

Abstract
Human African trypanosomiasis (HAT) is a parasitic disease, caused by the protozoan pathogen Trypanosoma brucei, which affects thousands every year and which is in need of new therapeutics. Herein we report the synthesis and assessment of a series of pyrrolidine and pyrazolone derivatives of human phosphodiesterase 4 (hPDE4) inhibitors for the assessment of their activity against the trypanosomal phosphodiesterase TbrPDEB1. The synthesized compounds showed weak potency against TbrPDEB1.
Keywords: Trypanosoma brucei, phosphodiesterases, TbrPDEB1, neglected tropical disease
Graphical Abstract
Human African trypanosomiasis (HAT), or African sleeping sickness, is a parasitic disease caused by the protozoan pathogen Trypanosoma brucei. HAT is predominantly found in poor, remote rural regions of sub-Saharan Africa. Currently, over 60 million people in 36 countries in sub-Saharan Africa are at risk, with approximately 10,000 infections annually.1 HAT is a neglected tropical disease (NTD), in that relatively little attention has been paid to finding new treatments.
Four drugs are approved for the treatment of HAT: pentamidine, suramin, eflornithine, and melarsoprol. The nifurtimox–eflornithine combination therapy (NECT) has also been approved for HAT which provides safety and dosing advantages over eflornithine monotherapy.2, 3 Though effective, these drugs are limited by serious and sometimes lethal side effects, inconvenient route of administration and increasing incidence of drug resistance.3, 4 Considering the economic and social burden that HAT produces, further work is needed in finding safe and effective drugs that are safer, simpler to administer and cheaper than those currently available. With this in mind, our research efforts have been directed towards repurposing established classes of inhibitors of druggable human targets to be inhibitors of essential parasite targets.5 Phosphodiesterases (PDEs) are a family of enzymes that regulate signal transduction mediated by cAMP and cGMP in the cell, and inhibition of these enzymes reduces cAMP or cGMP PDE degradation, thus affecting the physiological processes controlled by these second messengers. Numerous PDE inhibitors are used as therapeutics.6 Trypanosoma brucei expresses five PDEs, two of which, TbrPDEB1 and TbrPDEB2, have together been shown by RNAi to be essential for parasite proliferation.7 The catalytic domains of human PDEs share 30–35% sequence identity to those of the parasite enzymes TbrPDEB1 and TbrPDEB2,8 thus we reasoned that human PDE inhibitors repurposing could be a useful approach. We tested a range of human PDE (hPDE) inhibitors against TbrPDEB1 and B2, and we observed that the hPDE4 inhibitor piclamilast (1, Figure 1) inhibits both TbrPDEB1 and B2 and T. brucei brucei (Tbb) cell growth in vitro.9 Cilomilast (2), another commercialized hPDE4 inhibitor, and some of its analogs, have been found to inhibit TbrPDEB1.10 We observed the prototypical hPDE4 inhibitor rolipram (3) to be inactive against TbrPDEB1.
Figure 1.

Compounds previously studied as TbrPDEB1 inhibitors.
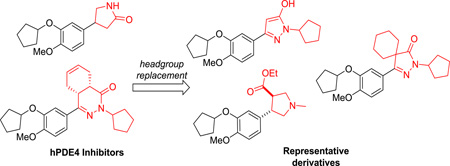
Another class of human PDE4 phthalizinones has also been pursued, leading to the discovery of compound 4, which is the most active TbrPDEB1/B2 inhibitor to date (IC50 3.98 nM and 6.0 nM respectively for TbrPDEB1 and B2).11 The related phtalazininone compound 5 shows an IC50 of 278 nM against TbrPDEB1.12 In addition, pyrazolone 6 was identified through a scaffold merging approach.13 Despite the obvious structural similarity between compounds 1–3, 5 and 6, their reported TbrPDEB1 IC50 values fall in a wide range. These molecules share the cyclopentyl-substituted catechol functionality, and differ in the region of the molecule (highlighted in red) pointing towards the catalytic metals in the binding site region. This suggested to us that this “headgroup” region must be a major driver of potency against TbrPDEB1. With this in mind, in this letter we report our attempt to find novel TbrPDEB1 inhibitors driven by the replacement of the pyrrolidinone moiety of compound 3 with different five-membered rings.
