

Structures of aspartate aminotransferases from eukaryotic pathogens were solved in complex with pyridoxal phosphate. The four structures show either the open or the closed conformation of the enzyme.
Keywords: aspartate aminotransferase, structural genomics, Seattle Structural Genomics Center for Infectious Disease, pyridoxalphosphate lysine, transferase, Giardia lamblia, Trypanosoma brucei, Leishmania major
Abstract
The structures of three aspartate aminotransferases (AATs) from eukaryotic pathogens were solved within the Seattle Structural Genomics Center for Infectious Disease (SSGCID). Both the open and closed conformations of AAT were observed. Pyridoxal phosphate was bound to the active site via a Schiff base to a conserved lysine. An active-site mutant showed that Trypanosoma brucei AAT still binds pyridoxal phosphate even in the absence of the tethering lysine. The structures highlight the challenges for the structure-based design of inhibitors targeting the active site, while showing options for inhibitor design targeting the N-terminal arm.
1. Introduction
Aspartate aminotransferases (AATs) catalyse the reversible transfer of an α-amino group between aspartate and glutamate, converting l-aspartate and 2-oxoglutamate to oxaloacetate and l-glutamate (EC 2.6.1.1; http://brenda-enzymes.info). The forward reaction is a crucial step in the degradation of the amino acid, while the reverse reaction synthesizes aspartate. The enzyme is dependent on the cofactor pyridoxal phosphate (PLP). During the reaction, the amino group is transferred to PLP, forming a pyridoxamine intermediate. PLP is anchored to the protein via a Schiff-base bond to a conserved lysine.
Two isoforms of AAT are known, a mitochondrial and a cytosolic isoform (Jensen & Gu, 1996 ▶), which share 49% sequence identity in humans. The three proteins used in this study are a mitochondrial AAT from Trypanosoma brucei (TbAAT-native and its active-site mutant TbAAT-K237A), a putative AAT from Leishmania major (LmAAT) and a cytoplasmic AAT from Giardia lamblia (GlAAT).
The mission of the US National Institute for Allergies and Infectious Disease (NIAID)-funded Seattle Structural Genomics Center for Infectious Disease (SSGCID) is to provide the scientific community with protein structures from NIAID category A–C agents, and from emerging and re-emerging infectious disease organisms, that are useful for drug or vaccine development or for better understanding the virulence or pathogenesis, or are markers of infection. The NIAID pathogen priority list ranks pathogens based on their risk level to public health in the US. Targets from the three organisms described here are regularly entered into the SSGCID pipeline: G. lamblia is the infective agent of giardiasis, T. brucei is the infective agent of African trypanosomiasis (sleeping sickness) and L. major is the infective agent of leishmaniasis.
Targets can qualify to enter the SSGCID pipeline in various ways (Phan et al., 2014 ▶): (i) databases such as the TDRtargets depository (Agüero et al., 2008 ▶) or DrugBank (Knox et al., 2011 ▶) provide sufficient evidence that a protein of interest is a potential drug target, (ii) the protein of interest is an ortholog of identified drug targets (Baugh et al., 2013 ▶) or (iii) targets are requested by the scientific community. T. brucei and L. major AAT were submitted as community requests from the developers of the TDRtargets depository, while G. lamblia AAT was selected owing to its similarity to protein targets in the DrugBank. Structural information on the targets is crucial for drug-development efforts, since, for instance, the three AATs described in this study only share between 30 and 40% sequence identity, while sequence identity in the active site is very high. For P. falciparum AAT it has been shown that the nonspecific inhibitor hydroxylamine interfers with the proliferation of the malaria parasite (Wrenger et al., 2011 ▶).
