

Abstract
Surface sediments are important systems for the removal of anthropogenically derived inorganic nitrogen in estuaries. They are often characterized by the presence of a microphytobenthos (MPB) biofilm, which can impact bacterial communities in underlying sediments for example by secretion of extracellular polymeric substances (EPS) and competition for nutrients (including nitrogen). Pyrosequencing and qPCR was performed on two intertidal surface sediments of the Westerschelde estuary characterized by a two-fold difference in MPB biomass but no difference in MPB composition. Doubling of MPB biomass was accompanied by a disproportionately (ten-fold) increase in total bacterial abundances while, unexpectedly, no difference in general community structure was observed, despite significantly lower bacterial richness and distinct community membership, mostly for non-abundant taxa. Denitrifier abundances corresponded likewise while community structure, both for nirS and nirK denitrifiers, remained unchanged, suggesting that competition with diatoms for nitrate is negligible at concentrations in the investigated sediments (appr. 1 mg/l NO3 -). This study indicates that MPB biomass increase has a general, significantly positive effect on total bacterial and denitrifier abundances, with stimulation or inhibition of specific bacterial groups that however do not result in a re-structured community.
Introduction
The rate of terrestrial nitrogen input has more than doubled in the past century, mostly through fossil fuel combustion and increased use of agricultural fertilizers [1, 2]. When it is not removed by biotic uptake or dissimilatory nitrate reduction in streams and rivers, excessive, anthropogenically-derived nitrogen ends up in estuaries and coastal areas [3], where it is implicated in eutrophication [4] that can generate an excessive biochemical oxygen demand resulting in hypoxic zones [5], and can promote harmful algal blooms [6]. Estuarine surface sediments, lying at the interface between the oxidized water column or atmosphere and the deeper reduced sediment, can serve as important removal sites for inorganic nitrogen [7]. Benthic dissimilatory nitrate reduction includes three primarily anoxic processes with varying importance [8–11]: nitrate can be retained in the system as biologically available ammonium via dissimilatory nitrate/nitrite reduction to ammonium (DNRA) or lost by reduction to a gaseous product via anaerobic ammonium oxidation (anammox) or denitrification, i.e. the respiratory reduction of nitrate to either the potent greenhouse gas nitrous oxide or the harmless dinitrogen gas. Denitrification is most likely to dominate in temperate coastal areas [11]. It is proposed to be favoured over DNRA with decreasing salinity [11, 12] and lower temperatures [9, 11], and outcompetes anammox in highly variable, eutrophic estuaries [13–15].
Denitrification is performed by a wide variety of phylogenetically unrelated microorganisms. Therefore, denitrifying communities are commonly characterized using the two genes encoding cytochrome cd1 and copper-containing nitrite reductases (nirS and nirK respectively) as a proxy [16]. These key enzymes convert fixed nitrogen to a gaseous form, as such distinguishing dissimilatory nitrate-reducing bacteria that produce nitrite as end-product from true denitrifiers. The two forms are functionally equivalent but structurally different and respond to different environmental drivers, most of which are currently still unknown [17, 18]. Recently, elaborate genome analyses showed that both nir genes are not mutually exclusive in a single organism [19]. However, the functionality of these two nitrite reductases in one organism remains to be demonstrated. NirS denitrifiers are more widespread, whereas nirK denitrifiers comprise more diverse taxa [18]. Despite the importance of denitrification in estuarine systems, little is known about the diversity and distribution of the two denitrifying guilds in these ecosystems and almost no attempts are made to target both, either due to unsuccessful amplification of nirK or the assumption that nirS denitrifiers are more important in situ because of their numerical dominance [20].
In addition to denitrifying microorganisms, biofilm-producing microphytobenthos (MPB) present in the uppermost mm of sediments can also influence benthic nitrate reduction. MPB metabolism decouples nitrification-denitrification through (i) competition for ammonium and nitrate between MPB and bacteria [21] and (ii) pH increase via CO2 removal from the pore water [22]. MPB inorganic nitrogen assimilation can even exceed N consumption via denitrification by one to two orders of magnitude [21, 23], depending on the in situ nitrate concentrations. Diatoms, which often dominate primary production in estuarine intertidal sediments, are also known to store nitrate intracellularly up to a few 100 mM [24–26], and even use DNRA as dark survival strategy, releasing ammonium to the environment. Thus, MPB intervene in nitrogen cycling but if and how they shape the denitrifier communities in situ is not known.
Besides for inorganic nitrogen, other types of algal-bacterial coupling exist in these complex estuarine ecosystem [27]. Photosynthetically fixed carbon by MPB is transferred to heterotrophic bacteria within hours, resulting in a quick use of labile biofilm DOC and hydrolysed EPS [28–32], while MPB can also produce cytotoxins that can inhibit bacterial growth [33]. These algal-bacterial interactions are species-specific, both for diatoms and bacteria [30, 34, 35]. However, the effects of increased MPB biomass on bacteria in underlying sediments, including higher bacterial enzymatic activity [35] and EPS production [36] but without or with only a small increase in total bacterial cell numbers [36–40], remains ambiguous.
Based on the current knowledge, we hypothesized that higher MPB biomass (i) does not affect total bacterial abundances, (ii) negatively impacts denitrifier abundances, (iii) results in different total bacterial and denitrifier community structure, and (iv) generates differential responses of nirK and nirS denitrifier communities. To investigate these hypotheses, we sampled estuarine sediments at the Paulina polder tidal flat (Westerschelde estuary, SW Netherlands), characterized by the presence of MPB biofilms stabilizing sediment surfaces [37, 41]. The eutrophied Westerschelde estuary has a nitrogen load of 5 × 109 mol N yr–1 [42] with nitrate being the predominant form of reactive nitrogen [43] and denitrification as the main nitrate removing process [44, 45]. Sediment samples solely differed in MPB biomass but not MPB composition. Abundance and diversity of the total bacterial community as well as both nirS and nirK denitrifying guilds were assessed using qPCR and 454 pyrosequencing. Abundant and non-abundant fractions of all three bacterial communities (i.e. all bacteria, nirK, nirS) were examined separately to also assess influences of the low-abundant fraction of the bacterial community on diversity parameters.
Experimental procedures
Sampling and analytical procedures
Samples were collected in October 2011 at the Paulina polder mudflat (51° 21' 24" N, 3° 42' 51" E) in collaboration with NIOZ, which provided the necessary permit for field sampling, issued by the ‘Provincie Zeeland, The Netherlands; Directie Ruimte, Milieu en Water’. A plexiglas corer (inner Ø 3.2 cm) was used to collect triplicate samples of bacterial communities in two muddy sediments. To assess the sole effect of MPB on total bacterial and denitrifier abundance and diversity, two adjacent (± 6 m), physico-chemically similar sediments with visually different MPB biofilm development (high (HBM) or low (LBM) biomass of MPB) were sampled (MPB biofilms are visible as a brown film on the sediment; Fig 1). In both LBM and HBM sediments, the three replicates were taken as close together as technical constraints would allow, i.e. within a 10 x 10 cm square (Fig 1). The sediment cores were sealed and kept at 4°C until further processing. In the lab, the upper cm of the sediment was sampled and stored in sterile falcon tubes at -20°C until DNA extraction. At each location three further cores (each in triplicate) were taken for nutrient analyses (inner Ø 6.2 cm), determination of total organic matter (TOM) and grain size (inner Ø 3.2 cm) and determination of extracellular polymeric substances (EPS) and chlorophyll a (inner Ø 3.2 cm). These additional cores were taken adjacent to the cores for bacterial community structure analysis. The upper one cm of these cores was immediately frozen at -80°C (for pigment analysis) or -20°C (for all other parameters) until further analysis. The samples were analysed for NO3 -/NO2 -/NH4 +/Si/PO4 - pore water concentrations (SANplus segmented flow analyzer, SKALAR), the total amount of organic matter (loss of mass after incineration at 500°C for 2 hours) and grain size distribution using laser diffraction (Malvern Mastersizer 2000). To assess MPB biomass in the sediment [46], chl a concentration was measured by HPLC analysis after pigment extraction using 10 ml 90% acetone—10% milliQ water solution [47]. Colloidal extracellular polymeric substances (cEPS) were determined by spectrophotometry using the phenol-sulfuric acid assay [48]. Furthermore, an additional single core was taken from each sediment type (inner Ø 6.2 cm, n = 1) to measure profiles of dissolved oxygen concentrations (vertical increments of 0.2 mm) and pH (increments of 1mm) in the laboratory in triplicate using Unisense microsensors (25 μm and 500 μm tip size for oxygen and pH respectively) and was used to determine the mm depth of the oxic-anoxic border and the geometric mean of the pH. Mean difference analysis using a t-test confirmed that both HBM and LBM sediments indeed only differed in parameters which could be related to MPB activity (i.e. chl a, EPS, phosphate, silicate), but not in other physical or chemical parameters (Table 1).
Fig 1. Geographical location of the Paulina tidal flat (Westerschelde estuary, SW Netherlands) and sampling design.
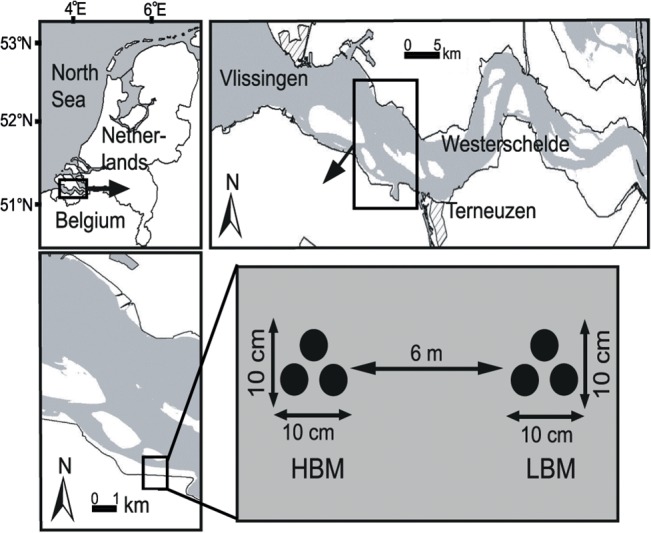
For both estuarine sediments types (HBM and LBM) triplicate samples were taken as close as technical constraints allowed. Additional cores for measuring physico-chemical parameters were taken in immediate vicinity of the sample cores (not shown on figure).
