

Abstract
Enterohaemorrhagic Escherichia coli (EHEC) O157:H7 infection in humans can cause acute haemorrhagic colitis and severe haemolytic uraemic syndrome. The role of enterohaemolysin (Ehx) in the pathogenesis of O157:H7-mediated disease in humans remains undefined. Recent studies have revealed the importance of the inflammatory response in O157:H7 pathogenesis in humans. We previously reported that Ehx markedly induced interleukin-1β (IL-1β) production in human macrophages. Here, we investigated the disparity in Ehx-induced IL-1β production between human and mouse macrophages and explored the underlying mechanism regarding the activation of NOD-like receptor family, pyrin domain containing 3 (NLRP3) inflammasomes. In contrast to the effects on human differentiated THP-1 cells and peripheral blood mononuclear cells, Ehx exerted no effect on IL-1β production in mouse macrophages and splenocytes because of a disparity in pro-IL-1β cleavage into mature IL-1β upon caspase-1 activation. Additionally, Ehx significantly contributed to O157:H7-induced ATP release from THP-1 cells, which was not detected in mouse macrophages. Confocal microscopy demonstrated that Ehx was a key inducer of cathepsin B release in THP-1 cells but not in mouse IC-21 cells upon O157:H7 challenge. Inhibitor experiments indicated that O157:H7-induced IL-1β production was largely dependent upon caspase-1 activation and partially dependent upon ATP signalling and cathepsin B release, which were both involved in NLRP3 activation. Moreover, inhibition of K+ efflux drastically diminished O157:H7-induced IL-1β production and cytotoxicity. The findings in this study may shed light on whether and how the Ehx contributes to the development of haemolytic uraemic syndrome in human O157:H7 infection.
Keywords: enterohaemolysin, enterohaemorrhagic Escherichia coli, human, inflammasomes, interleukin-1β, mouse
Introduction
Enterohaemorrhagic Escherichia coli (EHEC) O157:H7, one of the most common food-borne pathogens, is widely distributed in both humans and animals. EHEC O157:H7 infection in humans can cause acute haemorrhagic diarrhoea, haemorrhagic colitis and severe haemolytic uraemic syndrome (HUS).1 The pathogenesis of EHEC O157:H7-mediated diseases in humans is not fully understood and a main reason is the lack of an ideal animal model. There is no single animal model able to recapitulate all of the features of EHEC O157:H7 infection in humans, suggesting a difference in pathogenesis of EHEC O157:H7 between humans and other species.2 Although Shiga toxin (Stx) is considered a major virulence factor contributing to the development of HUS, the host inflammatory response has also been shown to be involved in the pathogenesis of Stx-induced vascular lesions in a mouse model.3 The inflammatory mediator interleukin-1β (IL-1β) has been found to enhance the expression of the glycosphingolipid receptor Gb3 on endothelial cells, which binds to Stx and promotes its cytotoxicity. This may partially explain the role of IL-1β and the host inflammatory response in HUS development.4 Supportive clinical data have also shown that the level of inflammatory cytokines in the serum of HUS patients is significantly increased compared with non-HUS patients.5,6 Of note, mice infected with EHEC do not develop HUS symptoms. Even the intraperitoneal administration of Stx fails to induce HUS development in a mouse model, unless lipopolysaccharide (LPS) is used in combination to induce the inflammatory response, which contributes to the pathogenesis of HUS in mice.7,8
Interleukin-1β plays a key role in initiating and maintaining the inflammatory response. Two distinct signals are required for the secretion of biologically active IL-1β. First, the 31 000 molecular weight, biologically inactive IL-1β precursor (pro-IL-1β) is synthesized via nuclear factor-κB activation, for example, by Toll-like receptors or cytokines. Second, inactive pro-IL-1β is processed to form a mature molecule with molecular weight 17 000, and released, which requires cleavage by cysteine protease caspase-1.9 Caspase-1 is activated by inflammasomes, cytoplasmic multiprotein complexes that are capable of sensing stress and danger signals.10 One of the most studied inflammasome complexes is NOD-like receptor family, pyrin domain containing 3 (NLRP3; also known as NALP3, cryopyrin, or CIAS1). The NLRP3 inflammasome can be activated in response to a number of diverse stimuli, including those of microbial, endogenous and exogenous origins. It has been reported that some bacterial pore-forming toxins can activate caspase-1 in an NLRP3-dependent manner.11,12 However, the common pathway by which these stimuli converge to activate NLRP3 remains unclear.
Enterohaemolysin (Ehx) is a repeat in toxin (RTX) cytotoxin encoded by a 93-kb plasmid (pO157) in EHEC O157:H7. It has been described as a pore-forming toxin with lytic and toxic effects on mammalian host cells. Along with the IL-1β level in the serum, the level of anti-Ehx in HUS patients is also significantly increased, indicating that Ehx may play a significant role in HUS caused by EHEC O157:H7. It has been reported that EHEC activates the NLRP3 inflammasome to induce IL-1β production.13 Our previous study further demonstrated that Ehx contributes to EHEC O157:H7-induced cytotoxicity and IL-1β production in human macrophages and that NLRP3 inflammasome activation is required for IL-1β production in response to EHEC O157:H7 infection.14 However, the sensors that are used may be specific to infectious agents and host species. For example, mouse and human cells infected with Francisella tularensis differ in their inflammatory response.15 Of note, pyrin is an important component in caspase-1 activation and IL-1β release in human cells challenged with Francisella novicida but plays no role in mouse macrophages.16,17 To confirm whether Ehx induces an inflammatory response to a similar extent in different hosts, we investigated the disparity in Ehx-induced cytotoxicity and IL-1β release in macrophages between human and mouse cells and explored the possible mechanisms involving the NLRP3 activation.
Materials and methods
Bacterial strains
The wild-type (WT) EHEC O157:H7 reference strain EDL933 was purchased from the American Type Culture Collection (ATCC 43895; Manassas, VA). The EDL933 ehxA deletion mutant (ΔehxA) and the strain with the ehxA gene complement (ΔehxA/pehxA) have been described previously.14 The commensal E. coli K-12 strain HB101 was purchased from ATCC (33694) and served as a comparison strain. All strains were confirmed by PCR.
Mice
C57BL/6 WT mice were obtained from Beijing Vital River Laboratory Animal Technology Co. Ltd (Beijing, China). All mice were housed in a specific pathogen-free facility in the China CDC Animal Centre. All animal studies were performed following the protocols approved by the Welfare & Ethical Inspection in Animal Experimentation Committee at China CDC.
Human cell culture, differentiation of monocytes to macrophages and PBMC isolation
The human monocyte cell line THP-1 was purchased from the ATCC (TIB-202). The cells were cultured in RPMI-1640 containing 10% heat-inactivated fetal bovine serum (FBS) (Life Technologies, Grand Island, NY) and seeded in 24-well culture plates, with 5 × 105 cells per well. The THP-1 cells were allowed to adhere for 48 hr with 1 nm PMA (Sigma-Aldrich, St Louis, MO), washed three times with PBS and cultured for one more day before infection. Human peripheral blood mononuclear cells (PBMCs) were isolated from whole blood obtained from the Beijing Red Cross Blood Centre (Beijing, China) using lymphocyte separation medium (MP Biomedicals, Santa Ana, CA), as previously described.18 The cells were cultured in RPMI-1640 containing 5% FBS and seeded in 24-well culture plates at 1 × 106 cells/ml.
