

MERS-CoV 3CLpro has been crystallized and X-ray crystal structures have been solved in three different crystal forms representing the free enzyme structure and the enzyme–product complex.
Keywords: MERS-CoV, coronavirus, main protease, 3CLpro
Abstract
Middle East respiratory syndrome coronavirus (MERS-CoV) is a highly pathogenic virus that causes severe respiratory illness accompanied by multi-organ dysfunction, resulting in a case fatality rate of approximately 40%. As found in other coronaviruses, the majority of the positive-stranded RNA MERS-CoV genome is translated into two polyproteins, one created by a ribosomal frameshift, that are cleaved at three sites by a papain-like protease and at 11 sites by a 3C-like protease (3CLpro). Since 3CLpro is essential for viral replication, it is a leading candidate for therapeutic intervention. To accelerate the development of 3CLpro inhibitors, three crystal structures of a catalytically inactive variant (C148A) of the MERS-CoV 3CLpro enzyme were determined. The aim was to co-crystallize the inactive enzyme with a peptide substrate. Fortuitously, however, in two of the structures the C-terminus of one protomer is bound in the active site of a neighboring molecule, providing a snapshot of an enzyme–product complex. In the third structure, two of the three protomers in the asymmetric unit form a homodimer similar to that of SARS-CoV 3CLpro; however, the third protomer adopts a radically different conformation that is likely to correspond to a crystallographic monomer, indicative of substantial structural plasticity in the enzyme. The results presented here provide a foundation for the structure-based design of small-molecule inhibitors of the MERS-CoV 3CLpro enzyme.
1. Introduction
Middle East respiratory syndrome coronavirus (MERS-CoV) was first reported in 2012 following isolation from a patient in Saudi Arabia (Zaki et al., 2012 ▶). MERS-CoV causes severe pneumonia (Falzarano et al., 2014 ▶; Cunha & Opal, 2014 ▶) reminiscent of the severe acute respiratory syndrome (SARS) outbreak of 2003, but cases of MERS-CoV exhibit a higher mortality rate than those of SARS-CoV (approximately 40% versus 10%). Although the number of new cases peaked in early 2014 (http://www.who.int/csr/disease/coronavirus_infections/archive_updates/en/; Holmes, 2014 ▶), the outbreak continues. The severity and rapid spread of MERS and SARS illustrate the need for the development of new therapeutics to combat known and emerging coronaviruses.
MERS-CoV belongs to the genus Betacoronavirus, which is divided into four clades: a–d. The clade b SARS coronavirus (SARS-CoV) is thought to have its reservoir in bats (Ge et al., 2013 ▶), with civets as an intermediate host facilitating human infection (Li et al., 2005 ▶). MERS-CoV belongs to Betacoronavirus clade c, along with the closely related bat coronaviruses HKU4 (BatCoV-HKU4) and HKU5 (Corman et al., 2014 ▶). A conspecific virus that shares 85% genome sequence identity with MERS-CoV has been isolated from the Neoromica capensis bat (Corman et al., 2014 ▶). Recent work showed that introduction of a clinical isolate of MERS-CoV into dromedary camels resulted in mild respiratory illness followed by persistent shedding of infectious virus from the upper respiratory tract (Adney et al., 2014 ▶). Taken together, these results suggest that MERS-CoV originated in bats, with camels serving as the carrier for human infection.
Coronaviruses, including MERS-CoV, SARS-CoV and the usually milder human coronaviruses (HCoV) HCoV-229E, HCoV-NL63 and HCoV-OC43, share a common organization of their polycistronic positive-strand RNA genomes. On the 5′ end of the MERS-CoV genome are the two large open reading frames (ORF1a and ORF1b) encoding nonstructural proteins (nsps), followed by genes encoding the spike, envelope, membrane and nucleocapsid structural proteins. The genomic mRNA of ORF1a is translated into the polyprotein pp1a. A longer polyprotein (pp1ab) is the product of a ribosomal frameshift that joins ORF1a together with ORF1b (van Boheemen et al., 2012 ▶). ORF1a encodes two proteases: a papain-like protease (PLpro) and a 3C-like ‘main’ protease (3CLpro). The 3CLpro, which in its essential role in viral replication is also called the ‘main protease’ (Mpro), processes the polyprotein at 11 cleavage sites (consensus: LQ↓A/S), including those flanking it (Ziebuhr et al., 2000 ▶; Anand et al., 2002 ▶; Hsu et al., 2005 ▶; van Boheemen et al., 2012 ▶; Li et al., 2010 ▶; Muramatsu et al., 2013 ▶; Stobart et al., 2013 ▶). The essential function and conservation among 3CLpros from different coronaviruses make the main protease an attractive drug target for currently known and future emerging coronaviruses (Anand et al., 2002 ▶, 2003 ▶, Zhao et al., 2013 ▶; Hilgenfeld, 2014 ▶). In contrast, the structural and accessory genes encoded towards the 3′ end of coronavirus genomes exhibit too much variability to serve as targets for broad anti-coronaviral agents (Yang et al., 2006 ▶).
Coronaviral 3CLpros are chymotrypsin-like proteases except that they use cysteine as the nucleophile in a catalytic dyad instead of serine in a catalytic triad (Anand et al., 2002 ▶). SARS-CoV 3CLpro exists in a monomer–dimer equilibrium in solution (Graziano et al., 2006 ▶), but the homodimer is the enzymatically active form (Chen et al., 2006 ▶; Shi & Song, 2006 ▶; Shi et al., 2008 ▶). Each monomer consists of three structural domains: domains I and II contain the catalytic site and chymotrypsin-like scaffold and are connected to a third C-terminal domain via a long loop (Yang et al., 2003 ▶; Shi et al., 2004 ▶; Tsai et al., 2010 ▶). In this study, we report the structure of a catalytically inactive variant (C148A) of MERS-CoV 3CLpro in three different crystal forms, each providing distinct biological insights.
2. Materials and methods
2.1. Cloning, expression and protein purification
Expression vectors were constructed by Gateway recombinational cloning (Life Technologies, Grand Island, New York, USA). The 3CLpro gene was amplified by polymerase chain reaction (PCR) from a cDNA clone constructed using total RNA isolated from MERS-CoV Jordan (primers: 5′-CAC CAG CGG TTT GGT GAA AAT GTC ACA TCC C-3′ and 5′-TTA CTA CTG CAT AAC CAC ACC CAT AAT CTG C-3′).
