

Abstract
In vivo measurement of neurotransmitters and modulators is now feasible with advanced proton magnetic resonance spectroscopy (1H-MRS) techniques. This review provides a basic tutorial of MRS, describes the methods available to measure brain glutamate, glutamine, γ-aminobutyric acid, glutathione, N-acetylaspartylglutamate, glycine, and serine at magnetic field strengths of 3Tesla or higher, and summarizes the neurochemical findings in schizophrenia. Overall, 1H-MRS holds great promise for producing biomarkers that can serve as treatment targets, prediction of disease onset, or illness exacerbation in schizophrenia and other brain diseases.
Keywords: magnetic resonance spectroscopy, glutamate, glutathione, GABA, schizophrenia
1.0 Introduction
Proton magnetic resonance spectroscopy (1H-MRS) is a noninvasive technique that allows the quantification of certain biochemical concentrations in vivo. In the brain, these biochemicals reflect a wide variety of mechanisms that range from cellular function and viability to neurotransmission. The information provided by 1H-MRS is useful for researchers to understand pathological processes and treatment effects in brain diseases such as schizophrenia. The majority of 1H-MRS studies of schizophrenia measured N-acetylaspartate (NAA, a marker of neuronal viability and function), choline + phosphocholine + glycerophosphocholine or choline-containing compounds (Cho, reflective of membrane turnover), and creatine+phosphocreatine (Cr, an index of energy metabolism), simply because these biochemicals yield the most prominent peaks in the 1H spectrum and therefore are the easiest to quantify. The consistent finding from 1H-MRS studies in schizophrenia is reduced frontal and temporal lobe NAA (Rowland et al., 2001; Steen et al., 2005; Brugger et al., 2011), plausibly reflecting neuronal dysfunction in these brain regions.
There are several neurochemicals that could prove fruitful in the elucidation of the pathophysiological processes that underlie schizophrenia and have potential to serve as biomarkers for treatment targets, prediction of disease onset, or exacerbation. However, reliable measurement of these biochemicals by 1H-MRS poses technical challenges because of their low concentrations and low sensitivity due to J-coupling which results in multiplet peaks in the spectrum as well as spectral overlap. Figure 1 provides an illustration of a proton spectrum and corresponding metabolites. Major advances have been made in measuring these biochemicals using 1H-MRS, and these advances include higher sensitivity and spectral dispersion due to higher magnetic field strengths, greater reliability due to improved MR scanner hardware, new optimized pulse sequences for spectral acquisition, and improved spectral quantification algorithms and commercial software packages. This review will focus on the latest single voxel 1H-MRS techniques and their applications for measuring such biochemicals as glutamate (Glu), glutamine (Gln), glutathione (GSH), γ-aminobutyric acid (GABA), N-acetylaspartylglutamate (NAAG), glycine, and serine in schizophrenia at field strengths of 3T or higher. Although this review focuses on schizophrenia, these 1H-MRS techniques would be useful in other psychiatric and neurological disorders.
Figure 1.
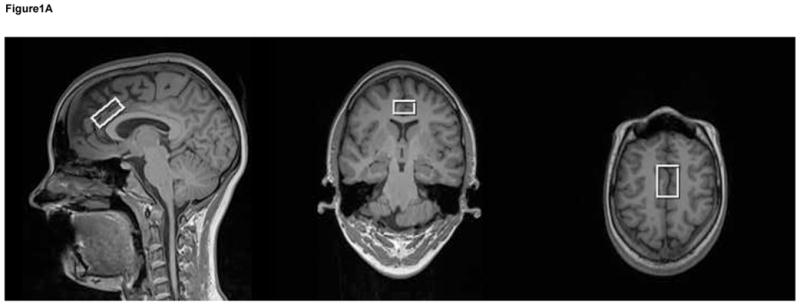
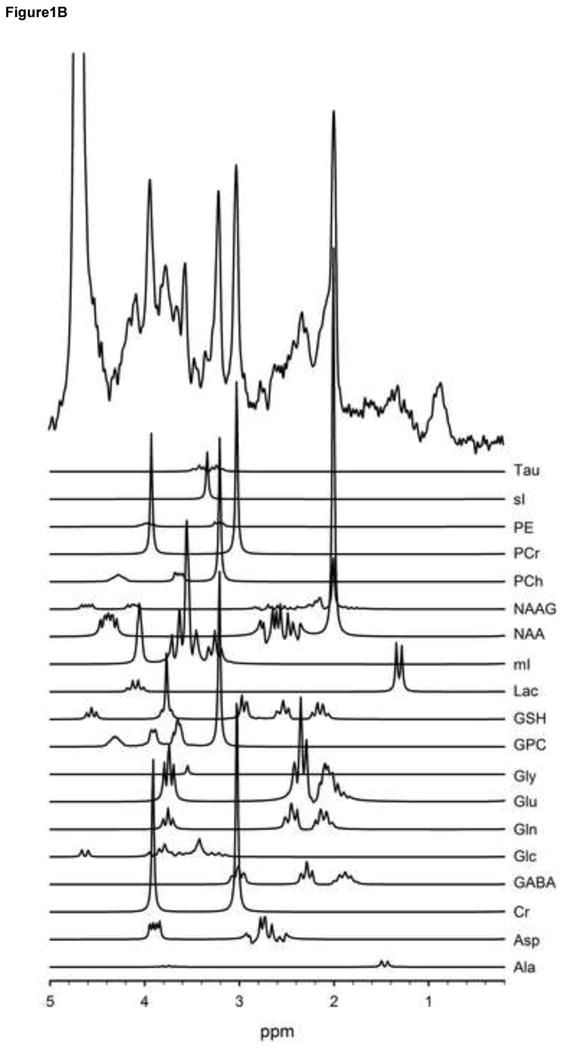
T1-weighted voxel images acquired at 3T with a MP-RAGE imaging sequence (TR/TE/TI=1900/3.45/900-ms, FOV 256 × 256 mm, 1-mm isotropic voxels, flip angle = 9°). These images show that the voxel was placed in the anterior cingulate (a). A very short echo time (TE) spectrum with corresponding plots of each metabolite in the spectrum shown below (b). These metabolites are included in the basis set used for quantification. The metabolites were simulated at a frequency of 123.56-MHz using a STEAM sequence (TR/TM/TE=2000/10/6.5ms).
2.0 1H-MRS Overview
2.1. 1H-MRS Signal
1H-MRS uses the same hardware equipment as standard magnetic resonance imaging (MRI), in which the radiofrequency (RF) coils and receiver channels are all tuned to the proton (1H) resonance frequency. The external magnetic field generated by the MR scanner is denoted by “B0”, and the strength typically ranges from 1.5 – 3 Tesla for clinical settings. An explanation of the basic magnetic resonance (MR) principles can be found in (Haacke et al., 1999; Liang et al., 2000; Brown and Semelka, 2010) while a detailed explanation of the principles of 1H-MRS can be found in (Drost et al., 2002; Zhu and Barker, 2011; De Graaf, 2007). NAA, Cho, and Cr yield the strongest signals in a normal 1H spectrum of the human brain, and therefore have been most extensively studied using 1H-MRS. However, with higher magnetic field strengths, glutamate and glutamine that have complex spectral patterns due to coupling between their 1H nuclei can also be quantified using short echo time (TE) pulse sequences (Gruetter et al., 1998; Petroff et al., 2000; Deelchand et al., 2010; Mekle et al., 2009; Wijtenburg and Knight-Scott, 2011; Mullins et al., 2008) or specifically tailored pulse sequences (Hurd et al., 2004; Schubert et al., 2004; Yang et al., 2008).
2.2 Spectroscopic Localization Techniques
There are two main types of spectroscopic acquisition techniques, single-voxel and spectroscopic imaging (SI) also known as chemical shift imaging (CSI). With single-voxel spectroscopy, the signal is acquired from a 3-dimensional volume called a “voxel” in a region of interest to produce a single spectrum. In contrast, SI is a combination of spectroscopy and imaging, whereby spectral information is acquired in a spatial matrix with corresponding spectra in multiple voxels. There are advantages and disadvantages to both techniques. Single-voxel spectroscopy is ideal for hypothesis-driven questions about specific regions, and to quantify metabolites that have short T2 relaxation or strongly coupled spin systems such as glutamate, glutamine, and GABA. Commonly used non-editing acquisition sequences include the Stimulated Echo Acquisition Mode (STEAM) and Point Resolved Excitation Spin-echo Sequence (PRESS) sequences (Frahm et al., 1987; Bottomley, 1987). There are two editing sequences commonly used to detect GABA: 2D JPRESS (Ryner et al., 1995) and the more frequently used Mescher-Garwood (MEGA)-PRESS sequence (Mescher et al., 1998). MEGA-PRESS combines the frequency-selective editing technique (MEGA) with a PRESS sequence and is employed for spectral editing of the metabolites with J-coupled spin systems, such as GABA (Terpstra et al., 2002), GSH (Terpstra et al., 2003), and NAAG (Edden et al., 2007). In recent years, improved editing efficiency of the traditional MEGA-PRESS sequence has been reported at higher magnetic fields (Edden and Barker, 2007; Kaiser et al., 2007). Compared to single voxel sequences, the spatial resolution is superior with SI and thus allows the investigator to assess more brain regions and smaller voxels that may allow a distinction between gray and white matter. However, SI is generally limited to the measurement of metabolites with longer T2 relaxation such as NAA, Cho, and Cr. Metabolites with shorter T2s may not be visible using long TE acquisition, which is typical for SI applications. Recent advances in the field do include the applications of short TE SI sequences (Otazo et al., 2007; Gruber et al., 2008), and for more detailed information regarding SI sequences, see (Posse et al., 2013; Zhu and Barker, 2011).
2.3 Spectroscopic Data Considerations
There are several important considerations for the evaluation and interpretation of proton spectroscopic data that include shimming, metabolite quantification, and reliability/reproducibility. An important factor when acquiring spectroscopic data is shimming. Shimming is the act of adjusting the currents of the shim coils in order to make the magnetic field homogeneous in the region of interest (ROI). A smaller line width is always desirable since it translates to better spectral resolution and therefore improved quantification. Often on 3T MR systems, a water peak line width of 12-Hz or less is most desirable, which indicates the acquired spectrum may be of sufficient quality for good quantification. It is common to report the shimming cutoff for inclusion in the data set.
Quantification is typically performed using commercially available software packages such as LCModel or jMRUI (Provencher, 1993; Ratiney et al., 2004) or in-house written programs. A common metric for the goodness of spectral fit is the Cramer Rao Lower Bounds (CRLB). While there is no set standard for CRLB cut-off, CRLBs less than or equal to 20% are generally considered to be of acceptable quality (Cavassila et al., 2001; Provencher, 2014). Generally, metabolites with lower concentrations have higher CRLBs due to the difficulty in separating the signal from other signals. This difficulty may lead to exclusion of data that may potentially bias the results. In these instances, a CRLBs cut-off greater than 20% such as 25% or 30% may be used as a compromise. Another consideration in quantification is that metabolites must be quantified with respect to a reference. The voxel water and Cr peaks are commonly used as references. The advantage of using Cr as a reference is that it enables comparisons to other studies since utilizing a ratio to Cr removes confounds from the data such as coil loading, scaling factors, and CSF proportion in the voxel. One disadvantage of using Cr as a reference is that Cr may be different across disease states including schizophrenia (Ongur et al., 2009) and changes with age (Danielsen and Ross, 1999). Therefore, alterations in metabolites that are referenced to Cr could actually be due to changes in Cr but not in the metabolite of interest. The use of water as a reference is preferable when metabolites such as NAA and Cr, often used in referencing, vary in different illnesses. There are two main methods for referencing to water that result in relative concentrations in institutional units or absolute concentrations in mmol/kg (Jansen et al., 2006). Relative concentrations are generated when each metabolite is referenced to the entire signal from the water in the voxel whereas absolute concentrations using an internal reference take into account the proportion of the water signal exclusively from the tissue compartment as well as the concentration of water and density of tissue types. There are advantages and disadvantages to water referencing (for review see (Gasparovic et al., 2006; Mullins et al., 2014)). Advantages include the ability to utilize the water reference for eddy-current correction and retention of all metabolite information since one metabolite is used in the ratio denominator resulting in lost information. Disadvantages of utilizing water as a reference include that water is acquired during a separate scan from the metabolites, potentially introducing patient motion into the dataset and increasing scan times, and that the water content can change with different pathologies such multiple sclerosis (Laule et al., 2004) and stroke (Gideon et al., 1999). Thus, it is highly recommended when using this methodology to perform correction for partial volume to remove effects of CSF from the water signal. Tissue composition correction or relaxation correction are also recommended and are reviewed elsewhere (for details see (Ernst et al., 1993; Gasparovic et al., 2006; Knight-Scott et al., 2003; Jansen et al., 2006). Another consideration in quantification when using shorter TEs is the macromolecule background signal. The macromolecule background is made up of broad resonances that are underneath the metabolites that visually appear to globally elevate the spectrum (Figure 2). These resonances are short T2s species and are usually not present at longer TEs since their signals are completely decayed. The specific constituents that contribute to the macromolecule background are unknown, but the characteristic pattern of macromolecule background has been shown in animal and human work (Behar and Ogino, 1993; Behar et al., 1994; Seeger et al., 2003; Gottschalk et al., 2008). There are different methods for accounting for macromolecule background in spectral quantification such as utilizing a program that already incorporates macromolecule handling into fitting such as LCModel or jMRUI or to collect a macromolecule spectrum via metabolite nulling for each participant and add it into the specific quantification program.
Figure 2.
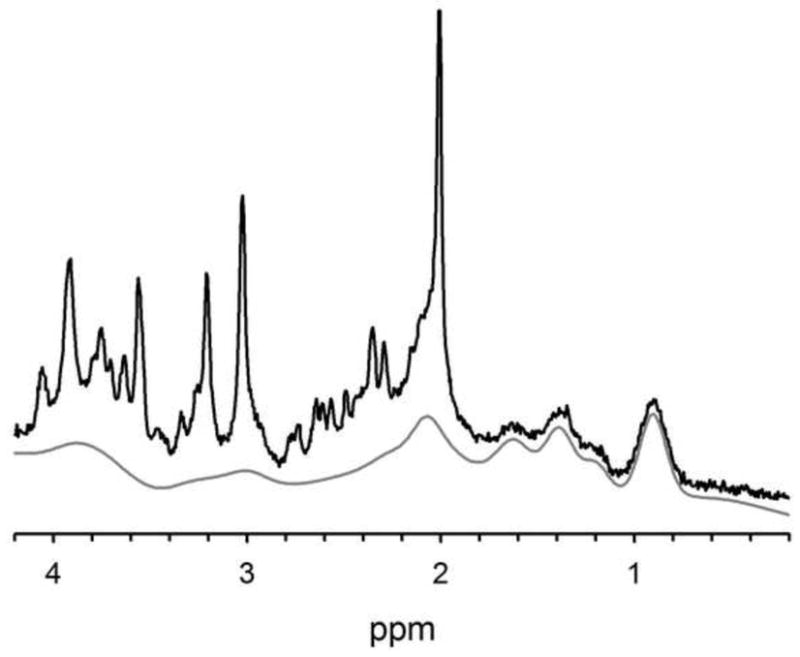
The macromolecule background is only present at shorter TEs due to its short T2 constituents, and it can be a complicating factor in quantification. This is an example short TE spectrum (black) with its macromolecule background fit spectrum shown underneath (gray) as fit by LCModel. These data were acquired at 3T using a very short TE phase rotation sequence (TR/TM/TE=2000/10/6.5-ms, 256 excitations, 2.5-kHz spectral width, 2048 complex points, and phases: ϕ1=135°, ϕ2=22.5°, ϕ13=112.5°, ϕADC=0°).
Assessments of reproducibility for spectroscopic acquisition sequences are also important. Standard sequences that are commercially available such as STEAM and PRESS are used extensively, and reproducibility measures on healthy populations have been well established. However, reliability assessments may be appropriate for applications to clinical populations in order to show feasibility and reproducibility such as (Mullins et al., 2003). When choosing a newly developed technique, it is important that reproducibility be established. For instance, more recent localization techniques such as MEGA-PRESS (Evans et al., 2010; Bogner et al., 2010; O’Gorman et al., 2011; Geramita et al., 2011), SPECIAL (Near et al., 2013), and PR-STEAM (Wijtenburg et al., 2013) demonstrated good reproducibility for historically difficult to detect metabolites such as GABA and glutamate.
3.0 1H-MRS in Schizophrenia
Due to the large number of studies examining glutamate and/or glutamine in SZ, the results will be summarized according to the population under investigation such as high risk (3.1.1), first degree relatives (3.1.2), first episode (3.1.3), chronic (3.1.4), unmedicated state (3.1.5), and anti-psychotic treatment effects (3.1.6). For the GABA, glutathione, NAAG, glycine, and serine sections, the paucity of studies examining these metabolites precludes separation into the different categories similar to the glutamate/glutamine section; thus, results in these sections will include discussions across all illness stages and anti-psychotic medication studies.