AATs are well described proteins: more than 100 crystal structures of AATs from various prokaryotic and eukaryotic organisms, variants of these protein and protein–ligand complexes have been deposited in the PDB. However, there is a distinct lack of AAT structures from parasites: only the structure of AAT from P. falciparum (PDB entry 3k7y; Wrenger et al., 2011 ▶) has been deposited in the PDB. For P. falciparum AAT a peptide inhibitor has been described that inhibits the pathogen protein but not the human protein. This study describes the crystallization and structures of AATs from three eukaryotic pathogens.
2. Materials and methods
2.1. Macromolecule production
The following genes were cloned into expression vectors using ligation-independent cloning (Aslanidis & de Jong, 1990 ▶). T. brucei brucei locus XP_828584 (UniProt Q385Q9) was used for TbAAT-native and TbAAT-K237A, L. major Fridelin locus XP_343989 (UniProt Q4FX34) for LmAAT and G. lamblia intestinalis locus XP_001709849 (UniProt A8B1V5) for GlAAT. The genes for TbAAT-native and GlAAT were cloned into expression vector pAVA0421, which provides an N-terminal His6 tag followed by a 3C protease cleavage site, while those for TbAAT-K237A and LmAAT were cloned into expression vector pBG1861, which provides a non-cleavable His6 tag prior to the ORF. The TbAAT-native construct contains two mutations compared with the database sequence: F147L and S275N. In order to create TbAAT-K237A, site-directed mutagenesis of TbAAT-native was performed using the primers GTTGCGCAGAGTTTCTCCGCGAACTTTGGCCTGTACG and CGTACAGGCCAAAGTTCGCGGAGAAACTCTGCGCAAC. In addition to the K237A mutation, Ser275 was mutated back to Asn275.
The genes for the proteins were expressed in Escherichia coli Rosetta BL21(DE3)R3 cells following standard SSGCID protocols as described in previous publications (Choi et al., 2011 ▶). Purification was performed using Ni–NTA affinity and size-exclusion chromatography following standard SSCID protocols (Bryan et al., 2011 ▶). The N-terminal C3-cleavable tag for TbAAT-native and GlAAT was not cleaved. The final buffer for all proteins was 25 mM HEPES pH 7.0, 500 mM NaCl, 5% glycerol, 2 mM DTT, 0.025% NaN3.
2.2. Crystallization
All crystallization experiments were set up as sitting-drop vapor-diffusion trials using XJR Junior crystallization trays (Rigaku Reagents) with drop sizes of 0.4 µl protein solution plus 0.4 µl well solution. Crystallization conditions were searched for using the commercial screens Wizard Classic 3 and 4, JCSG+ (Rigaku Reagents), Morpheus, PACT (Molecular Dimensions) and Index (Hampton Research). The trays were stored at 290 K. Crystallization conditions for the individual targets were as follows: TbAAT-native, Wizard Classic 3 and 4 condition B7 (20% PEG 3350, 200 mM ammonium nitrate), 27.7 mg ml−1 protein; TbAAT-K237A, JCSG+ condition G8 (20% PEG 3350, 150 mM dl-malic acid), 28.8 mg ml−1 protein; LmAAT, PACT condition E12 (20% PEG 3350, 200 mM sodium malonate pH 7.0), 19 mg ml−1 protein with 2.5 mM PLP; LmAAT-apo (not shown), Wizard Classic 3 and 4 condition H3 (3% MPD, 20% PEG 8000, 100 mM imidazole–HCl), 19.3 mg ml−1 protein; GlAAT, Index condition G3 (20% PEG 3350, 200 mM lithium sulfate, 100 mM bis-tris propane pH 6.5), 35 mg ml−1 protein.
Cryosolutions were prepared by adding 20% ethylene glycol to the reservoir solution. Crystals were incubated for several seconds in the cryosolution and were vitrified by plunging them into liquid nitrogen.
