

Abstract
A key component in Lewy body (LB) pathology in LB disorders is α‐synuclein phosphorylated at serine 129 (pαsyn). However, it is not known if increase in the level of biochemically measurable pαsyn precedes the presence of histologically identified Lewy‐type synucleinopathy (LTS). To gain sights into possible temporal sequence, we measured levels of pαsyn in cingulate and temporal cortices that develop LTS pathology at later stages of LB disorders. Brain homogenates from 128 autopsy cases including normal controls and subjects classified by Unified LTS histopathology staging system were studied. We found that biochemically measurable pαsyn levels in cingulate and temporal cortices were significantly increased at Unified stages III and IV. When pαsyn levels were compared between LTS density scores instead of Unified stages, significant increases were detected even as LTS density scores increased from 0 to 1 in olfactory bulb and substantia nigra. Therefore, our findings demonstrated that changes of pαsyn levels in cingulate and temporal cortices coincided with the early appearance of the LTS pathology in olfactory bulb and substantia nigra, even though histologically demonstrable LTS was lacking in the cortical region. Therefore, identifying the underlying mechanisms driving these changes could be crucial to understanding the pathogenesis of LB disorders.
Keywords: alpha synuclein, human brain, Lewy body disorder, Parkinson's disease, pathology staging, phosphorylation
INTRODUCTION
Soluble alpha synuclein (αsyn) protein is normally concentrated in presynaptic terminals in the brain (28). Deposition of aggregated αsyn in neuronal cytoplasm, processes and presynaptic terminals is believed to be a central event in the pathogenesis of Lewy body (LB) disorders, which include Parkinson's disease (PD), Alzheimer's disease with a restricted distribution of Lewy bodies (ADLB) and dementia with Lewy bodies (DLB) 4, 26, 44, 53. Subsequently, αsyn‐immunoreactive Lewy‐type synucleinopathy (LTS) has become a useful criterion for the neuropathological diagnosis of these heterogeneous diseases and it has been shown that the abundance and distribution of αsyn‐containing pathology is associated with the severity of LB disorders. Several staging systems have been developed to classify the stages of PD and DLB based on the regional brain distribution of LTS histopathology 9, 10, 11, 35, 36, 41, 50. Among these, the system developed by Braak and colleagues was specifically designed to classify PD subjects 10, 12. They proposed that αsyn pathology progresses in six sequential stages, beginning in the dorsal motor nucleus of vagus following a caudal‐to‐rostral direction in the order of medulla, pons, midbrain, nucleus basalis of Meynert, limbic regions and neocortex 10, 12. In the Braak's scheme, the involvement of olfactory bulb was thought to be inconsistently present, coincidental to medullary affectation. Another staging system was specifically devised for DLB by the Dementia with Lewy Bodies Consortium (third version), which grades αsyn‐immunoreactive pathologies in multiple brain regions to classify three types of Lewy pathology development: brain stem predominant, limbic/transitional and diffuse neocortical types 40, 41; in this system, there is no consideration of olfactory bulb involvement.
The efficacy of these classificatory systems has recently been assessed by several groups 8, 14, 20, 23, 29, 31. It has been reported that 6.3% to 47% of clinical PD cases did not follow the proscribed caudal‐to‐rostral pattern of LTS pathological development 13, 30, 46. A similar situation was found to exist for the DLB Consortium system where again, the dictated caudal‐to‐rostral progression was often not followed. Additionally, both systems failed to identify the presence of a relatively common olfactory bulb‐only stage, which may be the first stage common to all LB disorders (8). Because of these deficiencies, we proposed “Unified Lewy Body Staging System,” which can be used to classify all subjects regardless of types of LB disorders (Figure 1) (7). This system was established by the abundance and distribution of phosphorylated serine 129 alpha synuclein (pαsyn) immunoreactive pathologies in a large series of autopsy cases from the Banner Sun Health Research Institute Brain and Body Donation Program (7).
