

Cryptosporidium parvum is the primary protozoan parasite from the Cryptosporidium genus responsible for cryptosporidiosis in humans. Here, the 2.0 Å resolution structure of the divalent-cation tolerance protein CutA1 encoded in the genome of C. parvum Iowa II is described.
Keywords: Cryptosporidium parvum, divalent-cation tolerance protein, CutA1, cryptosporidiosis
Abstract
Cryptosporidiosis is an infectious disease caused by protozoan parasites of the Cryptosporidium genus. Infection is associated with mild to severe diarrhea that usually resolves spontaneously in healthy human adults, but may lead to severe complications in young children and in immunocompromised patients. The genome of C. parvum contains a gene, CUTA_CRYPI, that may play a role in regulating the intracellular concentration of copper, which is a toxic element in excess. Here, the crystal structure of this CutA1 protein, Cp-CutA1, is reported at 2.0 Å resolution. As observed for other CutA1 structures, the 117-residue protein is a trimer with a core ferrodoxin-like fold. Circular dichroism spectroscopy shows little, in any, unfolding of Cp-CutA1 up to 353 K. This robustness is corroborated by 1H–15N HSQC spectra at 333 K, which are characteristic of a folded protein, suggesting that NMR spectroscopy may be a useful tool to further probe the function of the CutA1 proteins. While robust, Cp-CutA1 is not as stable as the homologous protein from a hyperthermophile, perhaps owing to a wide β-bulge in β2 that protrudes Pro48 and Ser49 outside the β-sheet.
1. Introduction
Diarrhea accounts for over 800 000 deaths of children under the age of five each year (Liu et al., 2012 ▶), and one of the pathogens responsible for much of the diarrheal disease in low-income nations is the apicomplexan parasite Cryptosporidium (Kotloff et al., 2013 ▶). This genus of obligatory intracellular protozoa contains 16 recognized species, eight of which are known to infect humans (Sonia, 2011 ▶), with C. hominis and C. parvum being the most prevalent (Xiao & Ryan, 2004 ▶). Transmission of the disease occurs by the direct transfer of oocysts, the infective stage of the parasite, from host to host via the fecal–oral route or indirect transfer via the contamination of food or water (Ramirez et al., 2004 ▶). The disease is especially contagious because oocysts are resistant to common disinfectants and a single oocyst is sufficient for the disease to take root in susceptible hosts (Pereira et al., 2002 ▶). In humans, infection occurs in the microvillus border of gastrointestinal epithelium cells, usually by the invasion of sporozoites released by the oocysts, where the parasite passes through its developmental stages before completing the cycle with the release of oocysts into fecal material that is excreted (Ramirez et al., 2004 ▶). Cryptosporidiosis is usually self-limiting in healthy individuals, but may be fatal in immunocompromised patients (for example, those with HIV/AIDS) and malnourished children. The US Food and Drug Administration has only approved one drug, nitazoxanide, for the treatment of cryptosporidiosis; however, it is not effective for those people most vulnerable to the disease (Striepen, 2013 ▶). Consequently, new therapies need to be developed to combat this little-studied organism (Myler et al., 2009 ▶; Striepen, 2013 ▶).
While bacteria, plant and animal cells usually require trace amounts of heavy metals in order to function properly, these metals are often toxic in excess because they bind to essential cellular components or generate free radicals (Nies, 1999 ▶). Consequently, cells have evolved uptake, storage and secretion systems to tightly regulate the intracellular concentration of heavy-metal ions (Nelson, 1999 ▶). One heavy metal required in trace amounts is copper (Linder, 1991 ▶), and in microorganisms copper homeostasis is associated with two families of genes: cop and cut. The cop gene family is well understood, with the products of the four genes (copA, copB, copY and copZ) acting as active-transport efflux pumps (CopA and CopB) and pump regulators (CopY and CopZ) (Magani & Solioz, 2005 ▶). On the other hand, the cut gene family is less understood, with the functions of the gene products (cutA–cutF) not all clearly defined. Among the less defined cut gene products is CutA1 (Fong et al., 1995 ▶), an approximately 12 kDa cytoplasmic protein that is widespread among organisms, including humans (Yang et al., 2009 ▶). In Escherichia coli, genetic studies have associated CutA1 with divalent metal-ion tolerance (including copper; Fong et al., 1995 ▶). In mammals, CutA1 is found in the brain and has been associated with acetylcholine esterase trafficking and processing (Liang et al., 2009 ▶). In plants, the structure of CutA1 is homologous to the structure of the P-II nitrogen regulatory protein, suggesting that CutA1 may play a role in signal transduction (Arnesano et al., 2003 ▶). While the CutA1 protein from C. parvum has been identified as a potential drug target owing to its association with divalent metal-ion tolerance (Stacy et al., 2011 ▶), the determination of CutA1 structures from a wider variety of organisms may also assist in the disentanglement of the multiple functional roles attributed to this protein.