3.1 Glutamate and glutamine
Glutamate is the primary excitatory neurotransmitter in the adult brain and released in half of the synapses (Farber et al., 1998). After release into the synapse, glutamate is quickly transported to astrocytes where it is converted to glutamine by glutamine synthetase. Depending on the size of the synapse, number of transporters, and many other factors, removal of glutamate from the synaptic cleft can occur in sub-milliseconds to milliseconds (Danbolt, 2001). This quick transport and conversion helps prevent excitotoxicity. Glutamine is then transported to the neuron and is converted back to glutamate by glutaminase. Glutamate is synthesized from glucose (tricarboxylic cycle) and glutamine. In addition to its role as a neurotransmitter, glutamate also serves as a precursor for the synthesis of GABA and glutathione and as a building block in protein synthesis (Brosnan and Brosnan, 2013; Wu et al., 2004; Duarte and Gruetter, 2013; Mathews and Diamond, 2003). Due to these diverse roles, glutamate is found in multiple cell types, including neurons and glia, and is also found in the extracellular space. 1H-MRS cannot provide information regarding the amount of glutamate devoted to a specific function or how much is present in a specific cell types. The glutamate signal provided with 1H-MRS reflects the total amount of glutamate within a specified voxel of interest in the brain. This signal does not simply reflect glutamate neurotransmission, but most likely reflects multiple mechanisms and has been suggested to be an index of cortical excitability (Stagg et al., 2009; Stagg et al., 2011). Although a tight coupling between the neurotransmitter and metabolic glutamate pools has been suggested (Magistretti and Pellerin, 1999; Rothman et al., 1999), the functional relevance of total glutamate assessed through spectroscopy is not clear. It has been suggested that 20–30% of glutamate in tissue is not visible using MRS (Kauppinen et al., 1994; Maddock and Buonocore, 2012), and the MRS invisible glutamate may be part of the neurotransmitter pool (Kauppinen et al., 1994). Approximately 80% of brain glutamine takes part in the glutamatergic neurotransmission cycle (Magistretti and Pellerin, 1999; Rothman et al., 1999). Hence, glutamine concentration measured with 1H-MRS may be a good index of the turnover of glutamate involved in neurotransmission. Human brain tissue glutamate levels range from 5–15 mmol per kg wet weight (Danbolt, 2001), and glutamine levels range from 3.0–5.8 mmol per kg wet-weight (Govindaraju et al., 2000). The glutamate signal within the 1H spectrum occurs at 2.04, 2.11, 2.35, and 3.74 ppm and glutamine occurs at 2.11, 2.13, 2.44, and 3.76 ppm. There are several techniques available to measure glutamate and glutamine (Ramadan et al., 2013). Depending on the TE and the localization technique chosen, glutamate, glutamine, or glutamate+glutamine (Glx) may be reported. In schizophrenia research, glutamate and glutamine are predominantly measured using PRESS or STEAM localization techniques. Short TEs in the range of 20 to 35ms are chosen to minimize the signal losses due to J-coupling and T2 relaxation. Another TE often chosen is 80 ms since glutamate was shown to be distinguishable from glutamine (Schubert et al., 2004); however, a recent study suggests this TE may be optimal for glutamine detection (Hancu and Port, 2011). MEGA-PRESS and J-editing can also be used to measure Glx; however, the Glx level reported with these techniques consists of only the signal from the multiplets at 3.74 ppm. The advantage of Glx quantification from the edited spectrum is that there is no overlap with other spectral peaks making detection easier; however, a drawback is that the signal-noise ratio is much lower due to the longer TE typically used in these sequences.
There are several lines of evidence that implicate the glutamatergic system in the pathophysiology of schizophrenia. Dissociative anesthetics such as phencyclidine (PCP) and ketamine block the glutamate N-methyl-D-aspartate receptor (NMDAR), and the administration of these drugs produces a state in adults that is phenomenologically similar to schizophrenia (Rowland et al., 2005). Ketamine, when administered to people with stable schizophrenia, produces exacerbations of their unique psychotic content (Lahti et al., 2001) and may increase prefrontal glutamatergic metabolites (Rowland et al., 2005; Stone et al., 2012) similar to preclinical models (Moghaddam et al., 1997). Post-mortem work demonstrates alterations in glutamate receptors (Beneyto et al., 2007; Meador-Woodruff et al., 2003; Zavitsanou et al., 2002), and genetics studies show a link between genes for glutamatergic receptors and risk for schizophrenia (Winchester et al., 2014). These observations led to great interest in the role of glutamate in schizophrenia pathophysiology, as outlined in several previous articles (Poels et al., 2014a; Poels et al., 2014b; Egerton et al., 2012b; Marsman et al., 2013; Stone, 2009). Overall, there is a great emphasis on studying in vivo glutamate in those at risk for schizophrenia, first-episode and chronic phases, and the impact of antipsychotic medication treatment.
3.1.1 High Risk
Prior to onset of schizophrenia, there is a prodromal phase or “at-risk mental state” stage where individuals have experienced a decrease in global function, a first-degree relative with a psychotic disorder, and sub-threshold psychotic symptoms (Yung et al., 1996; Yung et al., 2007). These individuals are more likely to convert to a psychotic disorder than the general population (Fusar-Poli et al., 2012). Identifying biomarkers for those at greatest risk for psychosis is imperative for development of treatment interventions to potentially prevent or reduce the incidence of conversion to a psychotic disorder as well as reduce the symptom severity of those that do convert.
The majority of studies that examined frontal regions show no differences between high risk and healthy controls (Fusar-Poli et al., 2011; Valli et al., 2011; Natsubori et al., 2013), but there are two exceptions (Stone et al., 2009; Tibbo et al., 2004). Fusar-Poli et al. (2011) and Valli et al. observed no anterior cingulate glutamate differences between healthy controls and participants at risk for psychosis, who were antipsychotic medication naïve (Fusar-Poli et al., 2011; Valli et al., 2011). In a group of mixed antipsychotic medication use (10 on antipsychotics medications for less than 16 cumulative weeks and 14 were not taking antipsychotic medications), Natsubori et al. reported no differences in medial prefrontal cortex (including the anterior cingulate) Glx levels in ultra-high risk participants compared to controls (Natsubori et al., 2013). Stone et al. (2009) observed no glutamate differences in the anterior cingulate in at-risk participants compared to controls (Stone et al., 2009). In this study, 21 at-risk participants were not currently taking antipsychotic or antidepressant medications, while 6 at-risk participants were taking antipsychotic or antidepressant medications. However, Stone et al. (2009) found elevated glutamine levels in the anterior cingulate in this sample. Similarly, Tibbo et al. found elevated glutamate+glutamine relative to creatine in the right middle frontal region, which encompassed the anterior cingulate cortex (Tibbo et al., 2004).
In temporal regions, the majority of studies of participants at risk for psychosis report no alterations in glutamate or glutamine. Valli et al. observed no glutamate differences between antipsychotic medication naïve, at risk participants and controls in the left hippocampus (Valli et al., 2011). In a mixed antipsychotic medication use sample, Stone et al. (2009) found no glutamate or glutamine differences between groups in the left hippocampus (Stone et al., 2009). In addition, Wood et al. (2010) observed no Glx differences in either left or right medial temporal lobe voxels nor between participants who transitioned to psychosis and those that did not (Wood et al., 2010). Participants in the Wood et al. study did not take antipsychotic medications. However, Fusar-Poli et al. (2011) reported lower glutamate levels in antipsychotic medication naïve at risk participants in the left hippocampus compared to healthy controls at trend level (Fusar-Poli et al., 2011).
In the thalamus, decreased glutamate levels are commonly reported. Antipsychotic medication naïve participants who were at risk for developing a psychotic disorder had significantly lower thalamic glutamate levels compared to controls (Fusar-Poli et al., 2011). Valli et al. reported a trend level decrease in Glu levels in an antipsychotic medication naive at risk group compared to healthy controls in the left thalamus (Valli et al., 2011). In further support of these findings, Stone et al. (2009) also found decreased glutamate levels in the left thalamus in a mixed antipsychotic medication use, at-risk group compared to controls (Stone et al., 2009). In this same study, no glutamine levels differences were found between groups.
In the dorsal caudate nucleus, de la Fuente-Sandoval et al (2011) showed that 18 antipsychotic medication naïve, ultra high risk participants had elevated glutamate compared to healthy controls (de la Fuente-Sandoval et al., 2011). This same study reported no differences in the right cerebellum between groups. In a follow-up study, de la Fuente-Sandoval et al (2013a) reported that 7 out of 19 of these participants transitioned to a psychotic disorder within two years (de la Fuente-Sandoval et al., 2013a). Those that transitioned to a psychotic disorder had elevated baseline glutamate levels compared to controls; whereas, there were no glutamate differences between the 12 participants who did not convert to a psychotic disorder and healthy controls.
Thus, decreased thalamic glutamate levels and increased striatal glutamate levels are most often observed in participants at high-risk compared to healthy controls whereas the majority of studies in the frontal and temporal regions report no differences between groups.
3.1.2 First-Degree Relatives
There are two studies examining the neurochemistry of genetic high risk participants with a first-degree family member with schizophrenia. Lutkenhoff et al. found that co-twins (both monozygotic and dizygotic) of participants with schizophrenia had lower glutamate levels than healthy twin pairs across regions, which included mesial prefrontal gray matter, left prefrontal white matter, and left hippocampus (Lutkenhoff et al., 2010). In contrast, Purdon et al. found no Glu/Cr differences in the right and left frontal voxels in adults with a sibling with schizophrenia (Purdon et al., 2008). Thus, there appears to be no differences between first-degree relatives and healthy controls when studying one specific region, while one study suggests overall lower glutamate levels in relatives when combining results from multiple regions.
3.1.3 First Episode Schizophrenia
1H-MRS studies examining glutamate, separately from glutamine, in antipsychotic medication naive first episode schizophrenia participants reported no differences between groups in most regions except the striatum. A study by de la Fuente-Sandoval et al. (2011) showed elevated glutamate in the dorsal caudate but not the right cerebellum in antipsychotic medication naïve first episode schizophrenia participants compared to controls (de la Fuente-Sandoval et al., 2011). Several studies with overlapping samples reported no glutamate differences between medication naïve first-episode participants and healthy controls in the left anterior cingulate and left thalamus (Theberge et al., 2002; Theberge et al., 2007; Aoyama et al., 2011). In a cohort of minimally treated first episode schizophrenia participants, Bustillo et al. (2010) observed no differences in glutamate in voxels placed in the bilateral anterior cingulate, left frontal white matter, and left thalamus (Bustillo et al., 2010).
1H-MRS studies measuring glutamine consistently show elevations in the thalamus and mixed results in the anterior cingulate. In the left anterior cingulate cortex and left dorsomedial thalamus, Theberge et al. (2002) found elevated glutamine concentrations in 21 antipsychotic medication naïve people with first episode schizophrenia compared to 21 controls (Theberge et al., 2002). In overlapping samples with the Theberge et al. (2002) study, Theberge et al. (2007) and Aoyama et al. (2011) both found elevated left thalamic glutamine concentrations in first episode antipsychotic medication naïve schizophrenia participants (Theberge et al., 2007; Aoyama et al., 2011). These same two studies also observed elevated Gln in the left anterior cingulate. Using the sequence implemented by Theberge et al., Bustillo et al. (2010) observed no statistically significant differences between glutamine in the anterior cingulate cortex, which may be due to differences in voxel size (bilateral versus left) and participant age (these participants were older) (Bustillo et al., 2010).
However, compared to healthy controls, Bustillo et al. (2010) found elevations of Gln/Glu ratio in bilateral anterior cingulate cortex in a minimally treated schizophrenia group with a lifetime exposure to antipsychotic medications of less than three weeks (Bustillo et al., 2010). This group reported no glutamate or glutamine differences in thalamic or frontal white matter voxels between patients and controls. Since glutamine is a precursor of glutamate, Gln/Glu ratio may be a good measure of glutamatergic neurotransmission turnover (Bustillo et al., 2010; Ongur et al., 2008; Shirayama et al., 2010). Another study reported elevated Glx/tCr in the left basal ganglia of 16 first episode schizophrenia participants, and no differences in the frontal lobe or parieto-occipital lobe compared to controls (Goto et al., 2012). This study did not provide detailed information regarding the medication status of participants with these results or size and placement of the frontal lobe voxel so it is unclear if the frontal lobe voxel incorporated the anterior cingulate cortex.
In contrast, a study comparing controls with first-episode schizophrenia participants where 18 out of 19 were on antipsychotic medications found no differences in Glx levels in the medial prefrontal cortex, which included the anterior cingulate (Natsubori et al., 2013). In a study comparing medicated, treated with at least one antipsychotic medication, remitted and non-remitted first episode schizophrenia participants, Egerton et al. (2012) found anterior cingulate cortex Glu/Cr elevations in a group of first episode schizophrenia participants still experiencing symptoms despite receiving at least one antipsychotic medication compared to a group of first episode schizophrenia participants who were not experiencing symptoms and on at least one antipsychotic medication (Egerton et al., 2012a). This study found no Glx/Cr differences between groups in this region as well as no Glu/Cr or Glx/Cr differences in the left thalamus. The authors also found that higher Glu/Cr in the anterior cingulate cortex was associated with greater negative symptom severity and lower global functioning. Since a healthy control group was not included in the Egerton et al. study, it is difficult to gauge whether the Glu/Cr levels in the medicated, symptomatic group were higher than normal controls or whether Glu/Cr levels were reduced in medicated, non-symptomatic group.
Overall, in antipsychotic naïve participants experiencing their first episode, glutamate is elevated only in the striatum, and glutamine is elevated in the left thalamus with mixed results in the anterior cingulate. Longitudinal studies at first episode with administration of antipsychotic medications suggest a reduction in glutamate and glutamine levels over years.
3.1.4 Chronic Schizophrenia
Differences between chronic participants with schizophrenia (an illness duration of one year or longer) and healthy controls vary depending upon the region under investigation. In the anterior cingulate, two studies observed lower glutamate levels in chronic, medicated schizophrenia participants compared to controls (Tayoshi et al., 2009; Theberge et al., 2003). In addition, Theberge et al. (2003) also showed decreased glutamine in this population as well. In the medial prefrontal cortex (which contained the anterior cingulate), Natsubori et al. showed that chronic, medicated participants with schizophrenia had lower Glx than healthy controls, ultra-high risk participants, and first-episode schizophrenia participants (Natsubori et al., 2013). In this study, half of participants in the ultra-high risk group and the majority of participants in the first episode schizophrenia group were on antipsychotic medications. Rowland et al. (2013) reported a main effect of diagnostic group, which showed chronic, medicated schizophrenia participants had lower Glx levels compared to healthy controls across regions, which were the anterior cingulate cortex and centrum semiovale (Rowland et al., 2013). Lutkenhoff et al. showed that glutamate was significantly lower in medicated schizophrenia participants compared to healthy controls overall, but not in specific regions (Lutkenhoff et al., 2010). In an exploratory analysis, this study reported significantly reduced glutamate in adults with schizophrenia and their co-twin compared to healthy controls in the mesial prefrontal cortex. In another cohort, Chang et al. (2007) utilized an older sample of chronic, medicated schizophrenia participants with a mean age of 66.3 ± 7.2 years and observed elevated Glx in right prefrontal white matter (Chang et al., 2007). In contrast, several studies reported no differences in Glx, Glx/Cr, or Glu/Cr ratios between chronic, medicated schizophrenia participants and healthy controls in several regions, specifically the left and right rostral and dorsal anterior cingulate (Wood et al., 2007), bilateral dorsal anterior cingulate (Reid et al., 2010; Kraguljac et al., 2012), left middle prefrontal region (Rowland et al., 2009), left anterior cingulate or left frontal lobe (Jessen et al., 2013), and bilateral anterior cingulate (Ongur et al., 2010). Another study by Terpstra et al. (2005) reported no differences in glutamate or glutamine from the anterior cingulate between medicated adults with schizophrenia and controls; however, this study did not provide information regarding illness stage, illness duration, or type of medications currently being taken (Terpstra et al., 2005). In a mixed illness stage sample with the majority taking antipsychotic medications, Bustillo et al. (2014) showed that glutamine and Gln/Glu ratio are elevated in the dorsal anterior cingulate in adults with schizophrenia compared to controls, and in particular, that glutamine levels relates to severity of psychotic symptoms (Bustillo et al., 2014). Contrary to a meta-analysis (Marsman et al., 2013), Bustillo et al. (2014) observed increased glutamine with aging in both groups; thus, more comprehensive aging studies are needed to further investigate how these metabolites change with age. Interestingly, the studies that observed no Glx differences had younger cohorts on average with shorter illness durations than the Chang et al. (2007) or Rowland et al. (2013) studies, suggesting that there may be glutamatergic alterations later in the illness.
In temporal lobe regions, 1H-MRS results may depend on medication status and cohort age. Two studies examining the left hippocampus reported no Glx/Cr differences between healthy controls and chronic, medicated schizophrenia participants (Hutcheson et al., 2012; Kraguljac et al., 2012). In another medicated sample, Lutkenhoff et al. found no significant glutamate differences between groups in the hippocampal region (Lutkenhoff et al., 2010). In left medial temporal lobe white matter, another study with an older medicated cohort (mean age 66.3 ± 7.2 years old) showed elevated Glx in the chronic schizophrenia participants compared to healthy controls (Chang et al., 2007).
Four studies utilized 1H-MRS to examine neurochemistry of the parietal and occipital lobes. Rowland et al. (2009) showed no Glx differences between medicated schizophrenia participants and healthy controls in the left inferior parietal region while Ongur et al. (2010) observed no Glu/Cr differences between chronic, medicated participants and controls in the parietal-occipital cortex (Rowland et al., 2009; Ongur et al., 2010). Chang et al. (2007) showed elevated Glx in occipital white matter of chronic, older medicated schizophrenia participants (Chang et al., 2007). A third study showed no Glx/Cr differences between schizophrenia participants and controls in the calcarine sulci (Yoon et al., 2010). The cohort in this study comprised stable chronic and recent onset schizophrenia participants who had differing medication statuses, from unmedicated for at least one month to minimal antipsychotic medication exposure to medicated using antipsychotics.
Thus far, few studies reported results from subcortical regions. In the substantia nigra and the basal ganglia, two studies of chronic, medicated schizophrenia participants and healthy controls reported no differences glutamate or Glx/Cr (Tayoshi et al., 2009; Reid et al., 2013). In a study of the left dorsomedial thalamus, Theberge et al. (2003) observed elevated glutamine in chronic, medicated participants and healthy controls, but no Glu differences (Theberge et al., 2003).
Overall, in gray matter or mixed frontal, temporal, parietal, and occipital regions, results are mixed showing either no differences or lower glutamatergic metabolites in chronic SZ compared to healthy controls. In exclusively white matter regions and in the left thalamus, glutamatergic metabolites are elevated in chronic SZ compared to healthy controls.
3.1.5 Unmedicated State
To date, there are eight studies examining glutamate, glutamine, or Glx in unmedicated participants with schizophrenia. Six studies with exclusively first-episode participants with schizophrenia showed mixed regional results. In overlapping samples, Theberge et al. (2002), Theberge et al. (2007), and Aoyama et al. (2011) showed that antipsychotic medication naïve first-episode participants had elevated glutamine in the left anterior cingulate and left thalamus and showed no differences in glutamate in either region (Theberge et al., 2002; Theberge et al., 2007; Aoyama et al., 2011). Two studies reported elevated glutamate in the right dorsal caudate nucleus in antipsychotic medication naïve participants (de la Fuente-Sandoval et al., 2011; de la Fuente-Sandoval et al., 2013b) and either no differences in Glx levels (de la Fuente-Sandoval et al., 2011) or elevated Glx levels (de la Fuente-Sandoval et al., 2013b) compared to controls. In the right cerebellum, these studies reported either elevated glutamate and Glx levels (de la Fuente-Sandoval et al., 2013b) or no differences (de la Fuente-Sandoval et al., 2011) between first episode participants and controls. Goto et al. (2012) found no differences in Glx/Cr in the frontal or parieto-occipital lobe and elevated Glx/Cr in the basal ganglia (Goto et al., 2012). Overall, these data suggest higher glutamatergic metabolites in unmedicated, first-episode schizophrenia but caution is warranted since there is discrepancy among the specific glutamatergic metabolites quantified (glutamate, glutamine, or Glx) and brain regions.