As a starting point, we prepared racemic trans-3,4-disubstituted pyrrolidine analogues as an intended mimic of the pyrazolinone headgroup of 4 (Scheme 1). The sequence was initiated with Wittig olefination of aldehyde 8 to obtain the phenylacrylic acid ester 9. Iminium ylide cycloaddition of compound 9 with sarcosine and formaldehyde in refluxing toluene gave the corresponding trans-3,4-disubstituted N-methyl pyrrolidine ethyl ester 10a.14 A small library of amides (compounds 11a-k) has been prepared starting from the ester 10a by reaction with the corresponding aluminum amide, while LiOH hydrolysis of ester 10a gave the corresponding acid 10b. Demethylation of 10a with 1-chloroethyl chloroformate produced the pyrrolidine 12,15 which was converted to compounds 13a and 13b with methanesulfonyl chloride or acetyl chloride, respectively. Compounds 10, 11 and 13 were all found to be weak inhibitors of TbrPDEB1 (Table 1).
Scheme 1.

Synthesis of pyrrolidine derivatives. Reagents and conditions: (a) (Carbethoxymethylene)triphenylphosphorane, CH3CN, MW, 150 °C, 20 min; (b) formaldehyde, sarcosine, MgSO4, toluene, 170 °C, 24 h; (c) LiOH, H2O, MeOH, THF, rt, 2 h; (d) appropriate amine, Me3Al, Toluene, 80 °C, 12 h; (e) i. 1-Chloroethyl chloroformate, DMAP, 1,2-dichloroethane, reflux, overnight; ii. MeOH, reflux 4h; (f) acetyl chloride or methanesulfonyl chloride, DMAP, DMF, rt, overnight.
Table 1.
rac-(trans-3,4)-Disubstituted pyrrolidine analogs tested against TbrPDEB1
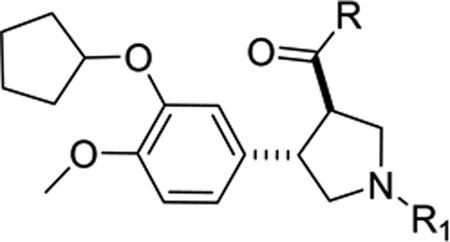 | |||
|---|---|---|---|
| Cpd | R | R1 | TbrPDEB1 (% inh)a |
| 10a | OCH2CH3 | CH3 | 8 ± 11 |
| 10b | OH | CH3 | 17 ± 9 |
| 11a | 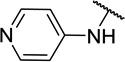 |
CH3 | 5 ± 5c |
| 11b | 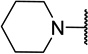 |
CH3 | 7 ± 5c |
| 11c | 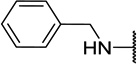 |
CH3 | 8 ± 0 |
| 11d | 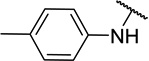 |
CH3 | 6 ± 6 |
| 11e | 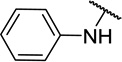 |
CH3 | 16 ± 11 |
| 11f | 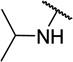 |
CH3 | 10 ± 5 |
| 11g | 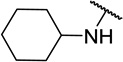 |
CH3 | 16 ± 6 |
| 11h | NHCH3 | CH3 | 8 ± 0 |
| 11i | NHCH2CH3 | CH3 | 5 ± 4 |
| 11j | CH3 | 1 ± 2 | |
| 11k | 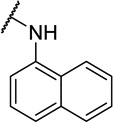 |
CH3 | 10 ± 3 |
| 13a | OCH2CH3 | SO2CH3 | 3 ± 4 |
| 13b | OCH2CH3 | COCH3 | 15 ± 0 |
Data shown are average of 2 replicate independent experiments. Compounds were tested at 10 µM concentrations.
n=1
Replicate of 3 independent experiments.
We then prepared two pyrazolone-based inhibitors (compounds 18a and 18b), as shown in Scheme 2. Acid 14 was converted to the corresponding acyl chloride with thionyl chloride and reacted with the lithium enolate of methyl acetate to provide 16. The β-keto-ester intermediate 16 was cyclized to the desired pyrazolol derivative 18a using hydrazine hydrochloride in refluxing acetic acid. Compound 18a was alkylated with bromocyclopentane to obtain compound 18b. Compounds 18a-b were also found to have little activity against TbrPDEB1 (Table 2).
