

Abstract
Cryptosporidium is the causative agent of a gastrointestinal disease, cryptosporidiosis, which is often fatal in immunocompromised individuals and children. Thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are essential enzymes in the folate biosynthesis pathway and are well established as drug targets in cancer, bacterial infections, and malaria. Cryptosporidium hominis has a bifunctional thymidylate synthase and dihydrofolate reductase enzyme, compared to separate enzymes in the host. We evaluated lead compound 1 from a novel series of antifolates, 2-amino-4-oxo-5-substituted pyrrolo[2,3-d]pyrimidines as an inhibitor of Cryptosporidium hominis thymidylate synthase with selectivity over the human enzyme. Complementing the enzyme inhibition compound 1 also has anti-cryptosporidial activity in cell culture. A crystal structure with compound 1 bound to the TS active site is discussed in terms of several van der Waals, hydrophobic and hydrogen bond interactions with the protein residues and the substrate analog 5-fluorodeoxyuridine monophosphate (TS), cofactor NADPH and inhibitor methotrexate (DHFR). Another crystal structure in complex with compound 1 bound in both the TS and DHFR active sites is also reported here. The crystal structures provide clues for analog design and for the design of ChTS-DHFR specific inhibitors.
Keywords: Thymidylate synthase, Cryptosporidium, Inhibitor, Dihydrofolate reductase, Crystal structure
Cryptosporidiosis, a protozoan gastrointestinal infection, continues to be a major reason for morbidity and mortality in immunocompromised individuals and young children1. Recent reports from the CDC estimate approximately 750,000 cases of cryptosporidiosis in United States each year2-4. Cryptosporidium is one of the four pathogens that cause most cases of moderate-to-severe diarrhea in infants and children in developing countries and was second to rotavirus as a cause of diarrheal morbidity and mortality in infants. Lack of effective treatment against cryptosporidiosis in immunocompromised individuals4-6 raises the need for development of effective yet less toxic drugs. Information from the crystal structure along with computational studies could guide the design and development of parasite specific inhibitors.
Thymidylate synthase (TS) and dihydrofolate reductase (DHFR) are essential enzymes in the folate biosynthesis pathway and are well established as drug targets in cancer, bacterial infections, and malaria. In Cryptosporidium hominis, TS and DHFR exist as a bifunctional enzyme. In human, these enzymes are on two separate polypeptide chains. In the presence of the cofactor 5,10-methylene tetrahydrofolate (CH2H4F), TS catalyzes reductive methylation of deoxyuridine monophosphate (dUMP) to form deoxythymine monophosphate (dTMP) and dihydrofolate (H2F). Dihydrofolate is then reduced to tetrahydrofolate in presence of NADPH by DHFR. In this study we report the x-ray crystal structures of Cryptosporidium hominis TS-DHFR (ChTS-DHFR) in complex with compound 1 (Scheme 1), an antifolate from a novel series of 2-amino-4-oxo-5-substituted pyrrolo[2,3-d]pyrimidines. In the first crystal structure, compound 1 is bound to the TS active site in the presence of 5-fluorodeoxyuridine monophosphate (FdUMP, a TS substrate analog) whereas methotrexate (MTX, a folate analog) and NADPH (DHFR cofactor) to the DHFR site. The second crystal structure reveals that compound 1 binds to both TS and DHFR active sites in the presence of FdUMP (a TS substrate analog) and NADPH (DHFR cofactor). Analysis of the interactions between the inhibitor, cofactors and the active site residues can be utilized to design parasite specific inhibitors.
Scheme 1.

Synthesis of compound 1.
Previous studies have evaluated compound 1 (2-amino-4-oxo-4,7-dihydro-pyrrolo[2,3-d]pyrimidine-methyl-phenyl-L-glutamic acid) on human and cryptosporidial forms of TS (hTS)7 and ChTS-DHFR8. In this study we evaluate its effect on ChTS-DHFR while comparing its activity on the human enzymes in combination with structural studies. While the synthesis of compound 1 has been reported earlier7, 9, a modified synthetic route (Scheme 1) is shown below that offers improvements in terms of more readily accessible precursors and shorter reaction times.
When tested against ChTS enzyme activity, compound 1 was found to be more potent on the parasite enzyme in comparison with the human enzyme. The ChTS catalytic activity was inhibited with an IC50 value of 0.38 ± 0.04 μM with a 5-fold selectivity with respect to hTS (IC50 value of 1.80 ± 0.45 μM). Compound 1 also inhibited DHFR enzyme activity with IC50 values of 0.049 ± 0.005 μM for ChDHFR and 0.64 ± 0.04 μM for hDHFR, respectively.
In this study, we also evaluated the anti-cryptosporidial activity of compound 1 on Cryptosporidium parvum sporozoites and in intracellular forms of the parasite in cell culture. Compound 1 significantly reduced parasite infection in cell culture, with a half maximal effective concentration ranging from 1 – 5 μM (Figure S5). Microscopically there were no morphological differences in treated and untreated cells. The ratios of dead and alive cells and ribosomal RNA levels in the treated and untreated cells were similar (data not shown). The detailed procedure for the cell culture assay is provided in the Supplementary data.