Two other unmedicated studies included participants with mixed previous exposure to antipsychotic medications. Each of these studies required participants to be unmedicated for a minimum of 14 days prior to the study. Kegeles et al. reported significantly elevated medial prefrontal cortex Glx in unmedicated participants with schizophrenia compared to healthy controls, and trend level elevations compared to medicated participants (Kegeles et al., 2012). In addition, this same study observed no Glx alterations between groups in the left dorsolateral prefrontal cortex in this same study. The unmedicated group was antipsychotic medication free for a mean of 21 ± 17 months, ranging from 4 days to 4 years. The schizophrenia group included both first-episode and chronic illness stage, with a mean duration of illness of 7 ± 7 years. Kraguljac et al. (2013) found elevated Glx/Cr in the left hippocampus of unmedicated schizophrenia participants compared to controls (Kraguljac et al., 2013). In this study, the schizophrenia cohort included eleven antipsychotic medication naïve participants and sixteen other participants who were medication free for a mean of 24.7 ± 45 months. In addition, this study included participants at varying illness stages, including first episode and chronic participants with a mean illness duration of 10.5 ± 8.7 years. Overall, statistical analyses in the Kraguljac et al. (2013) study showed that Glx/Cr from first episode, antipsychotic medication naïve, and chronic participants were not significantly different.
The majority of unmedicated studies reported increased glutamatergic metabolite levels. However, when considering the exceptions that found no differences in glutamatergic levels (Goto et al., 2012; de la Fuente-Sandoval et al., 2011) and the studies of antipsychotic medicated patients that reported higher glutamatergic levels (Chang et al., 2007; Aoyama et al., 2011; Egerton et al., 2012a), it is premature to conclude that glutamatergic levels are only higher in the unmedicated state. Inclusion of mixed illness stage studies appears to complicate interpretation since there is heterogeneity early in the illness, there are no studies of exclusively unmedicated, chronic participants, and the effect of illness duration and age on glutamate and glutamine levels later in the illness is unknown. More studies are needed in both unmedicated first-episode and chronic participants with lengthy illness durations, especially since recent data suggest that glutamatergic levels differ later in the illness. In particular, a meta-analysis suggested that medial frontal glutamate decreases across the illness (Marsman et al., 2013), and other data showed that glutamate levels decrease with age in healthy controls (Kaiser et al., 2005; Chang et al., 2009). In contrast, Chang et al. (2007) reported elevated Glx in white matter regions of medicated participants with mean illness durations of 43.1 ± 5.4 years (Chang et al., 2007), suggesting that there may be altered glutamatergic patterns in white versus gray matter in older adults with schizophrenia. Providing further support to an altered glutamatergic state later in the illness is that less severe psychopathology is observed and lower dosages of medication are required in older adults with schizophrenia (Jeste et al., 2003; Jeste et al., 2011). Thus, more off-medication research is needed to further our understanding glutamatergic levels throughout the illness.
3.1.6 Antipsychotic Treatment Effects
Antipsychotic medications are a first-line treatment for adults with schizophrenia, and several studies examined the effects of medication treatment both in the short term (4 weeks) and long term (80 months) on glutamate levels in several regions. After 6 months of atypical antipsychotic treatment in first episode schizophrenia participants, Goto et al. (2012) observed a decrease in frontal lobe Glx/Cr from baseline to the study end point (Goto et al., 2012). However, this study did not provide details regarding medication status of participants at baseline or voxel placement within the frontal lobe. Similarly, de la Fuente-Sandoval et al. (2013b) reported a decrease in glutamate levels in the right dorsal caudate nucleus after 4 weeks of treatment with risperidone in antipsychotic medication naïve first episode psychosis participants (de la Fuente-Sandoval et al., 2013b). At baseline, first-episode participants showed elevated glutamate levels compared to controls, and after treatment, this study found no differences in glutamate levels between groups. In the right cerebellar cortex, de la Fuente-Sandoval et al. (2013b) observed elevated glutamate levels in first episode psychosis participants compared to controls, and this effect remained after treatment with risperidone unlike the associative striatum (de la Fuente-Sandoval et al., 2013b). In another study that performed assessments at baseline on minimally treated first episode schizophrenia participants and at follow-up (mean of 7.3 months later) with participants on antipsychotic therapy, Bustillo et al. (2010) reported no differences in glutamate, glutamine, or Gln/Glu ratio between groups in bilateral anterior cingulate cortex, left frontal white matter, or left thalamus (Bustillo et al., 2010). Compared to a treatment as usual in an ultra-high risk group, Glx levels in participants receiving low dose lithium remained the same in the left or right medial temporal lobe (predominantly containing the hippocampus) over a three month interval (Berger et al., 2012).
In two follow-up studies (Theberge et al., 2007; Aoyama et al., 2011), antipsychotic medication naïve participants with first episode schizophrenia were treated and scanned at 10, 30, and 80 months after initial visit. At each time interval, the majority of schizophrenia participants were taking antipsychotic medications. In participants with schizophrenia, glutamine levels remained the same after 10 months of treatment in the left thalamus; however, glutamine levels at 30 months decreased significantly compared to glutamine levels at baseline and 10 months (Theberge et al., 2007). This study observed no differences in glutamine levels in the left anterior cingulate over these time intervals or for glutamate levels over time in either the left anterior cingulate or the left thalamus. After 80 months of treatment, Aoyama et al. (2011) reported that thalamic glutamine levels decreased compared to baseline at trend level, and that thalamic glutamate+glutamine levels decreased significantly compared to baseline (Aoyama et al., 2011). Thalamic glutamate+glutamine correlated negatively with social functioning despite treatment with antipsychotic medications. Comparisons at 80 months between groups showed no differences in glutamate or glutamine in either region.
Overall, these studies suggest that glutamate and glutamine levels decrease or remain stable with treatment depending upon the region examined and the variety of medications utilized, and that over a longer period of time, glutamate and glutamine levels decrease compared to baseline. The study by de la Fuente-Sandoval et al. (2013b) was the only study to examine the treatment effect of one antipsychotic medication whereas the other treatment studies utilized several different medications within their cohort. The de la Fuente-Sandoval et al. (2013b) study also examined spectroscopic differences following treatment after a short period of time compared to all other studies that ranged from 6 to 80 months. Longitudinal studies that more densely sample the time course of glutamatergic levels as a function of treatment would be informative.
3.1.7 Summary of Glutamatergic Alterations in Schizophrenia
Glutamate and glutamine levels depend upon illness stage, brain region, and medication status, and results of the studies previously discussed are summarized in Table 1. In the prodromal stage, thalamic glutamate levels are lower in at risk participants while in the striatum, glutamate is elevated in this same population. Frontal and temporal regions do not appear greatly affected in the prodromal stage. In antipsychotic naïve participants experiencing their first episode, glutamate is elevated in the striatum while increased glutamine and no glutamate differences are observed in the anterior cingulate and thalamus. At first episode, longitudinal studies with administration of antipsychotic medications suggest a reduction in glutamate and glutamine levels over years. Several studies with chronic participants with a longer duration of illness suggest that Glx is decreased in either gray matter or mixed regions. To date, there is one study that examined an older cohort of medicated adults with schizophrenia, and in this study, elevated Glx was observed in three white matter regions (Chang et al., 2007). More studies focused on old adults with schizophrenia who have longer durations of illness are needed to understand whether the glutamatergic system is altered late in life. Since studies in older adults with schizophrenia report less general psychopathology and the need for lower dosages of medication (Jeste et al., 2003), new information regarding the glutamatergic system would be useful and may directly inform treatment interventions.
Table 1.
Summary of Glutamatergic Findings in Ultra High Risk (UHR) or At Risk Mental State (ARMS), First Episode (FEP), and Chronic Participants with Schizophrenia at 3 T or Higher
| Stage of Illness | Region | Authors | Scanner & Method | Participants (SZ/HC) | Antipsychotic Medication Use | SZ Mean Age (yrs±std or yrs (range)) | Results (vs. HC) |
|---|---|---|---|---|---|---|---|
| UHR | right medial frontal region | Tibbo et al. (2004) | 3T STEAM (TE=20ms) | 20/22 | None | 16.4±1.99 | ↑ Glx/Cr in high risk adolescents |
| UHR | medial prefrontal cortex | Natsubori et al. (2013) | 3T STEAM (TE=15ms) | 24/26 | Yes | 21.7 (16–29) | no Glx diff. |
| ARMS | anterior cingulate | Stone et al. (2009) | 3T PRESS (TE=30ms) | 27/27 | Yes,g | 25±5 | no Glu diff. and ↑ Gln in ARMS |
| ARMS | anterior cingulate | Fusar-Poli et al. (2011) | 3T PRESS (TE=30ms) | 24/17 | None (naïve) | 26.6±5 | no Glu diff. |
| ARMS | anterior cingulate | Valli et al. (2011) | 3T PRESS (TE=30ms) | 22/16 | None (naïve) | 25.72±4.97 | no Glu diff. |
| UHR | left and right medial temporal lobe (mostly anterior hippocampus) | Wood et al. (2010) | 3T PRESS (TE=30ms) | 66/29 | None | 19.18±3.17 | no Glx diff. |
| ARMS | left hippocampus | Stone et al. (2009) | 3T PRESS (TE=30ms) | 27/27 | Yes,g | 25±5 | no Glu or Gln diff. |
| ARMS | left hippocampus | Fusar-Poli et al. (2011) | 3T PRESS (TE=30ms) | 24/17 | None (naïve) | 26.6±5 | ↓ Glu in ARMS (trend level) |
| ARMS | left hippocampus | Valli et al. (2011) | 3T PRESS (TE=30ms) | 22/16 | None (naïve) | 25.72±4.97 | no Glu diff. |
| UHR | left and right temporal lobe (largely anterior hippocampus) | Berger et al. (2012) | 3T PRESS (TE=30ms) | 11/10 | Baseline: None (naïve); Treatment: Low-Dose Lithium for 3 months | 20.74±3.32 | no Glx diff. between UHR on lithium and treatment as usual and UHR on treatment as usual only |
| UHR | right dorsal caudate nucleus | de la Fuente-Sandoval et al. (2011) | 3T PRESS (TE=35ms) | 18/40,a | None (naïve) | 19.56±3.46 | ↑ Glu and no Glx diff. in UHR; no diff. between UHR and FEP |
| ARMS | left thalamus | Stone et al. (2009) | 3T PRESS (TE=30ms) | 27/27 | Yes, g | 25±5 | ↓ Glu and no Gln diff in ARMS |
| ARMS | left thalamus | Fusar-Poli et al. (2011) | 3T PRESS (TE=30ms) | 24/17 | None (naïve) | 26.6±5 | ↓ Glu in ARMS (trend level) |
| ARMS | left thalamus | Valli et al. (2011) | 3T PRESS (TE=30ms) | 22/16 | None (naïve) | 25.72±4.97 | ↓ Glu in ARMS (trend level) |
| UHR | right cerebellum | de la Fuente-Sandoval et al. (2011) | 3T PRESS (TE=35ms) | 18/40,a | None (naïve) | 19.56±3.46 | no Glu and Glx diff.; no diff. between UHR and FEP |
| FEP | left anterior cingulate | Theberge et al. (2002) | 4T STEAM (TE=20ms) | 21/21 | None (naïve) | 26±7 | no Glu diff and ↑ Gln in FEP |
| FEP | left anterior cingulate | Theberge et al. (2007) | 4T STEAM (TE=20ms) | 16/16 | Baseline: none (naïve); 10 months: Yes; 30 months: Yes | baseline: 25±8, 10m: 25±8, 30m: 27±8 | no Glu diff at baseline, 10m, and 30m; ↑ Gln at baseline |
| FEP | left anterior cingulate | Aoyama et al. (2011) | 4T STEAM TE=20ms | 17/17 | Baseline: none (naïve); 10 months: Yes; 80 months: Yes | 32±9 | ↑ Gln at baseline and 80m in SZ; no Glu diff. |
| FEP | frontal lobe | Goto et al. (2012) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±11 | Baseline:no Glx/Cr diff.; 6m: ↓ Glx/Cr compared to baseline (trend level) |
| FEP | medial prefrontal cortex | Natsubori et al. (2013) | 3T STEAM (TE=15ms) | 19/19 | Yes | 25.4 (17–37) | no Glx diff. |
| FEP | bilateral anterior cingulate and left frontal white matter | Bustillo et al. (2010) | 4T STEAM (TE=20ms) | 14/10 | Baseline: minimal exposure (< 3weeks); Treatment: AP and f/u at 1, 6, and 12 months | 27.2±8.9 | no Glu and Gln diff. in both regions at baseline and following treatment; baseline: ↑Gln/Glu ratio in anterior cingulate of FEP |
| FEP | anterior cingulate cortex | Egerton et al. (2012) | 3T PRESS (TE=30ms) | 32/0,b | Yes | remission: 30±6; non-remission: 25±7 | ↑Glu/Cr in non-remission (symptomatic) versus remission; no Glx/Cr diff.; higher Glu/Cr was associated with greater negative symptom severity and lower global functioning |
| FEP | left thalamus | Egerton et al. (2012) | 3T PRESS (TE=30ms) | 32/0,b | Yes | remission: 30±6; non-remission: 25±7 | no Glu/Cr and Glx/Cr diff. |
| FEP | left medial thalamus | Theberge et al. (2002) | 4T STEAM (TE=20ms) | 21/21 | None (naïve) | 26±7 | no Glu diff and ↑ Gln in FEP |
| FEP | left thalamus | Theberge et al. (2007) | 4T STEAM (TE=20ms) | 16/16 | Baseline: none (naïve); 10 months: Yes; 30 months: Yes | baseline: 25±8, 10m: 25±8, 30m: 27±8 | No Glu diff. at baseline, 10m, and 30m; ↑ Gln at baseline; In SZ, Gln at 30m was reduced compared to baseline and 10m |
| FEP | left thalamus | Aoyama et al. (2011) | 4T STEAM TE=20ms | 17/17 | Baseline: none (naïve); 10 months: Yes; 80 months: Yes | 32±9 | ↑ Gln at baseline in SZ; no Glu diff.; ↓ Glx over 80m |
| FEP | left thalamus | Bustillo et al. (2010) | 4T STEAM (te=20ms) | 14/10 | Baseline: minimal exposure (< 3 weeks); Treatment: AP and f/u at 1, 6, and 12 months | 27.2±8.9 | no Glu and Gln diff. at baseline or following treatment |
| FEP | basal ganglia | Goto et al. (2012) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±11 | Baseline: ↑ Glx/Cr in SZ; 6m: no change Glx/Cr |
| FEP | right dorsal caudate nucleus | de la Fuente-Sandoval et al. (2011) | 3T PRESS (TE=35ms) | 18/40,a | None (naïve) | 23.44±4.93 | ↑ Glu and no Glx diff. in FEP; no diff. between UHR and FEP |
| FEP | right dorsal caudate nucleus | de la Fuente-Sandoval et al (2013a) | 3T PRESS (TE=35ms) | 19/26 | None (naïve) | not available | ↑ Glu in UHR who transitioned to psychotic disorder compared to UHR who did not transition and controls |
| FEP | right dorsal caudate nucleus | de la Fuente-Sandoval et al (2013b) | 3T PRESS (TE=35ms) | 28/18 | Baseline: none (naïve); Treatment: risperidone for 4 weeks | 26.58±8.49 | Baseline: ↑ Glu and ↑ Glx in FEP; Post-treatment (4-weeks): no Glu and Glx diff.; ↓ in Glu after 4-weeks was significant |
| FEP | parieto-occipital lobe | Goto et al. (2012) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±11 | Baseline: no Glx/Cr diff.; 6m: no change in Glx/Cr |
| FEP | right cerebellum | de la Fuente-Sandoval et al. (2011) | 3T PRESS (TE=35ms) | 18/40,a | None (naïve) | 23.44±4.93 | no Glu and Glx diff.; no diff. between UHR and FEP |
| FEP | right cerebellum | de la Fuente-Sandoval et al (2013b) | 3T PRESS (TE=35ms) | 28/18 | Baseline: none (naïve); Treatment: risperidone for 4 weeks | 26.58±8.49 | Baseline: ↑ Glu and ↑ Glx in FEP; Post-treatment (4-weeks): no Glu diff. and ↑ Glx remained |
| Chronic | left and right rostral and dorsal anterior cingulate | Wood et al. (2007) | 3T PRESS (TE=30ms) | 15/14 | Yes | 31.5±7.5 | no Glx diff. in either of the 4 voxels |
| Chronic | anterior cingulate | Tayoshi et al (2009) | 3T STEAM (TE=18ms) | 30/25 | Yes | 33.8±9.5 | ↓ Glu and no Gln diff in SZ, driven by diff. in males |
| Chronic | left anterior cingulate | Theberge et al. (2003) | 4T STEAM (TE=20ms) | 21/21 | Yes | 37±11 | ↓ Glu and ↓ Gln in SZ |
| Chronic | anterior cingulate cortex | Kraguljac et al. (2012) | 3T PRESS (TE=80ms) | 48/46 | Yes | 38.02±12.4 | no Glx/Cr diff. |
| Chronic | bilateral dorsal anterior cingulate cortex | Reid et al. (2010) | 3T PRESS (TE=80ms) | 28/25 | Yes | 40.4±13.1 | no Glx/Cr diff.; negative correlation between Glx/Cr and negative symptoms from BPRS |
| Chronic | left middle frontal | Rowland et al. (2009) | 3T PRESS (TE=35ms) | 20/10,c | Yes | Deficit: 43±6, Non-deficit: 40±6 | No Glx diff. |
| Chronic | mesial prefrontal gray matter and left prefrontal white matter | Lutkenhoff et al (2010) | 3T PRESS TE=30ms | 14/26,d | Yes | 48.8±11.5 | across frontal and temporal regions, ↓ Glu in SZ; exploratory analysis:↓Glu in mesial prefrontal cortex |
| Chronic | left and right prefrontal white matter | Chang et al. (2007) | 4T PRESS (TE=30ms) | 23/22 | Yes | 66.3±7.2 | ↑ Glx in right prefrontal white matter in SZ; overall, ↑ Glx across frontal, temporal, and occipital |
| Chronic | anterior cingulate and centrum semiovale | Rowland et al. (2013) | 3T PRESS (TE=35ms) | 21/20,e | Yes | YSZ: 30.2±6.6; OSZ: 51.1±4.0 | no Glx diff. in either region; overall effect of ↓ Glx in SZ |