2.3. Data collection and processing
Diffraction data sets for TbAAT-native, LmAAT and GlAAT were collected in-house with a Rigaku FR-E+ SuperBright generator using a Rigaku 944+ CCD detector. The diffraction data set for TbAAT-K237A was collected on the Life Sciences CAT (LS-CAT) beamline 21-ID-G at the Advanced Photon Source using a Rayonix MX-300 CCD detector. All data sets were collected at 100 K. The X-ray data were reduced with XDS/XSCALE (Kabsch, 2010 ▶). Details of the data collections are summarized in Table 1 ▶.
Table 1. Data collection and processing.
Values in parentheses are for the outer shell.
| Data set | TbAAT-native | TbAAT-K237A | LmAAT | GlAAT |
|---|---|---|---|---|
| Wavelength () | 1.5418 | 0.97856 | 1.5418 | 1.5418 |
| Crystal-to-detector distance (mm) | 50 | 175 | 50 | 50 |
| Rotation range per image () | 0.5 | 0.5 | 0.5 | 0.5 |
| Total rotation range () | 180 | 180 | 360 | 180 |
| Exposure time per image (s) | 30 | 1 | 30 | 30 |
| Space group | P21 | P21 | P21 | P21 |
| a, b, c () | 62.26, 96.25, 81.32 | 62.39, 96.66, 81.61 | 61.41, 93.67, 74.59 | 58.58, 101.15, 81.53 |
| , , () | 90, 111.62, 90 | 90, 111.03, 90 | 90, 106.57, 90 | 90, 90.62, 90 |
| Mosaicity () | 0.50 | 0.20 | 0.25 | 0.55 |
| Resolution range () | 502.30 (2.362.30) | 501.70 (1.741.70) | 501.85 (1.901.85) | 501.90 (1.951.90) |
| Total No. of reflections | 146263 (10489) | 372563 (27701) | 356493 (15370) | 121219 (8706) |
| No. of unique reflections | 39747 (2800) | 97248 (7108) | 68837 (5037) | 71707 (4842) |
| Completeness (%) | 98.0 (96.5) | 97.9 (97.0) | 99.6 (98.6) | 95.8 (87.3) |
| Multiplicity | 3.8 (3.8) | 3.8 (3.9) | 5.2 (3.1) | 2.7 (1.8) |
| I/(I) | 12.8 (3.1) | 16.1 (2.9) | 22.7 (2.3) | 9.9 (3.0) |
| R merge † | 0.085 (0.532) | 0.047 (0.517) | 0.054 (0.541) | 0.091 (0.336) |
| R meas ‡ | 0.100 (0.625) | 0.055 (0.599) | 0.060 (0.655) | 0.113 (0.475) |
| Overall B factor from Wilson plot (2) | 35.4 | 19.8 | 17.3 | 19.1 |
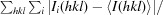
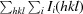
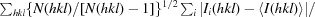
2.4. Structure solution and refinement
The following structures were used as search models in molecular-replacement searches for the various structures: PDB entry 4eff (Seattle Structural Genomics Center for Infectious Disease, unpublished work) for TbAAT-native, the TbAAT-native structure for TbAAT-K234A, and PDB entry 2cst (Malashkevich, Strokopytov et al., 1995 ▶) for LmAAT and GlAAT. Monomers of the search model were prepared using CHAINSAW (Stein, 2008 ▶) with the exception of the TbAAT-K234A structure. The structures were solved by molecular replacement using Phaser (McCoy et al., 2007 ▶) within CCP4 (Winn et al., 2011 ▶). Molecular replacement was followed by automatic model building with Buccaneer (Cowtan, 2006 ▶) for TbAAT-native or ARP/wARP (Langer et al., 2008 ▶). The model was completed by iterative cycles of reciprocal-space refinement with REFMAC (Murshudov et al., 2011 ▶) for TbAAT-native, LmAAT and GlAAT or with PHENIX (Adams et al., 2010 ▶) for TbAAT-K234A and manual real-space density fitting using Coot (Emsley & Cowtan, 2004 ▶). Based on the validation tools within Coot and based on MolProbity (Chen et al., 2010 ▶) analyses, all four structures have good stereochemistry (see Table 2 ▶). Some of the structures have Ramachandran outliers, with ‘minor’ outlier residues that are just beyond the border of the Ramachandran plot in Coot: Tyr242 in chains A and B, Tyr139 in chains A and B and Asn285 in chains A and B as minor outliers in TbAAT, Tyr242 in chains A and B, Tyr139 in chains A and B and Tyr274 in chain B as minor outliers in TbAAT-K234A, Ser109 in chains A and B as a clear outlier and Tyr159 in chains A and B and Gly44 in chain B as minor outliers in LmAAT and Ser10 in chains A and B, Asn146 in chains A and B, Ser302 in chains A and B, Tyr 261 in chains A and B and Ala227 in chain A as minor outliers in GlAAT. All of the outliers have well defined electron density and some of them are close to the ligand-binding site.