To construct the catalytically inactive C148A variant, a MERS-CoV 3C-like protease amplicon was first used as a PCR template with primers PE2635 (5′-GGC TCG GAG AAC CTG TAC TTC CAG AGC GGT TTG GTG AAA ATG TCA CAT-3′) and PE2636 (5′-GGG GAC CAC TTT GTA CAA GAA AGC TGG GTT ATT ACT GCA TAA CCA CAC CCA TAA TCT GC-3′), which added nucleotides encoding a tobacco etch virus (TEV) protease recognition site to the 5′ end of the MERS-CoV 3CLpro sequence. The product of the reaction was amplified in a second PCR with primers PE277 (5′-GGGG ACA AGT TTG TAC AAA AAA GCA GGC TCG GAG AAC CTG TAC TTC CAG-3′) and PE2636 to produce a product competent for Gateway cloning. The PCR product was recombined into donor vector pDONR221 to produce the entry vector pDN2482. The active-site cysteine (Cys148) was changed to an alanine with the QuikChange Lightning Site-Directed Mutagenesis Kit (Agilent, Santa Clara, California, USA) using primers PE2732 (5′-ACC AAC ACT ACC AGC AGA ACC ACA CAG AAA GGA ACC CTT A-3′) and PE2733 (5′-TAA GGG TTC CTT TCT GTG TGG TTC TGC TGG TAG TGT TGG T-3′) to produce the entry vector pDN2544. pDN2544 was recombined into the destination vector pDEST-527 (Protein Expression Laboratory, Leidos Biomedical Research Inc., Frederick, Maryland, USA) to produce pDN2551, an expression vector encoding a TEV protease-cleavable hexahistidine tag preceding MERS-CoV 3CLpro (residues 1–306; C148A). The protein was produced in Escherichia coli strain Rosetta 2(DE3) (EMD Millipore, Billerica, Massachusetts, USA). Cells were grown to mid-log phase at 310 K in LB broth containing 100 µg ml−1 ampicillin, 30 µg ml−1 chloramphenicol and 0.2% glucose. Overproduction of the fusion protein was induced with IPTG at a final concentration of 1 mM for 4 h at 303 K. The cells were pelleted by centrifugation and stored at 193 K.
For protein purification, all procedures were performed at 277–281 K. 5 g of E. coli cell paste were suspended in 150 ml buffer A (50 mM Tris, 200 mM NaCl, 25 mM imidazole pH 7.2). The cells were lysed with an APV-1000 homogenizer (Invensys APV Products, Albertslund, Denmark) at 69 MPa and centrifuged at 30 000g for 30 min. The supernatant was filtered through a 0.2 µm polyethersulfone membrane and applied onto a 5 ml HisTrap FF column (GE Healthcare Life Sciences, Pittsburgh, Pennsylvania, USA) equilibrated with buffer A. The column was washed to baseline with buffer A and eluted with a linear gradient of imidazole to 500 mM in buffer A. Fractions containing recombinant protein were pooled, concentrated using an Amicon YM10 membrane (EMD Millipore, Billerica, Massachusetts, USA), diluted to an imidazole concentration of about 25 mM with 50 mM Tris pH 7.2, 200 mM NaCl buffer and digested overnight at 277 K with His6-tagged TEV protease (Kapust et al., 2001 ▶; Tropea et al., 2009 ▶). TEV protease digestion, which removed the His6 affinity tag and amino acids encoded by sequences that facilitate Gateway cloning, resulted in a native protein product devoid of cloning artifacts. The digest was applied onto a 5 ml HisTrap FF column equilibrated in buffer A and recombinant protein emerged in the column effluent. The effluent was incubated overnight at 277 K with 10 mM dithiothreitol, concentrated using an Amicon YM10 membrane and applied onto a HiPrep 26/60 Sephacryl S-200 HR column (GE Healthcare Bio-Sciences Corporation) equilibrated with 25 mM Tris pH 7.2, 150 mM NaCl, 2 mM tris(2-carboxyethyl)phosphine buffer. The peak fractions were pooled and concentrated to about 20 mg ml−1 (as estimated at 280 nm using a molar extinction coefficient of 43 890 M −1 cm−1 derived using the ExPASy ProtParam tool (Artimo et al., 2012 ▶). Aliquots were flash-frozen with liquid nitrogen and stored at 193 K. The molecular weight of the product was confirmed by electrospray ionization mass spectroscopy.
2.2. Protein crystallization
Catalytically inactive (C148A) MERS-CoV 3CLpro (20.3 mg ml−1) was subjected to various crystallization screens including the MCSG Suite (Microlytic, Burlington, Massachusetts, USA) and Morpheus (Gorrec, 2009 ▶; Molecular Dimensions, Altamonte Springs, Florida, USA) using the sitting-drop vapor-diffusion method and a Gryphon crystallization robot (Art Robbins Inc., Sunnyvale, California, USA). Further optimization of the initial crystallization hits was performed by the hanging-drop vapor-diffusion method. Three different crystal forms were obtained. Crystal form I appeared from condition E10 of Morpheus by mixing 2 µl protein (20.3 mg ml−1) with 2 µl well solution [0.1 M Tris–Bicine pH 8.5, 0.03 M diethylene glycol, 0.03 M triethylene glycol, 0.03 M tetraethylene glycol, 0.03 M pentaethylene glycol, 10%(w/v) PEG 8000, 20%(v/v) ethylene glycol] and sealing the drop over 500 µl well solution. Crystal form II appeared under condition H10 from Morpheus [0.1 M Tris–Bicine pH 8.5, 0.02 M sodium l-glutamate, 0.02 M dl-alanine, 0.02 M glycine, 0.02 M dl-lysine–HCl, 0.02 M dl-serine, 10%(w/v) PEG 8000, 20%(v/v) ethylene glycol]. All stock reagents for crystallization conditions from the Morpheus Screen were obtained from Molecular Dimensions. Crystal form III was initially obtained from condition H1 of the MCSG 3 screen and was optimized by mixing 2 µl protein solution (20.3 mg ml−1) with 2 µl well solution [0.1 M HEPES pH 7.5, 0.2 M proline, 10%(w/v) polyethylene glycol 3350] and sealing over 500 µl well solution. All crystallization plates were incubated at 292 K and crystals generally appeared within 1–5 d. For data collection, crystal forms I and II were retrieved directly from the crystallization drop using a LithoLoop (Molecular Dimensions) and flash-cooled by plunging into liquid nitrogen without the need for additional cryoprotectant. Crystal form III was cryoprotected by transferring a crystal into a new drop consisting of well solution supplemented with 20%(v/v) polyethylene glycol 200, soaking for 1 min and flash-cooling by plunging into liquid nitrogen.