2. Materials and methods
2.1. Cloning, expression and purification
The Cp-CutA1 gene (CUTA_CRYPI/XP_627596.1) was amplified by PCR using the genomic DNA of C. parvum strain Iowa II and the oligonucleotide primers 5′-GGGTCCTGGTTCGATGATAAATTCAAACATGACGGAAAC-3′ (forward) and 5′-CTTGTTCGTGCTGTTTATTAGCTTCTAACTGTCTGGTTTACCC-3′ (reverse) (Invitrogen, Carlsbad, California, USA) containing ligation-independent cloning (LIC) primers (underlined). The amplified Cp-CutA1 gene was then inserted into the NruI/PmeI-digested expression vector AVA0421 by LIC, which provided a 21-residue tag (MAHHHHHHMGTLEAQTQGPGS–) at the N-terminus of the expressed protein (Choi et al., 2011 ▶). Stock solutions grown in LB medium (OD600 ≃ 0.8) were prepared from a single colony picked from a LB agar plate and were made ∼15%(v/v) in glycerol prior to aliquot storage (1 ml) at 193 K. Uniformly 15N-labeled Cp-CutA1 was prepared using an autoinduction minimal medium protocol (Studier, 2005 ▶) starting with a 20 ml LB culture (310 K) seeded with 1 ml of frozen glycerol stock. At an OD600 of ∼0.8, the 20 ml culture was directly transferred to 500 ml autoinduction minimal medium containing 15NH4Cl (1.4 mg ml−1), sodium diphosphate (7.1 mg ml−1), potassium monophosphate (6.8 mg ml−1), MgSO4 (120 µg ml−1), 1000× metals (1 ml per litre of medium) and 50× 5052 carbon source (20 ml per litre of medium) (Studier, 2005 ▶). The antibiotics chloramphenicol (34 µg ml−1) and ampicillin (150 µg ml−1) were included in the medium at all times. After ∼4 h of growth at 310 K, the culture was transferred to a 298 K shaker for overnight incubation. Approximately 16 h later, the cells were harvested by mild centrifugation and stored at 193 K. After thawing the frozen pellet, Cp-CutA1 was purified using two chromatography steps: immobilized metal-ion affinity (20 ml Ni-Agarose 6 FastFlow column (GE Healthcare, Piscataway, New Jersey, USA) and Superdex 75 gel filtration (HiLoad 26/60 column) (Buchko et al., 2013 ▶). The second chromatography step, in addition to removing minor impurities, exchanged Cp-CutA1 into the buffer used for the crystal screens and the NMR and CD studies: 100 mM NaCl, 20 mM Tris–HCl, 1.0 mM dithiothreitol pH 7.1. For the NMR study, the fraction containing Cp-CutA1 after the first chromatography step (∼40 ml) was exchanged into 3C protease cleavage buffer (150 mM NaCl, 20 mM Tris–HCl pH 7.6) by overnight dialysis (4 l) at 278 K. The protein was concentrated to ∼20 mg ml−1 (Amico Centriprep-10) and the N-terminal polyhistidine tag was removed by overnight incubation with 3C protease (1 µg per 50 µg of target protein) prior to the second chromatography step.
2.2. Circular dichroism spectroscopy
An Aviv Model 410 spectropolarimeter (Lakewood, New Jersey, USA) was used to collect circular dichroism data from an ∼0.05 mM Cp-CutA1 sample (uncleaved) in a quartz cell of 0.1 cm path length. The reported steady-state wavelength spectrum was the result of averaging two consecutive scans with a bandwidth of 1.0 nm and a time constant of 1.0 s. The final spectrum was processed by automatically line-smoothing the data using the Aviv software. A thermal denaturation curve was obtained by recording the ellipticity at 220 nm in 2.0 K intervals from 283 to 363 K.
2.3. NMR spectroscopy
Two-dimensional 1H–15N HSQC spectra for Cp-CutA1 (cleaved) were obtained at various temperatures on a Varian Inova 600 spectrometer equipped with a 1H{13C,15N,31P} pentaprobe and pulse-field gradients.
2.4. Crystallization
Hanging-drop vapor-diffusion crystallization trials were set up at room temperature (∼295 K) using the Precipitant Synergy sparse-matrix kit (Rigaku Reagents, Bainbridge, Washington, USA). Cp-CutA1 stock solution (uncleaved, 3.0 mg ml−1, 1.0 µl) was mixed with reservoir solution (1.0 µl) and equilibrated against reservoir solution (150 µl) in 48-well VDX48 plates (Hampton Research, Aliso Viejo, California, USA). The crystals used for X-ray data collection and structure determination were obtained from condition No. 48 (0.1 M HEPES pH 7.5, 30% PEG 1500, 20% PEG 400); they were cryoprotected with the reservoir solution supplemented with ∼15% glycerol, flash-cooled in liquid nitrogen and shipped to a synchrotron.