Table 1. Physico-chemical parameters of both estuarine sediments (HBM and LBM) (n = 3).
| Parameter | HBM | LBM |
|---|---|---|
| pH* | 6.69 ± 0.26 | 7.07 ± 0.19 |
| oxic-anoxic interface (mm depth) | 5.25 ± 0.99 | 6.51 ± 0.64 |
| TOM a (%) | 5.73 ± 0.67 | 4.65 ± 0.42 |
| Chl a * (μg/g dw) | 28.61 ± 1.61 | 13.26 ± 3.48 |
| EPS b ,* (mg/mg dry sediment) | 0.0004 ± 0.00015 | 0.0002 ± 0.00003 |
| % mud c | 51.26 ± 9.92 | 45.94 ± 5.08 |
| NH4 + d (μg/l) | 3 632.58 ± 434.83 | 4 445.93 ± 2870.13 |
| NO3 - d (μg/l) | 1 376.85 ± 281.89 | 761.33 ± 351.88 |
| NO2 - d (μg/l) | 10.17 ± 2.31 | 16.24 ± 4.51 |
| PO4 3- d ,* (μg/l) | 1 058.94 ± 66.53 | 1 918.8 ± 1276.08 |
| Si d ,* (μg/l) | 2 125.45 ± 622.9 | 4 356.73 ± 206.36 |
Significant differences (*) between both sediments (t-test, p ≤ 0.05) were detected using a t-test (p ≤ 0.05).; for PO4 3-, a non-parametric test (Mann-Whitney U) was performed. Reliability of significance testing was checked using the Levene’s p-value (>0.05). Millimeter depth till oxic-anoxic border and pH were excluded from statistical testing as no biological replicates were taken for these parameters.
a. TOM, total organic matter.
b. Extracellular polymeric substances.
c. Percentage mud (particle size < 63μm) determined using the Wenthworth grain size chart
d. Pore water concentrations.
DNA extraction
DNA was extracted in triplicate from each HBM and LBM biological replicate (only top 0–1cm) separately to account for both technical and biological variation using a slight modification of Boon et al. [49]. In brief, one gram of sediment, 750 μl 1x TE buffer (1 mM EDTA, 10 mM Tris-HCL, pH8) and 0.5 g of glass beads (Ø 0.1 mm) were added to a 2 ml Safe lock tube (Eppendorf). The mixture was vigorously shaken three times for 90 s using a bead beater (Mixer Mill MM200, Retsch) at a frequency of 30 Hz. Then, 150 μl of lyzosyme (6 mg /150 μl 1x TE buffer) was added and gently mixed for 5 minutes at room temperature. Subsequently, 40 μl of 20% SDS was added, and samples were again slowly mixed for 5 min at room temperature, with subsequent addition of 250 μl 8M ammonium acetate. The supernatant was collected after centrifugation at 7 000 rpm for 15 min at 4°C. A chloroform-isoamylalcohol (24:1) purification was performed, followed by centrifugation at 7 000 rpm for 15 min at 4°C. The aqueous phase was transferred to a new Safe lock tube, and 0.8 volume of isopropanol was added. The precipitation was performed for 1 h at -20°C. Finally, the pellet was obtained by centrifugation at 12 000 rpm for 25 min, washed in 70% ethanol (5 min at 12 000 rpm) and resolved in 50 μl of 1x TE buffer. DNA yields were determined using a Nanodrop 2000 spectrophotometer (Thermo, Scientific) and the quality was checked by gel electrophoresis. Finally, DNA triplicates of a single biological replicate were mixed together for further analysis.
Barcoded amplicon sequencing of nirK, nirS and 16S rRNA genes
Amplicon libraries were prepared using a two-step PCR procedure as recommended by Berry et al. [50] for the 16S rRNA, nirK and nirS genes. Six different multiplex identifiers (MIDs) were used to identify the different replicates per sediment type (S1 Table). Targeting the V3-V1 region of the 16S rRNA, 27 cycles of amplification were performed with the F19-38 (CTGGCTCAGGAYGAACGCTG [51]) / 518R (ATTACCGCGGCTGCTG [52]) primer set. Sequencing starting from the V3 region was selected as it was previously shown to result in good taxonomic assignment [53, 54]. Furthermore, the V1-V3 region is often targeted in sequencing projects and therefore well represented in publically available databases. In a second step, 2 μl of the PCR products of the first reaction was amplified in a 5 cycle PCR with the barcoded PCR primers including the 10 basepair-MID, a sequencing key and the sequencing primer (S1 Table). Each PCR reaction was performed in a 25 μl volume using 1.25 U FastStart High Fidelity Enzyme Blend (Roche), 0.1 μM primers, 0.2 mM dNTP mix and 0.1 mg bovine serum albumin (BSA, only used on the first round of PCR) and milliQ water. All reactions were performed in triplicate to take into account technical variation and were pooled before determination of PCR product quantity and quality. The thermal program consisted of an initial denaturation step of 95°C for 3 min, a cycling program of 94°C for 30 s, 57°C for 45 s, and 72°C for 120 s. Before sequencing, PCR amplicons were purified using the MinElute purification kit (Qiagen) and quantified with a fluorescent stain-based kit (Quant-iT Pico Green, Invitrogen). The quality of the PCR product was assessed on a Bioanalyzer 2100 using a high sensitivity DNA chip (Aligent). Amplicon libraries for the nirK and nirS genes using the primer pairs F1aCu (ATCATGGTSCTGCCGCG)—R3Cu (GCCTCGATCAGRTTGTGGTT) [55] and Cd1aF (GTSAACGTSAAGGARACSGG)—R3cd (GARTTCGGRTGSGTCTTGA) [56, 57] were prepared in a similar way as described above except that the number of cycles in the first PCR round was increased to 35 cycles. All amplicons were sequenced unidirectionally, starting from the forward primer for nirK and nirS and the reverse primer for 16S rRNA gene. Sequencing was performed on a GS FLX Titanium at the NXTGNT sequencing facility of Ghent University.
Sequence analyses
All sequence data were screened and de-multiplexed using default setting of MOTHUR v.1.30.1 [58]. Sequences containing homopolymers of more than 8 nucleotides, mismatches to the barcode (>1) and primers (>2) and sequences shorter than 200 bp were discarded. Subsequently, a MOTHUR-implemented version of pyronoise was used to further denoise the data. Chimera sequences were removed using the Uchime algorithm [59] and, in case of the 16S rRNA gene, potential chloroplast and mitochondrial sequences were also removed. The 16S rRNA gene sequences were aligned using the SILVA reference alignment (release 102) and binned into operational taxonomic units (OTUs) at a 97% gene sequence identity threshold. Sequences were classified using a MOTHUR formatted version of the RDP training set (v.9).
Prior to alignment, the nirK/nirS gene sequence data were checked for the presence of specific functional/conserved regions and screened using the HMM FRAME algorithm [60] included in the FunFrame pipeline [61] to detect and correct frameshift errors. The obtained HMM alignment scores were used for further quality filtering. The cytochrome d1 HMM from Pfam (accession PF02239.10) was used for the nirS gene sequence dataset and sequences with a HMM score ≥ 107 were retained. The nirK primers targeted a region that overlaps with two domains, plastocyanin-like 1 and plastocyanin-like 2, of the nirK gene and a HMM was designed covering the primer target region based on sequences obtained from the Fungene database using the HMMER3 program (Hmmer.org). NirK gene sequences with a HMM score ≥ 48 were retained and for both genes pairwise alignment was performed with the remaining sequences using MOTHUR v.1.30.1 [58]. Cut-offs for binning nirS and nirK gene sequences into OTUs were determined experimentally with the focus on only retaining functional diversity, i.e. binning identical amino acid sequences. A range of threshold distances (5–20%) was tested for both the nirK and nirS gene sequences using MOTHUR v.1.30.1 [58]. Subsequently, all OTU representative gene sequences per threshold distance were translated in silico and pairwise distance matrices of amino acid sequences of all OTU representatives were determined using MEGA 5.10 [62]. At a cut-off of 82% gene sequence identity, all pairwise distances of in silico translated AA sequences of all nirK OTU representatives were > 0, indicating that the sequences of all OTU representatives had a unique AA sequence (~functional diversity). In case of the nirS gene, a similarly obtained cut-off of 80% sequence identity was used for OTU binning. Maximum likelihood phylogenies for both genes were calculated using RAxML 7.4.2 [63, 64]. Protein BLAST searches with the OTU representatives were performed to determine the closest relatives using three different NCBI databases: a non-redundant protein database with/without uncultured/environmental sequences and the whole genome database. If possible, depending on alignable length and e-value, the first five hits were used for further analysis. Node confidence was determined using 1 000 bootstrap replicates.
Quantification of nirK, nirS and 16S rRNA genes
Quantitative real-time PCR (qPCR) of nirK, nirS and 16S rRNA genes was carried out using a Lightcycler 480 II (Roche, Applied Science). Standard curves were prepared from serial dilutions of linearized plasmid with the nirK gene from Alcaligenes faecalis LMG 1229T, nirS gene from Paracoccus sp. R-24615 and 16S rRNA gene from Flavobacterium swingsii LMG 25510, containing between 109 to 101 target gene copies calculated directly from the concentration of the extracted plasmid. DNA concentrations were determined using the Nanodrop 2000 spectrophotometer (Thermo, Scientific). The qPCR assays were carried out in a 20 μl reaction volume composed of SensiMix SYBR No-ROX (Bioline GmbH, Luckenwalde, Germany), 0.4 μM of each primer, 2.5 μl of template DNA (10 ng/μl), 0.1 mg BSA (not used in the nirS assay) and sterilized milliQ water. The same primers as for pyrosequencing were used and the thermal protocol can be found in S2 Table. All reactions per sample were performed in triplicate. Agarose gel electrophoresis, melting curve analysis, cloning and sequencing of the obtained amplicons indicated that the amplification was specific. PCR inhibition was determined by spiking sediment DNA with a known amount of standard DNA and corrected for according to Zaprasis et al. [65].
Statistical analysis
Chao1 estimations, Inverse Simpson diversity indices, rarefaction curves, Venn diagrams, community membership (Jaccard dissimilarity index) and structure (Bray Curtis dissimilarity index) were calculated using MOTHUR v.1.30.1 [58]. Mean differences between HBM and LBM sediment samples were tested using a t-test. Further, correlations between physico-chemical parameters and abundance data from the six collected samples were analysed using product moment correlations (Statistica 5.0, Statsoft 1984–1995). Homogeneities of variances were checked using the Levene’s test (p > 0.05). For PO4 3-, non-parametric Mann-Whitney U tests and Spearman’s rank correlations were used since the necessary assumptions for heterogeneity of variances and normal distributions were not met. Permutational analyses of variance (Permanova) were conducted using the Permanova add-on software for Primer v6 [66].
Nucleotide sequence accession numbers
Complete amplicon libraries of 16S rRNA, nirK and nirS gene sequences derived from barcoded amplicon sequencing were deposited in the Genbank SRA database under study accession number SRP035903.
Results
Sediment sampling and physico-chemical analysis
Two adjacent (± 6 m) sediments with visually different MPB biofilm development were sampled in triplicate. MPB communities in all HBM and LBM replicates were dominated mainly by Navicula spp. (Navicula arenaria var. rostellata, N. phyllepta and N. gregaria) and contained to a lesser degree also Gyrosigma fasciola, Amphora copulata and an unknown Nitzschia species. No significant differences could be observed between the HBM and LBM sediments in TOM, % mud content and pore water concentrations of NO3 -, NO2 - and NH4 + (Table 1). So, the only detected differences between HBM and LBM sediments were specifically related to the presence and activity of MPB: chlorophyll a, a proxy for MPB biomass, and EPS were significantly higher in the HBM sediments, while PO4 3- and Si were significantly lower (p < 0.05, Table 1).