2.3. X-ray data collection, structure solution and refinement
All X-ray diffraction data for crystal forms I and III were collected using a MAR345 detector mounted on a Rigaku MicroMax-007 HF high-intensity microfocus generator equipped with VariMax HF optics (Rigaku, The Woodlands, Texas, USA) and operated at 40 kV and 30 mA (λ = 1.5418 A). Crystals were held at 93 K. For crystal form I, 525 diffraction images were collected with an exposure time of 600 s per image, an oscillation angle of 0.5° and a crystal-to-detector distance of 200 mm. For crystal form III, 360 images were collected with an exposure time of 180 s per image, an oscillation angle of 0.5° and a crystal-to-detector distance of 150 mm. Diffraction data from crystal form II were collected remotely on the SER-CAT beamline 22-BM at the Advanced Photon Source, Argonne National Laboratory, Lemont, Illinois, USA. Using an X-ray wavelength of 1.0 Å and a MAR CCD 225 detector, 360 images were collected with an exposure time of 6 s per image, an oscillation angle of 0.5° and a crystal-to-detector distance of 125 mm. All X-ray diffraction data were integrated and scaled using HKL-3000 (Minor et al., 2006 ▶).
Firstly, the structure of MERS-CoV 3CLpro crystal form III was solved by molecular replacement using chain A of the main protease of coronavirus HKU4 (PDB entry 2yna; 81% sequence identity; Q. Ma, Y. Xiao & R. Hilgenfeld, unpublished work) as a search model, after stripping away all nonprotein atoms and changing non-identical residues to alanines. Molecular replacement was performed with MOLREP from the CCP4 suite (Vagin & Teplyakov, 2010 ▶; Winn et al., 2011 ▶). Two molecules (chains A and B) were located in the asymmetric unit using data to 2.5 Å resolution. The sequence for chains A and B could be fitted completely into the electron-density maps. A third molecule (chain C) was also found, but only residues 11–190 fitted well into the electron-density maps. Inspection of the initial electron-density maps after rigid-body refinement with REFMAC5 (Murshudov et al., 2011 ▶) revealed a large region of well defined 2mF o − DF c and mF o − DF c electron-density features for protein residues adjacent to residues 11–190 of chain C. This indicated that residues 191–306 of chain C, corresponding to domain III of MERS-CoV 3CLpro, had undergone a large rigid-body movement. Therefore, another round of molecular replacement was performed with MOLREP by fixing the positions of chains A, B and residues 11–190 of chain C and then using residues 200–306 of chain C as a search model. Inspection of the new electron-density maps revealed a good fit of residues 200–306, confirming the alternate conformation of this region of the protein in chain C. The model was refined after several rounds of manual rebuilding and inspection with Coot (Emsley et al., 2010 ▶), refinement with REFMAC5 and addition of water and other solvent molecules.
The structures of crystal forms I and II were subsequently solved by molecular replacement with MOLREP from the CCP4 suite of programs using chain A of crystal form III as a search model. Refinements for crystal form I were completed using PHENIX (Adams et al., 2011 ▶) and Coot, while the structures of crystal forms II and III were refined using REFMAC5. All structure validations were performed with MolProbity (Chen et al., 2010 ▶). Secondary-structure elements were assigned using phenix.ksdssp (Kabsch & Sander, 1983 ▶; Adams et al., 2011 ▶). Figures were prepared with PyMOL (v.1.5.0.4; Schrödinger). Structural alignments were performed with either PyMOL or PDBeFold (Krissinel & Henrick, 2004 ▶).
3. Results and discussion
3.1. Overall structure of MERS-CoV 3CLpro
The three different crystal forms (I, II and III) of catalytically inactive (C148A) MERS-CoV 3CLpro provide a structural view of three distinct states of the enzyme. Data-collection and refinement statistics for all three crystal forms are reported in Table 1 ▶. In all crystal forms a biological homodimer was observed that is similar to other 3CLpro enzymes such as those encoded by TGEV (Anand et al., 2002 ▶), HCoV-229E (Anand et al., 2003 ▶), SARS-CoV (Yang et al., 2003 ▶), IBV-CoV (Xue et al., 2008 ▶) and HCoV-HKU1 (Zhao et al., 2008 ▶) (Fig. 1 ▶) The two molecules of the homodimer are approximately perpendicular to one another. Each monomer is composed of a core chymotrypsin-like fold that is formed by two domains (domains I and II, residues 1–187), a connecting loop (residues 188–204) and a C-terminal α-helical domain (referred to as domain III; residues 205–306). The C-terminal domain mediates dimerization; it has been demonstrated to play a key role in controlling the dimer–monomer equilibrium in other 3CLpro family members (Anand et al., 2002 ▶; Shi et al., 2004 ▶, 2008 ▶; Shi & Song, 2006 ▶).
Table 1. X-ray diffraction data-collection and refinement statistics.