| Chronic | anterior cingulate | Ongur et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 21/19 | Yes | 39.0±10.8 | no Glu/Cr diff. |
| Chronic | left anterior cingulate cortex and left frontal lobe | Jessen et al. (2013) | 3T PRESS (TE=30ms) | 20/20 | Yes | 34.5±10.2 | no Gln/tCr and Glx/tCr diff. in both regions |
| Chronic | medial prefrontal cortex | Natsubori et al. (2013) | 3T STEAM (TE=15ms) | 25/28 | Yes | 32.7 (17–50) | ↓ Glx in SZ; ↓ Glx in chronic compared to UHR and FEP groups |
| Mixed | medial prefrontal cortex and left dorsolateral prefrontal cortex | Kegeles et al. (2012) | 3T J-edited (TE=68ms) | 32/22,f | Yes & No | unmedicated: 32±11, medicated: 32±10 | ↑ Glx in unmedicated SZ compared to HC in medial prefrontal cortex; ↑Glx in unmedicated versus medicated participants (trend level) in medial prefrontal cortex; no Glx diff between medicated SZ and HC in medial prefrontal cortex; no Glx diff. in dorsolateral prefrontal cortex between groups |
| Mixed | dorsal anterior cingulate | Bustillo et al. (2014) | 3T PRESS (TE=40ms) | 84/81 | Yes | 36.7±13.9 (16–64) | ↑ Gln in SZ and with aging (both groups); no Glu diff.; ↑ Gln/Glu |
| Chronic | left hippocampus | Hutcheson et al. 2012 | 3T PRESS (TE=80ms) | 39/41 | Yes | 36.7±12.2 | no Glx/Cr diff. |
| Chronic | left hippocampus | Kragljac et al. (2012) | 3T PRESS (TE=80ms) | 48/46 | Yes | 38.02±12.4 | no Glx/Cr diff. |
| Chronic | left hippocampus | Lutkenhoff et al (2010) | 3T PRESS TE=30ms | 14/26,d | Yes | 48.8±11.5 | No diff. in Glu; across frontal and temporal regions, ↓ Glu in SZ |
| Chronic | left and right medial temporal lobe white matter | Chang et al. (2007) | 4T PRESS (TE=30ms) | 23/22 | Yes | 66.3±7.2 | ↑ Glx in left medial temporal white matter in SZ; overall, ↑ Glx across frontal, temporal, and occipital |
| Mixed | left hippocampus | Kraguljac et al. (2013) | 3T PRESS (TE=80ms) | 27/27 | None (unmedicated at least 2 weeks) | 32.63±9.28 | ↑ Glx/Cr in SZ |
| Chronic | left thalamus | Theberge et al. (2003) | 4T STEAM (TE=20ms) | 21/21 | Yes | 37±11 | no Glu and ↑Gln in SZ |
| Chronic | left basal ganglia | Tayoshi et al (2009) | 3T STEAM (TE=18ms) | 30/25 | Yes | 33.8±9.5 | no Glu diff.; no Gln diff. (including both genders); Gln was lower in SZ females versus SZ males |
| Chronic | substantia nigra | Reid et al. (2013) | 3T PRESS (TE=80ms) | 35/22 | Yes | 37.9±12 | no Glx/Cr diff. |
| Chronic | left inferior parietal | Rowland et al. (2009) | 3T PRESS (TE=35ms) | 20/10,c | Yes | Deficit: 43±6, Non-deficit: 40±6 | no Glx diff. |
| Chronic | left and right occipital white matter | Chang et al. (2007) | 4T PRESS (TE=30ms) | 23/22 | Yes | 66.3±7.2 | ↑ Glx in SZ; overall, ↑ Glx across frontal, temporal, and occipital |
| Mixed | bilateral calcarine sulci | Yoon et al (2010) | 3T MEGA-PRESS (TE=68ms) | 13/13 | Yes | 27.5±8.8 | no Glx/Cr diff. |
| Chronic | parietal-occipital cortex | Ongur et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 21/19 | Yes | 39.0±10.8 | no Glu/Cr diff. |
| Unknown | anterior cingulate | Terpstra et al. (2005) | 4T STEAM (TE=5ms) | 13/9 | Yes | 26±5 | no Glu and Gln diff. |
| Relatives | |||||||
| Sibling | left and right frontal | Purdon et al. (2008) | 3T STEAM (TE=20ms) | 19/16 | None | 46.32±6.09 | no Glu/Cr and Glx/Cr diff. |
| Co-twin | mesial prefrontal cortex, left prefrontal white matter, left hippocampus | Lutkenhoff et al (2010) | 3T PRESS (TE=30ms) | 15/26,d | None | 49.5±10.0 | ↓ Glu overall (mesial prefrontal cortex, left prefrontal white matter, and left hippomcampus) |
| Child of Parent with SZ | right medial frontal region | Tibbo et al. (2004) | 3T STEAM (TE=20ms) | 20/22 | None | 16.4±1.99 | ↑ Glx/Cr in high risk adolescents |
↑ - increased, ↓ - decreased, Cr – creatine, diff – difference, Glu – glutamate, Gln – glutamine, Glx – glutamate+glutamine, HC – healthy controls, m – months, ms - millisecond
OHC – older healthy controls, OSZ, - older schizophrenia, SZ – schizophrenia, TE – echo time, YHC – younger healthy controls, YSZ – younger schizophrenia
one control group for comparison to UHR and FEP
16 patients in remission, 16 patients in non-remission
SZ group of 20: 10 deficit and 10 non-deficit
14 SZ, 15 co-twins, and same 26 HC (13 twin pairs)
YSZ: 11; YHC: 10; OSZ: 10; OHC: 10
16 unmedicated and 16 medicated SZ
6 on current AP or Antidepressant Medication (do not separate the two) types of meds)
3.2 Gamma-aminobutyric acid (GABA)
GABA is the primary inhibitory neurotransmitter in the brain and occurs at approximately 1 mmol/kg concentration. Altered GABAergic function is implicated in the pathophysiology of schizophrenia as evident through post-mortem studies (Hashimoto et al., 2008; Deng and Huang, 2006; Beneyto and Lewis, 2011) and challenge studies (Ahn et al., 2011; Taylor et al., 2013). Within the 1H spectrum, GABA peaks arise at 1.89, 2.28, and 3.01 ppm, and GABA concentrations typically range from 1.3–1.9 mM (Govindaraju et al., 2000). GABA levels are often reported relative to either water (GABA/W) or creatine (GABA/Cr). In addition, GABA is commonly measured with specifically tailored editing sequences such as MEGA-PRESS, which has been previously discussed. However, recent studies utilizing non-editing sequences with very short TEs demonstrate the ability to detect GABA as well (Near et al., 2013; Wijtenburg et al., 2013).
To date, seven studies utilized spectral editing to study GABA concentrations in schizophrenia (Table 2). In frontal regions, GABA levels differ based on illness stage and medication status. Goto et al. (2010) studied first-episode participants prior to and after treatment with antipsychotic medications and found no differences in GABA/Cr between controls and participants with SZ at baseline and after 6 months of treatment (Goto et al., 2010). In chronic, medicated participants with schizophrenia, Tayoshi et al. (2010) reported no differences in GABA levels between groups in the anterior cingulate, but this same study found a significant negative correlation between GABA levels and antipsychotic medication dose (Tayoshi et al., 2010). In a mixed illness stage sample, Kegeles et al. reported elevated GABA/W in unmedicated adults with schizophrenia compared to healthy controls in the medial prefrontal cortex, and no differences between medicated adults with schizophrenia and healthy controls in this same region (Kegeles et al., 2012). In addition, Kegeles et al. showed no differences in GABA/W levels between any of the three groups in the left dorsolateral prefrontal cortex. Rowland et al. (2013) reported no GABA/W group differences in the anterior cingulate or centrum semiovale, with no participants taking benzodiazepines or mood stabilizers. When broken down into age and diagnostic groups, this study reported lower GABA/W levels in older adults with schizophrenia (mean age of 51.1 ± 4.0 years old) compared to older healthy controls (Rowland et al., 2013). When comparing age groups by combining diagnoses, Rowland et al. (2013) observed higher GABA/W levels in younger participants compared to older participants across regions. In conjunction with a study by Gao et al. (2013), these data suggest that GABA levels may decrease due to age and illness (Rowland et al., 2013; Gao et al., 2013). In contrast, Ongur et al. (2010) showed elevated GABA/Cr ratios in the anterior cingulate cortex in chronic, medicated schizophrenia participants compared to healthy controls (Ongur et al., 2010). Ten adults with schizophrenia took anticonvulsants. When this sample was removed from analyses, Ongur et al. (2010) reported no GABA/Cr differenced between groups. Thus, concomitant usage of anticonvulsants and liberal criteria for GABA quantification (CRLB cutoff of 50%) may have influenced the results of this study.
Table 2.
Summary of GABA Findings in First Episode (FEP) and Chronic Participants with Schizophrenia at 3T or higher
| Stage of Illness | Region | Authors | Scanner & Method | Participants (SZ/HC) | Antipsychotic Medication Use | SZ Mean Age (yrs±std or yrs (range)) | Results (vs. HC) |
|---|---|---|---|---|---|---|---|
| FEP | frontal lobe | Goto et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±10.0 | Baseline: No GABA/Cr diff.; 6 months: No GABA/Cr diff. & no change |
| FEP | left basal ganglia | Goto et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±10.0 | Baseline: ↓ GABA/Cr in SZ; 6 months: ↓ GABA/Cr in SZ remained & no diff. between baseline and 6 months |
| FEP | parieto-occipital lobe | Goto et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 16/18 | Baseline: No Information; Treatment: AP for 6 months | 30±10.0 | Baseline: No GABA/Cr diff.; 6 months: No GABA/Cr diff. & no change |
| FEP | bilateral calcarine sulci | Keleman et al. (2013) | 3T MEGA-PRESS (TE=68ms) | 28/20 | Baseline: None (naïve); 8 weeks: Yes | 24.9±8.3 | Baseline: ↓GABA/Cr in SZ; 8 weeks: ↓ GABA/Cr in SZ |
| Chronic | anterior cingulate | Tayoshi et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 38/29 | Yes | 34.0±10.0 | No GABA diff; negative correlation between GABA level and antipsychotic dose |
| Chronic | anterior cingulate | Ongur et al. (2010) | 4T MEGA-PRESS (TE=68ms) | 21/19 | Yes | 39.0±10.08 | ↑ GABA/Cr in SZ |
| Mixed | medial prefrontal cortex and left dorsolateral prefrontal cortex | Kegeles et al. (2012) | 3T MEGA-PRESS (TE=68ms) | 32/22,a | Yes & No | unmedicated: 32±11, medicated: 32±10 | ↑ GABA/W in unmedicated versus HC in medial prefrontal cortex; no GABA/W diff in medicated versus HC in medial prefrontal cortex; no GABA/W diff in left dorsolateral prefrontal cortex |
| Chronic | anterior cingulate and centrum semiovale | Rowland et al. (2013) | 3T MEGA-PRESS (TE=68ms) | 21/20,b | Yes | YSZ: 30.2±6.6; OSZ: 51.1±4.0 | ↓ GABA/W in OSZ compared to OHC in anterior cingulate; No GABA/W diff in centrum semiovale; Younger group (SZ+HC) had higher GABA than older group (SZ+HC) across region |
| Chronic | left basal ganglia | Tayoshi et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 38/29 | Yes | 34.0±10.0 | No GABA diff; ↑ GABA in SZ taking typical APs compared to SZ taking atypical APs; positive correlation between GABA level and anticholinergic dose |
| Chronic | parieto-occipital cortex | Ongur et al. (2010) | 4T MEGA-PRESS (TE=68ms) | 21/19 | Yes | 39.0±10.08 | ↑ GABA/Cr in SZ |
| Mixed | bilateral calcarine sulci | Yoon et al. (2010) | 3T MEGA-PRESS (TE=68ms) | 13/13 | Yes | 27.5±8.8 | ↓ GABA/Cr in SZ |
↑ - increased, ↓ - decreased, AP – antipsychotic medications, Cr – creatine, diff –differences, GABA - γ-aminobutyric acid, HC – healthy controls, ms – millisecond
OHC – older healthy controls, OSZ - older schizophrenia, SZ – schizophrenia, TE – echo time, W – water, YHC – younger healthy controls, YSZ – younger schizophrenia
16 unmedicated SZ & 16 medicated SZ
YSZ: 11; YHC: 10; OSZ: 10; OHC: 10
Two studies examined GABA levels in the left basal ganglia of schizophrenia participants and controls. Goto et al. (2010) reported reduced GABA/Cr concentrations in the left basal ganglia in medicated subjects with early-stage schizophrenia compared to healthy control subjects at baseline and following six-months of antipsychotic treatment (Goto et al., 2010). Tayoshi et al. (2010) found no differences in GABA levels in the left basal ganglia between chronic, medicated participants and healthy controls (Tayoshi et al., 2010). When broken down according to typical versus atypical antipsychotic medication being taken, increased GABA was observed in participants taking typical antipsychotic medications versus atypical antipsychotic medications, and both groups included equal numbers of participants taking benzodiazepines.
In more posterior regions of the brain, results for GABA levels are mixed, most likely due to concomitant medication and mixture of illness stage. In early stage schizophrenia, Goto et al. (2010) reported no differences in GABA/Cr between medicated adults with schizophrenia and healthy controls in the parieto-occipital lobe (Goto et al., 2012). Keleman et al. (2013) found decreased GABA/Cr levels in the bilateral calcarine sulci of antipsychotic medication naïve, first-episode participants compared to controls, and this effect remained after 8 weeks of treatment with antipsychotic medications (Kelemen et al., 2013). Ongur et al. (2010) reported elevated GABA/Cr in the parieto-occipital cortex in chronic, medicated schizophrenia participants and healthy controls (Ongur et al., 2010). However, when participants who were taking concomitant anticonvulsants were removed from analyses, the effect was no longer significant. In another study by Yoon et al., GABA/Cr levels were decreased in the bilateral calcarine sulci (Yoon et al., 2010). The schizophrenia group included recent onset and chronic individuals, and the medication status of this group ranged from not taking antipsychotic medications for at least one month to medicated.
With advanced MR techniques, it is possible to measure both GABA and glutamate or glutamine within a study session and ratios of these may reflect excitatory and inhibitory neurotransmission balance. This may be useful since it is believed that an imbalance in excitatory and inhibitory neurotransmission occurs in schizophrenia (Lewis et al., 2012). To date, there have been no studies examining the ratios of GABA to glutamate or glutamine. Another approach to provide insight into excitatory-inhibitory balance is to investigate the relationship between GABA and glutamate or glutamine (i.e correlation analysis). However, caution is warranted if the metabolites are acquired with the same sequence and hence the same reference (water or Cr) is utilized which inflates r values and must be controlled (Maddock, 2014)
There have been four few studies that measured both GABA and glutamate metabolites within the same session. Ongur et al (2010), Yoon et al (2010), and Kegeles et al (2012) used spectral editing to quantify GABA and Glx relative to creatine or water whereas Rowland et al (2013) used spectral editing for GABA and single voxel spectroscopy for Glx, both referenced to water (Ongur et al., 2010; Yoon et al., 2010; Kegeles et al., 2012; Rowland et al., 2013). Based on approximate mean GABA/Glx or GABA/Glu ratios calculated from these studies, there appears to be an imbalance in the ratio of GABA to Glx or Glu in SZ participants versus healthy controls. In three of these studies, ratios of GABA/Glx or GABA/Glu are elevated in frontal and posterior brain regions in both medicated and unmedicated SZ subjects, consisting of either all chronic or mixed illness duration cohorts (Ongur et al., 2010; Kegeles et al., 2012; Rowland et al., 2013) whereas in one study, GABA/Glx is reduced in a mixed illness duration, medicated SZ cohort in the bilateral calcarine sulci (Yoon et al., 2010). It is important to note that it is not known whether these differences are significant between groups. In addition to ratios of GABA to Glx, Ongur et al (2010) and Kegeles et al (2012) studies performed correlation analyses between GABA and Glx or Glu measures and did not observe differences between healthy controls and participants with SZ. Thus, in terms of ratios of GABA to Glu or Glx, the few studies conducted thus far extend the potential for an inhibitory/exhibitory imbalance in SZ; however, more extensive research is needed to evaluate regional, illness stage, and medication differences.
In summary, the majority of studies focusing on GABA observed lower GABA levels than healthy controls regardless of region and illness stage. Interestingly, antipsychotic treatment had no effect on GABA levels in two studies of first-episode schizophrenia (Goto et al., 2010; Kelemen et al., 2013). Two other studies observed elevated GABA levels in frontal and posterior regions (Ongur et al., 2010; Kegeles et al., 2012), but when the effects of concomitant anticonvulsants were considered in the Ongur et al. (2010) study, the increased GABA/Cr levels were no longer significant. Since benzodiazepines and anticonvulsants are known to affect GABA(A) receptors and increase the observable concentration levels of GABA in the brain (Goddard et al., 2004), it is important to take the benzodiazepine and anticonvulsant medication usage into consideration when evaluating results for GABA. Thus, more studies with full concomitant medication disclosure are needed to fully determine GABA alterations throughout the illness.
3.3 Glutathione (GSH)
Glutathione (GSH) is a major intracellular antioxidant that protects the brain against oxidative stress and may play a role in the pathophysiology of schizophrenia (O’Donnell, 2011; Wu et al., 2013). Flatow et al. conducted a meta-analysis of serum and plasma oxidative stress markers measured in participants with schizophrenia and concluded that oxidative stress may be a potential biomarker of schizophrenia (Flatow et al., 2013). Further evidence stems from studies in adults with schizophrenia showing reduced GSH concentrations in CSF (Do et al., 2000), reduced GSH blood levels (Raffa et al., 2009), and reduced GSH concentrations in the post-mortem caudate nucleus (Yao et al., 2006) and pre-frontal cortex (Gawryluk et al., 2011). Flatow et al. also suggested oxidative stress abnormalities may be independent of antipsychotic medication status supported by the abnormalities observed in first episode participants (Flatow et al., 2013). Ballesteros et al. ruled out exogenous factors such as diet, smoking, and medication status from causing the lower GSH concentrations observed in adults with schizophrenia (Ballesteros et al., 2013).