Table 2. Structure solution and refinement.
Values in parentheses are for the outer shell.
| TbAAT-native | TbAAT-K237A | LmAAT | GlAAT | |
|---|---|---|---|---|
| Resolution range () | 502.30 (2.362.30) | 401.70 (1.721.70) | 501.85 (1.901.85) | 501.90 (1.951.90) |
| Completeness (%) | 98.1 | 97.9 | 99.7 | 95.8 |
| Cutoff | F > 0.000(F) | F > 0.000(F) | F > 0.000(F) | F > 0.000(F) |
| No. of reflections, working set | 38947 (2612) | 97154 (3036) | 68814 (4697) | 71615 (4613) |
| No. of reflections, test set | 1963 (129) | 4844 (147) | 3471 (244) | 3613 (21) |
| Final R cryst | 0.196 (0.300) | 0.153 (0.190) | 0.168 (0.274) | 0.174 (0.227) |
| Final R free | 0.245 (0.393) | 0.183 (0.230) | 0.213 (0.307) | 0.214 (0.270) |
| Cruickshank DPI | 0.33 | n/a | 0.14 | 0.15 |
| No. of non-H atoms | ||||
| Protein | 5820 | 5864 | 6239 | 6645 |
| Ion | 5 | 0 | 0 | 20 |
| Ligand | 48 | 32 | 48 | 32 |
| Water | 364 | 794 | 757 | 413 |
| Total | 6225 | 6709 | 7052 | 7562 |
| R.m.s. deviations | ||||
| Bonds () | 0.011 | 0.010 | 0.014 | 0.015 |
| Angles () | 1.38 | 1.16 | 1.56 | 1.43 |
| Average B factors (2) | ||||
| Protein | 29.6 | 28.8 | 19.0 | 12.6 |
| Ion | 49.3 | n/a | n/a | 15.0 |
| Ligand | 19.6 | 22.5 | 16.1 | 12.3 |
| Water | 27.5 | 36.2 | 27.5 | 14.8 |
| Ramachandran plot | ||||
| Most favoured (%) | 97.4 | 96.5 | 96.4 | 96.4 |
| Allowed (%) | 1.9 | 2.9 | 3.0 | 2.5 |
| PDB code | 4eu1 | 4w5k | 4h51 | 3meb |
The models contain almost the entire length of the protein: residues 6–388 in chain A and residues 8–388 in chain B for TbAAT-native, residues 9–388 in chains A and B for TbAAT-K342A, residues 7–399 in chain A and residues 7–409 in chain B for LmAAT and residues 1–426 in chains A and B for GlAAT. Refinement statistics are summarized in Table 2 ▶ along with the corresponding PDB codes.
3. Results and discussion
The structures of aspartarte aminotransferases from three eukaryotic pathogens were determined at medium to high resolution. All three AATs crystallize with a dimer in the asymmetric unit. The structures have the typical cashew-nut shape of ATTs, with an N-terminal arm, a small domain and a large domain (Ford et al., 1980 ▶; see Fig. 1 ▶ a). The three proteins share pairwise sequence identities of 35–40%. The structures superimpose well: the Cα-atom r.m.s.d.s between NCS mates range between 0.2 and 0.6 Å, while the Cα-atom r.m.s.d.s between various structures are between 1.3 and 1.4 Å, as determined by SSM (Krissinel & Henrick, 2004 ▶).