Total bacterial community diversity, estimated richness and structure
The rarefaction curves (S2 Fig) and the ratio observed:estimated 16S rRNA OTU richness for samples HBM1, HBM3, LBM1 and LBM2 (0.72–0.84, Table 2) indicated that the current sampling effort was almost sufficient to capture total bacterial diversity, but showed clear differences between HBM and LBM samples (0.72–0.76 vs. 0.84–0.85; Table 2). The 16S rRNA gene rarefaction curves of a replicate per sample type (HBM2 and LBM3) flattened very quickly in comparison with the other replicates (S2 Fig). As no such discrepancies were observed when using the same DNA material for nir gene sequencing and qPCR (see below), we suspect that the limited numbers of OTUs observed for HBM2 and LBM3 resulted from inconsistent emulsion PCR and therefore excluded both samples from further analyses. After removal of HBM2 and LBM3 sequences from the data set, a total number of 149 946 sequences were binned into 2 482 OTUs across all samples at a cut-off of 0.03. In total, 2.69% of all sequences were determined as chimera or chloroplast and removed from the data set. Both the observed and estimated OTU richness in LBM samples were significantly (p <0.05) higher than in HBM samples, with the majority of OTUs being non-abundant (i.e. relative abundance < 1%; Table 2). Singletons (832 OTUs) and doubletons (485 OTUs) together accounted for approximately half of the total number of OTUs. Given the higher OTU richness in the LBM sediments, % coverage was also significantly lower than in the HBM sediments (p < 0.05, Table 2). Despite this higher richness in LBM samples, the inverse Simpson diversity index did not significantly differ between the HBM and LBM sediments (Table 2).
Table 2. Overview of 16S rRNA (n = 2), nirK (n = 3) and nirS (n = 3) gene sequences derived from HBM and LBM estuarine sediments.
| Richness | Diversity | ||||||
|---|---|---|---|---|---|---|---|
| Target | Sample | # sequences | Library coverage a (%) | # OTUs observed b | # OTUs estimated c | Observed/estimated ratio | Inverse Simpson d |
| 16S rRNA | HBM1 | 31 415 | 99.57 | 779 (A:14-NA:765) | 1021 (927–1172) | 0.76 | 26.70 (26.07–27.36) |
| HBM3 | 37 298 | 99.5 | 821 (A:13-NA:808) | 1138 (1008–1347) | 0.72 | 34.83 (34.06–35.63) | |
| LBM1 | 42 512 | 98.86 | 1235 (A:16-NA:1219) | 1468 (1385–1578) | 0.84 | 24.71 (24.21–25.23) | |
| LBM2 | 38 721 | 99.02 | 1232 (A:15-NA:1217) | 1454 (1379–1554) | 0.85 | 33.76 (33.04–34.52) | |
| nirK | HBM1 | 13 510 | 99.88 | 59 (A: 11-NA:48) | 63 (57–89) | 0.94 | 4.30 (4.20–4.42) |
| HBM2 | 13 602 | 99.9 | 52 (A:7-NA:45) | 61 (57–80) | 0.85 | 4.67 (4.55–4.79) | |
| HBM3 | 9 028 | 99.84 | 59 (A:13-NA:46) | 65 (56–98) | 0.91 | 3.41 (3.32–3.51) | |
| LBM1 | 11 701 | 99.87 | 56 (A:5-NA:51) | 68 (59–101) | 0.87 | 4.05 (3.93–4.18) | |
| LBM2 | 9 779 | 99.82 | 54 (A:8-NA:46) | 68 (59–98) | 0.82 | 2.11 (2.06–2.16) | |
| LBM3 | 10 802 | 99.89 | 59 (A:10-NA:49) | 57 (54–75) | 0.95 | 3.50 (3.42–3.59) | |
| nirS | HBM1 | 6 365 | 99.64 | 58 (A:7-NA:51) | 71 (59–108) | 0.82 | 3.56 (3.48–3.65) |
| HBM2 | 16 427 | 99.76 | 61 (A:8-NA:53) | 59 (50–91) | 1.03 | 3.44 (3.38–3.50) | |
| HBM3 | 4 948 | 99.71 | 47 (A:7-NA:40) | 60 (51–93) | 0.78 | 1.98 (1.92–2.04) | |
| LBM1 | 6 948 | 99.69 | 57 (A:8-NA:49) | 65 (56–97) | 0.88 | 3.27 (3.20–3.34) | |
| LBM2 | 6 684 | 99.68 | 57 (A:7-NA:50) | 66 (56–97) | 0.86 | 2.31 (2.25–2.38) | |
| LBM3 | 17 548 | 99.68 | 72 (A:7-NA:65) | 70 (59–103) | 1.03 | 3.62 (3.56–3.68) | |
a. Good’s coverage estimates sampling completeness and calculates the probability that a randomly selected amplicon sequence from a sample has already been sequenced.
b. A, abundant OTUs (> 1% relative abundance); NA, non-abundant (< 1% relative abundance).
c. Chao1 richness with upper and lower 95% confidence intervals.
d. Inverse Simpson diversity index with upper and lower 95% confidence intervals.
Replicates of each sediment type shared only about half of their total number of OTUs (Fig 2A and 2B); this variation among replicates could be attributed to a high dissimilarity in non-abundant OTUs. Both sample types showed clear differences in community members, although not significant and again mostly among non-abundant OTUs (Table 3), with 78.5% of HBM OTUs and 85.8% of LBM OTUs being unique (Fig 2C). Nevertheless, community structures (Bray-Curtis dissimilarity index, which takes into account community membership as well as OTU relative abundances) did not show significant differences at any level (total community, abundant and non-abundant fraction) between both sample types (Table 3).
Fig 2. Distribution of 16S rRNA gene OTUs.
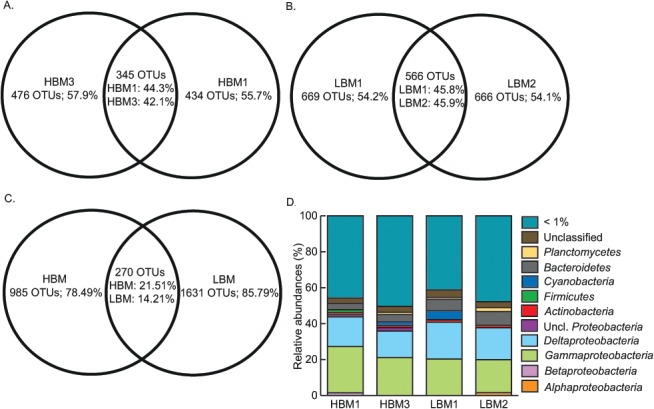
A-C, Venn diagrams representing the number of observed OTUs for the 16S rRNA gene. Comparisons are shown between (A) HBM replicates, (B) LBM replicates, (C) HBM (n = 2) and LBM (n = 2) samples. The number and percentage of unique and shared OTUs are given. D, The relative abundance of abundant 16S rRNA derived OTUs, grouped per phylum, from HBM (n = 2) and LBM (n = 2) sediment samples. Sequences were assigned to OTUs using sequence dissimilarity treshold of 3%. All OTUs with a relative abundance below 1% were grouped. Uncl. stands for unclassified.
Table 3. Dissimilarity in community membership (Jaccard) and structure (Bray-Curtis) between both estuarine sediment types (HBM and LBM) based on 16S rRNA, nirK and nirS genes.
| Jaccard | Bray-Curtis | ||
|---|---|---|---|
| All OTUs | 16S rRNA | 0.732897 | 0.299214 |
| nirK | 0.153846 | 0.185174 | |
| nirS | 0.255556 | 0.065105 | |
| Abundant OTUs | 16S rRNA | 0.357143 | 0.260035 |
| nirK | 0.411765 | 0.181191 | |
| nirS | 0.222222 | 0.0589 | |
| Non-abundant OTUs | 16S rRNA | 0.735594 | 0.402036 |
| nirK | 0.213333 | 0.430228 | |
| nirS | 0.277108 | 0.48913 |
Three different levels were assessed: total community (all), abundant and non-abundant fraction. An OTU was defined as abundant when its relative abundance was larger than 1%. Permanova analyses were performed to determine significant differences in community structure and membership.
Taxonomic diversity in HBM and LBM samples
The estuarine sediments harboured bacteria belonging to a broad range of known phyla, with approximately one third of the OTUs remaining unclassified (30.58% or 759 OTUs, Table 4). The majority of known OTUs belonged to the Proteobacteria (representing one third of all OTUs), Bacteroidetes, Planctomycetes, Actinobacteria, Acidobacteria and Verrucomicrobia. Candidate phyla OD1, OP11, SR1, TM7 and WS 3 were represented by only a few OTUs (Table 4). Twenty-one OTUs were found to have a relative abundance of more than 1% in at least one of the replicates of both sediment types. These abundant OTUs belonged to Alpha- (1 OTU), Beta- (1 OTU), Gamma- (6 OTUs), Delta- (4 OTUs) and unclassified Proteobacteria (1 OTU), Actinobacteria (1 OTU), Firmicutes (1 OTU), Cyanobacteria (1 OTU), Bacteroidetes (2 OTUs), Planctomycetes (1 OTU) and two OTUs with no taxonomic identification (Fig 2D). The phyla Armatimonadetes, Chlorobi, Spirochaetes and Tenericutes appeared to be unique to LBM samples, although they were only represented by one to five OTUs. A significant difference in the number of OTUs between HBM and LBM samples was found for five phyla, namely for Actinobacteria, Bacteroidetes, Proteobacteria (more specifically Alpha-, Gamma-, Delta- and unclassified), Spirochaetes, and Verrucomicrobia, as well as the unclassified fraction (p < 0.05) (Table 4). Eighteen bacterial taxa (families or genera within Alpha-, Gammaproteobacteria or Bacteroidetes) were detected that were previously described to harbour diatom-associated bacteria [34] (Fig 3).