Mouse cell culture, preparation of mouse macrophages and spleen cells
The mouse macrophage cell line IC-21 was purchased from the ATCC (TIB-186). Mouse bone marrow-derived macrophages (BMMs) were prepared from femurs and tibias of C57BL/6 mice and cultured for 6 days in 10% FBS/RPMI-1640 containing recombinant macrophage colony-stimulating factor (20 ng/ml; R&D Systems, Minneapolis, MN). The cell culture medium was replenished on day 3 and then the cells were harvested to be seeded in 24-well culture plates at 1 × 106 cells/ml on day 6. Mouse peritoneal exudate cells (PMs) were obtained by peritoneal lavage and enriched for macrophages using the method of Kumagai et al.19 Peritoneal macrophages prepared in this manner were at least 90% pure, as assessed by expression of F4/80 using flow cytometry. The macrophages were seeded in 24-well culture plates at 1 × 106 cells/ml. Single splenocytes were prepared using a cell strainer, and red blood cells were lysed using Gey's reagent. The splenocytes were washed and then cultured in RPMI-1640 medium supplemented with 5% FBS, 25 mmol/l HEPES, 2 mmol/l glutamine, 100 KU/l penicillin and 100 mg/l streptomycin (all from Gibco Invitrogen, Carlsbad, CA). The splenocytes were seeded in 24-well culture plates at 4 × 106 cells/ml and incubated at 37° in a 5% CO2 atmosphere. Animal handling and experimental procedures were approved by the Chinese Centre for Disease Control and Prevention Ethics Committee for the use of laboratory animals.
Bacterial infection
Bacteria were grown to the logarithmic growth phase in Luria–Bertoni broth at 37° with continuous shaking at 200 rpm. After being washed twice with PBS, bacteria were added in triplicate to the cell monolayer at a multiplicity of infection of 10 and incubated with reduced serum medium (GIBCO, Carlsbad, CA) at 37° in a 5% CO2 atmosphere. The human cells (PBMCs and THP-1 cells) and mouse cells (splenocytes, IC-21 cells, PMs and BMMs) were infected with the bacteria for 4 hr in the absence or presence of LPS (1 μg/ml). The LPS-primed cells treated with 5 mm ATP for 1 hr served as positive controls for mouse cells and LPS-stimulated PBMCs or THP-1 cells served as positive controls for human cells as mouse macrophages require two steps of activation to induce IL-1β. Commensal E. coli HB101 infection served as a comparison to EDL933. In some experiments, the human differentiated THP-1 cells were pre-incubated for 2 hr with 10 mm CA-074 Me (Calbiochem, San Diego, CA), 10 mm Z-YVAD-FMK (Alexis Biochemicals, Lörrach, Germany), 500 nm oATP (Merck, Darmstadt, Germany) and incubation with these chemicals was continued during infection. The reagents used had no effect on the bacterial growth or cell viability (data not shown).
Potassium (K+) efflux
We blocked K+ efflux by replacing normal culture medium with cell culture medium containing a high concentration of extracellular KCl (130 mm) at 2 hr before infection with the EDL933 strain.
Determination of IL-1β, ATP and lactate dehydrogenase levels
At the indicated time-points, the supernatants of the cell cultures were collected. Human or murine IL-1β concentrations in the cell-free supernatants were measured using commercially available sandwich ELISA kits (BD Biosciences, San Jose, CA; eBioscience, San Diego, CA). Lactate dehydrogenase release was quantified using a Cytotox96 Kit (Promega, Madison, WI) according to the manufacturer's instructions. The relative level of cytotoxicity was expressed as described previously,14 and ATP release from the cells into the supernatant was monitored using a bioluminescence assay kit (Molecular Probes, Eugene, OR).
Immunoblot analysis
Cell-free supernatants were concentrated using Amicon Ultra-4 10K Centrifugal Filter Devices (Millipore, Bedford, MA). Cell extracts and concentrated supernatants were separated using SDS–PAGE and then blotted. The membranes were first exposed to antibodies specific for IL-1β (no. sc-52012; Santa Cruz, CA), caspase-1 (no. sc-56036) or GAPDH (no. sc-137179; Santa Cruz, CA). The membranes were then incubated with secondary antibodies (IRDye 800-labelled anti-mouse IgG or anti-rabbit IgG; no. 610-132-121 and 611-132-002, respectively; Rockland, Gilbertsville, PA), and the proteins were detected using an Odyssey Infrared Imaging System (LI-COR, Lincoln, NE).
Confocal laser scanning microscopy
Human differentiated THP-1 cells and mouse IC-21 cells were left untreated or infected with bacteria. After 2 hr, the extracellular bacteria were removed by washing with PBS, and the cells were fixed with 3% paraformaldehyde (Sigma-Aldrich). The cells were permeabilized with 1% Triton X-100 (Sigma-Aldrich) and blocked with 5% goat serum (Invitrogen). Cathepsin B was stained by exposing cells to a specific antibody (ab58802; Abcam, Cambridge, UK), followed by incubation with AF546-conjugated anti-mouse IgG (Invitrogen). Host-cell actin was stained with phalloidin AF488 (Invitrogen), and confocal laser scanning microscopy was conducted using an LSM5 Pascal microscope (Zeiss, Oberkochen, Germany).
Statistical analysis
Statistical analysis was performed using one-way analysis of variance with Newman–Keuls post-testing. The correlation between the ATP level and IL-1β concentration in the supernatants of THP-1 cells infected with EHEC O157:H7 was assessed using Pearson's test and linear regression. Values of P < 0·05 were considered significant.
Results
Ehx-induced IL-1β production differs between human and mouse macrophages
Human PBMCs and differentiated THP-1 cells infected with EDL933 WT or ΔehxA/pehxA produced high levels of mature (p17) IL-1β at 4 hr post-infection (Fig.1a,b). EDL933 lacking ehxA (ΔehxA) induced a markedly lower IL-1β release (Fig.1a) compared with the WT and ΔehxA/pehxA. In contrast, there was no detectable IL-1β release from mouse splenocytes, IC-21, PMs, BMMs 4 hr after infection with EDL933 in the absence of LPS-priming. In the presence of LPS, all the above-mentioned mouse cells produced a similarly small amount of IL-1β after challenge with EDL933 WT or its mutants (Fig.1a), indicating that Ehx has no effect on IL-1β production in these mouse cells. As a comparison, HB101 only induced low levels of IL-1β release in both human cells and LPS-primed mouse cells.
Figure 1.