2.5. Data collection and structure refinement
Native X-ray data were collected from a single crystal on beamline X29A at the National Synchrotron Light Source, Brookhaven National Laboratory using an ADSC Quantum 315R CCD detector (Table 1 ▶). The data were reduced with XDS/XSCALE (Kabsch, 2010 ▶). The structure was solved by molecular replacement (MR) using Phaser (McCoy et al., 2007 ▶) from the CCP4 suite (Winn et al., 2011 ▶). The crystal structure of Thermus thermophilus HB8 CutA1 (monomer, chain A, PDB entry 1nza; Bagautdinov, 2014 ▶), a protein with 43% sequence identity to Cp-CutA1, was used as the MR search model after alignment modification in CHAINSAW (Stein, 2008 ▶). The structure was initially rebuilt with Buccaneer (Cowtan, 2006 ▶), followed by additional model building in ARP/wARP (Langer et al., 2008 ▶). The final model was produced after numerous iterative rounds of refinement in REFMAC v.5.6.0117 (Murshudov et al., 2011 ▶) and manual building in Coot (Emsley & Cowtan, 2004 ▶). Electron density was missing for the 21-residue polyhistidine tag plus the next six (chain A), ten (chain B) and 12 (chain C) residues at the N-terminus. At the C-terminus, electron density was missing for the terminal residue Ser117 in two of the three protomer chains (A and B). The final model deposited in the Protein Data Bank (PDB; Berman, 2008 ▶) under accession code 4e98 showed good geometry and fitness (Table 1 ▶) according to analysis with MolProbity (Chen et al., 2010 ▶).
Table 1. Summary of the diffraction data-collection and refinement statistics for Cp-CutA1.
Values in parentheses are for the highest resolution shell.
| Data collection | |
| X-ray source | NSLS beamline X29A |
| Detector | ADSC Q315R CCD |
| X-ray wavelength () | 1.075 |
| Crystal-to-detector distance (mm) | 250 |
| Rotation range per image () | 1.0 |
| Exposure time per image (s) | 0.4 |
| Total rotation range () | 270 |
| Temperature (K) | 100 |
| Data set | Native |
| Space group | C2 |
| Unit-cell parameters | |
| a () | 94.46 |
| b () | 55.59 |
| c () | 67.29 |
| = () | 90 |
| () | 108.21 |
| Mosaicity () | 0.55 |
| Matthews coefficient (3Da1) | 1.79 |
| Solvent content (%) | 31 |
| Resolution range () | 47.262.00 (2.052.00) |
| Mean I/(I) | 13.30 (3.58) |
| No. of observed reflections | 22551 (1521) |
| Completeness (%) | 98.9 (100) |
| Multiplicity | 5.5 (5.6) |
| R merge † (%) | 0.093 (0.472) |
| R meas ‡ (%) | 0.108 (0.559) |
| Overall B factor from Wilson plot (2) | 29.0 |
| Phasing | |
| Molecular-replacement model | 1nza |
| Refinement | |
| No. of non-H atoms | |
| Protein | 2540 |
| Water | 191 |
| Ions (Cl) | 3 |
| R work § (%) | 19.8 (23.0) |
| R free ¶ (%) | 24.7 (25.5) |
| R.m.s.d., bonds () | 0.014 |
| R.m.s.d., angles () | 1.568 |
| Average B factors (2) | |
| Protein | 22.4 |
| Water | 29.5 |
| Ions (Cl) | 58.6 |
| Model validation | |
| MolProbity Ramachandran analysis | |
| Most favored (%) | 98.7 |
| Additionally allowed (%) | 1.3 |
| MolProbity analysis | |
| Clash score, all atoms [percentile] | 4.07 [99th] |
| MolProbity score [percentile] | 1.52 [96th] |
| PDB identifier | 4e98 |
R
merge = 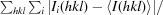
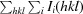 .
.
R
meas = 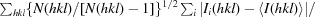 (Diederichs Karplus, 1997 ▶).
(Diederichs Karplus, 1997 ▶).
R
work = 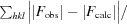
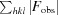 , where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated with the 95% of thereflections remaining after setting aside 5% for R
free.
, where F
obs and F
calc are the observed and calculated structure factors, respectively, calculated with the 95% of thereflections remaining after setting aside 5% for R
free.
R
free =
. The free R factor was calculated using 5% of the reflections which were omitted from the refinement.
Note that the 21 non-native residues at the N-terminus of Cp-CutA1 are numbered negatively in the PDB, starting with Met−21. The first non-native residue labeled Met0 in the PDB is Met1 in the native Cp-CutA1 primary amino acid sequence. Consequently, it is necessary to shift the numbering of PDB residues Met0–Ser116 up one number to obtain the native residue number: Met1–Ser117.