Figure 1.

Unified staging scheme. This Lewy body (LB) pathology classification system is based on a scoring system grading the anatomical distribution of phosphorylated α‐synuclein (pαsyn) published previously (7). The system proposed that the development and progression of pαsyn‐containing LB pathology is initiated in olfactory bulb as stage I, followed by brain stem‐predominant (stage IIa) or limbic‐predominant stages (stage IIb), then merged as both brainstem and limbic stages (stage III) and eventually advancing to neocortex (stage IV). The diseases which are likely to be observed at different stages are shown also in the scheme: PD = Parkinson's disease; ILBD = Incidental Lewy body disease; ADLB = Alzheimer's disease with Lewy bodies; DLB = dementia with Lewy bodies.
Although there are other serine and tyrosine residues on αsyn, phosphorylation at serine 129 is the most prominent pathological species in LB disorders 2, 21, 22. Under normal condition, the brain expression of pαsyn is very low, but in LB disorders, its levels are increased in synapse‐enriched fractions as well as in the hallmark pathologies, Lewy neurites and Lewy bodies 2, 43. Phosphorylation of αsyn promotes oligomerization, aggregation, neurotoxicity and formation of LTS pathology, and reduces the ability to regulate tyrosine hydroxylase 17, 25, 27, 33, 44, 45, 49, 52. In αsyn transgenic mouse model, enhancing phosphatase activity has been shown to reduce the levels of pαsyn and improve neuronal activity and motor performance (32).
In the present study, we explored the temporal and anatomical relationship between the presence of histologically identified pαsyn‐containing LTS and biochemically measured pαsyn. We hypothesized that increase in biochemically measurable serine 129 pαsyn precedes the presence of histologically identified LTS pathologies. The hypothesis was tested in cingulate and temporal cortices of 128 subjects (normal controls as well as cases from all LB stages). Because LTS pathology is absent or rare in the cingulate and temporal cortices at early disease stages, such as stages I and II, they can be used to determine the temporal relationship between the biochemical and histological changes of pαsyn occurring at early stages of the diseases.
We measured both pαsyn and the combined amounts of unmodified (nonphosphorylated) and phosphorylation‐modified αsyn, defined as total αsyn (tαsyn) and determine the ratios of pαsyn to tαsyn in individual samples. Based on this ratio, we determined whether increases in pαsyn levels were selective or due only to increases in total αsyn. We also determined if the increase in biochemically measurable pαsyn in cingulate and temporal cortical regions had already occurred when LTS histopathologies began to appear in olfactory bulb, and spread to substantia nigra and amygdala.
Our results showed a significant increase in the biochemical level of pαsyn when LTS density score in each given region becomes detectable, and the levels of pαsyn in cingulate and temporal cortices were significantly and selectively elevated when histological LTS density scores increased from 0 to 1 in either the olfactory bulb or the substantia nigra, even when LTS scores were zero in cingulate and temporal cortices. These results indicate that aberrant phosphorylation of αsyn is an early and thus possibly important phenomenon during the pathogenesis of the LB disorders.
MATERIALS AND METHODS
Human subjects for Brain Donation Program
Subjects who enrolled in the Brain and Body Donation Program (BBDP) of Banner Sun Health Research Institute (BSHRI) were consented according to the approved protocols of the Banner Health Institutional Review Board. Most subjects receive annual standardized batteries of neurological and neuropsychological assessments 1, 15. For those subjects lacking standardized antemortem evaluations, information was obtained, for some subjects, from a post‐mortem telephone interview with a contact, including an adaptation of the Clinical Dementia Rating Scale (CDR). The demographic features of the subjects used in this study are summarized in Table 1.
Table 1.