Values in parentheses are for the highest resolution shell.
| MERS-CoV 3CLpro, form I | MERS-CoV 3CLpro, form II | MERS-CoV 3CLpro, form III | |
|---|---|---|---|
| Data collection | |||
| X-ray source | MicroMax-007 HF | 22-BM, SER-CAT | MicroMax-007 HF |
| Wavelength () | 1.5418 | 1.0 | 1.5418 |
| Resolution () | 502.58 (2.622.58) | 501.55 (1.591.55) | 501.97 (2.021.97) |
| Space group | C2221 | C2 | P212121 |
| Unit-cell parameters | |||
| a () | 81.0 | 131.7 | 94.1 |
| b () | 168.5 | 91.4 | 120.4 |
| c () | 250.5 | 120.31 | 138.9 |
| = () | 90 | 90 | 90 |
| () | 90 | 106.6 | 90 |
| Total reflections | 404336 | 743771 | 751558 |
| Unique reflections | 53763 | 197587 | 107729 |
| Completeness (%) | 99.9 (99.7) | 99.9 (100) | 96.4 (93.1) |
| Multiplicity | 7.5 (5.3) | 3.8 (3.6) | 7.0 (5.1) |
| Mean I/(I) | 23.5 (2.0) | 27.1 (2.0) | 40.5 (2.2) |
| R merge † | 0.077 (0.646) | 0.058 (0.675) | 0.047 (0.775) |
| Refinement statistics | |||
| Resolution () | 46.22.58 | 501.55 | 501.97 |
| R work ‡ | 0.177 | 0.187 | 0.192 |
| R free ‡ | 0.217 | 0.215 | 0.226 |
| No. of atoms | |||
| Chain A | 2285 | 2598 | 2477 |
| Chain B | 2319 | 2487 | 2435 |
| Chain C | 2323 | 2506 | 2264 |
| Chain D | 2503 | ||
| Water | 284 | 1638 | 758 |
| Other solvent | 70 | 72 | 106 |
| Mean B factor (2) | |||
| Chain A | 50.7 | 16.7 | 31.0 |
| Chain B | 52.4 | 23.5 | 35.6 |
| Chain C | 47.2 | 19.0 | 42.6 |
| Chain D | 27.2 | ||
| Water | 46.6 | 35.8 | 45.6 |
| Other solvent | 62.2 | 29.5 | 57.9 |
| R.m.s. deviations from ideal geometry | |||
| Bond lengths () | 0.009 | 0.012 | 0.018 |
| Bond angles () | 1.2 | 1.4 | 1.5 |
| MolProbity analysis | |||
| All-atom clash score | 3.7 [99th percentile] | 6.2 [88th percentile] | 3.0 ]97th percentile] |
| Protein-geometry score | 1.6 [99th percentile] | 1.6 [81st percentile] | 1.6 [95th percentile] |
| Ramachandran plot | |||
| Favored | 97.0 | 98.1 | 97.9 |
| Allowed | 2.8 | 1.7 | 1.8 |
| Outliers | 0.2 | 0.2 | 0.3 |
| PDB entry | 4wmd | 4wme | 4wmf |
R
merge = 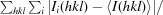
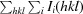 , where I(hkl) is the mean intensity of multiply recorded reflections.
, where I(hkl) is the mean intensity of multiply recorded reflections.
R = 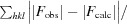
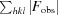 . R
free is the R value calculated for a randomly selected set of reflections that were not included in the refinement.
. R
free is the R value calculated for a randomly selected set of reflections that were not included in the refinement.
Figure 1.

The catalytically inactive MERS-CoV 3CLpro C148A homodimer as found in crystal form I. Protomer A is colored green and protomer B red. The residues forming the catalytic dyad are depicted as blue spheres.
Crystals of forms I, II and III belonged to space groups C2221, C2 and P212121, respectively. There are three protomers in the asymmetric unit of crystal form I. Two of them form a canonical homodimer (protomers A and B), while the third forms an analogous homodimer with a symmetry mate (protomers C and C′). There are no intermolecular interactions that mimic the binding of a peptide product in this crystal form. On the other hand, in both crystal forms II and III there is unambiguous electron density in the active site of protomer A that corresponds to the intercalated C-terminal tail residues of a neighboring protomer (Figs. 2 ▶ a and 2 ▶ b). The C-terminal residues Met301–Gln306 correspond to the P6–P1 sites of the autoprocessed product of the mature enzyme and therefore represent an enzyme–product complex. Surprisingly, in crystal form III, a significant shift in the orientation of domain III in protomer C, which inserts its C-terminal tail into the active site of protomer A, is observed (discussed below). Analysis of the crystal packing environment suggests that protomer C in crystal form III represents a crystallographic monomer, as it does not form a homodimer with any symmetry mate.
Figure 2.

(a) The C-terminal residues of protomer D (crystal form II), corresponding to the P6–P1 autoprocessed site of the mature enzyme fitted to the mF o − DF c electron-density maps shown (contour level of 3.0σ, green; 1.55 Å resolution) after a round of refinement with the C-terminal residues omitted from the model. (b) Illustration of the binding of the C-terminal tail (spheres) of protomer D (magenta ribbons) to the homodimer formed by protomer A (gray surface) and protomer B (cyan surface).
3.2. Comparison with structural homologs
The coordinates of MERS-Cov 3CLpro crystal form II were submitted to the PDBeFold server to search for structural homologs. The closest match was identified as the BatCoV-HKU4 main protease. Alignment of protomer A of MERS-CoV 3CLpro with protomer A of BatCoV-HKU4 (PDB entry 2yna) yields an r.m.s.d. of 0.7 Å over 270 Cα-atom pairs (81% sequence identity) when superimposed using the ‘super’ command in PyMOL. Alignment of the MERS-CoV and BatCoV-HKU4 3CLpro homodimers yields an r.m.s.d. of 0.8 Å over 552 Cα-atom pairs (Fig. 3 ▶ a). Superposition of MERS-CoV 3CLpro protomer A with protomer A from SARS-CoV 3CLpro (PDB entry 1uk3, 50% sequence identity; Yang et al., 2003 ▶) yields an r.m.s.d. of 1.9 Å over 258 Cα-atom pairs. When the structures of the two homodimers are aligned, the r.m.s.d. is 2.2 Å over 537 Cα-atom pairs (Fig. 3 ▶ a). Inspection of the superimposed homodimers reveals that the chymotrypsin-like cores (domains I and II) align very closely (r.m.s.d. of 0.9 Å over 164 Cα-atom pairs). When the domain III structures of MERS-CoVpro and SAR-CoV 3CLpro are aligned, the r.m.s.d. is higher (1.4 Å). The even higher r.m.s.d. that is obtained when the complete homodimers are superimposed (2.2 Å) reflects a small shift in the orientation of domain III (Fig. 3 ▶ b). There is a high degree of conservation of the residues that form the active site in the 3CLpro enzymes of MERS-CoV, BatCoV-HKU4 and SARS-CoV. The residues surrounding the P1′, P1 and P2 substrate-binding pockets are particularly well conserved, which may be advantageous for the design of broad-spectrum inhibitors targeting coronaviral 3CLpro enzymes (Fig. 4 ▶)
Figure 3.