3. Results and discussion
3.1. Crystal structure of Cp-CutA1
As shown in the cartoon representation in Fig. 1 ▶(a), Cp-CutA1 is a trimer composed of a circularly closed antiparallel β-sheet containing 15 β-strands. The sites of interprotomer β-strand hydrogen bonding are identified in Fig. 1 ▶(b), which shows a cartoon representation of a single protomer chain. On the front face of the trimer in Fig. 1 ▶(a) these interprotomer β-sheet interactions (shaded green) involve the edges of β2 with the opposite β2 edges in the two other symmetrically related protomers. On the opposite face of the trimer these interprotomer β-sheet interactions (shaded red) involve opposing residues in β4 and β5. Such intermolecular hydrogen-bonding interactions, in combination with associated hydrophobic interactions in the interior of the complex (Arnesano et al., 2003 ▶; Tanaka et al., 2004 ▶), support a trimer as the ‘biologically relevant’ unit. Assembly analysis with the PDBePISA server generates a complex significance score (CSS) of 0.787 and a buried surface area of 34.4%, values indicating that the trimer is thermodynamically stable (Krissinel & Henrick, 2007 ▶). The trimeric quaternary framework of Cp-CutA1 has been observed for CutA1 proteins from other organisms, including E. coli (Ec-CutA1; PDB entry 1naq; Arnesano et al., 2003 ▶), Rattus norvegicus (PDB entry 1osc; Arnesano et al., 2003 ▶), Thermotoga maritima (PDB entry 1kr4; Savchenko et al., 2004 ▶), Pyrococcus horikoshii (PDB entry 1j2v; Tanaka et al., 2004 ▶), Homo sapiens (PDB entry 2zfh; Bagautdinov et al., 2008 ▶), Shewanella sp. SIB1 (PDB entry 3ahp; Sato et al., 2011 ▶), Oryza sativa (PDB entry 2zom; Y. Kezuka, B. Bagautdinov, S. Katoh, Y. Ohtake, K. Yutani, T. Nonaka & E. Katoh, unpublished work) and Thermus thermophilus (PDB entry 1nza; Bagautdinov, 2014 ▶). Note that these solid-state observations translate to the solution state as the retention time of Cp-CutA1 on a size-exclusion column (data not shown) was characteristic of a trimer.
Figure 1.

(a) Cartoon representations of the trimer (a) and single protomer (b) in the crystal structure of Cp-CutA1 (PDB entry 4e98). The β-strands are colored gold, α-helices blue and 310-helices (η1) cyan, and the protomer chains are numbered 1–3. Two types of intertwined hydrogen bonds between the three protomers help to stabilize the trimeric structure. One type, colored green, involves dual interprotomer antiparallel hydrogen bonds between opposing residues in β2. The other type, colored firebrick red, involves single interprotomer antiparallel hydrogen bonds between opposing residues in β4 and β5. In chain 1 (b) the interprotomer positions of these intertwined hydrogen bonds are indicated by the numbers 2 and 3. The wide β-bulge in β2 is colored orange. (c) The primary amino acid sequence of Cp-CutA1 with the elements of secondary structure colored similar to the cartoon representations. Residues participating in the two types of interprotomer hydrogen bonds are highlighted in a green or red box and the residues of the β-bulge are highlighted in an orange box.
The structure of each monomer in the Cp-CutA1 trimer is similar, with a backbone-atom (N—Cα—C=O) root-mean-square deviation (r.m.s.d.) of between 0.22 to 0.30 Å between any pair of subunits (Ser13–Arg116). Fig. 1 ▶(b) shows a cartoon representation of the structure of a single protomer, with the elements of secondary structure labeled on the structure and also in the amino acid sequence of Cp-CutA1 (Fig. 1 ▶ c). Each monomer contains five β-strands, three α-helices and one 310-helix arranged in two domain-like parts. The major part is a ferredoxin-like fold (CATH No. 3.30.70.120; Savchenko et al., 2004 ▶) composed of a two-layer α/β sandwich, β1α1β2–β3α2β4, with the β-strands forming an antiparallel β-sheet, β2↑β3↓β1↑β4↓, and the two α-helices aligning antiparallel to each other on one face of the β-sheet. The minor part is composed of a short β-strand (β5) and a single α-helix (α3), with β5 forming an interprotomer antiparallel β-sheet opposite the C-terminal part of β4 (colored red in Fig. 1 ▶ b) and α3 aligned approximately orthogonal to α1 and α2.
3.2. Cp-CutA1 and copper binding
One of the functions attributed to the CutA1 proteins in microorganisms is a role in the regulation of copper and other divalent cations inside the cell (Fong et al., 1995 ▶). Such a function suggests cation binding to CutA1. While a number of CutA1 structures have been deposited in the PDB, only one, P. horikoshii CutA1 (PDB entry 1uku), has been solved with electron density identified for Cu2+ in the crystal lattice (Tanaka et al., 2004 ▶). In this CutA1–Cu2+ complex, the metal is octahedrally coordinated by six O atoms at a trimer–trimer interface with an aspartic acid side chain from each of the trimers and two water molecules nearly coplanar with the Cu2+ and with the main-chain carbonyl group of the adjacent lysine residue from each of the trimers (Lys49) roughly perpendicular to plane of the other O atoms. As shown in the sequence alignment in Fig. 2 ▶ (blue arrow), the aspartic acid position (Asp48 in P. horikoshii CutA1) is relatively conserved among species. At the equivalent position in Cp-CutA1 there is a glutamic acid residue instead, Glu63, with Asp62 next to it. As illustrated in Fig. 3 ▶(a), which shows a representation of the electrostatic potential at the solvent-accessible surface of Cp-CutA1, Asp62 and Glu63 are near the edge of one face on the surface of the trimer (black circles). Because P. horikoshii CutA1 is observed to precipitate in the presence of excess Cu2+, it has been postulated that the protein forms insoluble, face-to-face, Cu2+-mediated multimeric complexes as one CutA1 trimer interacts with three other CutA1 trimers (Tanaka et al., 2004 ▶). Cp-CutA1 may form multimeric complexes via the same mechanism because in the presence of molar excess Cu2+ it also precipitated out of solution (data not shown).