Demographic and neuropathological features of the patients grouped by Unified Staging System.
| Features/stages | Stage 0 | Stage I | Stage IIa | Stage IIb | Stage III | Stage IV |
|---|---|---|---|---|---|---|
| Number of subjects | 17 | 11 | 5 | 16 | 34 | 45 |
| Expired age (years) | 84.1 ± 1.5 | 85.8 ± 1.2 | 87.0 ± 1.4 | 81.6 ± 1.9 | 80.6 ± 1.2 | 78.8 ± 0.8 |
| Mean ± SE | *(0, I, IIa) | |||||
| Post‐mortem delay (PMI, hours) | 2.5 ± 0.1 | 3.0 ± 0.2 | 2.4 ± 0.3 | 4.3 ± 0.7 | 3.3 ± 0.2 | 3.0 ± 0.1 |
| Mean ± SE | *(0, I, IIa, III, IV) | |||||
| Rates of apoE ε4 carrier in each stage | 4/17 | 7/11 | 2/5 | 9/16 | 12/34 | 24/45 |
| Gender ratios (male to female) | 12/5 | 5/6 | 3/2 | 7/9 | 21/13 | 26/19 |
| Total amyloid plaque scores | 3.8 ± 1.0 | 12.5 ± 0.5 | 9.8 ± 1.6 | 13.3 ± 0.5 | 6.9 ± 0.9 | 10.8 ± 0.6 |
| Mean ± SE | *(I, IIb, IV) | *(0, III) | *(0, III) | *(I, IIb, IV) | *(0, III) | |
| Braak's scores | 2.8 ± 0.2 | 4.8 ± 0.4 | 3.7 ± 0.5 | 5.3 ± 0.2 | 2.9 ± 0.2 | 3.8 ± 0.2 |
| Mean ± SE | *(I, IIb, IV) | *(0, IIa, III, IV) | *(I, IIb) | *(0, IIa, III, IV) | *(I, IIb, IV) | *(0, I, IIb, III) |
| Substantia nigra depigmentation scores | 0.5 ± 0.2 | 1.5 ± 0.2 | 2.0 ± 0.3 | 0.9 ± 0.2 | 2.8 ± 0.1 | 2.4 ± 0.1 |
| Mean ± SE | *(I, III, IV) | *(0,IIb, III, IV) | *(III) | *(I, III, IV) | *(0, I, IIa, IIb) | *(0, I, IIb) |
Symbol * in front of parenthesis denotes statistical significance level at P < 0.05.
The Roman numerals within the parenthesis represent the stages that differed significantly with the stage indicated at the top row of this table.
An overview of the BSHRI Brain Donation Program (now the Brain and Body Donation Program, BBDP) has been published previously (5). Autopsy cases for this study were chosen by searching the BBDP database for cases in order to establish adequate subject numbers at each Unified stage. A secondary objective was to include subjects from all types of Lewy body disorders, including PD, DLB and ADLB. The diagnostic criteria used for AD, PD and DLB have been previously described (5). For AD and DLB, cases received the diagnosis if they were classified as “intermediate” or “high” probabilities in their respective classification schemes. Cases were classified as ADLB if they had AD and Lewy bodies in any brain region but failed to meet criteria for DLB or PD. The PD cases contained variable plaque and tangle scores and some of them met criteria for the diagnosis of AD. We called these cases PDAD. The cases, which did not contain AD pathology, were called PD when they were not demented and PDD when they were demented. Cases with DLB also often have AD pathology; in this study, 24 met intermediate or high criteria for AD while three did not. Subjects without dementia, Parkinsonism or neuropathological evidence of a LB disorder were chosen as control groups for the study.