Figure 1.

Open and closed conformations. (a) and (b) show dimers of TbAAT and LmAAT, respectively, in the same orientation. The surface rendition highlights the location of one of the protomers. The locations of the N-termini and the large and the small domains are indicated. The superposition of TbAAT and LmAAT (c) with chain A on the left as a reference highlights the open conformation of TbAAT and the closed conformation of LmAAT. (d) shows an extended superposition of TbAAT, LmAAT and GlAAT, along with open-conformation and closed-conformation chicken AAT (PDB entries 1oxo and 1oxp, respectively). The worm representation highlights the differences for chain B on the right. For simplicity, for chain A on the left only TbAAT-native and LmAAT are shown. This figure was created with CCP4mg (McNicholas et al., 2011 ▶).
Early work on chicken mitochondrial AATs showed that AATs can exist in two conformations, referred to as the open and closed conformations (Hohenester & Jansonius, 1994 ▶). The two conformations differ by a 10–15° rotation of the small domain. It has been suggested that the closed form represents the substrate-bound conformation (PDB entry 1oxp; Marković-Housley et al., 1996 ▶), while the open form represents the apo conformation (PDB entry 1oxo; Marković-Housley et al., 1996 ▶). Even though all four structures in this study were crystallized as the apo proteins, TbAAT-native and TbAAT-K237A showed the open conformation, while LmAAT and GlAAT resembled the closed conformation (Figs. 1 ▶ b and 1 ▶ c). For LmAAT, the structure does not provide a good explanation for the closed conformation. For GlAAT, which only has a partially occupied PLP, which was not added to the crystallization, one can speculate that a sulfate molecule in proximity to the PLP-binding site attracts Arg398 and induces the closed conformation.
The three proteins in their respective crystallization buffer appear to differ in affinity for the cofactor PLP. In TbAAT-native, PLP is covalently bound to Lys237 via a Schiff base. In TbAAT-K237A, this bond can no longer form. However, PLP is clearly bound to the active site. This is similar to what was described for a K258H mutant of chicken mitochondria AAT and E. coli AAT (Malashkevich, Jäger et al., 1995 ▶). The density for PLP is strong, and the occupancy of PLP was refined to 0.88 and 0.89 in the two chains, with B factors that were very similar to those for the environment (Fig. 2 ▶ a). It was not necessary to supplement the crystallization with PLP to obtain either of these structures. In contrast, LmAAP initially yielded an apo structure (1.95 Å resolution; data not shown) with only phosphate in the PLP-binding site. Co-crystallization of this protein with 2.5 mM PLP yielded isomorphous crystals with strong electron density for PLP. PLP binding in LmAAT is accompanied by ordering of Trp139, the side chain of which stacks against the PLP ring system. In GlAAT, PLP is only visible at partial occupancy (0.25). There is strong density in the location of the phosphate moiety of PLP; one can assume that this is a sulfate from the crystallization condition (200 mM Li2SO4).
Figure 2.

Active site. (a) compares the PLP-binding sites of TbAAT-native (thin lines) and TbAAT-K237A (thick lines). The σA-weighted 2F o − F c (blue) and F o − F c maps (green for positive, red for negative) for TbAAT-K237A are shown at 1σ and 3σ levels, respectively. PLP engages in a number of hydrogen bonds and hydrophobic interactions. These residues are labeled. The density for PLP in the K237A mutant is very clear. The two active sites are virtually identical. (b) shows a superposition of TbAAT (blue C atoms) and the open form of chicken mitochondria AAT (gray C atoms; PDB entry 1oxo) around the PLP-binding site. The structures are virtually identical. Mutations are subtle and are highlighted: for instance, ‘YW’ means that Tyr126 in TbAAT is Trp in chicken mitochondria AAT. The substrate-binding sites are also very similar. (c) is a rotated view of (b). In (d) LmAAT (orange C atoms), GlAAT (purple C atoms) and the closed form of chicken mitochondria AAT (gray C atoms; PDB entry 1oxp) are superimposed. Here again the differences are subtle. This figure was created with CCP4mg (McNicholas et al., 2011 ▶).