Figure 2.

ClustalW multiple amino acid sequence alignment, illustrated with ESPript (v.3; Robert & Gouet, 2014 ▶), of selected CutA1 proteins with structures deposited in the PDB: Cp-CutA1 from Cryptosporidium parvum (gi:3375883; PDB entry 4e98), Ec-CutA1 from Escherichia coli (gi:12933699; PDB entry 1naq), Rn-CutA1 from Rattus norvegicus (gi:294288; PDB entry 1osc), Tm-CutA1 from Thermotoga maritima (PDB entry 1kr4), Ph-CutA1 from Pyrococcus horikoshii (gi:1443316; PDB entry 1j2v), Hs-CutA1 from Homo sapiens (gi:51596; PDB entry 2zfh), Sh-CutA1 from Shewanella sp. SIB1 (3ahp) and Tt-CutA1 from Thermus thermophilus (gi:3168527; PDB entry 1nza). Above the alignment are the elements of secondary structure observed for Cp-CutA1. Below the alignments are blue, magenta and green arrows representing conserved sequence positions that may be relevant to copper binding.
Figure 3.

Comparison of the electrostatic potential at the solvent-accessible surface for Cp-CutA1 (PDB entry 4e98). The orientation in face 1 (a) is the same as in Fig. 1 ▶(a), with face 2 (b) being a 180° y-axis rotation of face 1. The view in (c) is a side projection generated by a 90° y-axis rotation of face 2 (b). The black circles represent the positions of negatively charged sites associated with copper binding in the P. horikoshii crystal structure (PDB entry 1uku). Red and blue represent negative and positive charge, respectively, while white is indicative of electronically neutral and hydrophobic character.
The coordination of copper exclusively by O atoms has only been observed for the P. horikoshii CutA1–Cu2+ crystal structure and there is still some debate regarding the biological significance of this complex. Firstly, as illustrated in Fig. 3 ▶ for Cp-CutA1, there are many pockets of negatively charged surfaces on the protein, especially on the edge of the disk-shaped structure (Fig. 3 ▶ c), and it is unclear why copper would bind to the Asp62/Glu63 site instead of the many other negatively charged sites if binding were mediated by negatively charged O atoms. Secondly, known copper-binding sites in proteins are built around cysteine and/or histidine residues (Opella et al., 2002 ▶; Rubino & Franz, 2012 ▶). While Cp-CutA1 contains a CXC motif instead of the more typical CXXC copper-binding motif in such sites, only the second Cys, Cys43, is highly conserved among the CutA1 sequences from various species (magenta arrow in Fig. 2 ▶). Cp-CutA1 also contains two histidine residues, His61 and His89; however, only the latter histidine (green arrow in Fig. 2 ▶) is highly conserved and they are both physically too distant from the CXC motif to be able to coordinate copper in the trimeric form. Moreover, the CXC motif is not surfaced-exposed in Cp-CutA1. Hence, the only way for the surface-exposed histidine residues to coordinate copper would be via intermolecular interactions, and this too could effect the precipitation of Cp-CutA1 observed in the presence of excess Cu2+. Thirdly, it may be that the Cu2+-binding sites in CutA1 proteins vary between species (Tanaka et al., 2004 ▶) because in Ec-CutA1 UV–visible, ESR and EXAFS analyses in the presence of Cu2+ supports a model in which the metal is bound to two histidine residues and one cysteine residue (with the fourth oxygen ligand being a water molecule or backbone carbonyl group; Arnesano et al., 2003 ▶). While this model is supported by the spatial proximity of these residues, Cys16, His83 and His84, in the Ec-CutA1 crystal structure, as illustrated in Fig. 2 ▶ only His84 is conserved among the CutA1 sequences from various species.
3.3. Thermal stability of Cp-CutA1
Circular dichroism spectroscopy is a useful method to rapidly probe the conformation and stability of proteins in solution because the CD signal is sensitive to small changes in the backbone of a protein (Woody, 1974 ▶). Fig. 4 ▶(a) shows the steady-state CD spectrum for Cp-CutA1 collected at 298 K. The major features of the spectrum are a double minimum at approximately 220 and 210 nm and an extrapolated maximum below 200 nm, features that are characteristic of α-helical secondary structure (Holzwarth & Doty, 1965 ▶; Greenfield, 2006 ▶). It is known from the crystal structure of Cp-CutA1 (Fig. 1 ▶) that the protein is approximately 32% α-helical and 41% β-sheet, with the latter element of secondary structure likely being responsible for the double minima not being equal in intensity.
Figure 4.

(a) Circular dichroism steady-state wavelength spectrum for Cp-CutA1 (∼0.05 mM) in CD buffer collected at 298 K. (b) The CD thermal melt for Cp-CutA1 obtained by measuring the ellipticity at 220 nm at 2.0 K intervals between 283 and 353 K.