GSH levels measured by 1H-MRS originate from resonances at 2.15, 2.55, 3.77 (glutamate-moiety), 2.93, 2.98, 4.56 (cysteine-moiety), and 3.77 (glycine-moiety) ppm. The concentration of GSH in the brain is approximately 2.0mM. To date, in vivo measurements of GSH concentrations are conducted either utilizing a specifically tailored pulse sequence or relying on improved MRI hardware by using standard localization sequences such as STEAM or PRESS. Using MEGA-PRESS, only the 2.95 ppm multiplet of GSH is observable and quantifiable whereas with PRESS or STEAM localization, the entire GSH spectrum is quantified. One previous study utilized a PRESS localization sequence to detect GSH in adults with schizophrenia (Wood et al., 2009), and a recent study by Wijtenburg et al. (2013) shows that GSH can be detected reproducibly in healthy controls without the use of spectral editing technique such as MEGA-PRESS (Wijtenburg et al., 2013).
Three 1H-MRS studies measured GSH in adults with schizophrenia (Table 3). In the frontal lobe, two studies measured GSH levels using MEGA-PRESS from medicated participants with schizophrenia and controls. Matsuzawa et al. observed no GSH differences between chronic participants with schizophrenia and controls in the posterior medial frontal cortex (Matsuzawa et al., 2008). Matsuzawa et al. also reported a negative correlation between medial prefrontal GSH concentrations and severity of negative symptoms, such that lower levels of GSH were associated with greater negative symptoms. Terpstra et al. (2005) reported no GSH differences in the anterior cingulate; however, this study noted that participants were medicated but did not provide information regarding illness stage or type of medication being currently taken (Terpstra et al., 2005). Interestingly, this study compared GSH quantification from a very short TE STEAM sequence and a GSH MEGA-PRESS sequence, and concluded that the techniques were comparable. Lastly, Woods et al. (2009) observed an increase in medial temporal lobe GSH in first-episode schizophrenia compared to control subjects (Wood et al., 2009). The medication status of the first-episode participants was mixed in that 13 participants were antipsychotic medication naïve while the remaining participants received at least one dose of atypical antipsychotic medication for a mean treatment duration of six days. Further investigation indicated that GSH increased only in those subjects with schizophrenia (approximately ½ of the sample) that were not responsive to niacin, and a non-responsive niacin skin test is related to increased oxidative stress. Despite the liberal criteria for inclusion of GSH metabolite fits (i.e. CRLB threshold of 50%), these exciting results provide important incentive for further investigation.
Table 3.
Summary of GSH Findings in First Episode (FEP) and Chronic Participants with Schizophrenia at 3T or higher
| Stage of Illness | Region | Authors | Scanner & Method | Participants (SZ/HC) | Antipsychotic Medication Use | SZ Mean Age (yrs ± std) | Results (vs. HC) |
|---|---|---|---|---|---|---|---|
| FEP | medial temporal lobe | Wood et al. (2009) | 3T PRESS (TE=30ms) | 30/18 | Yes | 19.4±3.4 | ↑ GSH in SZ |
| Chronic | posterior medial prefrontal cortex | Matsuzawa et al. (2008) | 3T MEGA-PRESS (TE=94ms) | 20/16 | Yes | 30.7±5.8 | No GSH diff.; negative correlation between GSH level and negative symptoms |
| Unknown | anterior cingulate | Terpstra et al. (2005) | 4T STEAM (TE=5ms) & MEGA-PRESS (TE=68ms) | 13/9 | Yes | 26±5 | No GSH diff. |
↑ - increased, diff. – difference, GSH – glutathione, HC - healthy controls, ms – millisecond, std – standard deviation, SZ – schizophrenia, TE – echo time, yrs - years
In the two studies at 3 T that provide participant illness status, it appears as though GSH is elevated earlier in the illness (with minimal medication exposure) in the temporal lobe, and this may relate to increased oxidative stress. Meanwhile, there are no observable differences in the posterior medial prefrontal cortex in chronic, medicated participants with schizophrenia. This suggests that earlier in the illness, GSH is elevated while compensatory mechanisms are still in place prior to chronic oxidative stress in the latter stage of the illness. This 1H-MRS data seems to further support prior findings in blood chemistry which show that GSH decreases from first episode to chronic (Ruiz-Litago et al., 2012). Overall, additional studies assessing GSH are needed to better characterize how GSH changes throughout the brain in different stages of the illness and whether these alterations are associated with any symptoms or outcomes.
3.4 N-acetylaspartylglutamate (NAAG)
NAAG is an abundant neuropeptide (0.5–3 mM) in the human brain, and is a highly selective agonist at the group II metabotropic glutamate subtype 3 receptor (mGluR3) that presynaptically inhibits glutamate release (Shave et al., 2001; Tsai, 2005; Wroblewska et al., 1997; Wroblewska et al., 2006; Neale et al., 2011; Bergeron and Coyle, 2012) and a NMDAR antagonist (Bergeron and Coyle, 2012). Post-mortem studies in schizophrenia report alterations in NAAG (Reynolds and Reynolds, 2011; Ghose et al., 2009), indicating that NAAG may play a pivotal role in the pathophysiology of schizophrenia. Additional evidence stems from studies utilizing the PCP drug model of schizophrenia revealing that NAAG peptidase inhibitors prevent the behavioral and physiological effects of NMDA receptor antagonism in rodents (Olszewski et al., 2008; Olszewski et al., 2012; Zuo et al., 2012).
Despite occurring at concentrations of 1.0–3.0 mM (Dringen, 2000; Govindaraju et al., 2000), NAAG has been exceedingly difficult to quantify with 1H-MRS since it has resonances at 2.04, 2.6, and 4.6 ppm which overlap with many other metabolites. NAAG’s concentration is higher in white matter versus gray matter (Pouwels and Frahm, 1997). Due to the fact that NAAG resonances overlap with many other metabolites, several different techniques are utilized to identify NAAG from NAA, glutamate, and other metabolites. Jessen et al. utilized a long TE PRESS sequence since simulations showed that contributions from overlapping resonances of glutamate and glutamine were minimized due to J-coupling and T2 relaxation (Jessen et al., 2013). Edden et al. (2007) optimized a MEGA-PRESS sequence specifically tailored to separate NAA from NAAG by employing a long TE of 140 ms and applying specific editing pulses that detect the 2.5 and 2.6 ppm resonances of NAA and NAAG, respectively (Edden et al., 2007). This method ensures separation of NAA and NAAG through editing, but the results suffer from a low signal-to-noise ratio. In addition, Zhang et al. utilized TE-Averaged PRESS and line shape deconvolution (Zhang et al., 2011). TE-Averaging acquires spectroscopic data at varying TEs and averages the data together. The resultant concentrations are typically lower than standard PRESS sequences due to T2 weighting and signal cancellation as a result of averaging across multiple TEs. The technique has been shown to enable detection of glutamate previously (Hurd et al., 2004). The purpose of line shape deconvolution is to improve the spectral resolution without sacrificing signal-to-noise ratio since standard techniques using Gaussian or exponential filters negatively impact spectral line widths. As a result, this technique will need additional steps in a spectroscopy processing pipeline. Thus, there are several techniques currently available to measure NAAG.
Recently, two studies quantified NAAG in vivo in schizophrenia (Table 4). Using a PRESS sequence at a TE of 140ms, Jessen et al. showed that NAAG was decreased at trend level in the left anterior cingulate of chronic, medicated patients with schizophrenia compared to healthy controls, but not in the left frontal lobe (Jessen et al., 2013). A negative correlation between NAAG levels and negative symptoms and total symptom score from the PANSS (Kay et al., 1987) was observed. Rowland et al. (2013) utilized the MEGA-PRESS editing technique and showed that NAAG decreased across age groups in the centrum semiovale in schizophrenia participants, but NAAG increased with age in healthy controls (Rowland et al., 2013). In younger schizophrenia participants, this study reported higher NAAG levels in the centrum semiovale compared to controls. In contrast, this same study found the opposite effect in the older cohort, suggesting that NAAG is higher earlier in the illness and declines over time. Rowland et al. (2013) did not observe a decrease in NAAG levels in the anterior cingulate that Jessen et al. did, and they reported a different directional relationship between NAAG and negative symptom severity. Direct comparisons between the studies are difficult due to different localization methodologies, quantification software packages, and brain regions assessed. Overall, these results suggest that NAAG levels change over the course of the illness, and further investigation is needed to determine whether NAAG levels are altered in first episode and whether NAAG peptidase inhibitors are beneficial for schizophrenia participants.
Table 4.
Summary of NAAG Findings in Chronic Participants with Schizophrenia at 3T or higher
| Stage of Illness | Region | Authors | Scanner & Method | Antipsychotic Medication Use | Participants (SZ/HC) | SZ Mean Age (yrs ± std) | Results (vs. HC) |
|---|---|---|---|---|---|---|---|
| Chronic | anterior cingulate & medial lateral part of left frontal lobe | Jessen et al. (2013) | 3T PRESS (TE=140ms) | Yes | 20/20 | 34.5±10.2 | ↑ NAAG in SZ (trend level) in anterior cingulate; ↑ NAAG/NAA in SZ in anterior cingulate; no NAAG diff. in left frontal lobe; negative correlation between NAAG from the frontal lobe and negative symptoms |
| Chronic | anterior cingulate & centrum semiovale | Rowland et al. (2013) | 3T MEGA-PRESS (TE=140ms) | Yes | 21/20,a | YSZ: 30.2±6.6; OSZ: 51.1±4.0 | No NAAG diff. in anterior cingulate; ↑ NAAG in younger SZ versus younger HC in centrum semiovale; ↓ NAAG in older SZ verus older HC in centrum semiovale; positive correlation with NAAG from centrum semiovale and negative symptom |
↑ - increased, ↓ - decreased, diff. – difference, HC – healthy controls, ms – millisecond, N-acetyalaspartylglutamate, OHC – older healthy controls, OSZ – older schizophrenia, SZ – schizophrenia, std – standard deviation, TE – echo time, yrs – years, YHC – younger healthy controls, YSZ – younger schizophrenia
YSZ: 11; YHC: 10; OSZ: 10; OHC: 10
3.5 Glycine
Glycine is an amino acid neurotransmitter that is an allosteric modulator of the glutamate NMDAR. Glycine acts on the glycine/D-serine site located on the NR1 subunit of the NMDAR. If NMDAR hypofunction is involved in the pathophysiology of schizophrenia, then a possible treatment target is the glycine system (Coyle and Tsai, 2004). Oral glycine administration improves negative symptoms in schizophrenia (Javitt et al., 1994; Heresco-Levy et al., 1996; Heresco-Levy et al., 1999; Heresco-Levy and Javitt, 2004) and has been explored as a possible drug target (Hashimoto, 2011; Cioffi, 2013). While a recent meta-analysis found no observable differences in serum glycine levels between healthy controls and adults with schizophrenia (Brouwer et al., 2013), a recent 1H-MRS study, in which healthy adult males were given two weeks of high dose oral glycine, showed elevated levels of Gly/Cr in 10 out of 11 participants in the occipital cortex (Kaufman et al., 2009). Even though it crosses the blood brain barrier poorly (Hashimoto et al., 2013), the ability to quantify cerebral glycine levels after treatment is exciting and is applicable to the study of schizophrenia.
Although two uncoupled methylene protons of glycine generate a simple singlet resonance at 3.55 ppm, the reliable detection of glycine in vivo with 1H-MRS is difficult. This is primarily due to its low concentration (0.4–1.0 mM) and the spectral overlap with myo-Inositol (mI), which usually yields much higher spectral resonances at 3.55 ppm. In order to reliably measure glycine with 1H-MRS, it is necessary to suppress the resonances of mI around 3.55 ppm. This can be achieved by exploiting the differences in spin coupling between the metabolites. The two glycine proton spins are uncoupled while the proton spins of mI are scalar coupled (Govindaraju et al., 2000). Metabolites with coupled spins undergo signal loss due to J-coupling and T2 relaxation, compared to metabolites with uncoupled spins that undergo signal loss due to T2 relaxation. Thus, at longer TEs, it is more feasible to separate glycine and mI than at short TEs (Govindaraju et al., 2000). TE-Averaging PRESS is a method to detect glycine (Kaufman et al., 2009; Prescot et al., 2006) where data are acquired over a range of TEs and then averaged together. While the signal from glycine and mI is expected to decrease due to T2 relaxation observed in the multiple TEs, the signal from mI is expected to be greatly reduced due to J-coupling, allowing detection of glycine. Another study by Choi et al. (2008) utilized multiple refocusing pulses at a long TE to suppress the mI signal (Choi et al., 2008). By using a long TE, both of these sequences remove the signal contributions from macromolecules from the spectral data. These techniques are employed at 3T and 4T, but at 7T, where the frequency separation between metabolites is improved, two other sequences are used to measure glycine in vivo: a long TE PRESS sequence (Choi et al., 2009b) and a short TE spin echo, full intensity acquired localized (SPECIAL) sequence (Gambarota et al., 2009). To date, there are no published reports of 1H-MRS glycine measurements in schizophrenia. Applying these techniques to study participants with schizophrenia would provide new information regarding cerebral glycine levels, insight into NMDAR function, an additional mechanism for monitoring treatment response, and overall, improve our understanding of the pathophysiology of schizophrenia.
3.6 Serine
Serine (Ser) is an amino acid in the brain acting as a co-agonist of glutamate transmission through the interactions with the glycine site of the NMDA receptor (Van Horn et al., 2013). Serine exists in two forms: L-serine and D-serine, and each isomer has similar chemical and physical properties (Bardaweel et al., 2014). While both forms exist in the brain, D-serine distribution is parallel to the distribution of NMDA glutamate receptors (Bardaweel et al., 2014), and D-serine binds three hundred times more strongly than L-serine at the NR1 subunit of NMDAR (Furukawa and Gouaux, 2003). Serum D-serine is typically reduced in adults with schizophrenia (Hashimoto et al., 2003; Calcia et al., 2012), and a recent meta-analysis found that serum levels of total serine are elevated in adults with schizophrenia (Brouwer et al., 2013). In addition, D-serine has been investigated as a monotherapy (Ermilov et al., 2013) and as adjunctive therapy (Heresco-Levy et al., 2005) in treatment-refractory adults with schizophrenia and found to be helpful in relieving negative symptoms.
The coupled proton spins of the CH2 and CH group of serine yield resonances at 3.98, 3.94, and 3.83 ppm (Govindaraju et al., 2000). Measurement of serine with 1H-MRS is significantly impaired by the spectral overlap with the prominent resonance of creatine (Cr) at 3.91 ppm as well as the low in vivo concentration of serine (around 0.4 mM), making quantification very difficult. To date, there are no 1H-MRS studies reporting serine at 3T or 4T; however, there is one study at 7T that was able to measure serine in healthy controls. Choi et al (2009a) utilized a constant-TE triple-refocusing difference editing method for detecting serine separate from Cr (Choi et al., 2009a). In this sequence, an additional 180° RF pulse was added to a PRESS sequence, and this RF pulse was spectrally-selective, in that it is tuned to 3.0 ppm and only excites resonances from 1.8 ppm to 4.2 ppm. This technique also utilized a subtraction method that maintains a fixed TE and varied sub-TEs in order to cancel out the overlapping creatine signal and provide the maximum signal difference for serine. Using a long TE attenuates the macromolecule signal, which is beneficial, but it also leads to significant signal loss due to T2 relaxation. This technique is not suitable for shorter TEs where the editing efficiency is poor and requires excellent water suppression due to serine’s proximity to the water peak within the spectrum. Since TE simulations for the maximal serine signal were performed at 7T, another simulation for detection of serine using the triple-refocusing technique would need to be conducted to determine the optimal TE and sub-TEs for 3T. To date, there have been no 1H-MRS schizophrenia studies that reported serine, but this technique by Choi et al. holds promise for acquisition at high fields, and would provide new and valuable information regarding NMDAR function in schizophrenia.
4.0 Discussion
In conclusion, 1H-MRS has provided invaluable information regarding the pathophysiology of schizophrenia. It has enhanced our understanding of metabolite differences between brain regions, stages of the illness, and pharmacological treatments, especially regarding glutamate, glutamine, and glutamate+glutamine. There is still much knowledge to be gained regarding historically difficult to detect metabolites, and currently, there are methods available to expand our understanding of the illness in terms of GABA, GSH, NAAG, glycine, and serine. Application of 1H-MRS to the study of schizophrenia has also elicited new questions regarding off-medication metabolite differences in participants with longer illness durations, effects of short and long term antipsychotic medication treatment on different brain regions, whether GABA and GSH levels change in different stages of the illness, and whether metabolite levels change later in life. Overall, 1H-MRS is a powerful technique that holds great promise for producing biomarkers that can serve as intervention targets and predict disease onset or illness exacerbation for schizophrenia and other brain diseases.
Highlights.