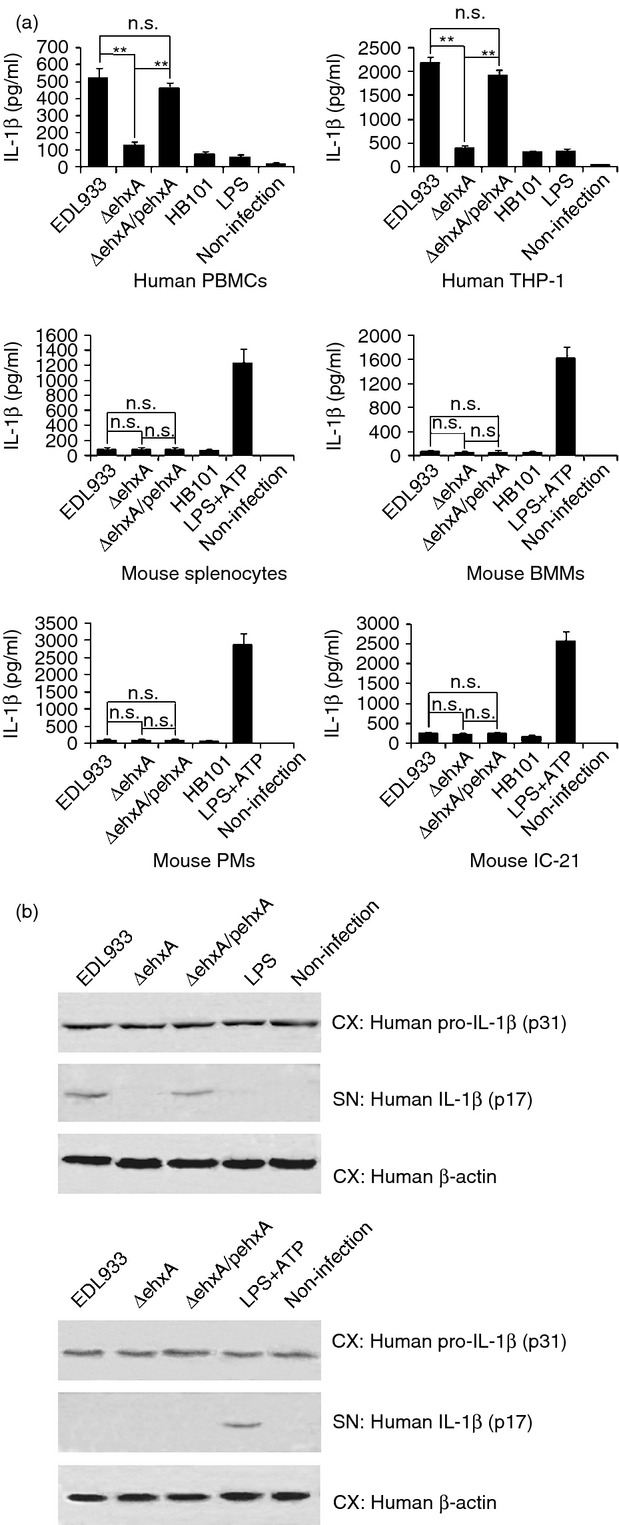
Effects of enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin on the production of interleukin-1β (IL-1β). Human peripheral blood mononuclear cells (PBMCs), differentiated human THP-1 cells, and lipopolysaccharide (LPS) -primed (1 μg/ml, 2 hr) mouse splenocytes, peritoneal exudate cells (PMs), bone marrow-derived macrophages (BMMs) and IC-21 cells were infected with EDL933 wild-type (WT), ΔehxA or ΔehxA/pehxA for 4 hr. LPS-primed mouse cells treated with ATP (5 mm, 1 hr) served as positive controls. The commensal E. coli HB101 served as a comparison strain. (a) The concentration of IL-1β in the supernatants was determined by ELISA. (b) The amount of IL-1β p17 and p31 in cell extracts (CX) and supernatants (SN) were visualized by Western blotting. (a) The results represent the mean ± SD of at least three independent experiments. Significant differences (**P < 0·01) are indicated. n.s., not significant (P > 0·05).
To confirm the difference in Ehx-induced IL-1β production between human and mouse macrophages, we further examined the production of pro-IL-1β (p31) in differentiated THP-1 cells and mouse PM lysates and mature IL-1β (p17) in both supernatants using immunoblotting after the cells were infected with the different bacterial strains. We found that all of these strains induced similar levels of pro-IL-1β production in the mouse PMs and human differentiated THP-1 cell lysates (Fig.1b). However, significantly higher levels of mature IL-1β were observed only in the supernatants from differentiated THP-1 cells but not in those from mouse PMs after infection with the ehxA-positive strains (Fig.1b). This indicates that Ehx may affect activation of mature IL-1β production in human THP-1-derived macrophages but not mouse macrophages.
Ehx-induced cytotoxicity differs between human and mouse macrophages
Lactate dehydrogenase release from human PBMCs and differentiated THP-1 cells infected with ΔehxA was significantly lower than that from the cells infected with the WT and ΔehxA/pehxA strains at 4 hr post infection, indicating that Ehx causes cytotoxicity in human PBMCs and THP-1-derived macrophages. In contrast, no significant difference in lactate dehydrogenase release was observed in mouse splenocytes, IC-21, PMs or BMMs infected with the three bacterial strains (Fig.2). These results suggest that there may be a difference in Ehx-induced cytotoxicity between macrophages of human and mouse origin.
Figure 2.
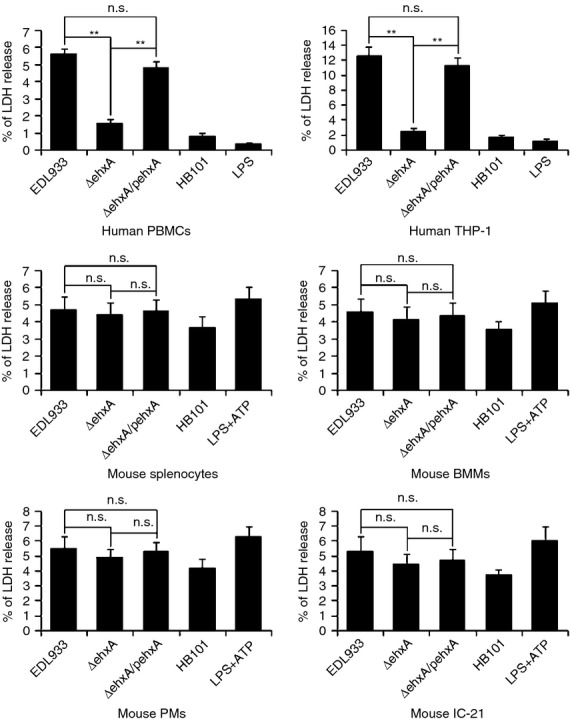
Cytotoxicity of enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin, as indicated by the release of lactate dehydrogenase (LDH). Human peripheral blood mononuclear cells (PBMCs), differentiated human THP-1 cells, mouse splenocytes, peritoneal macrophages (PMs), bone marrow-derived macrophages (BMMs) and IC-21 cells were infected with EDL933, ΔehxA or ΔehxA/pehxA. The commensal E. coli HB101 served as a comparison strain. The release of LDH was assessed at 4 hr post-infection. The results represent the mean ± SD of at least three independent experiments. Significant differences (**P < 0·01) are indicated. n.s., not significant (P > 0·05).