(a) Stereoview of the superimposed homodimers of MERS-CoV 3CLpro (crystal form II, green ribbons) and BatCoV-HKU4 (PDB entry 2yna, red ribbons). (b) Stereoview of the superimposed homodimers of MERS-CoV 3CLpro and SAR-CoV 3CLpro (PDB entry 1uk3, red ribbons; Yang et al., 2003 ▶).
Figure 4.

Sequence alignment of CoV 3CLpro enzymes from MERS-CoV, SARS-CoV, Tylonycteris bat coronavirus HKU4, Human coronavirus HKU1, Human coronavirus OC43, Human coronavirus NL63 and Human coronavirus 229E. Sequences were aligned using T-Coffee (Notredame et al., 2000 ▶) and the figure was prepared with ESPript3 (Robert & Gouet, 2014 ▶). The residues forming the catalytic dyad are highlighted with asterisks.
3.3. Details of the enzyme–product interactions
The fortuitous capture of an enzyme–product complex in crystal forms II and III at high resolution (1.55 and 1.97 Å, respectively) permits a detailed analysis of the intermolecular interactions and provides structural insight into substrate specificity and catalysis, complementing studies of other 3CLpro enzymes (Anand et al., 2002 ▶; Yang et al., 2003 ▶, 2006 ▶; Lee et al., 2005 ▶, 2007 ▶; Xue et al., 2008 ▶; Hilgenfeld, 2014 ▶). In crystal form II, residues Met301–Gln306 of protomer D are intercalated in the active site of protomer A. The interactions between the C-terminal peptide (product) residues and the active site are illustrated in Fig. 5 ▶(a). The S1 pocket, which is formed by residues Leu27, His41, Phe143–Ser150 and His166–Glu169, is occupied by the P1 residue Gln306, which is required for efficient processing by all coronavirus 3CLpro family members (Hegyi & Ziebuhr, 2002 ▶; Chuck et al., 2010 ▶, 2011 ▶). The side chain of Gln306 is held tightly in the S1 pocket near the catalytic dyad formed by His41 and Ala148 (Cys148 in the wild-type enzyme; Anand et al., 2002 ▶) via hydrogen bonds between (i) the P1 Gln306 N∊2 atom and the side-chain O∊1 atom of Glu169 (3.2 Å) and backbone carbonyl of Phe143 (3.1 Å), (ii) the Gln306 O∊1 atom and the His166 N∊2 atom (2.7 Å), (iii) the backbone carbonyl O atom of Gln306 and the N∊2 atom of His41 (3.0 Å) and (iv) the Gln306 OXT atom and the backbone amide of Gly146 (3.0 Å). Additionally, the main-chain amide N atom of Gln306 is hydrogen-bonded to the backbone carbonyl O atom of Gln167 (3.0 Å). The Ala148 Cβ atom is located 3.3 Å away from the backbone carbonyl C atom of Gln306, confirming that Cys148 would be appropriately positioned to act as the catalytic nucleophile in the active enzyme. Residue Ser1 from protomer B forms hydrogen bonds from its side-chain Oγ (2.8 Å) and backbone amide N (2.8 Å) atoms to the carboxylate side chain of Glu169 of protomer A, an interaction that is important for the maintenance of the biological homodimer structure (Anand et al., 2002 ▶; Yang et al., 2003 ▶; Xue et al., 2007 ▶; Cheng et al., 2010 ▶). Likewise, residue Ser1 from protomer A also forms analogous hydrogen-bond interactions with Glu169 in protomer B.
Figure 5.

(a) Stereoview of the hydrogen-bonding interactions (within 3.2 Å) between the C-terminal residues 301–306 of MERS-CoV 3CLpro protomer D (crystal form II, C atoms in green) and the active site of protomer A (C atoms in gray). Residue Ser1 (C atoms in yellow) is from protomer B of the homodimer. (b) Stereoview of the active-site residues from protomer A of the free enzyme form (crystal form I, C atoms in magenta) superimposed onto the active site of product-bound protomer A (crystal form II, C atoms in gray).
The P2 residue, Met305, is nestled into a hydrophobic pocket formed by His41, Gln167, Met168, Asp190, Lys191 and Gln192. In addition to hydrophobic contacts with neighboring side-chain residues, the backbone amide N atom of Met305 is hydrogen-bonded to the O∊1 atom of Gln192 (2.9 Å). Modeling of additional residues into the S2 pocket suggests that this site favors bulkier hydrophobic residues, in accord with the observed preference for leucine in this position of most natural processing sites in the MERS-CoV and SARS-CoV polyproteins (Chuck et al., 2010 ▶). The S3 site is occupied by Val304, the side-chain atoms of which occupy two alternate conformations in crystal form II. Val304 is surrounded by residues Met168, Glu169 and Gln192. Hydrogen-bonding interactions between the backbone amide N atom of Val304 and the backbone carbonyl O atom of Glu169 (3.0 Å) and between the backbone carbonyl of Val304 and the backbone amide N atom of Glu169 (2.9 Å) contribute additional stabilizing interactions. The P4 residue, Val303, is bound to the S4 site, which is formed by residues Gln192–Gln195, Met168, Glu169 and Leu170. The side chain of Val303 stacks against the hydrophobic side chain of Leu170. The S5 site is occupied by Gly302, which is held in place primarily by a water-mediated hydrogen bond to the Gly302 amide N atom (2.9 Å) and to the His194 Nδ1 atom (2.8 Å). Met301 begins to protrude into the solvent space and does not form any significant contacts with the active-site region other than stacking against the side chain of His194. Comparison of the active-site structure between the enzyme–product complex observed in crystal form II and those of the unbound structures from crystal form I illustrates that upon substrate/product binding, the residues forming the S1 pocket do not undergo any significant conformational shifts. Slight adjustments of the rotamers of side chains of residues His41, Gln192, Met168, Glu169 and His194 are observed, which are likely to facilitate substrate binding (Fig. 5 ▶ b)
3.4. An alternate conformation of MERS-CoV 3CLpro
A distinguishing feature of MERS-CoV 3CLpro crystal form III is the conformational change observed in protomer C. Although protomers A and B exhibit the canonical MERS-CoV 3CLpro homodimer structure, in order to insert its C-terminal tail into the active site of protomer A, protomer C has undergone a substantial conformational change. The core chymotrypsin-like part (domains I and II) of protomer C aligns well with those of protomers A or B (r.m.s.d. of 0.6 Å over 163 Cα-atom pairs; residues 11–190), but when domains I and II of the three protomers are aligned then domain III of protomer C occupies a very different position than it does in protomers A or B (Fig. 6 ▶ a). Conversely, if domain III of protomers A and C are superimposed then they align well (r.m.s.d. of 1.1 Å over 98 Cα-atom pairs; residues 200–306) but their chymotrypsin-like domains appear to have shifted relative to one another (not shown). Hence, the conformational change affects the relative orientation of the N- and C-terminal parts of the molecule but does not alter the conformations of the individual domains. The first ten residues in protomer C are disordered and the large shift in the orientation of domain III is mediated by a conformational change in the linker loop (Phe188–Ser204; residues His194–Val196 are disordered), in which it moves to cover the active site (Figs. 6 ▶ b ▶ and 6 ▶ c), potentially impeding access to substrates.