In an attempt to co-crystallize ChTS-DHFR with compound 1, multiple combinations of TS and DHFR site ligands were examined. Co-crystallization with compound 1 and FdUMP in the TS site and NADPH and MTX in DHFR site resulted in a crystal structure of 3.45 Å (Figure S1A, PDB code 4Q0D). Compound 1 bound to both the TS and DHFR active sites along with FdUMP and NADPH to yield a higher resolution (2.7 Å) structure (Figure S1B, PDB code 4Q0E). Detailed crystallization conditions are reported in the Supplementary data. The general ChTS-DHFR structure bound to compound 1 is similar in both structures with a root mean square deviation (RMSD) of 0.54 (Figure S2). In both structures, all residues from 3 to 521 except for residues 179 – 192 are clearly defined in the electron density, allowing all of the ligand binding sites of the structure to be visualized. When compound 1 is bound to the TS active site, the structure is essentially similar whether compound 1 or MTX is bound at the DHFR active site. Here we report both crystal structures as the higher resolution structure with compound 1 bound to both active sites allowed a more detailed analysis of important inhibitor-protein interactions.
Figure 1 shows 2mFo-Fc electron density maps of the active site region of ChTS bound to compound 1 revealing the positions of the FdUMP and compound 1 complex. Omit σA-weighted 2mFo-Fc electron density maps and data collection and refinement statistics are reported in the Supplementary data (Figure S3). The TS active site predominantly consists of hydrophobic residues, N256, A287, I315, W316, L399, F433, and M519 in addition to D518. At the TS site, compound 1 binds close to FdUMP, the pyrrolo[2,3-d]pyrimidine scaffold of compound 1 stacking with the pyrimidine ring of FdUMP. Several hydrophobic and van der Waals interactions are seen between compound 1 and L399, W316 and Y466 (Figure 2). The phenyl ring of compound 1 interacts with residues I315, F433 and M519. The non-conserved and unique residue A287 interacts with the glutamate tail of the inhibitor10. Four hydrogen bonds stabilize compound 1 optimally in the TS active site. The carbonyl O of D426 hydrogen bonds with N3 and the carbonyl O of N319 with N7 and the amino group of G430 hydrogen bonds with 4-oxo group of compound 1. The 2-amino group of compound 1 hydrogen bonds with the hydroxyl group of Y466 and carbonyl O of A520.
Figure 1.

2mFo-Fc election density maps for TS active site of ChTS-DHFR: FdUMP: compound 1 complex (A) with MTX and NADPH in the DHFR site (contour level at 1.0 σ) (B) with compound 1 and NADPH in the DHFR site (contour level at 1.3 σ).
Figure 2.
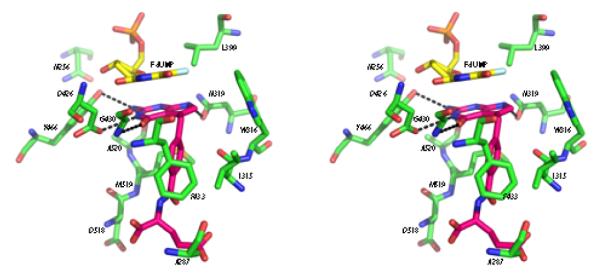
Stereo view of active site residues of ChTS (green) interacting with FdUMP (yellow) and compound 1 (pink). Hydrogen bonds (< 3.5 Å) are indicated as dashed lines.
Compound 1 has several hydrogen bonds and van der Waals interactions with the DHFR active site residues, binding close to the nicotinamide ring of NADPH (Figure S4). NADPH binds in an extended form making several hydrophobic and hydrogen bond interactions with the protein residues. The hydrophobic pocket consists of V9, A11, L25, I62 and T134 interacting with the pyrrolo[2,3-d]pyrimidine scaffold whereas T40 and F36 interact with the phenyl ring of compound 1. The carbonyl oxygens of the catalytic D32 hydrogen bond with N3 of compound 1 forming a fork that holds the inhibitor in optimal position, as seen in the case of H2F in ChTS-DHFR:H2F complex11. The amino group of V10 hydrogen bonds with N1 of compound 1 while the carboxyl of V9 interacts with N7 of compound 1. The N7 of compound 1 also interacts with the unique residue C113. The flexible glutamate tail of compound 1 is held in place by four hydrogen bonds to R70, S37 and K34. Though compound 1 binds to TS and DHFR active sites, the interactions and the conformation of the inhibitor in two sites is distinct in each case.