Histological methods
Neuropathological diagnosis was made from brain tissue blocks fixed with 4% neutral‐buffered formaldehyde and cut from both paraffin embedded and cryoprotected blocks of various brain regions. Tissue sections were processed with our standard histological procedure 6, 7. Histopathological grading of AD followed Braak neurofibrillary tangle staging and Consortium to Establish a Registry for Alzheimer's Disease (CERAD) templates (42). Microscopic observations of pαsyn‐stained histological slides were performed by a single experienced observer (TGB) without prior knowledge of the patient's medical record or neurological status. Each case was first staged according to the Unified Staging System for LB disorders using a standard set of brain sections stained with an immunohistochemical method for pαsyn as previously described (7). In this methodology, proteinase K pretreatment was used to unmask the epitopes for pαsyn in paraformaldehyde‐fixed brain tissues. For staging purposes, 10 brain regions were examined, including olfactory bulb and tract, anterior medulla, anterior and mid‐pons, mid‐amygdala and adjacent transentorhinal area, anterior cingulate cortex, middle temporal gyrus, middle frontal gyrus and inferior parietal lobule. The density of αsyn‐immunoreactive neuronal cytoplasmic inclusions as well as punctate and neuritic profiles was graded in each region according to the templates published by the DLB Consortium (41). The scores were assigned as none, sparse, moderate, frequent and very frequent. Based on these results, the autopsy cases were assigned to one of the following stages: I, IIa, IIb, III and IV, or 0 when there was no LB pathology present. Stage I is also called “olfactory bulb only;” stage IIa, “brain stem predominant;” stage IIb, “limbic predominant;” stage III, “brain stem and limbic;” and stage IV, “neocortical” as shown in Figure 1, (7).
Brain tissue homogenization and protein extraction
A collection of 128 autopsy cases received at the BBDP of BSHRI between years 1998 and 2008 with LB pathology classified as Unified stage 0 to IV were used for the study. The numbers of cases in each stage are stage 0, 17 normal control cases; stage I, 11 ADLB cases; stage IIa, one PDAD and four PD cases with mild cognitive impairment (MCI); stage IIb, one PDD and 15 ADLB cases; stage III, five PD, 12 PDD, eight PDAD and nine DLB cases; and stage IV, seven PDD, 10 PDAD and 28 DLB cases. Gray matter from cingulate and temporal cortices were dissected out from 1‐cm thick brain slices that were snap frozen between dry ice slabs at autopsy and then stored at −70 to −80°C. Samples were further cut into 2 mm cubes, weighed and homogenized with a sonicator on ice in four volumes of prechilled 1X radio‐immunoprecipitation assay (RIPA) buffer consisting of 25 mM Tris‐HCl (pH 7.6), 150 mM NaCl, 1% NP‐40, 1% sodium deoxycholate, 0.1% sodium dodecyl sulfate (SDS), 1X cocktail protease and phosphatase inhibitors and 1 mM EDTA (Pierce, Thermo Scientifics, Rockford, IL, USA). This buffer is commonly used for extraction of cytosolic, membrane‐associated and extracellular matrix‐associated proteins. After sonication, the extraction of proteins continued for 30 minutes on ice with gentle shaking. To remove unextracted nuclear components and debris, homogenates were centrifuged at 20 000 rpm for 30 minutes in a refrigerated table top centrifuge. A fraction of supernatant was removed for protein assay using Micro BCA™ method (Thermo Scientifics). The rest of the supernatant was aliquoted and stored at −80°C until preparation for Western blot procedure.
Western blotting
Frozen RIPA buffer extracts of cingulate and temporal cortical tissues were thawed and mixed with 4X lithium dodecyl sulfate (LDS) Western blot sample buffer (BD Biosciences, San Jose, CA, USA) and water in the absence of reducing reagent dithiothreitol to obtain protein concentration at 1 µg/µL in 1X LDS buffer. Samples are heated immediately before loading to NuPAGE® 10%–14% Bis‐Tris Mini gels (BD Biosciences). Ten µg of protein was loaded to each lane. The order of the samples was arranged to ensure that samples from cingulate and temporal cortices of the same autopsy case were loaded in adjacent lanes. We also made sure that samples loaded on the same gel contained samples from subjects of stage 0 to stage IV. Some of the Western blot samples were used as across‐gel loading and processing controls. These were used to assess the extent of discrepancy between gels at the end of procedure.