Table 4. Detection and distribution of bacterial phyla (or subdivisions of Proteobacteria).
| Phylum | Total | HBM | LBM | |
|---|---|---|---|---|
| % (# OTUs) | % (# OTUs) | % (# OTUs) | ||
| Acidobacteria | 3.63 (90) | 3.59 (45) | 4.10 (78) | |
| Actinobacteria | 4.67 (116) | 6.06 (76)* | 4.73 (90)* | |
| Armatimonadetes | 0.04 (1) | 0 (0) | 0.05 (1) | |
| Bacteroidetes | 9.31 (231) | 8.45 (106)* | 9.21 (175)* | |
| Chlamydiae | 0.16 (4) | 0.16 (2) | 0.16 (3) | |
| Chlorobi | 0.08 (2) | 0 (0) | 0.11 (2) | |
| Chloroflexi | 1.69 (42) | 1.83 (23) | 1.68 (32) | |
| Cyanobacteria | 0.73 (18) | 0.80 (10) | 0.58 (11) | |
| Deinococcus-Thermus | 0.04 (1) | 0.08 (1) | 0 (0) | |
| Firmicutes | 2.98 (74) | 3.19 (40) | 2.74 (52) | |
| Fusobacteria | 0.08 (2) | 0.08 (1) | 0.11 (2) | |
| OD1 | 1.49 (37) | 1.91 (24) | 1.10 (21) | |
| OP11 | 0.32 (8) | 0.32 (4) | 0.26 (5) | |
| Planctomycetes | 6.21 (154) | 4.06 (51) | 7.21 (137) | |
| Proteobacteria | Alphaproteobacteria | 5.04 (125) | 5.82 (73)* | 4.94 (94)* |
| Betaproteobacteria | 1.09 (27) | 1.59 (20) | 0.95 (18) | |
| Gammaproteobacteria | 14.67 (364) | 15.62 (196)* | 14.52 (276)* | |
| Deltaproteobacteria | 6.93 (172) | 6.69 (84)* | 7.63 (145)* | |
| Zetaproteobacteria | 0.04 (1) | 0.08 (1) | 0 (0) | |
| Epsilonproteobacteria | 0.08 (2) | 0.08 (1) | 0.11 (2) | |
| Unclassified | 2.54 (63) | 2.23 (28)* | 2.47 (47)* | |
| Spirochaetes | 0.20 (5) | 0 (0)* | 0.26 (5)* | |
| SR1 | 0.16 (4) | 0.08 (1) | 0.16 (3) | |
| Synergistetes | 0.12 (3) | 0.16 (2) | 0.05 (1) | |
| Tenericutes | 0.04 (1) | 0 (0) | 0.05 (1) | |
| TM7 | 1.09 (27) | 1.59 (20) | 1.00 (19) | |
| Verrucomicrobia | 5.48 (136) | 6.14 (77)* | 5.89 (112)* | |
| WS3 | 0.52 (13) | 0.64 (8) | 0.53 (10) | |
| Unclassified | 30.58 (759) | 28.76 (361)* | 29.41 (559)* |
Percentages OTU per phylum are given for both sediment types (Total) and for each sediment type separately (HBM and LBM), exact numbers are given between brackets. Phyla containing previously described diatom-associated bacteria are underlined. Significant differences (*) in the number of OTUs between HBM and LBM samples were detected using a t-test (p ≤ 0.05). Reliability of significance testing was checked using the Levene’s p-value (>0.05).
Fig 3. Overview of diatom-associated bacteria found in different phyla.

The inner tier represents diatom-associated bacterial taxa reported by Amin et al. (35). The outer tier depicts diatom-associated bacterial taxa found in our study, either previously reported (blue) or representing potentially new diatom- bacteria associations (red). The highest taxonomic identification of these taxa is shown. Diatom-bacteria associations were identified based on the difference in relative abundances of specific taxa (i.e. number of sequences per taxon) between HBM and LBM sediment samples.
Denitrifying guild diversity, estimated richness and structure
In total, 68 422 quality filtered nirK gene sequences, including 4 693 unique sequences, were assigned to 78 OTUs, with six singleton OTUs as and two doubletons. Comparable numbers of OTUs were found in HBM and LBM samples (Table 2). Rarefaction curves (S3A Fig) and the ratio observed:estimated OTU richness (0.82–0.94, Table 2) indicated that the sequencing effort was almost sufficient to cover the whole nirK diversity detectable with the applied primer set in both sediment types. HBM and LBM samples had similar richness and diversity values (Table 2), with both containing nine abundant OTUs and being dominated by OTUs 1 and 2 (S4A Fig). Although three biological replicates were taken from each sediment type, differences in relative abundances of abundant OTUs between replicates within sample types were as large as between sample types (S4A Fig), while non-abundant OTUs were very similar.
Looking at the in silico translated amino acid (AA) sequences of nirK, the majority of all OTU representatives clustered together with Alphaproteobacteria—spread over four clusters—and Betaproteobacteria (S5 Fig). One distinct cluster (supported by a high bootstrap value of 100%) did not contain known cultivated representatives. Sequences derived from Bacteriodetes and Firmicutes were included but proved unrelated to our OTU representatives. Most OTU representatives clustered together with sequences obtained from marine environments, with the exception of two OTUs (OTU 11 and 66) closely related to sequences from soil.
In total, 58 920 quality filtered nirS gene sequences, with 5 391 unique sequences, were assigned to 90 OTUs, with twelve singleton OTUs and eighteen doubletons. Comparable numbers of OTUs were found in HBM and LBM samples (Table 2). As for nirK, rarefaction curves (S3B Fig) and the ratio observed:estimated OTU richness (0.78–1.03, Table 2) indicated that the current sequencing effort was nearly sufficient to completely catalogue the nirS diversity detectable with the applied primer set in both sample types. HBM and LBM did not significantly differ in nirS gene richness and diversity. OTUs 1–3 dominated in both sediments, and nine OTUs were abundant (S4B Fig). In contrast to nirK, replicates of a single sediment type differed less in relative abundances of abundant OTUs (S4 Fig).
Looking at the in silico translated amino acid (AA) sequences of nirS, most OTU representatives clustered together with Alpha-, Beta and also Gammaproteobacteria (S6 Fig). Multiple clusters without cultured representatives were found, albeit not always supported by high bootstrap values. Most of the OTU representatives clustered together with sequences obtained from marine environments; eight OTUs resembled sequences derived from soil environments or activated sludge.
NirK and nirS community membership and structure were very similar between HBM and LBM samples (Table 3), with only a few unique OTU’s for each sediment type (Fig 4A and 4B). Similar trends were found when considering the abundant and non-abundant community levels separately (Table 3). However, it is interesting to note that OTUs 12 and 13 were exclusively present in the nirK abundant HBM community fraction (S4 Fig).
Fig 4. Venn diagrams representing number of observed OTUs for the nirK (A) and nirS (B) genes.
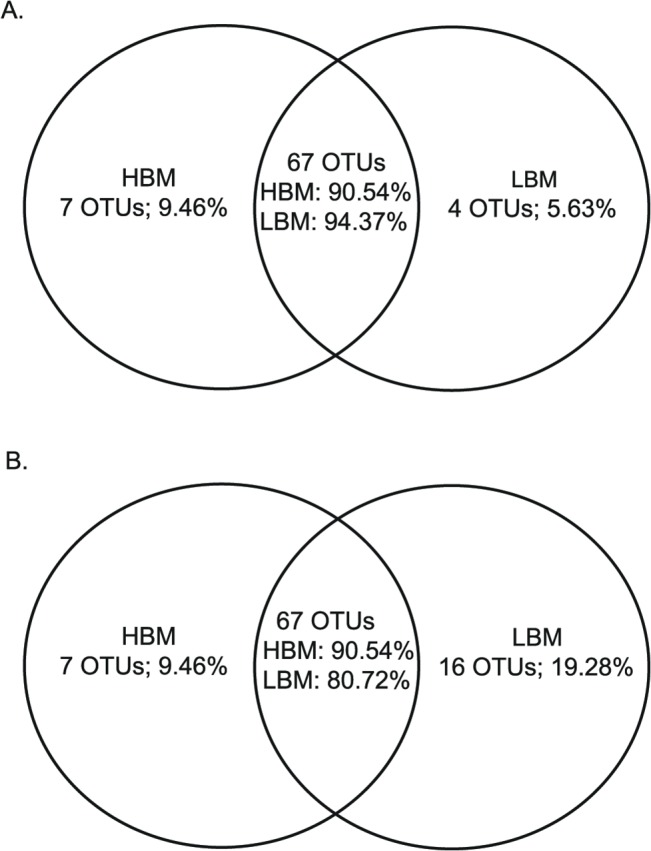
Comparison is shown between HBM and LBM samples (n = 3) for both genes. The number and percentage of unique and shared OTUs are given.
Quantification of the denitrifying guild abundance relative to total bacterial abundance
The abundances of both nirK and nirS denitrifiers and the total bacterial communities of HBM and LBM samples were determined via qPCR (Table 5). The PCR efficiency of the nirS assay (62.9%) was lower than for the other genes, probably due to the exclusion of BSA in this assay because of negative effects on the melting curves. Inhibition factors per gene type were experimentally determined and gene copy numbers were corrected accordingly. The level of inhibition for both nir genes was similar, but much lower than for the 16S rRNA gene. Total bacterial abundances ranged from 8.25 x 105 to 7.98 x 107 copies/g sediment. Final copy numbers of nirK and nirS ranged from 1.48 x 103 to 6.09 x 104 copies/g sediment and 3.66 x 104 to 3.35 x 105 copies/g sediment respectively (Table 5).
Table 5. Absolute and relative quantification of 16S rRNA, nirK and nirS genes in both estuarine sediment types (HBM and LBM).
| Sample | Absolute quantification | Relative quantification | |||
|---|---|---|---|---|---|
| 16S rRNA a gene | nirK b gene | nirS c gene | ratio | ratio | |
| copies / g sediment | copies / g sediment | copies / g sediment | nirK/16S rRNA | nirS/16S rRNA | |
| HBM1 | (6.68 ± 0.85) x 107 | (3.39 ± 0.38) x 104 | (2.93 ± 0.16) x 105 | 0.0005 | 0.0044 |
| HBM2 | (3.75 ± 0.13) x 107 | (5.40 ± 0.72) x 104 | (2.53 ± 0.18) x 105 | 0.0014 | 0.0068 |
| HBM3 | (7.98 ± 0.48) x 107 | (6.09 ± 0.70) x 104 | (3.35 ± 0.26) x 105 | 0.0008 | 0.0042 |
| LBM1 | (5.56 ± 0.64) x 106 | (1.48 ± 0.15) x 103 | (3.66 ± 0.60) x 104 | 0.0003 | 0.0066 |
| LBM2 | (5.19 ± 0.40) x 106 | (8.61 ± 0.36) x 103 | (6.00 ± 0.55) x 104 | 0.0017 | 0.0116 |
| LBM3 | (8.25 ± 0.24) x 105 | (2.72 ± 0.37) x 103 | (6.23 ± 0.14) x 104 | 0.0033 | 0.0755 |
Analyses was performed in triplicate for each biological replicate (n = 3). Gene copy numbers given were corrected for PCR inhibition.
a. Detection limit, 101 copies; PCR efficiency, 90.4%; standard curve R2, 0.99; inhibition factor range: 0.07–0.10.
b. Detection limit, 101 copies; PCR efficiency, 87%; standard curve R2, 0.99; inhibition factor range: 0.36–0.62.
c. Detection limit, 101 copies; PCR efficiency, 62.9%; standard curve R2, 0.99; inhibition factor range: 0.31–0.55.
Significant differences in the abundance of 16S rRNA, nirK and nirS genes were detected between HBM and LBM samples (p < 0.05), with the abundances of all three genes being a log unit higher in HBM samples. NirS genes were also consistently approximately a log unit more abundant than nirK genes (Table 5). For all three genes, a strong positive correlation was found with chl a and EPS whereas a significantly negative correlation was found for Si. Both are in agreement with the higher abundances of the all three genes in HBM sediments (p < 0.05, S3 Table). Furthermore, a positive correlation was found between 16S rRNA abundances and concentration NO3 - and nirK abundances and % TOM (p < 0.05, S3 Table).