In summary, we report compound 1 (2-amino-4-oxo-4,7-dihydro-pyrrolo[2,3-d]pyrimidine-methyl-phenyl-L-glutamic acid) as a potent inhibitor of ChTS with anti-cryptosporidial activity is cell culture. Though most residues in ChTS active site are conserved, there are two residues (A287 and S290) which are unique to ChTS. In other parasite species and also human, these residues are Phe and Gly, respectively12, 13. These residues have been shown to be important for optimal positioning of the cofactor CH2H4F and catalysis10. In addition to the van der Waals interaction between the glutamate tail of compound 1 and A287, the structural differences between ChTS and hTS can be exploited to design a parasite specific TS inhibitor. The strategy is to focus on improving the selectivity against human enzyme without compromising the activity against ChTS-DHFR by using the newly solved crystal structure coupled with computer-aided design. This strategy is greatly facilitated by the structural information of the complex with the lead compound 1, presented here. Promising anti-cryptosporidial activity was observed in parasite infected cells, although delivery of the compound through the parasitophorous vacuolar membranes requires new drug delivery strategies that are currently being explored in order to have better potency towards the intracellular parasites.
Supplementary Material
Acknowledgment
This work is supported by NIAID grant (AI083146) to KSA, NIAID grant (AI104334) to KMF, the Paul R Stalnaker MD Distinguished Professorship to ACW, NCI grant (CA152316) and the Duquesne University Adrian Van Kaam Chair in Scholarly Excellence to AG and NIH grant (GM32136) to WLJ.
Footnotes
Supplementary data
Full details on the enzyme assays, crystallography, experimental section, synthesis of compound 1 is provided. This material is available free of charge via the Internet at http://www.elsevier.com. Data for compound 1 have been deposited in the RCSB Protein Data Bank with codes 4Q0D and 4Q0E.
References
- 1.Kotloff KL, Nataro JP, Blackwelder WC, Nasrin D, Farag TH, Panchalingam S, Wu Y, Sow SO, Sur D, Breiman RF, Faruque AS, Zaidi AK, Saha D, Alonso PL, Tamboura B, Sanogo D, Onwuchekwa U, Manna B, Ramamurthy T, Kanungo S, Ochieng JB, Omore R, Oundo JO, Hossain A, Das SK, Ahmed S, Qureshi S, Quadri F, Adegbola RA, Antonio M, Hossain MJ, Akinsola A, Mandomando I, Nhampossa T, Acácio S, Biswas K, O’Reilly CE, Mintz ED, Berkeley LY, Muhsen K, Sommerfelt H, Robins-Browne RM, Levine MM. Lancet. 2013;382:209. [Google Scholar]
- 2.Feng Y, Tiao N, Li N, Hlavsa M, Xiao L. J Clin Microbiol. 2013 doi: 10.1128/JCM.02973-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Scallan E, Hoekstra RM, Angulo FJ, Tauxe RV, Widdowson MA, Roy SL, Jones JL, Griffin PM. Emerg Infect Dis. 2011;17:7. doi: 10.3201/eid1701.P11101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Checkley W, White AC, Jr., Jaganath D, Arrowood MJ, Chalmers RM, Chen X-M, Fayer R, Griffiths JK, Guerrant RL, Hedstrom L, Huston CD, Kotloff KL, Kang G, Mead JR, Miller M, Petri W, Jr., Priest JW, Roos DS, Striepen B, Thompson RA, Ward HD, Van Voorhis WC, Xiao L, Zhu G, Houpt ER. Lancet Infect Dis. 2014 doi: 10.1016/S1473-3099(14)70772-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Abubakar I, Aliyu SH, Arumugam C, Usman NK, Hunter PR. Br J Clin Pharmacol. 2007;63:387. doi: 10.1111/j.1365-2125.2007.02873.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gargala G, Delaunaya A, Lib X, Brasseurb P, Favennecb L, Balleta JJ. J Antimicrob Chemother. 2000;46:57. doi: 10.1093/jac/46.1.57. [DOI] [PubMed] [Google Scholar]
- 7.Aso K, Imai Y, Yukishige K, Ootsu K, Akimoto H. Chem. Pharm. Bull. 2001;49:1280. doi: 10.1248/cpb.49.1280. [DOI] [PubMed] [Google Scholar]
- 8.Kumar VP, Frey KM, Wang Y, Jain HK, Gangjee A, Anderson KS. Bioorganic & medicinal chemistry letters. 2013;23:5426. doi: 10.1016/j.bmcl.2013.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang YR,S, Ravindra MP, Hales E, Orr S, Cherian C, Hou Z, Matherly LH, Gangjee A. J. Med. Chem. 2013;56:10016. doi: 10.1021/jm401328u. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Doan LT, Martucci WE, Vargo MA, Atreya CE, Anderson KS. Biochemistry. 2007;46:8379. doi: 10.1021/bi700531r. [DOI] [PubMed] [Google Scholar]
- 11.Anderson AC. Acta Crystallogr Sect F Struct Biol Cryst Commun. 2005;61:258. doi: 10.1107/S1744309105002435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Martucci WE, Vargo MA, Anderson KS. Biochemistry. 2008;47:8902. doi: 10.1021/bi800466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Vásquez JR, Goozé L, Kim K, Gut J, Petersen C, Nelson RG. Mol Biochem Parasitol. 1996;79:153. doi: 10.1016/0166-6851(96)02647-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.