MRS is a powerful technique used to assess neurotransmitters and modulators in vivo
MRS techniques enable detection of Glu, GABA, GSH, NAAG, glycine, and serine
MRS studies of schizophrenia show alterations in Glu, GABA, GSH and NAAG
Metabolite differences depend on brain region, illness stage and medication status
MRS shows great promise for understanding glutamatergic systems in schizophrenia
Acknowledgments
The authors would like to thank NIH for support to: T32MH067533 (SAW) and R01MH094520 (LMR).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ahn K, Gil R, Seibyl J, Sewell RA, D’Souza DC. Probing GABA receptor function in schizophrenia with iomazenil. Neuropsychopharmacology. 2011;36:677–683. doi: 10.1038/npp.2010.198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aoyama N, Theberge J, Drost DJ, Manchanda R, Northcott S, Neufeld RW, Menon RS, Rajakumar N, Pavlosky WF, Densmore M, Schaefer B, Williamson PC. Grey matter and social functioning correlates of glutamatergic metabolite loss in schizophrenia. Br J Psychiatry. 2011;198:448–456. doi: 10.1192/bjp.bp.110.079608. [DOI] [PubMed] [Google Scholar]
- Ballesteros A, Jiang P, Summerfelt A, Du X, Chiappelli J, O’Donnell P, Kochunov P, Hong LE. No evidence of exogenous origin for the abnormal glutathione redox state in schizophrenia. Schizophr Res. 2013;146:184–189. doi: 10.1016/j.schres.2013.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bardaweel SK, Alzweiri M, Ishaqat AA. D-serine in neurobiology: CNS neurotransmission and neuromodulation. Can J Neurol Sci. 2014;41:164–176. doi: 10.1017/s031716710001653x. [DOI] [PubMed] [Google Scholar]
- Behar KL, Ogino T. Characterization of macromolecule resonances in the 1H NMR spectrum of rat brain. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 1993;30:38–44. doi: 10.1002/mrm.1910300107. [DOI] [PubMed] [Google Scholar]
- Behar KL, Rothman DL, Spencer DD, Petroff OA. Analysis of macromolecule resonances in 1H NMR spectra of human brain. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 1994;32:294–302. doi: 10.1002/mrm.1910320304. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Lewis DA. Insights into the neurodevelopmental origin of schizophrenia from postmortem studies of prefrontal cortical circuitry. Int J Dev Neurosci. 2011;29:295–304. doi: 10.1016/j.ijdevneu.2010.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger GE, Wood SJ, Ross M, Hamer CA, Wellard RM, Pell G, Phillips L, Nelson B, Amminger GP, Yung AR, Jackson G, Velakoulis D, Pantelis C, Manji H, McGorry PD. Neuroprotective effects of low-dose lithium in individuals at ultra-high risk for psychosis. A longitudinal MRI/MRS study. Curr Pharm Des. 2012;18:570–575. doi: 10.2174/138161212799316163. [DOI] [PubMed] [Google Scholar]
- Bergeron R, Coyle JT. NAAG, NMDA receptor and psychosis. Curr Med Chem. 2012;19:1360–1364. doi: 10.2174/092986712799462685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bogner W, Gruber S, Doelken M, Stadlbauer A, Ganslandt O, Boettcher U, Trattnig S, Doerfler A, Stefan H, Hammen T. In vivo quantification of intracerebral GABA by single-voxel (1)H-MRS-How reproducible are the results? Eur J Radiol. 2010;73:526–531. doi: 10.1016/j.ejrad.2009.01.014. [DOI] [PubMed] [Google Scholar]
- Bottomley PA. Spatial localization in NMR spectroscopy in vivo. Ann N Y Acad Sci. 1987;508:333–348. doi: 10.1111/j.1749-6632.1987.tb32915.x. [DOI] [PubMed] [Google Scholar]
- Brosnan JT, Brosnan ME. Glutamate: a truly functional amino acid. Amino Acids. 2013;45:413–418. doi: 10.1007/s00726-012-1280-4. [DOI] [PubMed] [Google Scholar]
- Brouwer A, Luykx JJ, van Boxmeer L, Bakker SC, Kahn RS. NMDA-receptor coagonists in serum, plasma, and cerebrospinal fluid of schizophrenia patients: a meta-analysis of case-control studies. Neurosci Biobehav Rev. 2013;37:1587–1596. doi: 10.1016/j.neubiorev.2013.06.007. [DOI] [PubMed] [Google Scholar]
- Brown MA, Semelka RC. MRI: basic principles and applications. 4. Wiley-Blackwell/John Wiley & Sons; Hoboken, N.J: 2010. [Google Scholar]
- Brugger S, Davis JM, Leucht S, Stone JM. Proton magnetic resonance spectroscopy and illness stage in schizophrenia--a systematic review and meta-analysis. Biol Psychiatry. 2011;69:495–503. doi: 10.1016/j.biopsych.2010.10.004. [DOI] [PubMed] [Google Scholar]
- Bustillo JR, Chen H, Jones T, Lemke N, Abbott C, Qualls C, Canive J, Gasparovic C. Increased Glutamine in Patients Undergoing Long-term Treatment for Schizophrenia: A Proton Magnetic Resonance Spectroscopy Study at 3 T. JAMA Psychiatry. 2014;71:265–272. doi: 10.1001/jamapsychiatry.2013.3939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bustillo JR, Rowland LM, Mullins P, Jung R, Chen H, Qualls C, Hammond R, Brooks WM, Lauriello J. 1H-MRS at 4 tesla in minimally treated early schizophrenia. Mol Psychiatry. 2010;15:629–636. doi: 10.1038/mp.2009.121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calcia MA, Madeira C, Alheira FV, Silva TC, Tannos FM, Vargas-Lopes C, Goldenstein N, Brasil MA, Ferreira ST, Panizzutti R. Plasma levels of D-serine in Brazilian individuals with schizophrenia. Schizophr Res. 2012;142:83–87. doi: 10.1016/j.schres.2012.09.014. [DOI] [PubMed] [Google Scholar]
- Cavassila S, Deval S, Huegen C, van Ormondt D, Graveron-Demilly D. Cramer-Rao bounds: an evaluation tool for quantitation. NMR in biomedicine. 2001;14:278–283. doi: 10.1002/nbm.701. [DOI] [PubMed] [Google Scholar]
- Chang L, Friedman J, Ernst T, Zhong K, Tsopelas ND, Davis K. Brain metabolite abnormalities in the white matter of elderly schizophrenic subjects: implication for glial dysfunction. Biol Psychiatry. 2007;62:1396–1404. doi: 10.1016/j.biopsych.2007.05.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang L, Jiang CS, Ernst T. Effects of age and sex on brain glutamate and other metabolites. Magnetic resonance imaging. 2009;27:142–145. doi: 10.1016/j.mri.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Bhardwaj PP, Seres P, Kalra S, Tibbo PG, Coupland NJ. Measurement of glycine in human brain by triple refocusing 1H-MRS in vivo at 3.0T. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2008;59:59–64. doi: 10.1002/mrm.21450. [DOI] [PubMed] [Google Scholar]
- Choi C, Dimitrov I, Douglas D, Zhao C, Hawesa H, Ghose S, Tamminga CA. In vivo detection of serine in the human brain by proton magnetic resonance spectroscopy (1H-MRS) at 7 Tesla. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2009a;62:1042–1046. doi: 10.1002/mrm.22079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi C, Douglas D, Hawesa H, Jindal A, Storey C, Dimitrov I. Measurement of glycine in human prefrontal brain by point-resolved spectroscopy at 7.0 tesla in vivo. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2009b;62:1305–1310. doi: 10.1002/mrm.22125. [DOI] [PubMed] [Google Scholar]
- Cioffi CL. Modulation of NMDA receptor function as a treatment for schizophrenia. Bioorg Med Chem Lett. 2013;23:5034–5044. doi: 10.1016/j.bmcl.2013.07.019. [DOI] [PubMed] [Google Scholar]
- Coyle JT, Tsai G. The NMDA receptor glycine modulatory site: a therapeutic target for improving cognition and reducing negative symptoms in schizophrenia. Psychopharmacology. 2004;174:32–38. doi: 10.1007/s00213-003-1709-2. [DOI] [PubMed] [Google Scholar]
- Danbolt NC. Glutamate uptake. Prog Neurobiol. 2001;65:1–105. doi: 10.1016/s0301-0082(00)00067-8. [DOI] [PubMed] [Google Scholar]
- Danielsen ER, Ross BD. Magnetic resonance spectroscopy diagnosis of neurological diseases. Marcel Dekker, Inc; New York: 1999. [Google Scholar]
- De Graaf RA. In vivo NMR spectroscopy: principles and techniques. 2. John Wiley & Sons; Chichester, West Sussex, England; Hoboken, NJ: 2007. [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Favila R, Stephano S, Graff-Guerrero A. Striatal glutamate and the conversion to psychosis: a prospective 1H-MRS imaging study. Int J Neuropsychopharmacol. 2013a;16:471–475. doi: 10.1017/S1461145712000314. [DOI] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Azcarraga M, Stephano S, Favila R, Diaz-Galvis L, Alvarado-Alanis P, Ramirez-Bermudez J, Graff-Guerrero A. Glutamate levels in the associative striatum before and after 4 weeks of antipsychotic treatment in first-episode psychosis: a longitudinal proton magnetic resonance spectroscopy study. JAMA Psychiatry. 2013b;70:1057–1066. doi: 10.1001/jamapsychiatry.2013.289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de la Fuente-Sandoval C, Leon-Ortiz P, Favila R, Stephano S, Mamo D, Ramirez-Bermudez J, Graff-Guerrero A. Higher levels of glutamate in the associative-striatum of subjects with prodromal symptoms of schizophrenia and patients with first-episode psychosis. Neuropsychopharmacology. 2011;36:1781–1791. doi: 10.1038/npp.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deelchand DK, Van de Moortele PF, Adriany G, Iltis I, Andersen P, Strupp JP, Vaughan JT, Ugurbil K, Henry PG. In vivo 1H NMR spectroscopy of the human brain at 9.4 T: initial results. J Magn Reson. 2010;206:74–80. doi: 10.1016/j.jmr.2010.06.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng C, Huang XF. Increased density of GABAA receptors in the superior temporal gyrus in schizophrenia. Exp Brain Res. 2006;168:587–590. doi: 10.1007/s00221-005-0290-9. [DOI] [PubMed] [Google Scholar]
- Do KQ, Trabesinger AH, Kirsten-Kruger M, Lauer CJ, Dydak U, Hell D, Holsboer F, Boesiger P, Cuenod M. Schizophrenia: glutathione deficit in cerebrospinal fluid and prefrontal cortex in vivo. Eur J Neurosci. 2000;12:3721–3728. doi: 10.1046/j.1460-9568.2000.00229.x. [DOI] [PubMed] [Google Scholar]
- Dringen R. Metabolism and functions of glutathione in brain. Prog Neurobiol. 2000;62:649–671. doi: 10.1016/s0301-0082(99)00060-x. [DOI] [PubMed] [Google Scholar]
- Drost DJ, Riddle WR, Clarke GD. Proton magnetic resonance spectroscopy in the brain: report of AAPM MR Task Group #9. Med Phys. 2002;29:2177–2197. doi: 10.1118/1.1501822. [DOI] [PubMed] [Google Scholar]
- Duarte JM, Gruetter R. Glutamatergic and GABAergic energy metabolism measured in the rat brain by (13) C NMR spectroscopy at 14.1 T. J Neurochem. 2013;126:579–590. doi: 10.1111/jnc.12333. [DOI] [PubMed] [Google Scholar]
- Edden RA, Barker PB. Spatial effects in the detection of gamma-aminobutyric acid: improved sensitivity at high fields using inner volume saturation. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2007;58:1276–1282. doi: 10.1002/mrm.21383. [DOI] [PubMed] [Google Scholar]
- Edden RA, Pomper MG, Barker PB. In vivo differentiation of N-acetyl aspartyl glutamate from N-acetyl aspartate at 3 Tesla. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2007;57:977–982. doi: 10.1002/mrm.21234. [DOI] [PubMed] [Google Scholar]
- Egerton A, Brugger S, Raffin M, Barker GJ, Lythgoe DJ, McGuire PK, Stone JM. Anterior cingulate glutamate levels related to clinical status following treatment in first-episode schizophrenia. Neuropsychopharmacology. 2012a;37:2515–2521. doi: 10.1038/npp.2012.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egerton A, Fusar-Poli P, Stone JM. Glutamate and psychosis risk. Curr Pharm Des. 2012b;18:466–478. doi: 10.2174/138161212799316244. [DOI] [PubMed] [Google Scholar]
- Ermilov M, Gelfin E, Levin R, Lichtenberg P, Hashimoto K, Javitt DC, Heresco-Levy U. A pilot double-blind comparison of d-serine and high-dose olanzapine in treatment-resistant patients with schizophrenia. Schizophr Res. 2013;150:604–605. doi: 10.1016/j.schres.2013.09.018. [DOI] [PubMed] [Google Scholar]
- Ernst T, Kreis R, Ross BD. Absolute quantitation of water and metabolites in the human brain. I. compartments and water. J Magn Reson. 1993;102:1–8. [Google Scholar]
- Evans CJ, McGonigle DJ, Edden RA. Diurnal stability of gamma-aminobutyric acid concentration in visual and sensorimotor cortex. J Magn Reson Imaging. 2010;31:204–209. doi: 10.1002/jmri.21996. [DOI] [PubMed] [Google Scholar]
- Farber NB, Newcomer JW, Olney JW. The glutamate synapse in neuropsychiatric disorders. Focus on schizophrenia and Alzheimer’s disease. Prog Brain Res. 1998;116:421–437. doi: 10.1016/s0079-6123(08)60453-7. [DOI] [PubMed] [Google Scholar]
- Flatow J, Buckley P, Miller BJ. Meta-analysis of oxidative stress in schizophrenia. Biol Psychiatry. 2013;74:400–409. doi: 10.1016/j.biopsych.2013.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frahm J, Merbolt J, Hanicke W. Localized proton spectroscopy using stimulated echoes. J Magn Reson. 1987;72:502–508. doi: 10.1002/mrm.1910170113. [DOI] [PubMed] [Google Scholar]
- Furukawa H, Gouaux E. Mechanisms of activation, inhibition and specificity: crystal structures of the NMDA receptor NR1 ligand-binding core. Embo J. 2003;22:2873–2885. doi: 10.1093/emboj/cdg303. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fusar-Poli P, Bonoldi I, Yung AR, Borgwardt S, Kempton MJ, Valmaggia L, Barale F, Caverzasi E, McGuire P. Predicting psychosis: meta-analysis of transition outcomes in individuals at high clinical risk. Arch Gen Psychiatry. 2012;69:220–229. doi: 10.1001/archgenpsychiatry.2011.1472. [DOI] [PubMed] [Google Scholar]
- Fusar-Poli P, Stone JM, Broome MR, Valli I, Mechelli A, McLean MA, Lythgoe DJ, O’Gorman RL, Barker GJ, McGuire PK. Thalamic glutamate levels as a predictor of cortical response during executive functioning in subjects at high risk for psychosis. Arch Gen Psychiatry. 2011;68:881–890. doi: 10.1001/archgenpsychiatry.2011.46. [DOI] [PubMed] [Google Scholar]
- Gambarota G, Mekle R, Xin L, Hergt M, van der Zwaag W, Krueger G, Gruetter R. In vivo measurement of glycine with short echo-time 1H MRS in human brain at 7 T. Magma. 2009;22:1–4. doi: 10.1007/s10334-008-0152-0. [DOI] [PubMed] [Google Scholar]
- Gao F, Edden RA, Li M, Puts NA, Wang G, Liu C, Zhao B, Wang H, Bai X, Zhao C, Wang X, Barker PB. Edited magnetic resonance spectroscopy detects an age-related decline in brain GABA levels. Neuroimage. 2013;78:75–82. doi: 10.1016/j.neuroimage.2013.04.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gasparovic C, Song T, Devier D, Bockholt HJ, Caprihan A, Mullins PG, Posse S, Jung RE, Morrison LA. Use of tissue water as a concentration reference for proton spectroscopic imaging. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2006;55:1219–1226. doi: 10.1002/mrm.20901. [DOI] [PubMed] [Google Scholar]
- Gawryluk JW, Wang JF, Andreazza AC, Shao L, Yatham LN, Young LT. Prefrontal cortex glutathione S-transferase levels in patients with bipolar disorder, major depression and schizophrenia. Int J Neuropsychopharmacol. 2011;14:1069–1074. doi: 10.1017/S1461145711000617. [DOI] [PubMed] [Google Scholar]
- Geramita M, van der Veen JW, Barnett AS, Savostyanova AA, Shen J, Weinberger DR, Marenco S. Reproducibility of prefrontal gamma-aminobutyric acid measurements with J-edited spectroscopy. NMR in biomedicine. 2011;24:1089–1098. doi: 10.1002/nbm.1662. [DOI] [PubMed] [Google Scholar]
- Ghose S, Chin R, Gallegos A, Roberts R, Coyle J, Tamminga C. Localization of NAAG-related gene expression deficits to the anterior hippocampus in schizophrenia. Schizophr Res. 2009;111:131–137. doi: 10.1016/j.schres.2009.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gideon P, Rosenbaum S, Sperling B, Petersen P. MR-visible brain water content in human acute stroke. Magnetic resonance imaging. 1999;17:301–304. doi: 10.1016/s0730-725x(98)00161-1. [DOI] [PubMed] [Google Scholar]
- Goddard AW, Mason GF, Appel M, Rothman DL, Gueorguieva R, Behar KL, Krystal JH. Impaired GABA neuronal response to acute benzodiazepine administration in panic disorder. Am J Psychiatry. 2004;161:2186–2193. doi: 10.1176/appi.ajp.161.12.2186. [DOI] [PubMed] [Google Scholar]
- Goto N, Yoshimura R, Kakeda S, Moriya J, Hori H, Hayashi K, Ikenouchi-Sugita A, Nakano-Umene W, Katsuki A, Nishimura J, Korogi Y, Nakamura J. No alterations of brain GABA after 6 months of treatment with atypical antipsychotic drugs in early-stage first-episode schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2010;34:1480–1483. doi: 10.1016/j.pnpbp.2010.08.007. [DOI] [PubMed] [Google Scholar]
- Goto N, Yoshimura R, Kakeda S, Nishimura J, Moriya J, Hayashi K, Katsuki A, Hori H, Umene-Nakano W, Ikenouchi-Sugita A, Korogi Y, Nakamura J. Six-month treatment with atypical antipsychotic drugs decreased frontal-lobe levels of glutamate plus glutamine in early-stage first-episode schizophrenia. Neuropsychiatr Dis Treat. 2012;8:119–122. doi: 10.2147/NDT.S25582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gottschalk M, Lamalle L, Segebarth C. Short-TE localised 1H MRS of the human brain at 3 T: quantification of the metabolite signals using two approaches to account for macromolecular signal contributions. NMR in biomedicine. 2008;21:507–517. doi: 10.1002/nbm.1219. [DOI] [PubMed] [Google Scholar]