Samples were electrophoresed at 200 V for 40 minutes using 2‐(N‐morpholino)ethanesulfonic acid (MES) buffer (BD Biosciences), and then transferred to nitrocellulose membrane at 30 V for 75 minutes. Blots were air‐dried before blocking with tris‐buffered saline consisting of 0.05% Tween® 20 (VWR International, West Chester, PA, USA) (TBS‐T) and 5% nonfat dry milk. A rabbit polyclonal antibody raised against pαsyn (S129P), characterized previously, was used for immunodetection 3, 7, 22. The specificity of the antibody was determined in a separate investigation (unpublished observation) by showing decreased immunoreactivity after treating the brain protein on the blots with phosphatase and after detection of pαsyn protein generated by co‐incubating αsyn with casein kinase.
After incubation with this antibody at 1:5000 dilution overnight at room temperature, blots were washed four times with TBS‐T and then probed with a goat anti‐rabbit IgG conjugated with horseradish peroxidase (HRP; Thermo Scientifics) at 1:20 000 concentration, and washed four times with TBS‐T before detection with Western Extended Dura chemiluminescence substrate for HRP (Thermo Scientifics).
Parallel blots were probed with a mouse anti‐αsyn antibody (No. 610786, BD Biosciences) at 1:1000 dilution. This antibody, raised against nonphosphorylated epitopes between amino acid 15–123, does not distinguish pαsyn from unmodified form, thus its immunoreactive products consisted of both phosphorylated and nonphosphorylated αsyn. We designated this measurement as tαsyn. After overnight incubation with primary antibody, anti‐mouse IgG conjugated with HRP (Thermo Scientifics) was used at 1:10 000. The level of β actin in each sample was detected in each blot with a mouse anti‐β actin (Clone AC‐15, Sigma, St. Louis, MO, USA) antibody at 1:5000. Finally, the immunoreactivities of the bands were detected with Western Extended Dura chemiluminescence kits.
Images of immunoreactive bands were captured using CCD camera in the FluorChem® Q (Cell Biosciences, Santa Clara, CA, USA) ChemiImager™. Images from three durations of exposure were taken for each protein, ranging from 5 s to 1.5 minute; and the intensity of individual band was quantified with the software provided with FluorChem® Q. The results of densitometry were compared between exposure durations for each protein in order to determine if the development of immunoreactivity was within linear range. Although three exposures were taken for each blot, only one set of densitometry results was used for normalization by β actin and calculation of the ratio of pαsyn to tαsyn.
Statistical analysis
Statistical analysis was performed with MedCalc software version 9.8 (Mariakerke, Belgium). The significance level for all comparisons was set at P < 0.05. One‐way analysis of variance (one‐way ANOVA) was used to analyze the stage‐related differences between pαsyn, total αsyn and their ratios in each brain region followed by Student Newman–Keuls pairwise comparisons. To assess the effects of pathological stages and brain regions on the levels of pαsyn, total αsyn and their ratios, two‐way ANOVA was used, followed by Student Newman–Keuls analysis for pairwise comparison. The F‐ratio is considered to be significant when P < 0.05. Spearman Rank correlation was used to relate the biochemical levels of pαsyn to pathological features including a standard set of inputs, namely, the LB‐related density scores from olfactory bulb, substantia nigra, amygdala, cingulate cortex, temporal cortex and frontal cortex.
RESULTS
Characteristics of the autopsy cases in this study
Demographic and neuropathological features
Demographic and neuropathological features including age at death, post‐mortem interval, frequency of the apolipoprotein Eε4 (ApoEε4) genotype, total amyloid plaque density scores, Braak's stage and substantia nigra pigmented neuron loss scores are shown in Table 1.