Disparity in Ehx-induced caspase-1 activation between human differentiated THP-1 cells and mouse macrophages
Our previous study showed that NLRP3 inflammasome activation is required for IL-1β production in response to EHEC O157:H7 infection. To determine if the caspase-1 and inflammasome pathway is involved in the difference in Ehx-induced IL-1β production between human and mouse cells, the activation of caspase-1 in infected differentiated THP-1 cells and mouse PMs was assessed by immunoblot analysis of the cleavage of caspase-1 to its active p10 subunit. The production of active p10 in differentiated THP-1 cells infected with WT or ΔehxA/pehxA was significantly higher than that in cells infected with the ΔehxA strain (Fig.3a), indicating the critical role of Ehx in triggering caspase-1 activation in differentiated THP-1 cells. Moreover, a caspase-1 inhibitor strongly diminished Ehx-induced IL-1β production, suggesting that Ehx triggers the release of IL-1β in differentiated THP-1 cells in a caspase-1-dependent way (Fig.3b). However, active p10 was undetectable in mouse macrophages infected with various strains (Fig.3a), suggesting that Ehx may play different roles in triggering caspase-1 activation in human and mouse cells, which in turn leads to differences in IL-1β production.
Figure 3.
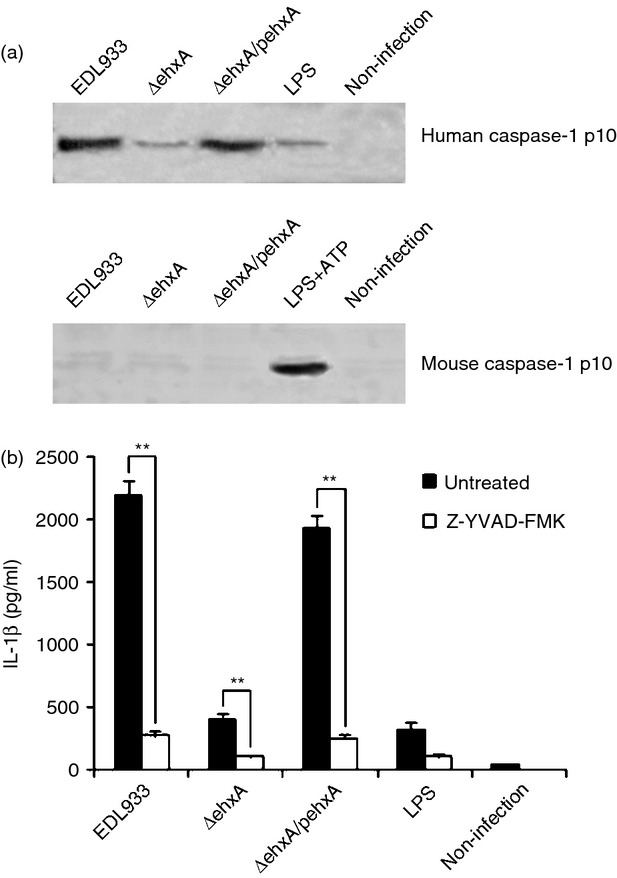
The role for caspase-1 in enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin-induced interleukin-1β (IL-1β) production. Differentiated human THP-1 cells and mouse peritoneal macrophages (PMs) were infected with EDL933, ΔehxA or ΔehxA/pehxA for 4 hr. (a) Lysates from the cells were analysed by immunoblotting directed against active caspase-1 p10. (b) The THP-1 cells were treated with 10 μm of the caspase-1 inhibitor Z-YVAD-FMK for 1 hr before infection with EHEC O157:H7. The IL-1β concentration in the supernatant was determined at 4 hr post-infection by ELISA. (b) The results represent the mean ± SD of three independent experiments. Significant differences (**P < 0·01) are indicated. n.s., not significant (P > 0·05).
Disparity in Ehx-induced ATP release between human differentiated THP-1 cells and mouse macrophages
Extracellular ATP has been shown to trigger NLRP3 activation via the ATP receptor P2X7. We next sought to determine whether the Ehx-induced ATP release in differentiated THP-1 cells differed from that of mouse macrophages. As shown in Fig.4(a), WT and ΔehxA/pehxA infection induced significantly higher ATP release into the culture supernatant of differentiated THP-1 cells compared with the ΔehxA strain, suggesting that ATP release into the supernatant may be involved in Ehx-induced IL-1β secretion in differentiated THP-1 cells. However, the ATP concentration in the supernatant of mouse PMs treated with each of the strains was undetectable, implying that the similar, low level of IL-1β production may partially be the result of the lack of ATP release by mouse macrophages treated with each strain.
Figure 4.
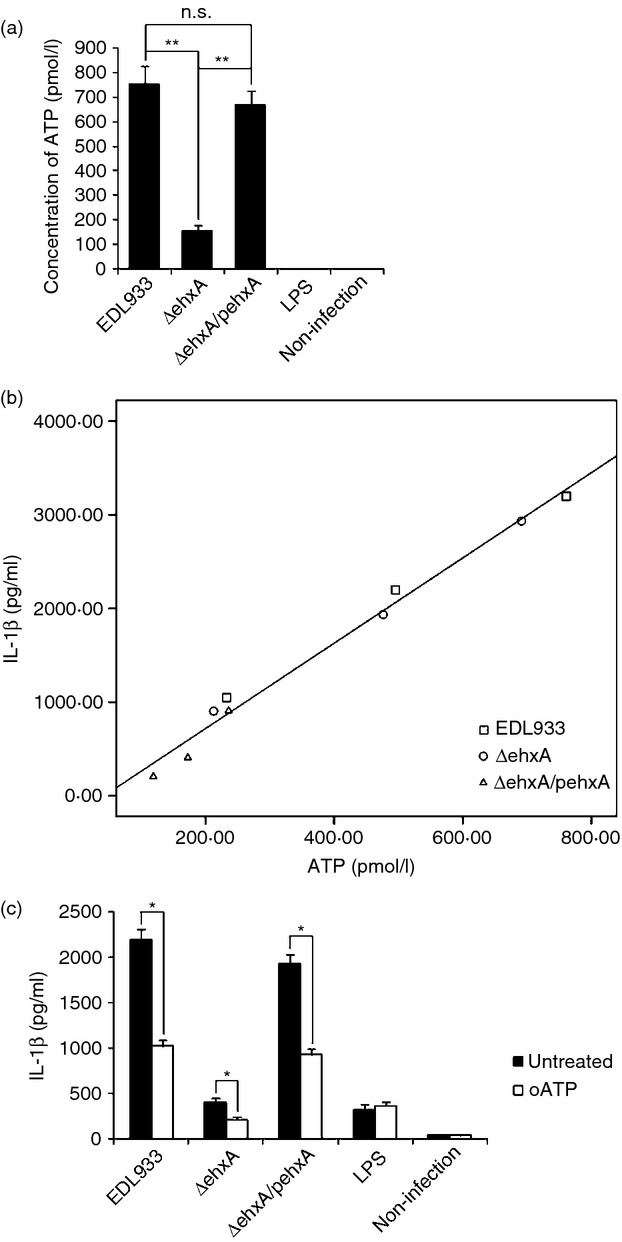
Role of ATP release and the purinergic signalling pathway in enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin-induced interleukin-1β (IL-1β) production by THP-1 cells. Differentiated human THP-1 cells were infected with EDL933, ΔehxA or ΔehxA/pehxA. (a) ATP release from the differentiated THP-1 cells at 4 hr post-infection. (b) Positive correlation between ATP release and IL-1β production in the supernatant of THP-1 cells infected with EHEC O157:H7. (c) The differentiated THP-1 cells were treated with 500 nm of the ATP inhibitor oATP for 2 hr before infection. The IL-1β concentration in the supernatant was determined at 4 hr post-infection by ELISA. (a, c) The results represent the mean ± SD of three independent experiments. Significant differences (*P < 0·05, **P < 0·01) are indicated. n.s., not significant (P > 0·05).