Figure 6.

(a) Stereoview of the superimposed structures of MERS-CoV 3CLpro crystal form III protomer A (green ribbons) and protomer C (magenta ribbons). (b, c) Surface representations of protomer A (b) and protomer C (c) with domains I and II colored gray, the linker loop (residues 188–204) cyan, domain III magenta, the oxyanion loop (residues 143–148) blue and the S1 binding pocket green.
The four molecules found in the asymmetric unit of crystal form II exist as two canonical homodimers (AB and CD), but the C-terminal tail of protomer D is inserted into the active site of protomer A. Therefore, the distortion observed in protomer C of crystal form III is not a necessary prerequisite for the intermolecular interaction that mimics an enzyme–product complex. The distortion of protomer C in crystal form III is probably tolerated because it does not form a canonical 3CLpro homodimer with a neighboring symmetry mate. A similar situation was observed in the crystal structure of infectious bronchitis virus IBV-CoV 3CLpro (PDB entry 2q6d; 40% sequence identity; Xue et al., 2008 ▶). In the case of IBV-CoV 3CLpro three molecules were found in the asymmetric unit, with protomers A and B forming a homodimer and the C-terminal tail of protomer C inserted into the active site of protomer A. When domains I and II in protomers A and C were aligned, they were found to have very similar conformations (r.m.s.d. of 1.0 Å over 171 Cα-atom pairs), but substantial differences were observed in the orientation of domain III in the two molecules; namely, a 5 Å shift of domain III away from domains I and II. The authors claimed that protomer C represents a novel monomeric form of IBV-CoV 3CLpro that was induced by binding of the C-terminus in the active site of the homodimer. Structural alignment of domains I and II of MERS-CoV protomer C from crystal form III with protomer C from IBV-CoV yields an r.m.s.d. of 1.2 Å over 161 Cα-atom pairs (residues 1–193). However, there is a significant shift in the orientation of domain III between the two homologs (Fig. 7 ▶ a). One difference is that the entire linker region in the IBV-CoV homolog could be modeled into electron density, whereas MERS-CoV 3CLpro residues 194–196 are disordered, resulting in different conformations of the linker loops in the two homologs. Additionally, we do not observe the oxyanion loop (residues 143–148) adopting a 310-helix as seen the IBV-CoV 3CLpro structure. This is likely to be due to differences in the conformation of loop residues 276–293 in the two structures. The larger shift in the position of domain III in the MERS-CoV 3CLpro structure than occurs in the structure of IBV-CoV 3CLpro causes these loop residues to come into close contact with the oxyanion loop in MERS-CoV 3CLpro. As a result, a hydrogen bond is formed between the backbone carbonyl of Leu287 and the side-chain Ser142 Oγ atom, which may prevent the formation of a 310-helix.
Figure 7.

(a) Stereoview of the structure of MERS-CoV 3CLpro protomer C (crystal form III, magenta ribbons) superimposed on the structure of IBV-CoV 3CLpro protomer C (PDB entry 2q6d, red ribbons; Xue et al., 2008 ▶). (b) Stereoview of the structure of MERS-CoV 3CLpro protomer C (crystal form III, magenta ribbons) superimposed on the structure of the SARS-CoV 3CLpro G11A monomer (PDB entry 2pwx, cyan ribbons; Chen et al., 2008 ▶).
Previous studies with variants of the SARS-CoV 3CLpro enzyme in which the residues involved in dimerization were altered revealed that certain amino-acid substitutions, such as G11A and R289A, cause a structural shift in 3CLpro that disrupts dimerization and gives rise to a shift in the orientation of domain III similar to what we observe in the case of protomer C in MERS-CoV 3CLpro crystal form III (Fig. 7 ▶ b; Chen et al., 2008 ▶; Shi et al., 2008 ▶; Hu et al., 2009 ▶; Barrila et al., 2010 ▶). Prior studies of monomeric forms of other 3CLpro enzymes revealed that there is very little or no activity in this state (Shi & Song, 2006 ▶; Shi et al., 2011 ▶; Chen et al., 2008 ▶). The significant structural flexibility found in the interdomain linker loop region suggests that there may be significant structural plasticity in 3CLpro enzymes that allows the shift between dimeric and monomeric forms. Indeed, prior studies of SARS-CoV 3CLpro protease demonstrated that truncations of the linker loop between the chymotrypsin-like domain and domain III gave rise to a significant reduction in enzymatic activity, confirming that the proper orientation of the linker between domains I/II and domain III is important (Tsai et al., 2010 ▶). Although protomer C of MERS-CoV 3CLpro crystal form III exhibits a large change in the orientation of domain III similar to what was observed in both IBV-CoV 3CLpro and engineered monomers of SARS-CoV 3CLpro, experimental insight into the enzymatic activity of this form is currently lacking. Therefore, more studies need to be conducted to determine whether this conformation is a crystallographic artifact or a monomeric form of the enzyme that is also populated in solution to some degree.