The PLP-binding site is well defined; see Fig. 2 ▶(a) for the electron density for TbAAT-K237A and for a superposition of the TbAAT and TbAAT-K237A structures. The phosphate group of PLP engages in hydrogen bonds to a variety of amino acids: Tyr54 OH, Gly92 N, Thr93 Oγ and N, Ser234 Oγ and Arg245 Nη1 and Nη2. The pyridoxal ring is engaged in hydrogen bonds to Asp201 Oδ1 and Oδ2, Asn173 Nδ2 and Tyr204 OH, and makes hydrophobic interactions with the side chains of Tyr120 and Ala203. Similar interactions occur in the structures of LmAAT and GlAAT.
The environment around PLP is highly conserved between various species. In Figs. 2 ▶(b), 2 ▶(c) and 2 ▶(d) we compare the PLP-binding and substrate-binding sites of the parasite AATs with those of chicken mitochondria AAT in the respective open or closed form. The pairwise r.m.s.d. between chain A of the parasite AATs and chicken mitochondria AAT is around 1 Å. Fig. 2 ▶(b) compares the PLP-binding site of TbAAT with that of the open form of chicken mitochondria AAT, which are virtually identical. The mutations are very subtle. The substrate-binding pockets compared are very conserved as well. Fig. 2 ▶(c) shows a superposition of TbAAT with the open-form chicken mitochondria AAT, while in Fig. 2 ▶(d) models of LmAAT, GlAAT and the closed form of chicken mitochondria AAT are superimposed. The high degree of structural conservation will pose a significant challenge to the design of specific inhibitors that target the active site.
The N-terminal arm of AATs, which wraps around the other subunit of the dimer, has been described to modulate the activity of P. falciparum AAT (Wrenger et al., 2011 ▶). Since the sequence of the N-terminus is more divergent than the very conserved active site, the N-terminus has been suggested as a target for inhibitor design (Wrenger et al., 2011 ▶). The three AATs presented display a variety of lengths in the N-terminus. LmAAT is 17 residues longer than TbAAT and nine residues longer than GlAAT. Only for GlAAT is the full N-terminus visible in the structure, while the three other structures have interpretable electron density starting between residues 6 and 9. Hence, the N-terminus wraps around the other part of the dimer only in LmAAT and GlAAT (Figs. 1 ▶ a and 1 ▶ b). The conformation of the N-termini of LmAAT, GlAAT and chicken mitochondria AAT are distinctly different (Fig. 1 ▶ d). This might render the N-terminal arm as a useful target for the design of specific inhibitors.
Supplementary Material
PDB reference: L. major AAT, 4h51
PDB reference: T. brucei AAT, 4eu1
PDB reference: TK237A mutant, 4w5k
PDB reference: G. lamblia AAT, 3meb
Acknowledgments
This project has been funded in whole with Federal funds from the National Institute of Allergy and Infectious Diseases, National Institutes of Health, Department of Health and Human Services under Contract No. HHSN272201200025C.
References
- Adams, P. D. et al. (2010). Acta Cryst. D66, 213–221.