Scheme 2.

Synthesis of pyrazolone derivatives. Reagents and conditions: (a) SOCl2, DMF, 90 °C, 3 h; (b) MeOAc, LDA, THF, −78 °C then rt, 1 h; (c) 1,5-diiodopentane, K2CO3, DMF, 90 °C, overnight; (d) hydrazine hydrochloride, acetic acid, 120 °C, 2 h; (e) hydrazine hydrochloride, DIPEA, BuOH, 120 °C, 3 h; (f) alkyl bromide, K2CO3, DMF, 70 °C, 2 h; (g) NaH, alkyl bromide, DMF, 60 °C, 3 h.
Table 2.
Pyrazololo analogs tested against TbrPDEB1
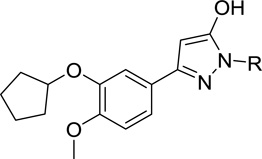 | |||
|---|---|---|---|
| Cpd | R | R1 | TbrPDEB1 (% inh)a |
| 18a | H | 0 | |
| 18b | 5 ± 3 | ||
Data shown are average of 2 replicate independent experiments. Compounds were tested at 10 µM concentrations.
We looked towards increasing the size of the headgroup region, looking to mimic the size and shape of compounds 4–7 more closely, and therefore prepared the spirocyclic compounds 21a-d (Scheme 2). The β-keto-ester 16 was first alkylated with 1,5-diiodopentane to give compound 19 which cyclized to 21a when treated with hydrazine. This could be N-alkylated with various alkyl bromides to give compounds 21b-d.
Besides preparing the cyclopentyl-substituted catechol analogs, we noted that improved T brucei cellular potency had been reported for 7 (Tbb EC50: 6.3 µM) when compared to 6 (>64 µM).13 Thus, we prepared the benzyl-substituted analogs 22a-d. Compounds 21 and 22 were tested against TbrPDEB1 (Table 3), and, despite the structural similarity these compounds and the known actives 6 and 7, we found that these analogs had very little ability to inhibit TbrPDEB1. We can conclude, based on this and on our previous attempts to explore structural variations around compound 1 that the SAR is extraordinarily tight for this class of compounds.9, 12 With this in mind, our efforts are focused on obtaining a better understanding of the subtle structural features needed for an optimal enzyme inhibition.
Table 3.
Spiro pyrazolone analogs tested against TbrPDEB1
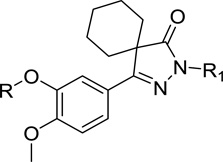 | |||
|---|---|---|---|
| Cpd | R | R1 | TbrPDEB1 (% inh)a |
| 21a | H | 11 ± 10 | |
| 21b | 18 ± 2 | ||
| 21c | 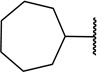 |
9 ± 4b | |
| 21d | 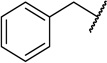 |
9 ± 6b | |
| 22a | 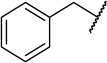 |
H | 14 ± 20 |
| 22b | 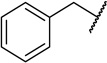 |
11 ±10b | |
| 22c | 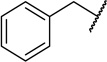 |
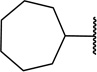 |
7 ± 5 |
| 22d | 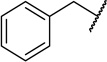 |
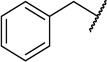 |
10 ± 3 |
Data shown are average of 2 replicate independent experiments. Compounds were tested at 10 µM concentrations.
Replicate of 3 independent experiments.
Supplementary Material
Acknowledgments
We acknowledge funding from the National Institutes of Health (R01AI082577).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Supplementary data
The synthetic preparation and characterization of new compounds, along with biological assay conditions, and a recapitulation of the tables from the manuscript that include compound registry numbers may be found in the Supplementary data. All screening data reported in this paper is freely available on the Collaborative Drug Discovery database (www.collaborativedrug.com).
References
- 1. http://apps.who.int/iris/bitstream/10665/77950/1/9789241564540_eng.pdf.