Discussion
In intertidal sediments of the Westerschelde estuary, a two-fold increase in biomass (i.e. chl a) of otherwise compositionally identical MPB communities coincided with a disproportionate (ten-fold) increase in both total bacterial and denitrifying community abundances in underlying sediments (Table 5). This contrasts with previous studies that reported correlations between MPB biomass and bacterial abundances with a more moderate quantitative influence (two to three-fold), but, unlike here, these studies did not exclude variation of other environmental factors non-related to MPB (e.g. salinity, temperature, seasonality) [38–40]. Unexpectedly, this two-fold increase in MPB biomass also corresponded with a ten-fold increase in denitrifier abundance. Potential competition with diatoms for nitrate appeared negligible at the NO3 - concentration in the investigated sediments (appr. 1 mg/l, i.e. 10x higher than in sediments with comparable MPB biomass with competition [21, 23]). Thus, microphytobenthos was no specific determinant of denitrifying guilds but rather influenced the bacterial population more generally. This agrees with the facultative nature of the denitrification capacity, and the assumption that the population distribution of denitrifiers is determined by their general ability to compete for natural carbon substrates under aerobic conditions [67]. Unfortunately, our results could not ascertain whether the MPB directly affected the bacterial abundance or whether (identical) non-measured variables influenced both MPB and bacteria simultaneously. Estuarine MPB biofilms can be extremely variable in time (seasonality) and space (from cm to km), with complex interactions of biotic and abiotic parameters responsible for their patchiness [68]. Tidal height, sediment type and hydrodynamism [69, 70], and intraspecies competition [71, 72] are unlikely causes as both samples, from the same sediment type, were taken at the same tidal height and both biofilms contained the same MPB species. However, grazing of MPB, alone or with bacteria, by higher organisms such as grazing benthic deposit feeding invertebrates like harpacticoid copepods [73], (bacterivorous) nematodes [74, 75], polychaetes and bivalves [76] is a plausible factor.
Besides with chl a, total bacterial abundances correlated with other parameters related to MPB activity, namely EPS, produced by both diatoms and bacteria [37] and a potential nutrient source for bacteria and diatoms [29, 31, 41], and Si, a major limiting nutrient for diatoms and hence an important factor controlling primary production [77]. Taxon-specific effects of diatom-derived EPS (i.e. a shift to algal organic matter degrading taxa) could explain the significant decreases in number of OTUs in Actinobacteria, Bacteroidetes, Proteobacteria, Spirochaetes, and Verrucomicrobia (Table 4) in HBM compared to LBM as well as the big, albeit not significant, difference in community membership, specifically for non-abundant taxa (Table 3). The same effects could also be the result of potential bactericidal effects of MPB, not measured in this study (e.g. by polyunsaturated aldehydes (PUAs) [33]). In addition to a shift in number of OTUs, diatom-derived EPS (and PUA) might also explain the increase in total bacterial abundances in HBM samples, as specialist EPS-degrading bacteria are specifically favoured by these additional nutrient sources [27, 29, 78]. Using the relative abundances of specific taxa (i.e. number of sequences per taxon) as a proxy, our data also suggest that growth of other than the typical diatom-associated bacteria [34] appears to have been facilitated (Fig 3). Specifically the phyla Betaproteobacteria (Oxalobacteraceae, Comamonadaceae, Neisseriaceae, Nitrosomonadaceae), Firmicutes (Streptococcaceae, Lactobacillaceae, Clostridales incertae sedis XI), and the candidate division TM7 demonstrated a more than 30% increase in their sequence abundances in HBM compared to LBM sediment (S4 Table). Bacteroidetes and Gammaproteobacteria, the two phyla that are already known to contain taxa able to assimilate diatom-derived EPS [27] as well as to thrive in the presence of PUAs [78], were dominant in both sediment types (if unclassified phyla are ignored), but specific known diatom-associated bacterial genera [34] within both phyla (Pseudoalteromonas, Maribacter and Winogradskyella) were only represented by few sequences (<40 sequences) in HBM samples. The only exception was Pseudomonas [34] well represented (appr. 5000 sequences) and with an increase of over 30% in its relative abundance in HBM sediments. However, considering the relative increase in sequence abundance, Pseudomonas and the above mentioned taxa could not have been solely responsible for the observed tenfold increase in total bacterial abundance. To our surprise, despite all these pronounced differences between both sample types, the Bray-Curtis index for community structure remained unaltered due to the dominant influence of relative abundances of abundant OTUs.
Temporal and spatial heterogeneity can be considerable at the micro- and mesoscale in natural environments, and especially in marine sediments [79], stressing the importance of replication. Biological replicates are often pooled before analyses, resulting in loss of spatial and experimental variability [80]. In our study, replicate environmental samples were not pooled and a remarkable degree of variability between replicates of a single sediment type was observed, sharing less than 50% of their OTUs (Fig 2A and 2B). These differences probably result from the patchiness of the sediments that were sampled and/or incomplete sequencing, indicated by the 16S rRNA gene rarefaction curves, resulting in a snapshot of a subset of especially non-abundant taxa. Unfortunately, due to technical issues only two replicates per sampling site in the 16S rRNA gene sequences analysis were analysed, resulting in a considerable loss of information. Next to patchiness, this variability might also result from stochastic processes as explained by the neutral community assembly theory [81]. Despite containing bacteria belonging to different OTUs, calculated richness and diversity indices were similar for different replicates in both sample types (Table 2), which is in agreement with Bowen et al. [82] who reported similar estimates of richness and diversity when comparing individual and homogenized replicate sediment samples of a salt marsh. Only 14–21% of OTUs were shared between both sediment types, again with the non-abundant fraction being responsible for most of the differences in community members (Fig 2, Table 3). Abundant and non-abundant fractions of bacterial communities clearly have a distinct influence on community composition analysis with the non-abundant fraction as major determining factor for phylogenetic diversity [83]. The potential role of these non-abundant taxa remains unclear although it has been suggested that these non-abundant taxa are disproportionally active in comparison to more abundant taxa [81, 84] or can serve as a microbial seed bank [85, 86].
The two-fold increase in MPB biomass was accompanied by an ten-fold increase of denitrifier abundances, while community structure remained unchanged. NirK and nirS gene abundances correlated well with chl a, Si and EPS concentration. The only available comparable study also showed a positive correlation of MPB biomass with nirK but not with nirS [87], albeit at much lower concentration of chl a (ng instead of μg/g dry weight) than here. Interestingly, nirK and not nirS abundances correlated with levels of organic matter, despite this feature not being significantly different among HBM and LBM samples. This observation confirms differential responses to environmental variables by both nir communities [17, 88], and organic matter as important driver of nirK denitrifier abundances [89, 90]. Other differential drivers (temperature, salinity and concentration of O2, NO3 -, NO2 -, NH4 +) of denitrifier guilds composition [17, 87, 91–94] could not be confirmed here because of absence of correlation or the spatial proximity of the sediment types. In congruence with the few other reports on denitrifier marker genes in estuarine sediments [10, 87, 95], nirS genes were one order of magnitude more abundant than nirK genes, regardless of sample type. Nevertheless, establishing the in situ importance of either nir populations in this system requires further studies acquiring gene transcript data and activity measurements [84, 85, 96, 97].
Our nir sequences had a close match with sequences from estuarine and marine sediments [20, 95, 98–101] as well as from soil and activated sludge [102, 103], suggesting that the Westerschelde estuary has both tidal and terrestrial influences. They were affiliated with sequences derived from Alpha-, Beta- and Gammaproteobacteria which is similar to previous studies using the same primer sets [104, 105]. However, we need to keep in mind the limitation of the PCR based approach to evaluate denitrifier diversity and abundance. It is widely recognized that available primer sets only target part of the denitrifier population because of high sequence divergence in these molecular markers [104, 106, 107]. With a rough calculation of the potentially undetected functional diversity for nirK based on data from the present study we want to put this relevant but often ignored issue into context. All retrieved nirK sequences (assigned to approximately 60 OTUs, see Table 2) were related to those from Alpha- and Betaproteobacteria, with both taxa together responsible for a little over 6% of the total bacterial community diversity (based on number of 16S rRNA OTUs). Let’s assume the following: (i) all Proteobacterial nirK OTUs were detected (although this seems highly unlikely considering the reported difference in amplification success among denitrifiers, even within the same genera [108, 109] and reports of Delta-, Gamma- and Epsilonproteobacteria harbouring nirK which were not picked up in this study), (ii) other phyla harbouring nirK denitrifiers are restricted to Firmicutes and Bacteroidetes, accounting for over 12% of the community diversity (whole genome studies confirm that denitrifiers from both phyla almost exclusively contain nirK but also demonstrate that other phyla do as well), and (iii) the ratio denitrifiers/non-denitrifiers is stable over all phylogenetic lineages, then the undetected nirK denitrifier diversity is actually double of what we could detect (120 OTU undetected vs 60 detected). So at best, our study considered one third of this specific denitrifier guild when addressing our research questions. Currently, only shotgun metagenomics can completely uncover in situ functional diversity and overcome the limitations of PCR-based sequencing which is essential for further identification of the actual key denitrifiers. Bioinformatic approaches to resolve the bottleneck of detecting target genes in short-read metagenomic libraries are currently under development [110] making it a valid alternative for future phylogenetic studies.
In conclusion, our study indicated that a disproportional tenfold increase in bacterial cell numbers in the sediments coincided with a doubling in biomass of MPB. A similar correlation between MPB biomass and both nirK and nirS denitrifiers is evidence for the lack of competition for nitrate between denitrifiers and MPB and suggests that MPB are a general determinant of bacterial populations. No causal relation between MPB biomass and bacterial abundance could be inferred as no plausible environmental variable accounting for the doubling in MPB biomass could be deduced from this dataset. Surprisingly, although bacterial abundance did increase tenfold, no significant differences in total bacterial community structure between both sediment types could be detected.
Supporting Information
(EPS)
(EPS)
(EPS)
Sequences were assigned to OTUs using sequence dissimilarity tresholds of 18% and 20% respectively. All OTUs with a relative abundance below 1% were grouped.
(EPS)
The tree is based on in silico translated amino-acid sequences. One representative per OTU is shown. Environmental sequences were included from marine sediments (black dot), soil (grey dot), urethral sample (discontinuous black circle), activated sludge (black circle) and MFC cathode (grey circle). Sequences only represented by an accession number are unknown environmental sequences. Values in parentheses represent the number of sequences that are present from that OTU in a particular replicate. Red colored boxes indicate sequences affiliated to Alphaproteobacterial nirK sequences and the blue box represent sequences affiliated to Betaproteobacterial nirK sequences. The percentages of replicates trees (n = 1000) in which the OTUs clustered together, are shown next to the branches. Bootstrap values below 50% are not shown.
(EPS)
The tree is based on in silico translated amino-acid sequences. One representative per OTU is shown. Environmental sequences were included from marine sediments (black dot), soil (grey dot), activated sludge (black circle), MFC cathode (grey circle) and a membrane reactor (striped dot). Sequences only represented by an accession number are unknown environmental sequences. Values in parentheses represent the number of sequences that are present from that OTU in a particular replicate. Red colored boxes indicate sequences affiliated to Alphaproteobacterial nirS sequences, blue boxes represent sequences affiliated to Betaproteobacterial nirS sequences, and the green box represents sequences derived from Gammaproteobacteria nirS sequences. The percentages of replicates trees (n = 1000) in which the OTUs clustered together, are shown next to the branches. Bootstrap values below 50% are not shown.
(EPS)
(XLSX)
(XLSX)
Product moment correlation coefficients are given, significant correlations are indicated in bold. Millimeter depth till oxic-anoxic border and pH were excluded from correlation analyses as no biological replicates were taken for these parameters.
(XLSX)
The number of OTUs and sequences in HBM and LMB samples are given.