4. Conclusion
In summary, we have determined three crystal structures of MERS-CoV 3CLpro representing the free enzyme, an enzyme–product comple and a crystallographic monomer arising from a conformational change in the linker loop that results in a large shift in the orientation of domain III. The enzyme–product complex reveals the structural basis of substrate recognition by MERS-CoV 3CLpro on the N-terminal side of the scissile bond. The high degree of conservation between the active sites of coronavirus 3CLpro enzymes, particularly in their S2, S1 and S1′ pockets, suggests that broad-spectrum coronaviral 3CLpro inhibitors can be developed. This objective will be facilitated by determining additional structures of 3CLpro enzymes alone and in complex with substrates and inhibitors.
Supplementary Material
PDB reference: MERS-CoV 3CL protease, 4wmd
PDB reference: 4wme
PDB reference: 4wmf
Acknowledgments
This project has been funded in whole or in part with Federal funds from the Frederick National Laboratory for Cancer Research, National Institutes of Health under contract HHSN261200800001E and the Intramural Research Program of the NIH, National Cancer Institute, Center for Cancer Research. The content of this publication does not necessarily reflect the views or policies of the Department of Health and Human Services, nor does the mention of trade names, commercial products or organizations imply endorsement by the US Government. Additional funding came from a National Interagency Confederation for Biological Research Collaborative Project Award Program to DN. We are grateful to our collaborators at the United States Army Research Institute of Infectious Diseases (R. Ulrich and colleagues) for providing us with a MERS-CoV 3CLpro PCR amplicon. We thank the Biophysics Resource in the Structural Biophysics Laboratory, NCI at Frederick for use of the LC/ESMS and dynamic light-scattering instruments. X-ray diffraction data were collected at the Southeast Regional Collaborative Access Team (SER-CAT) beamline 22-BM of the Advanced Photon Source, Argonne National Laboratory. Supporting institutions may be found at http://www.ser-cat.org/members.html. Use of the Advanced Photon Source was supported by the US Department of Energy, Office of Science, Office of Basic Energy Sciences under contract No. W-31-109-Eng-38.
References
- Adams, P. D. et al. (2011). Methods, 55, 94–106.
- Adney, D. R., van Doremalen, N., Brown, V. R., Bushmaker, T., Scott, D., de Wit, E., Bowen, R. A. & Munster, V. J. (2014). Emerg. Infect. Dis. 20, 1999–2005. [DOI] [PMC free article] [PubMed]
- Anand, K., Palm, G. J., Mesters, J. R., Siddell, S. G., Ziebuhr, J. & Hilgenfeld, R. (2002). EMBO J. 21, 3213–3224. [DOI] [PMC free article] [PubMed]
- Anand, K., Ziebuhr, J., Wadhwani, P., Mesters, J. R. & Hilgenfeld, R. (2003). Science, 300, 1763–1767. [DOI] [PubMed]
- Artimo, P. et al. (2012). Nucleic Acids Res. 40, W597–W603. [DOI] [PMC free article] [PubMed]
- Barrila, J., Gabelli, S. B., Bacha, U., Amzel, L. M. & Freire, E. (2010). Biochemistry, 49, 4308–4317. [DOI] [PMC free article] [PubMed]
- Boheemen, S. van, de Graaf, M., Lauber, C., Bestebroer, T. M., Raj, V. S., Zaki, A. M., Osterhaus, A. D., Haagmans, B. L., Gorbalenya, A. E., Snijder, E. J. & Fouchier, R. A. (2012). MBio, 3, e00473-12. [DOI] [PMC free article] [PubMed]
- Chen, H., Wei, P., Huang, C., Tan, L., Liu, Y. & Lai, L. (2006). J. Biol. Chem. 281, 13894–13898. [DOI] [PMC free article] [PubMed]
- Chen, S., Hu, T., Zhang, J., Chen, J., Chen, K., Ding, J., Jiang, H. & Shen, X. (2008). J. Biol. Chem. 283, 554–564. [DOI] [PMC free article] [PubMed]
- Chen, V. B., Arendall, W. B., Headd, J. J., Keedy, D. A., Immormino, R. M., Kapral, G. J., Murray, L. W., Richardson, J. S. & Richardson, D. C. (2010). Acta Cryst. D66, 12–21. [DOI] [PMC free article] [PubMed]
- Cheng, S.-C., Chang, G.-G. & Chou, C.-Y. (2010). Biophys. J. 98, 1327–1336. [DOI] [PMC free article] [PubMed]
- Chuck, C.-P., Chong, L.-T., Chen, C., Chow, H.-F., Wan, D. C.-C. & Wong, K.-B. (2010). PLoS One, 5, e13197. [DOI] [PMC free article] [PubMed]
- Chuck, C.-P., Chow, H.-F., Wan, D. C.-C. & Wong, K.-B. (2011). PLoS One, 6, e27228. [DOI] [PMC free article] [PubMed]
- Corman, V. M., Ithete, N. L., Richards, L. R., Schoeman, M. C., Preiser, W., Drosten, C. & Drexler, J. F. (2014). J. Virol. 88, 11297–11303. [DOI] [PMC free article] [PubMed]
- Cunha, C. B. & Opal, S. M. (2014). Virulence, 5, 650–654. [DOI] [PMC free article] [PubMed]
- Emsley, P., Lohkamp, B., Scott, W. G. & Cowtan, K. (2010). Acta Cryst. D66, 486–501. [DOI] [PMC free article] [PubMed]
- Falzarano, D. et al. (2014). PLoS Pathog. 10, e1004250. [DOI] [PMC free article] [PubMed]
- Ge, X.-Y. et al. (2013). Nature (London), 503, 535–538.