- 2.Priotto G, Kasparian S, Mutombo W, Ngouama D, Ghorashian S, Arnold U, Ghabri S, Baudin E, Buard V, Kazadi-Kyanza S, Ilunga M, Mutangala W, Pohlig G, Schmid C, Karunakara U, Torreele E, Kande V. Lancet. 2009;374:56. doi: 10.1016/S0140-6736(09)61117-X. [DOI] [PubMed] [Google Scholar]
- 3.Babokhov P, Sanyaolu AO, Oyibo WA, Fagbenro-Beyioku AF, Iriemenam NC. Pathog. Glob. Health. 2013;107:242. doi: 10.1179/2047773213Y.0000000105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bouteille B, Oukem O, Bisser S, Dumas M. Fundam. Clin. Pharmacol. 2003;17:171. doi: 10.1046/j.1472-8206.2003.00167.x. [DOI] [PubMed] [Google Scholar]
- 5.Pollastri MP, Campbell RK. Future Med. Chem. 2011;3:1307. doi: 10.4155/fmc.11.92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Bender AT, Beavo JA. Pharmacol. Rev. 2006;58:488. doi: 10.1124/pr.58.3.5. [DOI] [PubMed] [Google Scholar]
- 7.Oberholzer M, Marti G, Baresic M, Kunz S, Hemphill A, Seebeck T. FASEB J. 2007;21:720. doi: 10.1096/fj.06-6818com. [DOI] [PubMed] [Google Scholar]
- 8.Kunz S, Kloeckner T, Essen LO, Seebeck T, Boshart M. Eur. J. Biochem. 2004;271:637. doi: 10.1111/j.1432-1033.2003.03967.x. [DOI] [PubMed] [Google Scholar]
- 9.Bland ND, Wang C, Tallman C, Gustafson AE, Wang Z, Ashton TD, Ochiana SO, McAllister G, Cotter K, Fang AP, Gechijian L, Garceau N, Gangurde R, Ortenberg R, Ondrechen MJ, Campbell RK, Pollastri MP. J. Med. Chem. 2011;54:8188. doi: 10.1021/jm201148s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Amata E, Bland ND, Hoyt CT, Settimo L, Campbell RK, Pollastri MP. Bioorg. Med. Chem. Lett. 2014;24:4084. doi: 10.1016/j.bmcl.2014.07.063. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.de Koning HP, Gould MK, Sterk GJ, Tenor H, Kunz S, Luginbuehl E, Seebeck T. J. Infect. Dis. 2012;206:229. doi: 10.1093/infdis/jir857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Woodring JL, Bland ND, Ochiana SO, Campbell RK, Pollastri MP. Bioorg. Med. Chem. Lett. 2013;23:5971. doi: 10.1016/j.bmcl.2013.08.057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Orrling KM, Jansen C, Vu XL, Balmer V, Bregy P, Shanmugham A, England P, Bailey D, Cos P, Maes L, Adams E, van den Bogaart E, Chatelain E, Ioset JR, van de Stolpe A, Zorg S, Veerman J, Seebeck T, Sterk GJ, de Esch IJ, Leurs R. J. Med. Chem. 2012;55:8745. doi: 10.1021/jm301059b. [DOI] [PubMed] [Google Scholar]
- 14.Wong JC, Tang G, Wu X, Liang C, Zhang Z, Guo L, Peng Z, Zhang W, Lin X, Wang Z, Mei J, Chen J, Pan S, Zhang N, Liu Y, Zhou M, Feng L, Zhao W, Li S, Zhang C, Zhang M, Rong Y, Jin TG, Zhang X, Ren S, Ji Y, Zhao R, She J, Ren Y, Xu C, Chen D, Cai J, Shan S, Pan D, Ning Z, Lu X, Chen T, He Y, Chen L. J. Med. Chem. 2012;55:8903. doi: 10.1021/jm3011838. [DOI] [PubMed] [Google Scholar]
- 15.Marrazzo A, Cobos EJ, Parenti C, Arico G, Marrazzo G, Ronsisvalle S, Pasquinucci L, Prezzavento O, Colabufo NA, Contino M, Gonzalez LG, Scoto GM, Ronsisvalle G. J. Med. Chem. 2011;54:3669. doi: 10.1021/jm200144j. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.