The CutA1 proteins are known to be stable at high temperatures (Sato et al., 2011 ▶), with the CutA1 protein from P. horikoshii, a hyperthermophile, having a denaturation temperature of 423 K (Tanaka et al., 2006 ▶). To determine whether Cp-CutA1 was also stable to high temperatures, the ellipticity at 210 nm was measured as a function of temperature between 283 and 353 K and the results are shown in Fig. 4 ▶(b). Clearly, there is little, if any, change in the ellipticity at 210 nm over this temperature range, indicating that Cp-CutA1 is also stable to high temperatures.
3.4. High temperature 1H–15N HSQC spectra of Cp-CutA1
The observation from the CD thermal melt showing that the structure of Cp-CutA1 changes little up to a temperature of 353 K suggests that it may be possible to study this protein in solution at high temperatures using NMR spectroscopy. The advantages of studying the protein at high temperature are improved spectral line shape and better magnetization transfer in three-dimensional backbone assignment experiments for the ∼40 kDa trimer. Fig. 5 ▶(a) shows a TROSY 1H–15N HSQC spectrum for Cp-CutA1, with the N-terminal polyhistidine tag removed, at 313 K. While the intensities of the side-chain resonances (red box) are weak and variable, the rest of the spectrum features amide cross peaks of near-uniform intensity that are well dispersed in both the proton and nitrogen dimensions. These later features are characteristic of a folded protein (Yee et al., 2002 ▶) and have been reported for Ec-CutA1 at 298 K (Arnesano et al., 2003 ▶). The amino acid sequence of Cp-CutA1 contains 117 native residues plus four non-native residues (GPGS–) at the N-terminus after digestion with 3C protease. 116 backbone amide cross peaks are expected in the 1H–15N HSQC spectrum [121 − (the N-terminal residue + four proline residues)] and 102 cross peaks are observed, indicating that at most only 14 amide cross peaks are missing (a few may overlap). Given that the interfacial regions between adjacent protomers are largely composed of an intertwined β-sheet, the missing amide resonances are likely not at protomer interfaces, but instead in flexible, surface-exposed regions of the protein (the many loops).
Figure 5.

TROSY 1H–15N HSQC spectra of an ∼1 mM sample of Cp-CutA1 in NMR buffer (100 mM sodium chloride, 20 mM Tris–HCl, 1 mM DTT pH 7.0) collected at 313 K (a) and 333 K (b) at a 1H resonance frequency of 600 MHz. The red box primarily contains cross peaks for side-chain resonances.
Fig. 5 ▶(b) shows a TROSY 1H–15N HSQC spectrum for Cp-CutA1 at 333 K. The faint side-chain resonances that were present in the red box in Fig. 5 ▶(ba) have completely disappeared. Otherwise, except for the disappearance of eight additional amide cross peaks, the remaining 96 amide cross peaks are still near-uniform in intensity and well dispersed in both the proton and nitrogen dimensions, indicating that the protein is still largely folded. It may be possible to intimately follow the thermally induced unfolding of Cp-CutA1 at the residue level by assigning its 1H–15N HSQC spectrum at low temperature and performing a ‘temperature titration’ to determine the sequential order of disappearance of amide cross peaks. Such assignments will also enable chemical shift perturbation studies to assist in the identification of ligand-binding surfaces on the protein (Buchko et al., 1999 ▶; Zuiderweg, 2002 ▶), such as the copper-binding site.
Note that when performing the CD temperature experiment on an ∼0.05 mM sample, there was no visible evidence of protein precipitation in the CD cell (which would have resulted in less negative ellipticity values). At an ∼1 mM concentration in the NMR tube, there was evidence of some protein precipitation after 3 h at 333 K. These observations suggest that heating may induce concentration-dependent aggregation.
3.5. A β-bulge in β2 that may trigger denaturation
It has been postulated that the differences in the thermostability of CutA1 proteins may be related to features in β2 because CutA1 proteins from thermophiles contain no kink in β2, while those from mesophiles contain a β2 kink and in those from psychrophiles β2 is divided into two short β-strands (Sato et al., 2011 ▶). Fig. 6 ▶ illustrates the backbone hydrogen-bonding pattern between antiparallel β-strands 2 and 3 in Cp-CutA1 and highlights a wide β-bulge (Craveur et al., 2013 ▶) that protrudes Pro48 and Ser49 in β2 (orange circle) outside the β-sheet. While Cp-CutA1 is stable to high temperatures, it likely not as thermally stable as the CutA1 protein from the hyperthermophile P. horikoshii (Tanaka et al., 2006 ▶) because there was evidence of protein precipitation at high protein concentrations at 333 K. Furthermore, the P. horikoshii CutA1 trimer is also stable to SDS, running as a trimer on SDS–PAGE, while Cp-CutA1 runs primarily as a monomer (data not shown). The trigger for this unfolding may be the β-bulges in β2, which are centered near each other in the middle of one face of the trimeric protein (colored orange in Fig. 1 ▶ a).
Figure 6.