- Gorrec, F. (2009). J. Appl. Cryst. 42, 1035–1042. [DOI] [PMC free article] [PubMed]
- Graziano, V., McGrath, W. J., Yang, L. & Mangel, W. F. (2006). Biochemistry, 45, 14632–14641. [DOI] [PubMed]
- Hegyi, A. & Ziebuhr, J. (2002). J. Gen. Virol. 83, 595–599. [DOI] [PubMed]
- Hilgenfeld, R. (2014). FEBS J. 281, 4085–4096. [DOI] [PMC free article] [PubMed]
- Holmes, D. (2014). Lancet, 383, 1793. [DOI] [PMC free article] [PubMed]
- Hsu, M.-F., Kuo, C.-J., Chang, K.-T., Chang, H.-C., Chou, C.-C., Ko, T.-P., Shr, H.-L., Chang, G.-G., Wang, A. H.-J. & Liang, P.-H. (2005). J. Biol. Chem. 280, 31257–31266. [DOI] [PMC free article] [PubMed]
- Hu, T., Zhang, Y., Li, L., Wang, K., Chen, S., Chen, J., Ding, J., Jiang, H. & Shen, X. (2009). Virology, 388, 324–334. [DOI] [PMC free article] [PubMed]
- Kabsch, W. & Sander, C. (1983). Biopolymers, 22, 2577–2637. [DOI] [PubMed]
- Kapust, R. B., Tözsér, J., Fox, J. D., Anderson, D. E., Cherry, S., Copeland, T. D. & Waugh, D. S. (2001). Protein Eng. 14, 993–1000. [DOI] [PubMed]
- Krissinel, E. & Henrick, K. (2004). Acta Cryst. D60, 2256–2268. [DOI] [PubMed]
- Lee, T.-W., Cherney, M. M., Huitema, C., Liu, J., James, K. E., Powers, J. C., Eltis, L. D. & James, M. N. G. (2005). J. Mol. Biol. 353, 1137–1151. [DOI] [PMC free article] [PubMed]
- Lee, T.-W., Cherney, M. M., Liu, J., James, K. E., Powers, J. C., Eltis, L. D. & James, M. N. G. (2007). J. Mol. Biol. 366, 916–932. [DOI] [PMC free article] [PubMed]
- Li, C., Qi, Y., Teng, X., Yang, Z., Wei, P., Zhang, C., Tan, L., Zhou, L., Liu, Y. & Lai, L. (2010). J. Biol. Chem. 285, 28134–28140. [DOI] [PMC free article] [PubMed]
- Li, W. et al. (2005). Science, 310, 676–679. [DOI] [PubMed]
- Minor, W., Cymborowski, M., Otwinowski, Z. & Chruszcz, M. (2006). Acta Cryst. D62, 859–866. [DOI] [PubMed]
- Muramatsu, T., Kim, Y. T., Nishii, W., Terada, T., Shirouzu, M. & Yokoyama, S. (2013). FEBS J. 280, 2002–2013. [DOI] [PMC free article] [PubMed]
- Murshudov, G. N., Skubák, P., Lebedev, A. A., Pannu, N. S., Steiner, R. A., Nicholls, R. A., Winn, M. D., Long, F. & Vagin, A. A. (2011). Acta Cryst. D67, 355–367. [DOI] [PMC free article] [PubMed]
- Notredame, C., Higgins, D. G. & Heringa, J. (2000). J. Mol. Biol. 302, 205–217. [DOI] [PubMed]
- Robert, X. & Gouet, P. (2014). Nucleic Acids Res. 42, W320–W324. [DOI] [PMC free article] [PubMed]
- Shi, J., Han, N., Lim, L., Lua, S., Sivaraman, J., Wang, L., Mu, Y. & Song, J. (2011). PLoS Comput. Biol. 7, e1001084. [DOI] [PMC free article] [PubMed]
- Shi, J., Sivaraman, J. & Song, J. (2008). J. Virol. 82, 4620–4629. [DOI] [PMC free article] [PubMed]
- Shi, J. & Song, J. (2006). FEBS J. 273, 1035–1045. [DOI] [PMC free article] [PubMed]
- Shi, J., Wei, Z. & Song, J. (2004). J. Biol. Chem. 279, 24765–24773. [DOI] [PMC free article] [PubMed]
- Stobart, C. C., Sexton, N. R., Munjal, H., Lu, X., Molland, K. L., Tomar, S., Mesecar, A. D. & Denison, M. R. (2013). J. Virol. 87, 12611–12618. [DOI] [PMC free article] [PubMed]
- Tropea, J. E., Cherry, S. & Waugh, D. S. (2009). Methods Mol. Biol. 498, 297–307. [DOI] [PubMed]
- Tsai, M.-Y., Chang, W.-H., Liang, J.-Y., Lin, L.-L., Chang, G.-G. & Chang, H.-P. (2010). J. Biochem. 148, 349–358. [DOI] [PMC free article] [PubMed]
- Vagin, A. & Teplyakov, A. (2010). Acta Cryst. D66, 22–25. [DOI] [PubMed]
- Winn, M. D. et al. (2011). Acta Cryst. D67, 235–242.
- Xue, X., Yang, H., Shen, W., Zhao, Q., Li, J., Yang, K., Chen, C., Jin, Y., Bartlam, M. & Rao, Z. (2007). J. Mol. Biol. 366, 965–975. [DOI] [PMC free article] [PubMed]
- Xue, X., Yu, H., Yang, H., Xue, F., Wu, Z., Shen, W., Li, J., Zhou, Z., Ding, Y., Zhao, Q., Zhang, X. C., Liao, M., Bartlam, M. & Rao, Z. (2008). J. Virol. 82, 2515–2527. [DOI] [PMC free article] [PubMed]
- Yang, H., Bartlam, M. & Rao, Z. (2006). Curr. Pharm. Des. 12, 4573–4590. [DOI] [PubMed]
- Yang, H., Yang, M., Ding, Y., Liu, Y., Lou, Z., Zhou, Z., Sun, L., Mo, L., Ye, S., Pang, H., Gao, G. F., Anand, K., Bartlam, M., Hilgenfeld, R. & Rao, Z. (2003). Proc. Natl Acad. Sci. USA, 100, 13190–13195. [DOI] [PMC free article] [PubMed]
- Zaki, A. M., van Boheemen, S., Bestebroer, T. M., Osterhaus, A. D. & Fouchier, R. A. (2012). N. Engl. J. Med. 367, 1814–1820. [DOI] [PubMed]
- Zhao, Q., Li, S., Xue, F., Zou, Y., Chen, C., Bartlam, M. & Rao, Z. (2008). J. Virol. 82, 8647–8655. [DOI] [PMC free article] [PubMed]
- Zhao, Q., Weber, E. & Yang, H. (2013). Recent Pat. Anti-Infect. Drug Discov. 8, 150–156. [DOI] [PubMed]
- Ziebuhr, J., Snijder, E. J. & Gorbalenya, A. E. (2000). J. Gen. Virol. 81, 853–879. [DOI] [PubMed]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
PDB reference: MERS-CoV 3CL protease, 4wmd
PDB reference: 4wme
PDB reference: 4wmf