- Govindaraju V, Young K, Maudsley AA. Proton NMR chemical shifts and coupling constants for brain metabolites. NMR Biomed. 2000;13:129–153. doi: 10.1002/1099-1492(200005)13:3<129::aid-nbm619>3.0.co;2-v. [DOI] [PubMed] [Google Scholar]
- Gruber S, Pinker K, Riederer F, Chmelik M, Stadlbauer A, Bittsansky M, Mlynarik V, Frey R, Serles W, Bodamer O, Moser E. Metabolic changes in the normal ageing brain: consistent findings from short and long echo time proton spectroscopy. Eur J Radiol. 2008;68:320–327. doi: 10.1016/j.ejrad.2007.08.038. [DOI] [PubMed] [Google Scholar]
- Gruetter R, Weisdorf SA, Rajanayagan V, Terpstra M, Merkle H, Truwit CL, Garwood M, Nyberg SL, Ugurbil K. Resolution improvements in in vivo 1H NMR spectra with increased magnetic field strength. J Magn Reson. 1998;135:260–264. doi: 10.1006/jmre.1998.1542. [DOI] [PubMed] [Google Scholar]
- Haacke EM, Brown RW, Thompson MR, Venkatesan R. Magnetic resonance imaging: physical principles and sequence design. Wiley; New York: 1999. [Google Scholar]
- Hancu I, Port J. The case of the missing glutamine. NMR in biomedicine. 2011;24:529–535. doi: 10.1002/nbm.1620. [DOI] [PubMed] [Google Scholar]
- Hashimoto K. Glycine transporter-1: a new potential therapeutic target for schizophrenia. Curr Pharm Des. 2011;17:112–120. doi: 10.2174/138161211795049598. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Fukushima T, Shimizu E, Komatsu N, Watanabe H, Shinoda N, Nakazato M, Kumakiri C, Okada S, Hasegawa H, Imai K, Iyo M. Decreased serum levels of D-serine in patients with schizophrenia: evidence in support of the N-methyl-D-aspartate receptor hypofunction hypothesis of schizophrenia. Arch Gen Psychiatry. 2003;60:572–576. doi: 10.1001/archpsyc.60.6.572. [DOI] [PubMed] [Google Scholar]
- Hashimoto K, Malchow B, Falkai P, Schmitt A. Glutamate modulators as potential therapeutic drugs in schizophrenia and affective disorders. Eur Arch Psychiatry Clin Neurosci. 2013;263:367–377. doi: 10.1007/s00406-013-0399-y. [DOI] [PubMed] [Google Scholar]
- Hashimoto T, Arion D, Unger T, Maldonado-Aviles JG, Morris HM, Volk DW, Mirnics K, Lewis DA. Alterations in GABA-related transcriptome in the dorsolateral prefrontal cortex of subjects with schizophrenia. Mol Psychiatry. 2008;13:147–161. doi: 10.1038/sj.mp.4002011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC. Comparative effects of glycine and D-cycloserine on persistent negative symptoms in schizophrenia: a retrospective analysis. Schizophr Res. 2004;66:89–96. doi: 10.1016/S0920-9964(03)00129-4. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Ebstein R, Vass A, Lichtenberg P, Bar G, Catinari S, Ermilov M. D-serine efficacy as add-on pharmacotherapy to risperidone and olanzapine for treatment-refractory schizophrenia. Biol Psychiatry. 2005;57:577–585. doi: 10.1016/j.biopsych.2004.12.037. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Horowitz A, Kelly D. Double-blind, placebo-controlled, crossover trial of glycine adjuvant therapy for treatment-resistant schizophrenia. Br J Psychiatry. 1996;169:610–617. doi: 10.1192/bjp.169.5.610. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Ermilov M, Mordel C, Silipo G, Lichtenstein M. Efficacy of high-dose glycine in the treatment of enduring negative symptoms of schizophrenia. Arch Gen Psychiatry. 1999;56:29–36. doi: 10.1001/archpsyc.56.1.29. [DOI] [PubMed] [Google Scholar]
- Hurd R, Sailasuta N, Srinivasan R, Vigneron DB, Pelletier D, Nelson SJ. Measurement of brain glutamate using TE-averaged PRESS at 3T. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2004;51:435–440. doi: 10.1002/mrm.20007. [DOI] [PubMed] [Google Scholar]
- Hutcheson NL, Reid MA, White DM, Kraguljac NV, Avsar KB, Bolding MS, Knowlton RC, den Hollander JA, Lahti AC. Multimodal analysis of the hippocampus in schizophrenia using proton magnetic resonance spectroscopy and functional magnetic resonance imaging. Schizophr Res. 2012;140:136–142. doi: 10.1016/j.schres.2012.06.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jansen JF, Backes WH, Nicolay K, Kooi ME. 1H MR spectroscopy of the brain: absolute quantification of metabolites. Radiology. 2006;240:318–332. doi: 10.1148/radiol.2402050314. [DOI] [PubMed] [Google Scholar]
- Javitt DC, Zylberman I, Zukin SR, Heresco-Levy U, Lindenmayer JP. Amelioration of negative symptoms in schizophrenia by glycine. Am J Psychiatry. 1994;151:1234–1236. doi: 10.1176/ajp.151.8.1234. [DOI] [PubMed] [Google Scholar]
- Jessen F, Fingerhut N, Sprinkart AM, Kuhn KU, Petrovsky N, Maier W, Schild HH, Block W, Wagner M, Traber F. N-acetylaspartylglutamate (NAAG) and N-acetylaspartate (NAA) in patients with schizophrenia. Schizophrenia bulletin. 2013;39:197–205. doi: 10.1093/schbul/sbr127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jeste DV, Twamley EW, Eyler Zorrilla LT, Golshan S, Patterson TL, Palmer BW. Aging and outcome in schizophrenia. Acta Psychiatr Scand. 2003;107:336–343. doi: 10.1034/j.1600-0447.2003.01434.x. [DOI] [PubMed] [Google Scholar]
- Jeste DV, Wolkowitz OM, Palmer BW. Divergent trajectories of physical, cognitive, and psychosocial aging in schizophrenia. Schizophrenia bulletin. 2011;37:451–455. doi: 10.1093/schbul/sbr026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser LG, Schuff N, Cashdollar N, Weiner MW. Age-related glutamate and glutamine concentration changes in normal human brain: 1H MR spectroscopy study at 4 T. Neurobiol Aging. 2005;26:665–672. doi: 10.1016/j.neurobiolaging.2004.07.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaiser LG, Young K, Matson GB. Elimination of spatial interference in PRESS-localized editing spectroscopy. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2007;58:813–818. doi: 10.1002/mrm.21407. [DOI] [PubMed] [Google Scholar]
- Kaufman MJ, Prescot AP, Ongur D, Evins AE, Barros TL, Medeiros CL, Covell J, Wang L, Fava M, Renshaw PF. Oral glycine administration increases brain glycine/creatine ratios in men: a proton magnetic resonance spectroscopy study. Psychiatry Res. 2009;173:143–149. doi: 10.1016/j.pscychresns.2009.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kauppinen RA, Pirttila TR, Auriola SO, Williams SR. Compartmentation of cerebral glutamate in situ as detected by 1H/13C n.m.r. Biochem J. 1994;298 (Pt 1):121–127. doi: 10.1042/bj2980121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kay SR, Fiszbein A, Opler LA. The positive and negative syndrome scale (PANSS) for schizophrenia. Schizophrenia bulletin. 1987;13:261–276. doi: 10.1093/schbul/13.2.261. [DOI] [PubMed] [Google Scholar]
- Kegeles LS, Mao X, Stanford AD, Girgis R, Ojeil N, Xu X, Gil R, Slifstein M, Abi-Dargham A, Lisanby SH, Shungu DC. Elevated prefrontal cortex gamma-aminobutyric acid and glutamate-glutamine levels in schizophrenia measured in vivo with proton magnetic resonance spectroscopy. Arch Gen Psychiatry. 2012;69:449–459. doi: 10.1001/archgenpsychiatry.2011.1519. [DOI] [PubMed] [Google Scholar]
- Kelemen O, Kiss I, Benedek G, Keri S. Perceptual and cognitive effects of antipsychotics in first-episode schizophrenia: the potential impact of GABA concentration in the visual cortex. Prog Neuropsychopharmacol Biol Psychiatry. 2013;47:13–19. doi: 10.1016/j.pnpbp.2013.07.024. [DOI] [PubMed] [Google Scholar]
- Knight-Scott J, Haley AP, Rossmiller SR, Farace E, Mai VM, Christopher JM, Manning CA, Simnad VI, Siragy HM. Molality as a unit of measure for expressing 1H MRS brain metabolite concentrations in vivo. Magnetic resonance imaging. 2003;21:787–797. doi: 10.1016/s0730-725x(03)00179-6. [DOI] [PubMed] [Google Scholar]
- Kraguljac NV, Reid MA, White DM, den Hollander J, Lahti AC. Regional decoupling of N-acetyl-aspartate and glutamate in schizophrenia. Neuropsychopharmacology. 2012;37:2635–2642. doi: 10.1038/npp.2012.126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kraguljac NV, White DM, Reid MA, Lahti AC. Increased hippocampal glutamate and volumetric deficits in unmedicated patients with schizophrenia. JAMA Psychiatry. 2013;70:1294–1302. doi: 10.1001/jamapsychiatry.2013.2437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lahti AC, Weiler MA, Tamara Michaelidis BA, Parwani A, Tamminga CA. Effects of ketamine in normal and schizophrenic volunteers. Neuropsychopharmacology. 2001;25:455–467. doi: 10.1016/S0893-133X(01)00243-3. [DOI] [PubMed] [Google Scholar]
- Laule C, Vavasour IM, Moore GR, Oger J, Li DK, Paty DW, MacKay AL. Water content and myelin water fraction in multiple sclerosis. A T2 relaxation study. J Neurol. 2004;251:284–293. doi: 10.1007/s00415-004-0306-6. [DOI] [PubMed] [Google Scholar]
- Lewis DA, Curley AA, Glausier JR, Volk DW. Cortical parvalbumin interneurons and cognitive dysfunction in schizophrenia. Trends Neurosci. 2012;35:57–67. doi: 10.1016/j.tins.2011.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z-P, Lauterbur PC. Principles of magnetic resonance imaging: a signal processing perspective. SPIE Optical Engineering Press; IEEE Press; Bellingham, Wash. New York: 2000. IEEE Engineering in Medicine and Biology Society. [Google Scholar]
- Lutkenhoff ES, van Erp TG, Thomas MA, Therman S, Manninen M, Huttunen MO, Kaprio J, Lonnqvist J, O’Neill J, Cannon TD. Proton MRS in twin pairs discordant for schizophrenia. Mol Psychiatry. 2010;15:308–318. doi: 10.1038/mp.2008.87. [DOI] [PubMed] [Google Scholar]
- Maddock R. The problem of spurious correlations between pairs of brain metabolite values measured in the same voxel with magnetic resonance spectroscopy. JAMA Psychiatry. 2014;71:338–339. doi: 10.1001/jamapsychiatry.2013.4343. [DOI] [PubMed] [Google Scholar]
- Maddock RJ, Buonocore MH. MR spectroscopic studies of the brain in psychiatric disorders. Curr Top Behav Neurosci. 2012;11:199–251. doi: 10.1007/7854_2011_197. [DOI] [PubMed] [Google Scholar]
- Magistretti PJ, Pellerin L. Cellular mechanisms of brain energy metabolism and their relevance to functional brain imaging. Philos Trans R Soc Lond B Biol Sci. 1999;354:1155–1163. doi: 10.1098/rstb.1999.0471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsman A, van den Heuvel MP, Klomp DW, Kahn RS, Luijten PR, Hulshoff Pol HE. Glutamate in schizophrenia: a focused review and meta-analysis of (1)H-MRS studies. Schizophrenia bulletin. 2013;39:120–129. doi: 10.1093/schbul/sbr069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews GC, Diamond JS. Neuronal glutamate uptake Contributes to GABA synthesis and inhibitory synaptic strength. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2003;23:2040–2048. doi: 10.1523/JNEUROSCI.23-06-02040.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuzawa D, Obata T, Shirayama Y, Nonaka H, Kanazawa Y, Yoshitome E, Takanashi J, Matsuda T, Shimizu E, Ikehira H, Iyo M, Hashimoto K. Negative correlation between brain glutathione level and negative symptoms in schizophrenia: a 3T 1H-MRS study. PLoS One. 2008;3:e1944. doi: 10.1371/journal.pone.0001944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meador-Woodruff JH, Clinton SM, Beneyto M, McCullumsmith RE. Molecular abnormalities of the glutamate synapse in the thalamus in schizophrenia. Ann N Y Acad Sci. 2003;1003:75–93. doi: 10.1196/annals.1300.005. [DOI] [PubMed] [Google Scholar]
- Mekle R, Mlynarik V, Gambarota G, Hergt M, Krueger G, Gruetter R. MR spectroscopy of the human brain with enhanced signal intensity at ultrashort echo times on a clinical platform at 3T and 7T. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2009;61:1279–1285. doi: 10.1002/mrm.21961. [DOI] [PubMed] [Google Scholar]
- Mescher M, Merkle H, Kirsch J, Garwood M, Gruetter R. Simultaneous in vivo spectral editing and water suppression. NMR in biomedicine. 1998;11:266–272. doi: 10.1002/(sici)1099-1492(199810)11:6<266::aid-nbm530>3.0.co;2-j. [DOI] [PubMed] [Google Scholar]
- Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. The Journal of neuroscience: the official journal of the Society for Neuroscience. 1997;17:2921–2927. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins PG, Chen H, Xu J, Caprihan A, Gasparovic C. Comparative reliability of proton spectroscopy techniques designed to improve detection of J-coupled metabolites. Magn Reson Med. 2008;60:964–969. doi: 10.1002/mrm.21696. [DOI] [PubMed] [Google Scholar]
- Mullins PG, McGonigle DJ, O’Gorman RL, Puts NA, Vidyasagar R, Evans CJ, Edden RA. Current practice in the use of MEGA-PRESS spectroscopy for the detection of GABA. Neuroimage. 2014;86:43–52. doi: 10.1016/j.neuroimage.2012.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mullins PG, Rowland L, Bustillo J, Bedrick EJ, Lauriello J, Brooks WM. Reproducibility of 1H-MRS measurements in schizophrenic patients. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2003;50:704–707. doi: 10.1002/mrm.10598. [DOI] [PubMed] [Google Scholar]
- Natsubori T, Inoue H, Abe O, Takano Y, Iwashiro N, Aoki Y, Koike S, Yahata N, Katsura M, Gonoi W, Sasaki H, Takao H, Kasai K, Yamasue H. Reduced Frontal Glutamate + Glutamine and N-Acetylaspartate Levels in Patients With Chronic Schizophrenia but not in Those at Clinical High Risk for Psychosis or With First-Episode Schizophrenia. Schizophrenia bulletin. 2013 doi: 10.1093/schbul/sbt124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neale JH, Olszewski RT, Zuo D, Janczura KJ, Profaci CP, Lavin KM, Madore JC, Bzdega T. Advances in understanding the peptide neurotransmitter NAAG and appearance of a new member of the NAAG neuropeptide family. J Neurochem. 2011;118:490–498. doi: 10.1111/j.1471-4159.2011.07338.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Near J, Andersson J, Maron E, Mekle R, Gruetter R, Cowen P, Jezzard P. Unedited in vivo detection and quantification of gamma-aminobutyric acid in the occipital cortex using short-TE MRS at 3 T. NMR Biomed. 2013 doi: 10.1002/nbm.2960. [DOI] [PubMed] [Google Scholar]
- O’Donnell P. Adolescent onset of cortical disinhibition in schizophrenia: insights from animal models. Schizophrenia bulletin. 2011;37:484–492. doi: 10.1093/schbul/sbr028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O’Gorman RL, Michels L, Edden RA, Murdoch JB, Martin E. In vivo detection of GABA and glutamate with MEGA-PRESS: reproducibility and gender effects. J Magn Reson Imaging. 2011;33:1262–1267. doi: 10.1002/jmri.22520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski RT, Janczura KJ, Ball SR, Madore JC, Lavin KM, Lee JC, Lee MJ, Der EK, Hark TJ, Farago PR, Profaci CP, Bzdega T, Neale JH. NAAG peptidase inhibitors block cognitive deficit induced by MK-801 and motor activation induced by d-amphetamine in animal models of schizophrenia. Transl Psychiatry. 2012;2:e145. doi: 10.1038/tp.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olszewski RT, Wegorzewska MM, Monteiro AC, Krolikowski KA, Zhou J, Kozikowski AP, Long K, Mastropaolo J, Deutsch SI, Neale JH. Phencyclidine and dizocilpine induced behaviors reduced by N-acetylaspartylglutamate peptidase inhibition via metabotropic glutamate receptors. Biol Psychiatry. 2008;63:86–91. doi: 10.1016/j.biopsych.2007.04.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongur D, Jensen JE, Prescot AP, Stork C, Lundy M, Cohen BM, Renshaw PF. Abnormal glutamatergic neurotransmission and neuronal-glial interactions in acute mania. Biol Psychiatry. 2008;64:718–726. doi: 10.1016/j.biopsych.2008.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongur D, Prescot AP, Jensen JE, Cohen BM, Renshaw PF. Creatine abnormalities in schizophrenia and bipolar disorder. Psychiatry Res. 2009;172:44–48. doi: 10.1016/j.pscychresns.2008.06.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ongur D, Prescot AP, McCarthy J, Cohen BM, Renshaw PF. Elevated gamma-aminobutyric acid levels in chronic schizophrenia. Biol Psychiatry. 2010;68:667–670. doi: 10.1016/j.biopsych.2010.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Otazo R, Tsai SY, Lin FH, Posse S. Accelerated short-TE 3D proton echo-planar spectroscopic imaging using 2D-SENSE with a 32-channel array coil. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2007;58:1107–1116. doi: 10.1002/mrm.21426. [DOI] [PubMed] [Google Scholar]
- Petroff OA, Mattson RH, Rothman DL. Proton MRS: GABA and glutamate. Adv Neurol. 2000;83:261–271. [PubMed] [Google Scholar]
- Poels EM, Kegeles LS, Kantrowitz JT, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR. Glutamatergic abnormalities in schizophrenia: a review of proton MRS findings. Schizophr Res. 2014a;152:325–332. doi: 10.1016/j.schres.2013.12.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poels EM, Kegeles LS, Kantrowitz JT, Slifstein M, Javitt DC, Lieberman JA, Abi-Dargham A, Girgis RR. Imaging glutamate in schizophrenia: review of findings and implications for drug discovery. Mol Psychiatry. 2014b;19:20–29. doi: 10.1038/mp.2013.136. [DOI] [PubMed] [Google Scholar]