(XLSX)
Acknowledgments
We thank Dirk Van Gansbeke and Bart Beuselinck for performing the physico-chemical analyses. We also thank NIOZ for facilitating the sampling campaign.
Data Availability
All sequence data files are available in the Genbank SRA database (study accession number SRP035903).
Funding Statement
This work was supported by the Special Research Fund Ghent University (BOF-UGent projects 01G01911 and 0J28410) and Belgian Science Policy Office (BelSPO) project CCAMBIO. KH and CVC are funded by the Fund for Scientific Research (FWO), Flanders for a position as postdoctoral research fellows (FWO- 12BO812N; FWO-1.2.380.11.N.00). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Vitousek PM, Aber JD, Howarth RW, Likens GE, Matson PA, Schindler DW, et al. Human alteration of the global nitrogen cycle: Sources and consequences. Ecological Applications. 1997;7(3):737–50. 10.2307/2269431 . [DOI] [Google Scholar]
- 2. Canfield DE, Glazer AN, Falkowski PG. The Evolution and Future of Earth's Nitrogen Cycle. Science. 2010;330(6001):192–6. 10.1126/science.1186120 . [DOI] [PubMed] [Google Scholar]
- 3. Turner RE, Rabalais NN. Linking landscape and water quality in the Mississippi river basin for 200 years. Bioscience. 2003;53(6):563–72. 10.1641/0006-3568(2003)053[0563:llawqi]2.0.co;2 . [DOI] [Google Scholar]
- 4. Paerl HW, Valdes LM, Peierls BL, Adolf JE, Harding LW. Anthropogenic and climatic influences on the eutrophication of large estuarine ecosystems. Limnology and Oceanography. 2006;51(1):448–62. . [Google Scholar]
- 5. Diaz RJ, Rosenberg R. Spreading dead zones and consequences for marine ecosystems. Science. 2008;321(5891):926–9. 10.1126/science.1156401 . [DOI] [PubMed] [Google Scholar]
- 6. Anderson DM, Glibert PM, Burkholder JM. Harmful algal blooms and eutrophication: Nutrient sources, composition, and consequences. Estuaries. 2002;25(4B):704–26. 10.1007/bf02804901 . [DOI] [Google Scholar]
- 7. Seitzinger S. Denitrification in freshwater and coastal marine ecosystems: ecological and geochemical significance. Limnol Oceanogr. 1988;33(4 part 2):702–24. [Google Scholar]
- 8. Dong LF, Smith CJ, Papaspyrou S, Stott A, Osborn AM, Nedwell DB. Changes in benthic denitrification, nitrate ammonification, and anammox process rates and nitrate and nitrite reductase gene abundances along an estuarine nutrient gradient (the Colne estuary, United Kingdom). Applied and Environmental Microbiology. 2009;75(10):3171–9. 10.1128/aem.02511-08 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Dong LF, Sobey MN, Smith CJ, Rusmana I, Phillips W, Stott A, et al. Dissimilatory reduction of nitrate to ammonium, not denitrification or anammox, dominates benthic nitrate reduction in tropical estuaries. Limnology and Oceanography. 2011;56(1):279–91. 10.4319/lo.2011.56.1.0279 . [DOI] [Google Scholar]
- 10. Smith CJ, Nedwell DB, Dong LF, Osborn AM. Diversity and abundance of nitrate reductase genes (narG and napA), nitrite reductase genes (nirS and nrfA), and their transcripts in estuarine sediments. Applied and Environmental Microbiology. 2007;73(11):3612–22. 10.1128/aem.02894-06 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Giblin AE, Tobias CR, Song B, Weston N, Banta GT, Rivera-Monroy VH. The importance of dissimilatory nitrate reduction to ammonium (DNRA) in the nitrogen cycle of coastal ecosystems. Oceanography. 2013;26(3):124–31. . [Google Scholar]
- 12. Gardner WS, McCarthy MJ. Nitrogen dynamics at the sediment-water interface in shallow, sub-tropical Florida Bay: why denitrification efficiency may decrease with increased eutrophication. Biogeochemistry. 2009;95(2–3):185–98. 10.1007/s10533-009-9329-5 . [DOI] [Google Scholar]
- 13. Trimmer M, Nicholls JC, Deflandre B. Anaerobic ammonium oxidation measured in sediments along the Thames estuary, United Kingdom. Applied and Environmental Microbiology. 2003;69(11):6447–54. 10.1128/aem.69.11.6447-6454.2003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Rich JJ, Dale OR, Song B, Ward BB. Anaerobic ammonium oxidation (Anammox) in Chesapeake Bay sediments. Microbial Ecology. 2008;55(2):311–20. 10.1007/s00248-007-9277-3 . [DOI] [PubMed] [Google Scholar]
- 15. Dale OR, Tobias CR, Song BK. Biogeographical distribution of diverse anaerobic ammonium oxidizing (anammox) bacteria in Cape Fear River Estuary. Environmental Microbiology. 2009;11(5):1194–207. 10.1111/j.1462-2920.2008.01850.x . [DOI] [PubMed] [Google Scholar]
- 16. Philippot L, Hallin S. Finding the missing link between diversity and activity using denitrifying bacteria as a model functional community. Current Opinion in Microbiology. 2005;8(3):234–9. 10.1016/j.mib.2005.04.003 . [DOI] [PubMed] [Google Scholar]
- 17. Jones CM, Hallin S. Ecological and evolutionary factors underlying global and local assembly of denitrifier communities. Isme Journal. 2010;4(5):633–41. 10.1038/ismej.2009.152 . [DOI] [PubMed] [Google Scholar]
- 18. Zumft WG. Cell biology and molecular basis of denitrification. Microbiology and Molecular Biology Reviews. 1997;61(4):533–616. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Graf DRH, Jones CM, Hallin S. Intergenomic comparisons highlight modularity of the denitrification pathway and underpin the importance of community structure for N2O emissions. PLOS One. 2014;9(12). 10.1371/journal.pone.0114118 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Francis CA, O'Mullan GD, Cornwell JC, Ward BB. Transitions in nirS-type denitrifier diversity, community composition, and biogeochemical activity along the Chesepeake Bay estuary Frontiers in microbiology. 2013;4:237 10.3389/fmicb.2013.00237 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Hochard S, Pinazo C, Grenz C, Evans JLB, Pringault O. Impact of microphytobenthos on the sediment biogeochemical cycles: A modeling approach. Ecological Modelling. 2010;221(13–14):1687–701. 10.1016/j.ecolmodel.2010.04.002 . [DOI] [Google Scholar]
- 22. Bartoli M, Castaldelli G, Nizzoli D, Viaroli P. Benthic primary production and bacterial denitrification in a Mediterranean eutrophic coastal lagoon. Journal of Experimental Marine Biology and Ecology. 2012;438:41–51. 10.1016/j.jembe.2012.09.011 . [DOI] [Google Scholar]
- 23. Sundback K, Miles A. Balance between denitrification and microalgal incorporation of nitrogen in microtidal sediments, NE Kattegat. Aquat Microb Ecol. 2000;22(3):291–300. 10.3354/ame022291 . [DOI] [Google Scholar]
- 24. Kamp A, de Beer D, Nitsch JL, Lavik G, Stief P. Diatoms respire nitrate to survive dark and anoxic conditions. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(14):5649–54. 10.1073/pnas.1015744108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lomas MW, Glibert PM. Comparisons of nitrate uptake, storage, and reduction in marine diatoms and flagellates. Journal of Phycology. 2000;36(5):903–13. 10.1046/j.1529-8817.2000.99029.x . [DOI] [Google Scholar]
- 26. Stief P, Kamp A, de Beer D. Role of diatoms in the spatial temporal distribution of intracellular nitrate in intertidal sediment. PLOS One. 2013;8(9):e73257 10.1371/journal.pone.0073257 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Haynes K, Hofmann TA, Smith CJ, Ball AS, Underwood GJC, Osborn AM. Diatom-derived carbohydrates as factors affecting bacterial community composition in estuarine sediments. Applied and Environmental Microbiology. 2007;73(19):6112–24. 10.1128/aem.00551-07 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Risgaard-Petersen N. Coupled nitrification-denitrification in autotrophic and heterotrophic estuarine sediments: On the influence of benthic microalgae. Limnology and Oceanography. 2003;48(1):93–105. . [Google Scholar]
- 29. Taylor JD, McKew BA, Kuhl A, McGenity TJ, Underwood GJC. Microphytobenthic extracellular polymeric substances (EPS) in intertidal sediments fuel both generalist and specialist EPS-degrading bacteria. Limnology and Oceanography. 2013;58(4):1463–80. 10.4319/lo.2013.58.4.1463 . [DOI] [Google Scholar]
- 30. Bellinger BJ, Underwood GJC, Ziegler SE, Gretz MR. Significance of diatom-derived polymers in carbon flow dynamics within estuarine biofilms determined through isotopic enrichment. Aquat Microb Ecol. 2009;55(2):169–87. 10.3354/ame01287 . [DOI] [Google Scholar]
- 31. McKew BA, Dumbrell AJ, Taylor JD, McGenity TJ, Underwood GJC. Differences between aerobic and anaerobic degradation of microphytobenthic biofilm-derived organic matter within intertidal sediments. Fems Microbiology Ecology. 2013;84(3):495–509. 10.1111/1574-6941.12077 . [DOI] [PubMed] [Google Scholar]
- 32. Middelburg JJ, Barranguet C, Boschker HTS, Herman PMJ, Moens T, Heip CHR. The fate of intertidal microphytobenthos carbon: An in situ C-13-labeling study. Limnology and Oceanography. 2000;45(6):1224–34. . [Google Scholar]
- 33. Ianora A, Miralto A. Toxigenic effects of diatoms on grazers, phytoplankton and other microbes: a review. Ecotoxicology. 2010;19(3):493–511. 10.1007/s10646-009-0434-y . [DOI] [PubMed] [Google Scholar]
- 34. Amin SA, Parker MS, Armbrust EV. Interactions between Diatoms and Bacteria. Microbiology and Molecular Biology Reviews. 2012;76(3):667–84. 10.1128/mmbr.00007-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Hanlon ARM, Bellinger B, Haynes K, Xiao G, Hofmann TA, Gretz MR, et al. Dynamics of extracellular polymeric substance (EPS) production and loss in an estuarine, diatom-dominated, microalgal biofilm over a tidal emersion-immersion period. Limnology and Oceanography. 2006;51(1):79–93. . [Google Scholar]
- 36. Yallop ML, Paterson DM, Wellsbury P. Interrelationships between rates of microbial production, exopolymer production, microbial biomass, and sediment stability in biofilms of intertidal sediments. Microbial Ecology. 2000;39(2):116–27. 10.1007/s002489900186 . [DOI] [PubMed] [Google Scholar]
- 37. Lubarsky HV, Hubas C, Chocholek M, Larson F, Manz W, Paterson DM, et al. The stabilisation potential of individual and mixed assemblages of natural bacteria and microalgae. PLOS One. 2010;5(11):e13794 10.1371/journal.pone.0013794 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Orvain F, De Crignis M, Guizien K, Lefebvre S, Mallet C, Takahashi E, et al. Tidal and seasonal effects on the short-term temporal patterns of bacteria, microphytobenthos and exopolymers in natural intertidal biofilms (Brouage, France). Journal of Sea Research. 2014; 10.1016/j.seares.2014.02.018 [DOI]
- 39. Belzile C, Brugel S, Nozais C, Gratton Y, Demers S. Variations of the abundance and nucleic acid content of heterotrophic bacteria in Beaufort Shelf waters during winter and spring. Journal of Marine Systems. 2008;74(3–4):946–56. 10.1016/j.jmarsys.2007.12.010 . [DOI] [Google Scholar]