Next, the analysis of correlation between ATP concentration and IL-1β production in the supernatants of differentiated THP-1 cells infected with each strain at 2, 4 and 6 hr post-infection was conducted to better understand the relationship between ATP release and IL-1β production induced by Ehx. A significant positive correlation was confirmed between IL-1β production and ATP release in differentiated THP-1 cells (Fig.4b).
To study whether the effect of Ehx is mediated through ATP release and purinergic signalling, we inhibited ATP signalling using the purinergic receptor antagonist oxidized ATP (oATP). As demonstrated in Fig.3(c), oATP significantly reduced Ehx-induced IL-1β release, suggesting that ATP action is involved in the EhX-induced inflammasome activation.
Role of cathepsin B in Ehx-induced IL-1β production in human differentiated THP-1 cells
It has been demonstrated that the NLRP3 inflammasome is activated either by lysosomal damage induced by phagocytosis or the subsequent release of cathepsin B. To determine whether cathepsin B plays a role in Ehx-induced IL-1β release, we determined cathepsin B in both differentiated THP-1 cells and mouse IC-21 cells using immunofluorescence staining and confocal microscopy. Using an anti-cathepsin B antibody, we found splotchy staining in both unchallenged THP-1 macrophages and IC-21 cells. In infected differentiated THP-1 cells, obviously diminished, punctate cathepsin B staining in cells infected with WT or ΔehxA/pehxA was observed compared with ΔehxA-infected cells (Fig.5a), which strongly suggests that Ehx leads to cathepsin B release from phagoendosomes into the cytosol and consequent activation in human macrophages. However, no difference was found in cathepsin B staining among the IC-21 cells infected with the three stains, indicating that Ehx has no effect on cathepsin B release in mouse macrophages.
Figure 5.
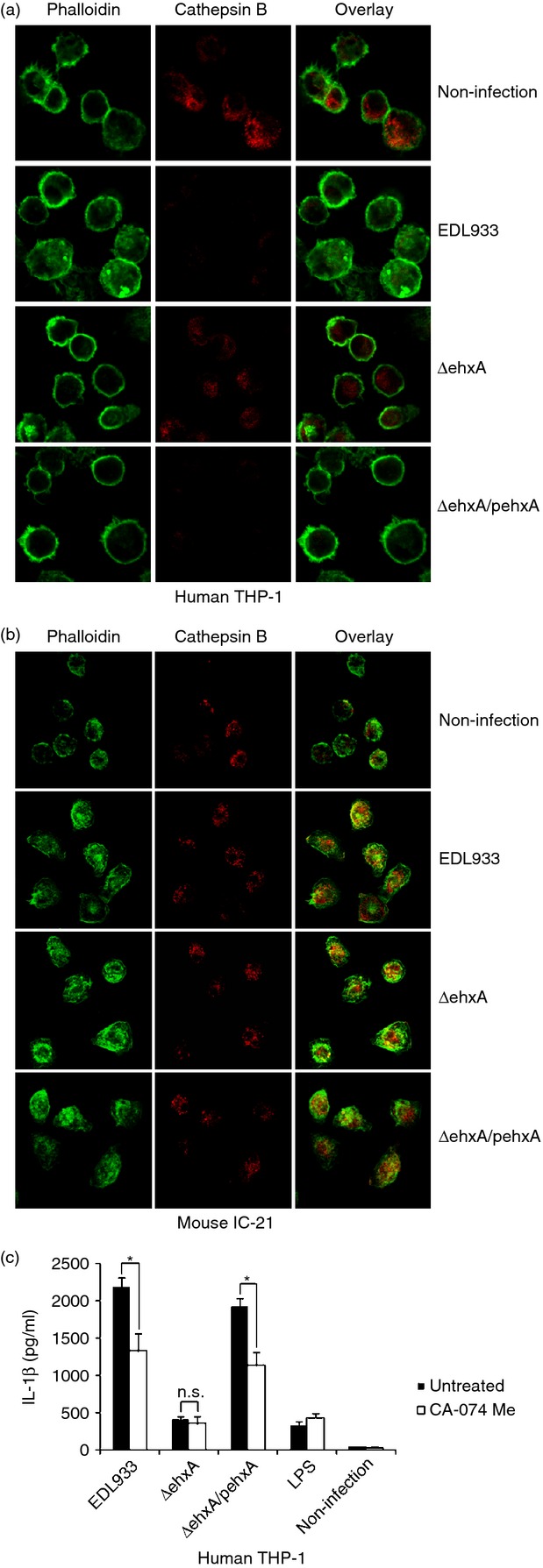
The role of cathepsin B in enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin-induced interleukin-1β (IL-1β) production by human THP-1 cells and mouse IC-21 cells. Cells were left untreated or were infected with EDL933, ΔehxA or ΔehxA/pehxA for 2 hr. Cathepsin B was visualized by confocal laser scanning microscopy after staining with a specific antibody. Actin was stained with phalloidin AF488. (a) Cathepsin B in human THP-1 cells. (b) Cathepsin B in mouse IC-21 cells. (c) The THP-1 cells were treated with 10 μm of the cathepsin B inhibitor CA-074Me for 1 hr before infection with EHEC O157:H7. The IL-1β concentration in the supernatant was determined 4 hr post-infection by ELISA. (c) The results represent the mean ± SD of three independent experiments. Significant differences (*P < 0·05) are indicated. n.s., not significant (P > 0·05).
Furthermore, we sought to assess the role of cathepsin B in activating IL-1β production of human THP-1 cells in response to EHEC O157:H7. As shown in Fig.5(b), blockade of cathepsin B activity with the inhibitor CA-074 significantly attenuated IL-1β production in differentiated THP-1 cells infected with WT or ΔehxA/pehxA but not ΔehxA, suggesting that cathepsin B is involved in the IL-1β secretion pathway in human macrophages in response to Ehx stimulation.
Role of potassium (K+) efflux in Ehx-induced cytotoxicity and IL-1β production in human differentiated THP-1 cells
Previous studies have demonstrated that high extracellular K+ blocks IL-1β release. To investigate the role of K+ efflux in Ehx-induced cytotoxicity and IL-1β production in human differentiated THP-1 cells, we blocked K+ efflux by replacing normal culture medium with cell culture medium containing high concentrations of KCl before infection with EDL933 strains, and found that blocking K+ efflux almost completely inhibited IL-1β production (Fig.6a) and reduced the cytotoxicity induced by EHEC O157:H7 (Fig.6b). These results demonstrated that K+ efflux plays a key role in EHEC O157:H7-induced cytotoxicity and IL-1β production in differentiated THP-1 cells.