- Posse S, Otazo R, Dager SR, Alger J. MR spectroscopic imaging: principles and recent advances. Journal of magnetic resonance imaging: JMRI. 2013;37:1301–1325. doi: 10.1002/jmri.23945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pouwels PJ, Frahm J. Differential distribution of NAA and NAAG in human brain as determined by quantitative localized proton MRS. NMR in biomedicine. 1997;10:73–78. doi: 10.1002/(sici)1099-1492(199704)10:2<73::aid-nbm448>3.0.co;2-4. [DOI] [PubMed] [Google Scholar]
- Prescot AP, de BFB, Wang L, Brown J, Jensen JE, Kaufman MJ, Renshaw PF. In vivo detection of brain glycine with echo-time-averaged (1)H magnetic resonance spectroscopy at 4.0 T. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2006;55:681–686. doi: 10.1002/mrm.20807. [DOI] [PubMed] [Google Scholar]
- Provencher SW. Estimation of metabolite concentrations from localized in vivo proton NMR spectra. Magn Reson Med. 1993;30:672–679. doi: 10.1002/mrm.1910300604. [DOI] [PubMed] [Google Scholar]
- Provencher SW. LCModel & LCMgui User’s Manual. 2014. [Google Scholar]
- Purdon SE, Valiakalayil A, Hanstock CC, Seres P, Tibbo P. Elevated 3T proton MRS glutamate levels associated with poor Continuous Performance Test (CPT-0X) scores and genetic risk for schizophrenia. Schizophr Res. 2008;99:218–224. doi: 10.1016/j.schres.2007.11.028. [DOI] [PubMed] [Google Scholar]
- Raffa M, Mechri A, Othman LB, Fendri C, Gaha L, Kerkeni A. Decreased glutathione levels and antioxidant enzyme activities in untreated and treated schizophrenic patients. Prog Neuropsychopharmacol Biol Psychiatry. 2009;33:1178–1183. doi: 10.1016/j.pnpbp.2009.06.018. [DOI] [PubMed] [Google Scholar]
- Ramadan S, Lin A, Stanwell P. Glutamate and glutamine: a review of in vivo MRS in the human brain. NMR in biomedicine. 2013;26:1630–1646. doi: 10.1002/nbm.3045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratiney H, Coenradie Y, Cavassila S, van Ormondt D, Graveron-Demilly D. Time-domain quantitation of 1H short echo-time signals: background accommodation. Magma. 2004;16:284–296. doi: 10.1007/s10334-004-0037-9. [DOI] [PubMed] [Google Scholar]
- Reid MA, Kraguljac NV, Avsar KB, White DM, den Hollander JA, Lahti AC. Proton magnetic resonance spectroscopy of the substantia nigra in schizophrenia. Schizophr Res. 2013;147:348–354. doi: 10.1016/j.schres.2013.04.036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid MA, Stoeckel LE, White DM, Avsar KB, Bolding MS, Akella NS, Knowlton RC, den Hollander JA, Lahti AC. Assessments of function and biochemistry of the anterior cingulate cortex in schizophrenia. Biol Psychiatry. 2010;68:625–633. doi: 10.1016/j.biopsych.2010.04.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds LM, Reynolds GP. Differential regional N-acetylaspartate deficits in postmortem brain in schizophrenia, bipolar disorder and major depressive disorder. J Psychiatr Res. 2011;45:54–59. doi: 10.1016/j.jpsychires.2010.05.001. [DOI] [PubMed] [Google Scholar]
- Rothman DL, Sibson NR, Hyder F, Shen J, Behar KL, Shulman RG. In vivo nuclear magnetic resonance spectroscopy studies of the relationship between the glutamate-glutamine neurotransmitter cycle and functional neuroenergetics. Philos Trans R Soc Lond B Biol Sci. 1999;354:1165–1177. doi: 10.1098/rstb.1999.0472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland L, Bustillo JR, Lauriello J. Proton magnetic resonance spectroscopy (H-MRS) studies of schizophrenia. Semin Clin Neuropsychiatry. 2001;6:121–130. doi: 10.1053/scnp.2001.21838. [DOI] [PubMed] [Google Scholar]
- Rowland LM, Bustillo JR, Mullins PG, Jung RE, Lenroot R, Landgraf E, Barrow R, Yeo R, Lauriello J, Brooks WM. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–396. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- Rowland LM, Kontson K, West J, Edden RA, Zhu H, Wijtenburg SA, Holcomb HH, Barker PB. In vivo measurements of glutamate, GABA, and NAAG in schizophrenia. Schizophrenia bulletin. 2013;39:1096–1104. doi: 10.1093/schbul/sbs092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rowland LM, Spieker EA, Francis A, Barker PB, Carpenter WT, Buchanan RW. White matter alterations in deficit schizophrenia. Neuropsychopharmacology. 2009;34:1514–1522. doi: 10.1038/npp.2008.207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruiz-Litago F, Seco J, Echevarria E, Martinez-Cengotitabengoa M, Gil J, Irazusta J, Gonzalez-Pinto AM. Adaptive response in the antioxidant defence system in the course and outcome in first-episode schizophrenia patients: a 12-months follow-up study. Psychiatry Res. 2012;200:218–222. doi: 10.1016/j.psychres.2012.07.024. [DOI] [PubMed] [Google Scholar]
- Ryner LN, Sorenson JA, Thomas MA. Localized 2D J-resolved 1H MR spectroscopy: strong coupling effects in vitro and in vivo. Magnetic resonance imaging. 1995;13:853–869. doi: 10.1016/0730-725x(95)00031-b. [DOI] [PubMed] [Google Scholar]
- Schubert F, Gallinat J, Seifert F, Rinneberg H. Glutamate concentrations in human brain using single voxel proton magnetic resonance spectroscopy at 3 Tesla. Neuroimage. 2004;21:1762–1771. doi: 10.1016/j.neuroimage.2003.11.014. [DOI] [PubMed] [Google Scholar]
- Seeger U, Klose U, Mader I, Grodd W, Nagele T. Parameterized evaluation of macromolecules and lipids in proton MR spectroscopy of brain diseases. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2003;49:19–28. doi: 10.1002/mrm.10332. [DOI] [PubMed] [Google Scholar]
- Shave E, Pliss L, Lawrance ML, FitzGibbon T, Stastny F, Balcar VJ. Regional distribution and pharmacological characteristics of [3H]N-acetyl-aspartyl-glutamate (NAAG) binding sites in rat brain. Neurochem Int. 2001;38:53–62. doi: 10.1016/s0197-0186(00)00045-0. [DOI] [PubMed] [Google Scholar]
- Shirayama Y, Obata T, Matsuzawa D, Nonaka H, Kanazawa Y, Yoshitome E, Ikehira H, Hashimoto K, Iyo M. Specific metabolites in the medial prefrontal cortex are associated with the neurocognitive deficits in schizophrenia: a preliminary study. Neuroimage. 2010;49:2783–2790. doi: 10.1016/j.neuroimage.2009.10.031. [DOI] [PubMed] [Google Scholar]
- Stagg CJ, Bestmann S, Constantinescu AO, Moreno LM, Allman C, Mekle R, Woolrich M, Near J, Johansen-Berg H, Rothwell JC. Relationship between physiological measures of excitability and levels of glutamate and GABA in the human motor cortex. The Journal of physiology. 2011;589:5845–5855. doi: 10.1113/jphysiol.2011.216978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stagg CJ, Wylezinska M, Matthews PM, Johansen-Berg H, Jezzard P, Rothwell JC, Bestmann S. Neurochemical effects of theta burst stimulation as assessed by magnetic resonance spectroscopy. J Neurophysiol. 2009;101:2872–2877. doi: 10.1152/jn.91060.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steen RG, Hamer RM, Lieberman JA. Measurement of brain metabolites by 1H magnetic resonance spectroscopy in patients with schizophrenia: a systematic review and meta-analysis. Neuropsychopharmacology. 2005;30:1949–1962. doi: 10.1038/sj.npp.1300850. [DOI] [PubMed] [Google Scholar]
- Stone JM. Imaging the glutamate system in humans: relevance to drug discovery for schizophrenia. Curr Pharm Des. 2009;15:2594–2602. doi: 10.2174/138161209788957438. [DOI] [PubMed] [Google Scholar]
- Stone JM, Day F, Tsagaraki H, Valli I, McLean MA, Lythgoe DJ, O’Gorman RL, Barker GJ, McGuire PK. Glutamate dysfunction in people with prodromal symptoms of psychosis: relationship to gray matter volume. Biol Psychiatry. 2009;66:533–539. doi: 10.1016/j.biopsych.2009.05.006. [DOI] [PubMed] [Google Scholar]
- Stone JM, Dietrich C, Edden R, Mehta MA, De Simoni S, Reed LJ, Krystal JH, Nutt D, Barker GJ. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–665. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Taylor SF, Demeter E, Phan KL, Tso IF, Welsh RC. Abnormal GABAergic function and negative affect in schizophrenia. Neuropsychopharmacology. 2013 doi: 10.1038/npp.2013.300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayoshi S, Nakataki M, Sumitani S, Taniguchi K, Shibuya-Tayoshi S, Numata S, Iga J, Ueno S, Harada M, Ohmori T. GABA concentration in schizophrenia patients and the effects of antipsychotic medication: a proton magnetic resonance spectroscopy study. Schizophr Res. 2010;117:83–91. doi: 10.1016/j.schres.2009.11.011. [DOI] [PubMed] [Google Scholar]
- Tayoshi S, Sumitani S, Taniguchi K, Shibuya-Tayoshi S, Numata S, Iga J, Nakataki M, Ueno S, Harada M, Ohmori T. Metabolite changes and gender differences in schizophrenia using 3-Tesla proton magnetic resonance spectroscopy (1H-MRS) Schizophr Res. 2009;108:69–77. doi: 10.1016/j.schres.2008.11.014. [DOI] [PubMed] [Google Scholar]
- Terpstra M, Henry PG, Gruetter R. Measurement of reduced glutathione (GSH) in human brain using LCModel analysis of difference-edited spectra. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2003;50:19–23. doi: 10.1002/mrm.10499. [DOI] [PubMed] [Google Scholar]
- Terpstra M, Ugurbil K, Gruetter R. Direct in vivo measurement of human cerebral GABA concentration using MEGA-editing at 7 Tesla. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2002;47:1009–1012. doi: 10.1002/mrm.10146. [DOI] [PubMed] [Google Scholar]
- Terpstra M, Vaughan TJ, Ugurbil K, Lim KO, Schulz SC, Gruetter R. Validation of glutathione quantitation from STEAM spectra against edited 1H NMR spectroscopy at 4T: application to schizophrenia. Magma. 2005;18:276–282. doi: 10.1007/s10334-005-0012-0. [DOI] [PubMed] [Google Scholar]
- Theberge J, Al-Semaan Y, Williamson PC, Menon RS, Neufeld RW, Rajakumar N, Schaefer B, Densmore M, Drost DJ. Glutamate and glutamine in the anterior cingulate and thalamus of medicated patients with chronic schizophrenia and healthy comparison subjects measured with 4.0-T proton MRS. Am J Psychiatry. 2003;160:2231–2233. doi: 10.1176/appi.ajp.160.12.2231. [DOI] [PubMed] [Google Scholar]
- Theberge J, Bartha R, Drost DJ, Menon RS, Malla A, Takhar J, Neufeld RW, Rogers J, Pavlosky W, Schaefer B, Densmore M, Al-Semaan Y, Williamson PC. Glutamate and glutamine measured with 4.0 T proton MRS in never-treated patients with schizophrenia and healthy volunteers. Am J Psychiatry. 2002;159:1944–1946. doi: 10.1176/appi.ajp.159.11.1944. [DOI] [PubMed] [Google Scholar]
- Theberge J, Williamson KE, Aoyama N, Drost DJ, Manchanda R, Malla AK, Northcott S, Menon RS, Neufeld RW, Rajakumar N, Pavlosky W, Densmore M, Schaefer B, Williamson PC. Longitudinal grey-matter and glutamatergic losses in first-episode schizophrenia. Br J Psychiatry. 2007;191:325–334. doi: 10.1192/bjp.bp.106.033670. [DOI] [PubMed] [Google Scholar]
- Tibbo P, Hanstock C, Valiakalayil A, Allen P. 3-T proton MRS investigation of glutamate and glutamine in adolescents at high genetic risk for schizophrenia. Am J Psychiatry. 2004;161:1116–1118. doi: 10.1176/appi.ajp.161.6.1116. [DOI] [PubMed] [Google Scholar]
- Tsai SJ. Central N-acetyl aspartylglutamate deficit: a possible pathogenesis of schizophrenia. Med Sci Monit. 2005;11:HY39–45. [PubMed] [Google Scholar]
- Valli I, Stone J, Mechelli A, Bhattacharyya S, Raffin M, Allen P, Fusar-Poli P, Lythgoe D, O’Gorman R, Seal M, McGuire P. Altered medial temporal activation related to local glutamate levels in subjects with prodromal signs of psychosis. Biol Psychiatry. 2011;69:97–99. doi: 10.1016/j.biopsych.2010.08.033. [DOI] [PubMed] [Google Scholar]
- Van Horn MR, Sild M, Ruthazer ES. D-serine as a gliotransmitter and its roles in brain development and disease. Front Cell Neurosci. 2013;7:39. doi: 10.3389/fncel.2013.00039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijtenburg SA, Gaston FE, Spieker EA, Korenic SA, Kochunov P, Hong LE, Rowland LM. Reproducibility of phase rotation STEAM at 3T: Focus on glutathione. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2013 doi: 10.1002/mrm.24959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wijtenburg SA, Knight-Scott J. Very short echo time improves the precision of glutamate detection at 3T in 1H magnetic resonance spectroscopy. J Magn Reson Imaging. 2011;34:645–652. doi: 10.1002/jmri.22638. [DOI] [PubMed] [Google Scholar]
- Winchester CL, Pratt JA, Morris BJ. Risk genes for schizophrenia: Translational opportunities for drug discovery. Pharmacol Ther. 2014 doi: 10.1016/j.pharmthera.2014.02.003. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Berger GE, Wellard RM, Proffitt TM, McConchie M, Berk M, McGorry PD, Pantelis C. Medial temporal lobe glutathione concentration in first episode psychosis: a 1H-MRS investigation. Neurobiol Dis. 2009;33:354–357. doi: 10.1016/j.nbd.2008.11.018. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Kennedy D, Phillips LJ, Seal ML, Yucel M, Nelson B, Yung AR, Jackson G, McGorry PD, Velakoulis D, Pantelis C. Hippocampal pathology in individuals at ultra-high risk for psychosis: a multi-modal magnetic resonance study. Neuroimage. 2010;52:62–68. doi: 10.1016/j.neuroimage.2010.04.012. [DOI] [PubMed] [Google Scholar]
- Wood SJ, Yucel M, Wellard RM, Harrison BJ, Clarke K, Fornito A, Velakoulis D, Pantelis C. Evidence for neuronal dysfunction in the anterior cingulate of patients with schizophrenia: a proton magnetic resonance spectroscopy study at 3 T. Schizophr Res. 2007;94:328–331. doi: 10.1016/j.schres.2007.05.008. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wegorzewska IN, Bzdega T, Olszewski RT, Neale JH. Differential negative coupling of type 3 metabotropic glutamate receptor to cyclic GMP levels in neurons and astrocytes. J Neurochem. 2006;96:1071–1077. doi: 10.1111/j.1471-4159.2005.03569.x. [DOI] [PubMed] [Google Scholar]
- Wroblewska B, Wroblewski JT, Pshenichkin S, Surin A, Sullivan SE, Neale JH. N-acetylaspartylglutamate selectively activates mGluR3 receptors in transfected cells. J Neurochem. 1997;69:174–181. doi: 10.1046/j.1471-4159.1997.69010174.x. [DOI] [PubMed] [Google Scholar]
- Wu G, Fang YZ, Yang S, Lupton JR, Turner ND. Glutathione metabolism and its implications for health. J Nutr. 2004;134:489–492. doi: 10.1093/jn/134.3.489. [DOI] [PubMed] [Google Scholar]
- Wu JQ, Kosten TR, Zhang XY. Free radicals, antioxidant defense systems, and schizophrenia. Prog Neuropsychopharmacol Biol Psychiatry. 2013;46:200–206. doi: 10.1016/j.pnpbp.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Yang S, Hu J, Kou Z, Yang Y. Spectral simplification for resolved glutamate and glutamine measurement using a standard STEAM sequence with optimized timing parameters at 3, 4, 4.7, 7, and 9.4T. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2008;59:236–244. doi: 10.1002/mrm.21463. [DOI] [PubMed] [Google Scholar]
- Yao JK, Leonard S, Reddy R. Altered glutathione redox state in schizophrenia. Dis Markers. 2006;22:83–93. doi: 10.1155/2006/248387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoon JH, Maddock RJ, Rokem A, Silver MA, Minzenberg MJ, Ragland JD, Carter CS. GABA concentration is reduced in visual cortex in schizophrenia and correlates with orientation-specific surround suppression. The Journal of neuroscience: the official journal of the Society for Neuroscience. 2010;30:3777–3781. doi: 10.1523/JNEUROSCI.6158-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yung AR, McGorry PD, McFarlane CA, Jackson HJ, Patton GC, Rakkar A. Monitoring and care of young people at incipient risk of psychosis. Schizophrenia bulletin. 1996;22:283–303. doi: 10.1093/schbul/22.2.283. [DOI] [PubMed] [Google Scholar]
- Yung AR, Yuen HP, Berger G, Francey S, Hung TC, Nelson B, Phillips L, McGorry P. Declining transition rate in ultra high risk (prodromal) services: dilution or reduction of risk? Schizophrenia bulletin. 2007;33:673–681. doi: 10.1093/schbul/sbm015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zavitsanou K, Ward PB, Huang XF. Selective alterations in ionotropic glutamate receptors in the anterior cingulate cortex in schizophrenia. Neuropsychopharmacology. 2002;27:826–833. doi: 10.1016/S0893-133X(02)00347-0. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Li S, Marenco S, Shen J. Quantitative measurement of N-acetyl-aspartyl-glutamate at 3 T using TE-averaged PRESS spectroscopy and regularized lineshape deconvolution. Magnetic resonance in medicine: official journal of the Society of Magnetic Resonance in Medicine / Society of Magnetic Resonance in Medicine. 2011;66:307–313. doi: 10.1002/mrm.23029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu H, Barker PB. MR spectroscopy and spectroscopic imaging of the brain. Methods Mol Biol. 2011;711:203–226. doi: 10.1007/978-1-61737-992-5_9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zuo D, Bzdega T, Olszewski RT, Moffett JR, Neale JH. Effects of N-acetylaspartylglutamate (NAAG) peptidase inhibition on release of glutamate and dopamine in prefrontal cortex and nucleus accumbens in phencyclidine model of schizophrenia. J Biol Chem. 2012;287:21773–21782. doi: 10.1074/jbc.M112.363226. [DOI] [PMC free article] [PubMed] [Google Scholar]