- 40. Gurung TB, Kagami M, Yoshida T, Urabe J. Relative importance of biotic and abiotic factors affecting bacterial abundance in Lake Biwa: an empirical analysis. Limnology 2001;2:19–28. [Google Scholar]
- 41. Stal LJ. Microphytobenthos as a biogeomorphological force in intertidal sediment stabilization. Ecological Engineering. 2010;36(2):236–45. 10.1016/j.ecoleng.2008.12.032 . [DOI] [Google Scholar]
- 42. Soetaert K, Herman PMJ. Nitrogen dynamics in the Westerschelde estuary (SW Netherlands) estimated by means of the ecosystem model MOSES. Hydrobiologia. 1995;311(1–3):225–46. 10.1007/bf00008583 . [DOI] [Google Scholar]
- 43. Soetaert K, Middelburg JJ, Heip C, Meire P, Van Damme S, Maris T. Long-term change in dissolved inorganic nutrients in the heterotrophic Scheldt estuary (Belgium, The Netherlands). Limnology and Oceanography. 2006;51(1):409–23. . [Google Scholar]
- 44. Dahnke K, Moneta A, Veuger B, Soetaert K, Middelburg JJ. Balance of assimilative and dissimilative nitrogen processes in a diatom-rich tidal flat sediment. Biogeosciences. 2012;9(10):4059–70. 10.5194/bg-9-4059-2012 . [DOI] [Google Scholar]
- 45. Van Colen C, Rossi F, Montserrat F, Andersson MGI, Gribsholt B, Herman PMJ, et al. Organism-sediment interactions govern post-hypoxia recovery of ecosystem functioning. PLOS One. 2012;7(11):e49795 10.1371/journal.pone.0049795 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Jeffrey SW, Mantoura RFC, Wright SW. Phytoplankton pigments in oceanography United Nations Educational and Scientific Organization, Paris: 1997. [Google Scholar]
- 47. Wright SW, Jeffrey SW. High-resolution HPLC system for chlorophylls and carotenoids of marine phytoplankton In:Jeffrey S.W.,Mantoura R.F.C., Wright S.W.(Eds.),Phytoplankton Pigments in Oceanography.Scorand Unesco; 1997:343–61. [Google Scholar]
- 48. Dubois M, Gilles KA, Hamilton JK, Rebers PA, Smith F. Colorimetric method for determination of sugars and related substances. Analytical Chemistry. 1956;28(3):350–6. 10.1021/ac60111a017 . [DOI] [Google Scholar]
- 49. Boon N, Goris J, De Vos P, Verstraete W, Top EM. Bioaugmentation of activated sludge by an indigenous 3-chloroaniline-degrading Comamonas testosteroni strain, I2gfp. Applied and Environmental Microbiology. 2000;66(7):2906–13. 10.1128/aem.66.7.2906-2913.2000 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Berry D, Ben Mahfoudh K, Wagner M, Loy A. Barcoded primers used in multiplex amplicon pyrosequencing bias amplification. Applied and Environmental Microbiology. 2011;77(21):7846–9. 10.1128/aem.05220-11 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Koort J, Murros A, Coenye T, Eerola S, Vandamme P, Sukura A, et al. Lactobacillus oligofermentans sp nov., associated with spoilage of modified-atmosphere-packaged poultry products. Applied and Environmental Microbiology. 2005;71(8):4400–6. 10.1128/aem.71.8.4400-4406.2005 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Yu ZT, Morrison M. Comparisons of different hypervariable regions of rrs genes for use in fingerprinting of microbial communities by PCR-denaturing gradient gel electrophoresis. Applied and Environmental Microbiology. 2004;70(8):4800–6. 10.1128/aem.70.8.4800-4806.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Huse SM, Dethlefsen L, Huber JA, Welch DM, Relman DA, Sogin ML. Exploring Microbial Diversity and Taxonomy Using SSU rRNA Hypervariable Tag Sequencing. PLOS Genetics. 2008;4(11). doi: e1000255 10.1371/journal.pgen.1000255 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Mizrahi-Man O DE, Gilad Y. Taxonomic classification of bacterial 16S rRNA genes using short sequencing reads: evaluation of effective study designs. PLOS One. 2013;8((1)):e53608 10.1371/journal.pone.0053608 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Hallin S, Lindgren PE. PCR detection of genes encoding nitrite reductase in denitrifying bacteria. Applied and Environmental Microbiology. 1999;65(4):1652–7. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Throback IN, Enwall K, Jarvis A, Hallin S. Reassessing PCR primers targeting nirS, nirK and nosZ genes for community surveys of denitrifying bacteria with DGGE. Fems Microbiology Ecology. 2004;49(3):401–17. 10.1016/j.femsec.2004.04.011 . [DOI] [PubMed] [Google Scholar]
- 57. Michotey V, Mejean V, Bonin P. Comparison of methods for quantification of cytochrome cd(1)-denitrifying bacteria in environmental marine samples. Applied and Environmental Microbiology. 2000;66(4):1564–71. 10.1128/aem.66.4.1564-1571.2000 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58. Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, et al. Introducing mothur: open-source, platform-independent, community-supported software for describing and comparing microbial communities. Applied and Environmental Microbiology. 2009;75(23):7537–41. 10.1128/aem.01541-09 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Edgar RC, Haas BJ, Clemente JC, Quince C, Knight R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics. 2011;27(16):2194–200. 10.1093/bioinformatics/btr381 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Zhang Y, Sun YN. HMM-FRAME: accurate protein domain classification for metagenomic sequences containing frameshift errors. Bmc Bioinformatics. 2011;12:198 10.1186/471-2105-12-198 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Weisman D, Yasuda M, Bowen JL. FunFrame: functional gene ecological analysis pipeline. Bioinformatics. 2013;29(9):1212–4. 10.1093/bioinformatics/btt123 . [DOI] [PubMed] [Google Scholar]
- 62. Kumar S, Nei M, Dudley J, Tamura K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Briefings in Bioinformatics. 2008;9(4):299–306. 10.1093/bib/bbn017 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Stamatakis A. RAxML-VI-HPC: Maximum likelihood-based phylogenetic analyses with thousands of taxa and mixed models. Bioinformatics. 2006;22(21):2688–90. 10.1093/bioinformatics/btl446 . [DOI] [PubMed] [Google Scholar]
- 64.Ott M, Zola J, Aluru S, Stamatakis A, ACM. Large-scale maximum likelihood-based phylogenetic analysis on the IBM BlueGene/L. 2007 Acm/Ieee Sc07 Conference. 2010:466–76. .
- 65. Zaprasis A, Liu YJ, Liu SJ, Drake HL, Horn MA. Abundance of novel and diverse tfdA-like genes, encoding putative phenoxyalkanoic acid herbicide-degrading dioxygenases, in soil. Applied and Environmental Microbiology. 2010;76(1):119–28. 10.1128/aem.01727-09 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Anderson MJ, Gorley RN, Clarcke KR. Permanova+ for primer: guide to software and 366 statistical methods Primer-e: Plymouth, UK: 2008. [Google Scholar]
- 67.Tiedje JM. Ecology of denitrification and dissimilatory nitrate reduction to ammonium. Environmental microbiology of anaerobes. 1988:179–244.
- 68. Jesus B, Brotas V, Marani M, Paterson DM. Spatial dynamics of microphytobenthos determined by PAM fluorescence. Estuarine Coastal and Shelf Science. 2005;65(1–2):30–42. 10.1016/j.ecss.2005.05.005 . [DOI] [Google Scholar]
- 69. Jesus B, Brotas V, Ribeiro L, Mendes CR, Cartaxana P, Paterson DM. Adaptations of microphytobenthos assemblages to sediment type and tidal position. Cont Shelf Res. 2009;29(13):1624–34. 10.1016/j.csr.2009.05.006 . [DOI] [Google Scholar]
- 70. Fonseca ALD, Machado ED, Brandini FP, Brandini N. Microphytobenthic biomass on a subtropical intertidal flat of Paranagua Bay (Southern Brazil): spatio-temporal distribution and the influence of environmental conditions. Brazilian Journal of Oceanography. 2013;61(2):83–92. . [Google Scholar]
- 71. Seuront L, Spilmont N. Self-organized criticality in intertidal microphytobenthos patch patterns. Physica a-Statistical Mechanics and Its Applications. 2002;313(3–4):513–39. 10.1016/s0378-4371(02)00989-5 . [DOI] [Google Scholar]
- 72. Brito A, Newton A, Tett P, Fernandes TF. Temporal and spatial variability of microphytobenthos in a shallow lagoon: Ria Formosa (Portugal). Estuarine Coastal and Shelf Science. 2009;83(1):67–76. 10.1016/j.ecss.2009.03.023 . [DOI] [Google Scholar]
- 73. BuffanDubau E, deWit R, Castel J. Feeding selectivity of the harpacticoid copepod Canuella perplexa in benthic muddy environments demonstrated by HPLC analyses of chlorin and carotenoid pigments. Marine Ecology Progress Series. 1996;137(1–3):71–82. 10.3354/meps137071 . [DOI] [Google Scholar]
- 74. Moens T, Vincx M. Observations on the feeding ecology of estuarine nematodes. Journal of the Marine Biological Association of the United Kingdom. 1997;77(1):211–27. . [Google Scholar]
- 75. Moens T, Verbeeck L, de Maeyer A, Swings J, Vincx M. Selective attraction of marine bacterivorous nematodes to their bacterial food. Marine Ecology Progress Series. 1999;176:165–78. 10.3354/meps176165 . [DOI] [Google Scholar]
- 76. Van Colen C, Underwood GJC, Serodio J, Paterson DM. Ecology of intertidal microbial biofilms: Mechanisms, patterns and future research needs. Journal of Sea Research. 2014;92:2–5. 10.1016/j.seares.2014.07.003 . [DOI] [Google Scholar]
- 77. Martin-Jezequel V, Hildebrand M, Brzezinski MA. Silicon metabolism in diatoms: Implications for growth. Journal of Phycology. 2000;36(5):821–40. 10.1046/j.1529-8817.2000.00019.x . [DOI] [Google Scholar]
- 78. Ribalet F, Intertaglia L, Lebaron P, Casotti R. Differential effect of three polyunsaturated aldehydes on marine bacterial isolates. Aquatic Toxicology. 2008;86(2):249–55. 10.1016/j.aquatox.2007.11.005 . [DOI] [PubMed] [Google Scholar]
- 79. Lozupone CA, Knight R. Global patterns in bacterial diversity. Proceedings of the National Academy of Sciences of the United States of America. 2007;104(27):11436–40. 10.1073/pnas.0611525104 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80. Prosser JI. Replicate or lie. Environmental Microbiology. 2010;12(7):1806–10. 10.1111/j.1462-2920.2010.02201.x . [DOI] [PubMed] [Google Scholar]