Figure 6.
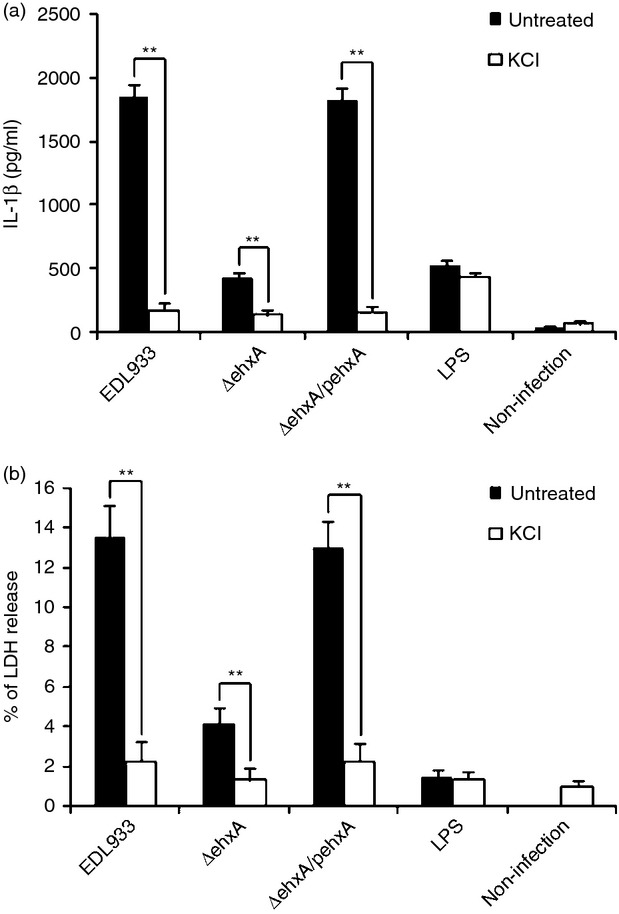
The role of K+ efflux in enterohaemorrhagic Escherichia coli (EHEC) O157:H7 enterohaemolysin-induced interleukin-1β (IL-1β) production and cytotoxicity in THP-1 cells. Human THP-1 cells were infected with EHEC O157:H7 for 4 hr in the presence of 140 mm K+. (a) The IL-1β concentration in the supernatants was determined 4 hr post-infection by ELISA. (b) Cytotoxicity was evaluated by the release of lactate dehydrogenase (LDH) from the THP-1 cells, which was assayed after 4 hr of exposure to EHEC O157:H7. The results represent the mean ± SD of three independent experiments. Significant differences (**P < 0·01) are indicated. n.s., not significant (P > 0·05).
Discussion
Information on the pathogenesis of EHEC O157:H7 infection is very limited, largely because of the lack of ideal animal models capable of recapitulating all the features of EHEC O157:H7 infection in humans. It is well known that naturally infected mice are not a good model with which to mimic the various pathological changes in human EHEC infection.20 Though some mouse models have been developed for EHEC O157:H7 infection including axenic mouse (no indigenous intestinal flora) or streptomycin-treated mouse (reduced normal flora),21 no naturally occurring HUS-like diseases have been described in mice.
Recent studies demonstrated that EHEC-infected mice treated with inflammatory cytokines showed systemic disease, neurological manifestations and glomerular lesions, suggesting that an inflammatory response is essential for HUS development in O157:H7 infection.22 It has been suggested that IL-1β may play an important role in HUS through increased expression of Gb3, the receptor for Stx on endothelial cells, and allowing increased binding of Stx.23 In this study, we found that EHEC O157:H7 Ehx played a critical role in inducing cytotoxicity and IL-1β production in human macrophages but not in C57BL/6 mouse macrophages, suggesting that the effect of EHEC O157:H7 Ehx on macrophages is species specific. Hence, the fact that human macrophages are more sensitive to Ehx than mouse macrophages may at least in part account for why the conventional C57BL/6 mouse is not a good model for HUS of EHEC infection. Taken together, we postulate that the difference in the pathogenicity of O157:H7 infection between humans and mice may be partially due to the disparity between the two species in effect of Ehx on IL-1β production.
Our previous study confirmed that NLRP3/apoptosis-associated speck-like protein containing a caspase recruitment domain/caspase-1 is required for EHEC O157:H7-induced IL-1β production in differentiated THP-1 cells.14 In this study, we found that Ehx contributed to caspase-1 activation in differentiated THP-1 cells, whereas no effect was observed in mouse macrophages. We postulated that the difference in Ehx-driven NLRP3 activation between human and mouse cells might account for the disparity in macrophage IL-1β production between the two species. A recent study reported that EHEC EDL933 induced higher levels of IL-1β production in LPS-primed mouse macrophages than ours using a higher challenge dose (multiplicities of infection 25 versus 5) and longer challenge time (8 versus 4 hr). Although the study identified that EHEC nucleic acids mediated NLRP3 inflammasome-dependent responses, it also demonstrated that EHEC-specific virulence factors including Ehx were dispensable for NLRP3 activation, which is consistent with our findings.24 We propose that mouse macrophages are much less sensitive than human macrophages in terms of NLRP3 activation in response to EHEC infection because of the differential response of human and mouse macrophages to Ehx. There is an increasing body of evidence showing a difference between the human and mouse genes related to inflammasome activation. For example, CARD8 is present in the human genome and seems to be part of the NLRP3 inflammasome but is not found in the mouse genome.25
At least three distinct pathways including potassium (K+) efflux, lysosomal destabilization and release of cathepsin B, and generation of reactive oxygen species have been proposed to be involved in NLRP3 activation.26
Pioneering studies have proposed that K+ efflux is responsible for the maturation of pro-IL-1β.27,28 In this study, when we inhibited K+ efflux with a high concentration of extracellular K+, IL-1β production by differentiated THP-1 cells in response to EHEC O157:H7 infection was largely blocked, suggesting that K+ efflux may play a key role in Ehx-induced IL-1β production. Extracellular ATP stimulates the purogenic P2X7 ATP-gated ion channel leading to K+ efflux, which is necessary for inflammasome activation. Cellular stimulation triggers ATP release and subsequent activation of purinergic receptors at the cell surface.29 Some studies indicated that certain non-nucleotide inflammasome activators may interact directly with purinergic receptors, which excludes the role of ATP release in a one-step model of inflammasome activation.30,31 However, in the present study Ehx was found to contribute to endogenous ATP release and inhibition of ATP signalling partially suppressed/impaired IL-1β production in differentiated THP-1 cells. Our results suggest that Ehx-induced ATP release may be involved in regulating IL-1β production but not the only pathway mediating NLRP3 activation triggered by Ehx in differentiated THP-1 cells. In contrast, Ehx did not have the same effect on ATP release in mouse macrophages as it did in THP-1 cells. In addition, some studies revealed that K+ is involved in the activation of NLRP3 inflammasome by an array of stimuli. For example, some bacterial pore-forming toxins were reported to cause K+ efflux by permeabilizing the plasma membrane, forming pores and activating NLRP3 independently of P2X7R.30–33 Hence, we propose that the ATP signalling activation induced by Ehx and the pores formed directly by Ehx may contribute to K+ efflux from THP-1 cells and result in the consequent activation of NLRP3. Another interesting finding is that a high concentration of extracellular K+ also suppresses EHEC-induced cytotoxicity. One possible mechanism is that prevention of K+ efflux contributes to maintaining the physiological environment and reducing NLRP3 inflammasome activation, which leads to a less inflammatory pathological response with less cytotoxicity.