- Agüero, F. et al. (2008). Nature Rev. Drug Discov. 7, 900–907. [DOI] [PMC free article] [PubMed]
- Aslanidis, C. & de Jong, P. J. (1990). Nucleic Acids Res. 18, 6069–6074. [DOI] [PMC free article] [PubMed]
- Baugh, L. et al. (2013). PLoS One, 8, e53851. [DOI] [PMC free article] [PubMed]
- Bryan, C. M., Bhandari, J., Napuli, A. J., Leibly, D. J., Choi, R., Kelley, A., Van Voorhis, W. C., Edwards, T. E. & Stewart, L. J. (2011). Acta Cryst. F67, 1010–1014. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Choi, R., Kelley, A., Leibly, D., Nakazawa Hewitt, S., Napuli, A. & Van Voorhis, W. (2011). Acta Cryst. F67, 998–1005. [DOI] [PMC free article] [PubMed]
- Cowtan, K. (2006). Acta Cryst. D62, 1002–1011. [DOI] [PubMed]
- Diederichs, K. & Karplus, P. A. (1997). Nature Struct. Mol. Biol. 4, 269–275. [DOI] [PubMed]
- Emsley, P. & Cowtan, K. (2004). Acta Cryst. D60, 2126–2132. [DOI] [PubMed]
- Ford, G. C., Eichele, G. & Jansonius, J. N. (1980). Proc. Natl Acad. Sci. USA, 77, 2559–2563. [DOI] [PMC free article] [PubMed]
- Hohenester, E. & Jansonius, J. N. (1994). J. Mol. Biol. 236, 963–968. [PubMed]
- Jensen, R. A. & Gu, W. (1996). J. Bacteriol. 178, 2161–2171. [DOI] [PMC free article] [PubMed]
- Kabsch, W. (2010). Acta Cryst. D66, 125–132. [DOI] [PMC free article] [PubMed]
- Knox, C., Law, V., Jewison, T., Liu, P., Ly, S., Frolkis, A., Pon, A., Banco, K., Mak, C., Neveu, V., Djoumbou, Y., Eisner, R., Guo, A. C. & Wishart, D. S. (2011). Nucleic Acids Res. 39, D1035–D1041. [DOI] [PMC free article] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Langer, G., Cohen, S. X., Lamzin, V. S. & Perrakis, A. (2008). Nature Protoc. 3, 1171–1179. [DOI] [PMC free article] [PubMed]
- Malashkevich, V. N., Jäger, J., Ziak, M., Sauder, U., Gehring, H., Christen, P. & Jansonius, J. N. (1995). Biochemistry, 34, 405–414. [DOI] [PubMed]
- Malashkevich, V. N., Strokopytov, B. V., Borisov, V. V., Dauter, Z., Wilson, K. S. & Torchinsky, Y. M. (1995). J. Mol. Biol. 247, 111–124. [DOI] [PubMed]
- Marković-Housley, Z., Schirmer, T., Hohenester, E., Khomutov, A. R., Khomutov, R. M., Karpeisky, M. Y., Sandmeier, E., Christen, P. & Jansonius, J. N. (1996). Eur. J. Biochem. 236, 1025–1032. [DOI] [PubMed]
- McCoy, A. J., Grosse-Kunstleve, R. W., Adams, P. D., Winn, M. D., Storoni, L. C. & Read, R. J. (2007). J. Appl. Cryst. 40, 658–674. [DOI] [PMC free article] [PubMed]
- McNicholas, S., Potterton, E., Wilson, K. S. & Noble, M. E. M. (2011). Acta Cryst. D67, 386–394. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Phan, I. Q. H., Stacy, R. & Myler, P. J. (2014). Methods Mol. Biol. 1140, 53–59. [DOI] [PMC free article] [PubMed]
- Stein, N. (2008). J. Appl. Cryst. 41, 641–643.
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Wrenger, C., Müller, I. B., Schifferdecker, A. J., Jain, R., Jordanova, R. & Groves, M. R. (2011). J. Mol. Biol. 405, 956–971. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: L. major AAT, 4h51
PDB reference: T. brucei AAT, 4eu1
PDB reference: TK237A mutant, 4w5k
PDB reference: G. lamblia AAT, 3meb