- 81. Nemergut DR, Schmidt SK, Fukami T, O'Neill SP, Bilinski TM, Stanish LF, et al. Patterns and processes of microbial community assembly. Microbiology and Molecular Biology Reviews. 2013;77(3):342–56. 10.1128/mmbr.00051-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82. Bowen JL, Morrison HG, Hobbie JE, Sogin ML. Salt marsh sediment diversity: a test of the variability of the rare biosphere among environmental replicates. Isme Journal. 2012;6(11):2014–23. 10.1038/ismej.2012.47 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83. Sogin ML, Morrison HG, Huber JA, Mark Welch D, Huse SM, Neal PR, et al. Microbial diversity in the deep sea and the underexplored "rare biosphere". Proceedings of the National Academy of Sciences of the United States of America. 2006;103(32):12115–20. 10.1073/pnas.0605127103 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84. Campbell BJ, Yu LY, Heidelberg JF, Kirchman DL. Activity of abundant and rare bacteria in a coastal ocean. Proceedings of the National Academy of Sciences of the United States of America. 2011;108(31):12776–81. 10.1073/pnas.1101405108 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85. Jones SE, Lennon JT. Dormancy contributes to the maintenance of microbial diversity. Proceedings of the National Academy of Sciences of the United States of America. 2010;107(13):5881–6. 10.1073/pnas.0912765107 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86. Lennon JT, Jones SE. Microbial seed banks: the ecological and evolutionary implications of dormancy. Nat Rev Microbiol. 2011;9(2):119–30. 10.1038/nrmicro2504 . [DOI] [PubMed] [Google Scholar]
- 87. Abell GCJ, Revill AT, Smith C, Bissett AP, Volkman JK, Robert SS. Archaeal ammonia oxidizers and nirS-type denitrifiers dominate sediment nitrifying and denitrifying populations in a subtropical macrotidal estuary. Isme Journal. 2010;4(2):286–300. 10.1038/ismej.2009.105 . [DOI] [PubMed] [Google Scholar]
- 88. Smith JM, Ogram A. Genetic and functional variation in denitrifier populations along a short-term restoration chronosequence. Applied and Environmental Microbiology. 2008;74(18):5615–20. 10.1128/aem.00349-08 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89. Kandeler E, Deiglmayr K, Tscherko D, Bru D, Philippot L. Abundance of narG, nirS, nirK, and nosZ genes of denitrifying bacteria during primary successions of a glacier foreland. Applied and Environmental Microbiology. 2006;72(9):5957–62. 10.1128/aem.00439-06 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90. Barta J, Melichova T, Vanek D, Picek T, Santruckova H. Effect of pH and dissolved organic matter on the abundance of nirK and nirS denitrifiers in spruce forest soil. Biogeochemistry. 2010;101(1–3):123–32. 10.1007/s10533-010-9430-9 . [DOI] [Google Scholar]
- 91. Liu XD, Tiquia SM, Holguin G, Wu LY, Nold SC, Devol AH, et al. Molecular diversity of denitrifying genes in continental margin sediments within the oxygen-deficient zone off the Pacific coast of Mexico. Applied and Environmental Microbiology. 2003;69(6):3549–60. 10.1128/aem.69.6.3549-3560.2003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92. Taroncher-Oldenburg G, Griner EM, Francis CA, Ward BB. Oligonucleotide microarray for the study of functional gene diversity in the nitrogen cycle in the environment. Applied and Environmental Microbiology. 2003;69(2):1159–71. 10.1128/aem.69.2.1159-1171.2003 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93. Jayakumar DA, Francis CA, Naqvi SWA, Ward BB. Diversity of nitrite reductase genes (nirS) in the denitrifying water column of the coastal Arabian Sea. Aquat Microb Ecol. 2004;34(1):69–78. 10.3354/ame034069 . [DOI] [Google Scholar]
- 94. Hannig M, Braker G, Dippner J, Jurgens K. Linking denitrifier community structure and prevalent biogeochemical parameters in the pelagial of the central Baltic Proper (Baltic Sea). Fems Microbiology Ecology. 2006;57(2):260–71. 10.1111/j.1574-6941.2006.00116.x . [DOI] [PubMed] [Google Scholar]
- 95. Mosier AC, Francis CA. Denitrifier abundance and activity across the San Francisco Bay estuary. Environ Microbiol Rep. 2010;2(5):667–76. 10.1111/j.1758-2229.2010.00156.x . [DOI] [PubMed] [Google Scholar]
- 96. Bodelier PLE, Meima-Franke M, Hordijk CA, Steenbergh AK, Hefting MM, Bodrossy L, et al. Microbial minorities modulate methane consumption through niche partitioning. Isme Journal. 2013;7(11):2214–28. 10.1038/ismej.2013.99 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97. Hunt DE, Lin YJ, Church MJ, Karl DM, Tringe SG, Izzo LK, et al. Relationship between abundance and specific activity of bacterioplankton in open ocean surface waters. Applied and Environmental Microbiology. 2013;79(1):177–84. 10.1128/aem.02155-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98. Santoro AE, Boehm AB, Francis CA. Denitrifier community composition along a nitrate and salinity gradient in a coastal aquifer. Applied and Environmental Microbiology. 2006;72(3):2102–9. 10.1128/aem.72.3.2102-2109.2006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99. Sokoll S, Holtappels M, Lam P, Collins G, Schluter M, Lavik G, et al. Benthic nitrogen loss in the Arabian Sea off Pakistan. Frontiers in microbiology. 2012;3(395):395 10.3389/fmicb.2012.00395 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100. Nogales B, Timmis KN, Nedwell DB, Osborn AM. Detection and diversity of expressed denitrification genes in estuarine sediments after reverse transcription-PCR amplification from mRNA. Applied and Environmental Microbiology. 2002;68(10):5017–25. 10.1128/aem.68.10.5017-5025.2002 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101. Dang HY, Wang CY, Li J, Li TG, Tian F, Jin W, et al. Diversity and distribution of sediment nirS-encoding bacterial assemblages in response to environmental gradients in the eutrophied Jiaozhou bay, China. Microbial Ecology. 2009;58(1):161–9. 10.1007/s00248-008-9469-5 . [DOI] [PubMed] [Google Scholar]
- 102. Hallin S, Throback IN, Dicksved J, Pell M. Metabolic profiles and genetic diversity of denitrifying communities in activated sludge after addition of methanol or ethanol. Applied and Environmental Microbiology. 2006;72(8):5445–52. 10.1128/aem.00809-06 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103. Hashimoto T, Koga M, Masaoka Y. Advantages of a diluted nutrient broth medium for isolating N-2-producing denitrifying bacteria of alpha-Proteobacteria in surface and subsurface upland soils. Soil Science and Plant Nutrition. 2009;55(5):647–59. 10.1111/j.1747-0765.2009.00404.x . [DOI] [Google Scholar]
- 104. Palmer K, Biasi C, Horn MA. Contrasting denitrifier communities relate to contrasting N2O emission patterns from acidic peat soils in arctic tundra. Isme Journal. 2012;6(5):1058–77. 10.1038/ismej.2011.172 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105. Depkat-Jakob PS, Brown GG, Tsai SM, Horn MA, Drake HL. Emission of nitrous oxide and dinitrogen by diverse earthworm families from Brazil and resolution of associated denitrifying and nitrate-dissimilating taxa. Fems Microbiology Ecology. 2013;83(2):375–91. 10.1111/j.1574-6941.2012.01476.x . [DOI] [PubMed] [Google Scholar]
- 106. Penton CR, Johnson TA, Quensen JF III, Iwai S, cole JR, Tiedje JM. Functional genes to assess nitrogen cycling and aromatic hydrocarbon degradation: primers and processing matter. Frontiers in microbiology. 2013;4:279 10.3389/fmicb.2013.00279 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107. Green SJ, Prakash O, Gihring TM, Akob DM, Jasrotia P, Jardine PM, et al. Denitrifying bacteria isolated from terrestrial subsurface sediments exposed to mixed-waste contamination. Applied and Environmental Microbiology. 2010;76(10):3244–54. 10.1128/aem.03069-09 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108. Heylen K, Gevers D, Vanparys B, Wittebolle L, Geets J, Boon N, et al. The incidence of nirS and nirK and their genetic heterogeneity in cultivated denitrifiers. Environmental Microbiology. 2006;8(11):2012–21. 10.1111/j.1462-2920.2006.01081.x . [DOI] [PubMed] [Google Scholar]
- 109. Heylen K, Vanparys B, Gevers D, Wittebolle L, Boon N, De Vos P. Nitric oxide reductase (norB) gene sequence analysis reveals discrepancies with nitrite reductase (nir) gene phylogeny in cultivated denitrifiers. Environmental Microbiology. 2007;9(4):1072–7. 10.1111/j.1462-2920.2006.01194.x . [DOI] [PubMed] [Google Scholar]
- 110. Orellana LH, Rodriguez-R LM, Higgins S, Chee-Sanford JC, Sanford RA, Ritalahti KM, et al. Detecting nitrous oxide reductase (nosZ) genes in soil metagenomes: method development and implications for the nitrogen cycle. Mbio. 2014;5(3). 10.1128/mBio.01193-14 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(EPS)
(EPS)
(EPS)
Sequences were assigned to OTUs using sequence dissimilarity tresholds of 18% and 20% respectively. All OTUs with a relative abundance below 1% were grouped.
(EPS)
The tree is based on in silico translated amino-acid sequences. One representative per OTU is shown. Environmental sequences were included from marine sediments (black dot), soil (grey dot), urethral sample (discontinuous black circle), activated sludge (black circle) and MFC cathode (grey circle). Sequences only represented by an accession number are unknown environmental sequences. Values in parentheses represent the number of sequences that are present from that OTU in a particular replicate. Red colored boxes indicate sequences affiliated to Alphaproteobacterial nirK sequences and the blue box represent sequences affiliated to Betaproteobacterial nirK sequences. The percentages of replicates trees (n = 1000) in which the OTUs clustered together, are shown next to the branches. Bootstrap values below 50% are not shown.
(EPS)
The tree is based on in silico translated amino-acid sequences. One representative per OTU is shown. Environmental sequences were included from marine sediments (black dot), soil (grey dot), activated sludge (black circle), MFC cathode (grey circle) and a membrane reactor (striped dot). Sequences only represented by an accession number are unknown environmental sequences. Values in parentheses represent the number of sequences that are present from that OTU in a particular replicate. Red colored boxes indicate sequences affiliated to Alphaproteobacterial nirS sequences, blue boxes represent sequences affiliated to Betaproteobacterial nirS sequences, and the green box represents sequences derived from Gammaproteobacteria nirS sequences. The percentages of replicates trees (n = 1000) in which the OTUs clustered together, are shown next to the branches. Bootstrap values below 50% are not shown.
(EPS)
(XLSX)
(XLSX)
Product moment correlation coefficients are given, significant correlations are indicated in bold. Millimeter depth till oxic-anoxic border and pH were excluded from correlation analyses as no biological replicates were taken for these parameters.
(XLSX)
The number of OTUs and sequences in HBM and LMB samples are given.
(XLSX)
Data Availability Statement
All sequence data files are available in the Genbank SRA database (study accession number SRP035903).