It has also been reported that cathepsin B-induced lysosomal damage acts as a trigger of the NLRP3 inflammasome and cathepsin B inhibitors prevent the activation of caspase-1 and IL-1β secretion induced by certain microbes.34–36 In this study, different effects of Ehx on cathepsin B release were found in human and mouse macrophages. We demonstrated that NLRP3-mediated IL-1β production induced by EHEC was partially attributable to Ehx-mediated cathepsin B release in THP-1 cells. However, we cannot conclude a direct role of Ehx in activating cathepsin B release. First, the membrane pores that are formed by Ehx may also allow other extracellular particulate structures to access the lysosome. Second, Ehx might mediate phagosomal destabilization, which is a possible activator triggering cathepsin B release.37
In this study, we did not measure reactive oxygen species production in human cells. However, the difference in Ehx-induced cytotoxicity between human and mouse cells may cause a disparity in reactive oxygen species generation between species which is known to affect NLRP3 activation.
In summary, in this study we have demonstrated the disparity between human and mouse macrophages in NLRP3-driven IL-1β production in response to EHEC O157:H7 Ehx. In human cells, Ehx may induce IL-1β production by affecting NLRP3 activation, which is mediated through altered K+ efflux and cathepsin B release. This difference may provide mechanistic insight helping to understand the differential response to EHEC O157:H7 infection between humans and mice. However, it cannot be ruled out that other mechanisms might be involved. For example, Vibrio vulnificus hemolysin-induced NLRP3 activation has been shown to require nuclear factor-κB activation via Toll-like receptors.38 Further study is required to investigate the recognition mechanism of macrophage membrane receptors that underlies the Ehx-induced species-specific activation of NLRP3.
Acknowledgments
This work was supported by grants from the National Natural Science Foundation of China (Nos. 81371761; 81371762). The authors thank Drs Yinyan Sun and Ting Li (National Institute of Biological Science, Beijing, China) for their technical assistance.
Glossary
Abbreviations:
- BMMs
bone marrow-derived macrophages
- EHEC
enterohaemorrhagic Escherichia coli
- Ehx
enterohaemolysin
- FBS
fetal bovine serum
- HUS
haemolytic uraemic syndrome
- IL
interleukin
- LPS
lipopolysaccharides
- NLR
NOD-like receptor
- PBMCs
peripheral blood mononuclear cells
- PMs
peritoneal exudate cells
- Stx
Shiga toxin
- WT
wild-type
Disclosure
The authors declare no competing financial interests.
Reference
- Nataro JP, Kaper JB. Diarrheagenic Escherichia coli. Clin Microbiol Rev. 1998;11:142–201. doi: 10.1128/cmr.11.1.142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melton-Celsa AR, O'Brien AD. Animal models for STEC-mediated disease. Methods Mol Med. 2003;73:291–305. doi: 10.1385/1-59259-316-x:291. [DOI] [PubMed] [Google Scholar]
- Inward CD, Varagunam M, Adu D, Milford DV, Taylor CM. Cytokines in haemolytic uraemic syndrome associated with verocytotoxin-producing Escherichia coli infection. Arch Dis Child. 1997;77:145–7. doi: 10.1136/adc.77.2.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van de Kar NC, Monnens LA, Karmali MA, van Hinsbergh VW. Tumor necrosis factor and interleukin-1 induce expression of the verocytotoxin receptor globotriaosylceramide on human endothelial cells: implications for the pathogenesis of the hemolytic uremic syndrome. Blood. 1992;80:2755–64. [PubMed] [Google Scholar]
- Litalien C, Proulx F, Mariscalco MM, et al. Circulating inflammatory cytokine levels in hemolytic uremic syndrome. Pediatr Nephrol. 1999;13:840–5. doi: 10.1007/s004670050712. [DOI] [PubMed] [Google Scholar]
- Westerholt S, Pieper AK, Griebel M, Volk HD, Hartung T, Oberhoffer R. Characterization of the cytokine immune response in children who have experienced an episode of typical hemolytic-uremic syndrome. Clin Diagn Lab Immunol. 2003;10:1090–5. doi: 10.1128/CDLI.10.6.1090-1095.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keepers TR, Psotka MA, Gross LK, Obrig TG. A murine model of HUS: Shiga toxin with lipopolysaccharide mimics the renal damage and physiologic response of human disease. J Am Soc Nephrol. 2006;17:3404–14. doi: 10.1681/ASN.2006050419. [DOI] [PubMed] [Google Scholar]
- Ikeda M, Ito S, Honda M. Hemolytic uremic syndrome induced by lipopolysaccharide and Shiga-like toxin. Pediatr Nephrol. 2004;19:485–9. doi: 10.1007/s00467-003-1395-7. [DOI] [PubMed] [Google Scholar]
- Thornberry NA, Bull HG, Calaycay JR, et al. A novel heterodimeric cysteine protease is required for interleukin-1β processing in monocytes. Nature. 1992;356:768–74. doi: 10.1038/356768a0. [DOI] [PubMed] [Google Scholar]
- Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-β. Mol Cell. 2002;10:417–26. doi: 10.1016/s1097-2765(02)00599-3. [DOI] [PubMed] [Google Scholar]
- Mariathasan S, Weiss DS, Newton K, et al. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature. 2006;440:228–32. doi: 10.1038/nature04515. [DOI] [PubMed] [Google Scholar]
- Gurcel L, Abrami L, Girardin S, Tschopp J, van der Goot FG. Caspase-1 activation of lipid metabolic pathways in response to bacterial pore-forming toxins promotes cell survival. Cell. 2006;126:1135–45. doi: 10.1016/j.cell.2006.07.033. [DOI] [PubMed] [Google Scholar]
- Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, Leong JM, Fitzgerald KA. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–19. doi: 10.1016/j.cell.2012.07.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X, Cheng Y, Xiong Y, et al. Enterohemorrhagic Escherichia coli specific enterohemolysin induced IL-1β in human macrophages and EHEC-induced IL-1β required activation of NLRP3 inflammasome. PLoS One. 2012;7:e50288. doi: 10.1371/journal.pone.0050288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger CE, Forestal CA, Italo JK, Benach JL, Furie MB. The live vaccine strain of Francisella tularensis replicates in human and murine macrophages but induces only the human cells to secrete proinflammatory cytokines. J Leukoc Biol. 2005;77:893–7. doi: 10.1189/jlb.1104637. [DOI] [PubMed] [Google Scholar]