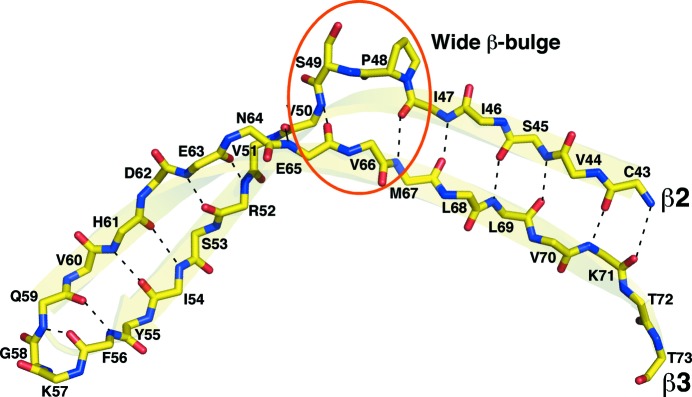
Backbone hydrogen-bonding pattern observed between antiparallel β-strands 2 and 3 in Cp-CutA1 (PDB entry 4e98), highlighting the Pro48–Ser49 wide β-bulge (orange circle) in β2. Interstrand hydrogen bonds are indicated by dashed black lines and the main-chain backbone atoms are colored as follows: oxygen, red; nitrogen, blue; carbon, yellow.
4. Conclusions
In general, the crystal structure of Cp-CutA1 is similar to the structures reported for other eukaryotic and prokaryotic CutA1 proteins: a trimer with a core ferrodoxin-like fold. While electron density was not observed for any divalent cation in the Cp-CutA1 crystal lattice, analysis of the Cp-CutA1 structure suggests that copper binding may occur through intermolecular binding at negatively charged surfaces on the protein or perhaps at intermolecular surface-exposed histidines. The temperature profiles obtained by circular dichroism spectroscopy suggest that the protein is stable up to 353 K at low protein concentrations (∼0.05 mM). While the temperature stability was confirmed by NMR experiments at high temperature, there was evidence of precipitation at the higher protein concentrations (∼1 mM), suggesting a concentration dependence of the stability of the protein at higher temperatures. The quality of the 1H–15N HSQC spectra at higher temperatures suggest that NMR spectroscopy could be used to assist our understanding of the biological function of CutA1 (Barrett et al., 2013 ▶). Such information, together with the structure provided for Cp-CutA1 here, may facilitate rational structure-based drug design targeting C. parvum.
Supplementary Material
PDB reference: divalent-cation tolerance protein, 4e98
Acknowledgments
This research was funded by the National Institute of Allergy and Infectious Diseases under Federal Contract Nos. HHSN2722001200025C and HHSN272200700057C. The SSGCID internal ID for Cp-CutA1 is CrpA.01087.a. Part of the research was conducted at the W. R. Wiley Environmental Molecular Sciences Laboratory, a national scientific user facility sponsored by the US Department of Energy’s Office of Biological and Environmental Research program located at Pacific Northwest National Laboratory (PNNL). Battelle operates PNNL for the US Department of Energy. The assistance of the X29A beamline scientists at the National Synchrotron Light Source at Brookhaven National Laboratory is appreciated. Support for beamline X29A at the National Synchrotron Light Source comes principally from the Offices of Biological and Environmental Research and of Basic Energy Sciences of the US Department of Energy and from the National Center for Research Resources of the National Institutes of Health.
References
- Arnesano, F., Banci, L., Benvenuti, M., Bertini, I., Calderone, V., Mangani, S. & Viezzoli, M. S. (2003). J. Biol. Chem. 278, 45999–46006. [DOI] [PubMed]
- Bagautdinov, B. (2014). Acta Cryst. F70, 404–413. [DOI] [PMC free article] [PubMed]
- Bagautdinov, B., Matsuura, Y., Bagautdinova, S., Kunishima, N. & Yutani, K. (2008). Acta Cryst. F64, 351–357. [DOI] [PMC free article] [PubMed]
- Barrett, P. J., Chen, J., Cho, M.-K., Kim, J.-H., Lu, Z., Mathew, S., Peng, D., Song, Y., Van Horn, W. D., Zhuang, T., Sönnichsen, F. D. & Sanders, C. R. (2013). Biochemistry, 52, 1303–1320. [DOI] [PMC free article] [PubMed]
- Berman, H. M. (2008). Acta Cryst. A64, 88–95. [DOI] [PubMed]
- Buchko, G. W., Abendroth, J., Robinson, H., Zhang, A., Hewitt, S. N., Edwards, T. E., Van Voorhis, W. C. & Myler, P. J. (2013). J. Struct. Funct. Genet. 14, 47–57. [DOI] [PMC free article] [PubMed]
- Buchko, G. W., Daughdrill, G. W., de Lorimier, R., Rao B, K., Isern, N. G., Lingbeck, J. M., Taylor, J. S., Wold, M. S., Gochin, M., Spicer, L. D., Lowry, D. F. & Kennedy, M. A. (1999). Biochemistry, 38, 15116–15128. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choi, R., Kelley, A., Leibly, D., Nakazawa Hewitt, S., Napuli, A. & Van Voorhis, W. C. (2011). Acta Cryst. F67, 998–1005. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Craveur, P., Joseph, A. P., Rebehmed, J. & de Brevern, A. G. (2013). Protein Sci. 22, 1366–1378. [DOI] [PMC free article] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Biol. 4, 269–275. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Fong, S. T., Camakaris, J. & Lee, B. T. (1995). Mol. Microbiol. 15, 1127–1137. [DOI] [PubMed]
- Greenfield, N. J. (2006). Nature Protoc. 1, 2527–2535. [DOI] [PMC free article] [PubMed]
- Holzwarth, G. M. & Doty, P. (1965). J. Am. Chem. Soc. 87, 218–228. [DOI] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Kotloff, K. L. et al. (2013). Lancet, 382, 209–222. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2007). J. Mol. Biol. 372, 774–797. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Liang, D., Nunes-Tavares, N., Xie, H. W., Carvalho, S., Bon, S. & Massoulié, J. (2009). J. Biol. Chem. 284, 5195–5207. [DOI] [PubMed]
- Linder, M. C. (1991). Biochemistry of Copper, edited by E. Frieden, pp. 1–13. New York: Plenum Press.
