, Shin J, Ellis L, Morrison J, Ip W, Dror Y, et al. Shwachman syndrome: phenotypic manifestations of sibling sets and isolated cases in a large patient cohort are similar. J Pediatr. 1999 Jul;135(1):81–8. doi: 10.1016/s0022-3476(99)70332-x. [DOI] [PubMed] [Google Scholar]
- 85.Woloszynek JR, Rothbaum RJ, Rawls AS, Minx PJ, Wilson RK, Mason PJ, et al. Mutations of the SBDS gene are present in most patients with Shwachman-Diamond syndrome. Blood. 2004 Dec 1;104(12):3588–90. doi: 10.1182/blood-2004-04-1516. [DOI] [PubMed] [Google Scholar]
- 86.Wong TE, Calicchio ML, Fleming MD, Shimamura A, Harris MH. SBDS protein expression patterns in the bone marrow. Pediatr Blood Cancer. 2010 Sep;55(3):546–9. doi: 10.1002/pbc.22573. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dror Y, Freedman MH. Shwachman-Diamond syndrome: An inherited preleukemic bone marrow failure disorder with aberrant hematopoietic progenitors and faulty marrow microenvironment. Blood. 1999;94(9):3048–54. [PubMed] [Google Scholar]
- 88.Minelli A, Maserati E, Nicolis E, Zecca M, Sainati L, Longoni D, et al. The isochromosome i(7)(q10) carrying c.258+2t>c mutation of the SBDS gene does not promote development of myeloid malignancies in patients with Shwachman syndrome. Leukemia. 2009 Apr;23(4):708–11. doi: 10.1038/leu.2008.369. [DOI] [PubMed] [Google Scholar]
- 89.Pressato B, Valli R, Marletta C, Mare L, Montalbano G, Lo Curto F, et al. Deletion of chromosome 20 in bone marrow of patients with Shwachman-Diamond syndrome, loss of the EIF6 gene and benign prognosis. Br J Haematol. 2012 May;157(4):503–5. doi: 10.1111/j.1365-2141.2012.09033.x. [DOI] [PubMed] [Google Scholar]
- 90.Toiviainen-Salo S, Pitkanen O, Holmstrom M, Koikkalainen J, Lotjonen J, Lauerma K, et al. Myocardial function in patients with Shwachman-Diamond syndrome: aspects to consider before stem cell transplantation. Pediatr Blood Cancer. 2008 Oct;51(4):461–7. doi: 10.1002/pbc.21686. [DOI] [PubMed] [Google Scholar]
- 91.Cesaro S, Oneto R, Messina C, Gibson BE, Buzyn A, Steward C, et al. Haematopoietic stem cell transplantation for Shwachman-Diamond disease: a study from the European Group for blood and marrow transplantation. Br J Haematol. 2005 Oct;131(2):231–6. doi: 10.1111/j.1365-2141.2005.05758.x. [DOI] [PubMed] [Google Scholar]
- 92.Ballmaier M, Germeshausen M. Congenital amegakaryocytic thrombocytopenia: clinical presentation, diagnosis, and treatment. Semin Thromb Hemost. 2011 Sep;37(6):673–81. doi: 10.1055/s-0031-1291377. [DOI] [PubMed] [Google Scholar]
- 93.Jongmans MC, Kuiper RP, Carmichael CL, Wilkins EJ, Dors N, Carmagnac A, et al. Novel RUNX1 mutations in familial platelet disorder with enhanced risk for acute myeloid leukemia: clues for improved identification of the FPD/AML syndrome. Leukemia. 2010 Jan;24(1):242–6. doi: 10.1038/leu.2009.210. [DOI] [PubMed] [Google Scholar]
- 94.Spinner MA, Sanchez LA, Hsu AP, Shaw PA, Zerbe CS, Calvo KR, et al. GATA2 deficiency: a protean disorder of hematopoiesis, lymphatics, and immunity. Blood. 2014 Feb 6;123(6):809–21. doi: 10.1182/blood-2013-07-515528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Alter BP. Inherited bone marrow failure syndromes. In: Nathan DG, Orkin SH, Ginsburg D, Look AT, editors. Nathans and Oski's Hematology of infancy and childhood. sixth. Philadephia: Saunders; pp. 2003pp. 281–365. [Google Scholar]
- 96.Willig TN, Niemeyer CM, Leblanc T, Tiemann C, Robert A, Budde J, et al. Identification of new prognosis factors from the clinical and epidemiologic analysis of a registry of 229 Diamond-Blackfan anemia patients. DBA group of Societe d'Hematologie et d'Immunologie Pediatrique (SHIP), Gesellshaft fur Padiatrische Onkologie und Hamatologie (GPOH), and the European Society for Pediatric Hematology and Immunology (ESPHI) Pediatr Res. 1999 Nov;46(5):553–61. doi: 10.1203/00006450-199911000-00011. [DOI] [PubMed] [Google Scholar]
- 97.Ball SE, McGuckin CP, Jenkins G, Gordon-Smith EC. Diamond-Blackfan anaemia in the U.K.: analysis of 80 cases from a 20-year birth cohort. Br J Haematol. 1996 Sep;94(4):645–53. doi: 10.1046/j.1365-2141.1996.d01-1839.x. [DOI] [PubMed] [Google Scholar]
- 98.Ramenghi U, Garelli E, Valtolina S, Campagnoli MF, Timeus F, Crescenzio N, et al. Diamond-Blackfan anaemia in the Italian population. Br J Haematol. 1999 Mar;104(4):841–8. doi: 10.1046/j.1365-2141.1999.01267.x. [DOI] [PubMed] [Google Scholar]
- 99.Cipolli M, D'Orazio C, Delmarco A, Marchesini C, Miano A, Mastella G. Shwachman's syndrome: pathomorphosis and long-term outcome. J Pediatr Gastroenterol Nutr. 1999 Sep;29(3):265–72. doi: 10.1097/00005176-199909000-00006. [DOI] [PubMed] [Google Scholar]
- 100.Myers KC, Bolyard AA, Otto B, Wong TE, Jones AT, Harris RE, et al. Variable Clinical Presentation of Shwachman-Diamond Syndrome: Update from the North American Shwachman-Diamond Syndrome Registry. J Pediatr. 2013 Dec 31; doi: 10.1016/j.jpeds.2013.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]