- Gavrilin MA, Mitra S, Seshadri S, Nateri J, Berhe F, Hall MW, Wewers MD. Pyrin critical to macrophage IL-1β response to Francisella challenge. J Immunol. 2009;182:7982–9. doi: 10.4049/jimmunol.0803073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atianand MK, Duffy EB, Shah A, Kar S, Malik M, Harton JA. Francisella tularensis reveals a disparity between human and mouse NLRP3 inflammasome activation. J Biol Chem. 2011;286:39033–42. doi: 10.1074/jbc.M111.244079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng H, Luo X, Segura M, Sun H, Ye C, Gottschalk M, Xu J. The role of toll-like receptors in the pathogenesis of Streptococcus suis. Vet Microbiol. 2012;156:147–56. doi: 10.1016/j.vetmic.2011.10.014. [DOI] [PubMed] [Google Scholar]
- Kumagai K, Itoh K, Hinuma S, Tada M. Pretreatment of plastic Petri dishes with fetal calf serum. A simple method for macrophage isolation. J Immunol Methods. 1979;29:17–25. doi: 10.1016/0022-1759(79)90121-2. [DOI] [PubMed] [Google Scholar]
- Conlan JW, Perry MB. Susceptibility of three strains of conventional adult mice to intestinal colonization by an isolate of Escherichia coli O157:H7. Can J Microbiol. 1998;44:800–5. doi: 10.1139/cjm-44-8-800. [DOI] [PubMed] [Google Scholar]
- Calderon Toledo C, Rogers TJ, Svensson M, Tati R, Fischer H, Svanborg C, Karpman D. Shiga toxin-mediated disease in MyD88-deficient mice infected with Escherichia coli O157:H7. Am J Pathol. 2008;173:1428–39. doi: 10.2353/ajpath.2008.071218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Isogai E, Isogai H, Kimura K, Hayashi S, Kubota T, Fujii N, Takeshi K. Role of tumor necrosis factor α in gnotobiotic mice infected with an Escherichia coli O157:H7 strain. Infect Immun. 1998;66:197–202. doi: 10.1128/iai.66.1.197-202.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Louise CB, Obrig TG. Shiga toxin-associated hemolytic-uremic syndrome: combined cytotoxic effects of Shiga toxin, interleukin-1β, and tumor necrosis factor α on human vascular endothelial cells in vitro. Infect Immun. 1991;59:4173–9. doi: 10.1128/iai.59.11.4173-4179.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kailasan Vanaja S, Rathinam VA, Atianand MK, Kalantari P, Skehan B, Fitzgerald KA, Leong JM. Bacterial RNA:DNA hybrids are activators of the NLRP3 inflammasome. Proc Natl Acad Sci USA. 2014;111:7765–70. doi: 10.1073/pnas.1400075111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrilli V, Papin S, Tschopp J. The inflammasome. Curr Biol. 2005;15:R581. doi: 10.1016/j.cub.2005.07.049. [DOI] [PubMed] [Google Scholar]
- van de Veerdonk FL, Netea MG, Dinarello CA, Joosten LA. Inflammasome activation and IL-1β and IL-18 processing during infection. Trends Immunol. 2011;32:110–6. doi: 10.1016/j.it.2011.01.003. [DOI] [PubMed] [Google Scholar]
- Muñoz-Planillo R, Kuffa P, Martínez-Colón G, Smith BL, Rajendiran TM, Núñez G. K⁺ efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity. 2013;38:1142–53. doi: 10.1016/j.immuni.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pétrilli V, Papin S, Dostert C, Mayor A, Martinon F, Tschopp J. Activation of the NALP3 inflammasome is triggered by low intracellular potassium concentration. Cell Death Differ. 2007;14:1583–9. doi: 10.1038/sj.cdd.4402195. [DOI] [PubMed] [Google Scholar]
- Piccini A, Carta S, Tassi S, Lasiglié D, Fossati G, Rubartelli A. ATP is released by monocytes stimulated with pathogen-sensing receptor ligands and induces IL-1β and IL-18 secretion in an autocrine way. Proc Natl Acad Sci USA. 2008;105:8067–72. doi: 10.1073/pnas.0709684105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walev I, Martin E, Jonas D, Mohamadzadeh M, Müller-Klieser W, Kunz L, Bhakdi S. Staphylococcal α-toxin kills human keratinocytes by permeabilizing the plasma membrane for monovalent ions. Infect Immun. 1993;61:4972–9. doi: 10.1128/iai.61.12.4972-4979.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harder J, Franchi L, Muñoz-Planillo R, Park JH, Reimer T, Núñez G. Activation of the Nlrp3 inflammasome by Streptococcus pyogenes requires streptolysin O and NF-κB activation but proceeds independently of TLR signaling and P2X7 receptor. J Immunol. 2009;183:5823–9. doi: 10.4049/jimmunol.0900444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fiser R, Masin J, Bumba L, et al. Calcium influx rescues adenylate cyclase-hemolysin from rapid cell membrane removal and enables phagocyte permeabilization by toxin pores. PLoS Pathog. 2012;8:e1002580. doi: 10.1371/journal.ppat.1002580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Munoz-Planillo R, Franchi L, Miller LS, Nunez G. A critical role for hemolysins and bacterial lipoproteins in Staphylococcus aureus-induced activation of the Nlrp3 inflammasome. J Immunol. 2009;183:3942–8. doi: 10.4049/jimmunol.0900729. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duncan JA, Gao X, Huang MT, et al. Neisseria gonorrhoeae activates the proteinase cathepsin B to mediate the signaling activities of the NLRP3 and ASC-containing inflammasome. J Immunol. 2009;182:6460–9. doi: 10.4049/jimmunol.0802696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu J, Thomas LM, Watkins SC, Franchi L, Núñez G, Salter RD. Cholesterol-dependent cytolysins induce rapid release of mature IL-1β from murine macrophages in a NLRP3 inflammasome and cathepsin B-dependent manner. J Leukoc Biol. 2009;86:1227–38. doi: 10.1189/jlb.0309164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hentze H, Lin XY, Choi MS, Porter AG. Critical role for cathepsin B in mediating caspase-1-dependent interleukin-18 maturation and caspase-1-independent necrosis triggered by the microbial toxin nigericin. Cell Death Differ. 2003;10:956–68. doi: 10.1038/sj.cdd.4401264. [DOI] [PubMed] [Google Scholar]
- Meixenberger K, Pache F, Eitel J, et al. Listeria monocytogenes-infected human peripheral blood mononuclear cells produce IL-1β, depending on listeriolysin O and NLRP3. J Immunol. 2010;184:922–30. doi: 10.4049/jimmunol.0901346. [DOI] [PubMed] [Google Scholar]
- Toma C, Higa N, Koizumi Y, et al. Pathogenic Vibrio activate NLRP3 inflammasome via cytotoxins and TLR/nucleotide-binding oligomerization domain-mediated NF-κB signaling. J Immunol. 2010;184:5287–97. doi: 10.4049/jimmunol.0903536. [DOI] [PubMed] [Google Scholar]