- Liu, L., Johnson, H. L., Cousens, S., Perin, J., Scott, S., Lawn, J. E., Rudan, I., Campbell, H., Cibulskis, R., Li, M., Mathers, C. & Black, R. E. (2012). Lancet, 379, 2151–2161. [DOI] [PubMed]
- Magani, D. & Solioz, M. (2005). Biometals, 18, 407–412. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Myler, P. J., Stacy, R., Stewart, L. J., Staker, B. L., Van Voorhis, W. C. & Buchko, G. W. (2009). Infect. Disord. Drug Targets, 9, 493–506. [DOI] [PMC free article] [PubMed]
- Nelson, N. (1999). EMBO J. 18, 4361–4371. [DOI] [PMC free article] [PubMed]
- Nies, D. H. (1999). Appl. Microbiol. Biotechnol. 51, 730–750. [DOI] [PubMed]
- Opella, S. J., DeSilva, T. M. & Veglia, G. (2002). Curr. Opin. Chem. Biol. 6, 217–223. [DOI] [PubMed]
- Pereira, S. J., Ramirez, N. E., Xiao, L. & Ward, L. A. (2002). J. Infect. Dis. 186, 715–718. [DOI] [PubMed]
- Ramirez, N. E., Ward, L. A. & Sreevatsan, S. (2004). Microbes Infect. 6, 773–785. [DOI] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Rubino, J. T. & Franz, K. J. (2012). J. Inorg. Chem. 107, 129–143. [DOI] [PubMed]
- Sato, A., Yokotani, S., Tadokoro, T., Tanaka, S., Angkawidjaja, C., Koga, Y., Takano, K. & Kanaya, S. (2011). J. Synchrotron Rad. 18, 6–10. [DOI] [PMC free article] [PubMed]
- Savchenko, A., Skarina, T., Evdokimova, E., Watson, J. D., Laskowski, R., Arrowsmith, C. H., Edwards, A. M., Joachimiak, A. & Zhang, R.-G. (2004). Proteins, 54, 162–165. [DOI] [PMC free article] [PubMed]
- Sonia, A. (2011). Microbes, Viruses and Parasites in AIDS Process, edited by C. Zajec, pp. 289–306. Rijeka: InTech.
- Stacy, R., Begley, D. W., Phan, I., Staker, B. L., Van Voorhis, W. C., Varani, G., Buchko, G. W., Stewart, L. J. & Myler, P. J. (2011). Acta Cryst. F67, 979–984. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Striepen, B. (2013). Nature (London), 503, 189–191. [DOI] [PubMed]
- Studier, F. W. (2005). Protein Expr. Purif. 41, 207–234. [DOI] [PubMed]
- Tanaka, T., Sawano, M., Ogasawara, K., Sakaguchi, Y., Bagautdinov, B., Katoh, E., Kuroishi, C., Shinkai, A., Yokoyama, S. & Yutani, K. (2006). FEBS Letts. 580, 4224–4230. [DOI] [PubMed]
- Tanaka, Y., Tsumoto, K., Nakanishi, T., Yasutake, Y., Sakai, N., Yao, M., Tanaka, I. & Kumagai, I. (2004). FEBS Letts. 556, 167–174. [DOI] [PubMed]
- Winn, M. D. (2011). Acta Cryst. D67, 235–242.
- Woody, R. W. (1974). Peptides, Polypeptides and Proteins, edited by E. R. Blout, F. A. Bovey, M. Goodman & N. Lotan, pp. 338–348. New York: John Wiley & Sons.
- Xiao, L. & Ryan, U. M. (2004). Curr. Opin. Infect. Dis. 17, 483–490. [DOI] [PubMed]
- Yang, J., Yang, H., Yan, L., Yang, L. & Yu, L. (2009). Mol. Biol. Rep. 36, 63–69. [DOI] [PubMed]
- Yee, A. et al. (2002). Proc. Natl Acad. Sci. USA, 99, 1825–1830.
- Zuiderweg, E. R. (2002). Biochemistry, 41, 1–7. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: divalent-cation tolerance protein, 4e98