Subjects at Unified stage IV died at younger age (mean ± SE = 78 ± 0.8) than subjects at stages 0, I and IIa (P < 0.05). Post‐mortem intervals (PMI) were significantly longer for subjects at Unified stage IIb but did not otherwise differ between groups. The frequency of ApoE ε4 allele carriers was not significantly different between subjects at different Unified stages. The substantia nigra pigmented neuron loss score was significantly different from controls for all groups except for those in stage IIb. In terms of AD‐related pathology, total plaque scores were not significantly different between stages I, IIb and IV, but were significantly higher than stages 0 and III. Braak's stages were not significantly different between Unified stages I and IIb, but were significantly higher than Unified stages 0, IIa, III and IV.
Lewy‐type synucleinopathy (LTS) histopathological density scores in various brain regions
A grading system with a scale of 0 to 4 was used to assess the abundance of pαsyn‐immunoreactive Lewy‐type synucleinopathy (LTS) in olfactory bulb, brain stems, amygdala, trans‐entorhinal cortex, cingulate cortex and neocortical cortical regions, as described in previous publication (41). The means and standard errors of density scores of pαsyn‐immunoreactive LTS in 10 brain regions are listed according to Unified stages of this studied population in Table 2.
Table 2.
Lewy pathology density scores in 10 brain regions of the studied patients grouped by Unified stages [mean ± standard errors (subject numbers)].
| Unified stages/brain regions | I | IIa | IIb | III | IV |
|---|---|---|---|---|---|
| Olfactory bulb | 1.71 ± 0.19 (11) | 1.40 ± 0.68 (5) | 3.31 ± 0.18 (16) | 2.17 ± 0.24 (30) | 2.92 ± 0.20 (39) |
| Substantia nigra | 0.00 ± 0.00 (11) | 0.50 ± 0.29 (5) | 0.50 ± 0.18 (16) | 2.27 ± 0.18 (33) | 2.67 ± 0.13 (42) |
| Pons | 0.00 ± 0.00 (11) | 0.75 ± 0.48 (4) | 0.25 ± 0.18 (12) | 2.74 ± 0.18 (34) | 2.78 ± 0.16 (40) |
| Medulla | 0.00 ± 0.00 (11) | 1.00 ± 0.70 (4) | 0.83 ± 0.21 (12) | 2.88 ± 0.19 (32) | 2.81 ± 0.17 (42) |
| Amygdala | 0.00 ± 0.00 (11) | 0.60 ± 0.40 (5) | 3.60 ± 0.13 (15) | 3.44 ± 0.11 (32) | 3.73 ± 0.09 (41) |
| Trans‐entorhinal cortex | 0.00 ± 0.00 (11) | 0.40 ± 0.40 (5) | 2.93 ± 0.31 (14) | 2.36 ± 0.17 (33) | 3.31 ± 0.13 (42) |
| Cingulate cortex | 0.00 ± 0.00 (11) | 0.20 ± 0.20 (5) | 0.60 ± 0.13 (15) | 1.88 ± 0.13 (32) | 3.11 ± 0.12 (44) |
| Temporal cortex | 0.00 ± 0.00 (11) | 0.00 ± 0.00 (5) | 0.33 ± 0.13 (15) | 0.97 ± 0.05 (32) | 2.50 ± 0.12 (44) |
| Parietal cortex | 0.00 ± 0.00 (11) | 0.00 ± 0.00 (5) | 0.06 ± 0.06 (15) | 0.77 ± 0.09 (34) | 1.73 ± 0.11 (45) |
| Frontal cortex | 0.00 ± 0.00 (11) | 0.00 ± 0.00 (5) | 0.06 ± 0.06 (15) | 0.77 ± 0.07 (34) | 1.67 ± 0.11 (45) |
We examined the pαsyn‐immunoreactive LTS density scores in cingulate and temporal cortex, the two brain regions that we studied with biochemical methods. At stage IIb, the density scores were lower than 1 in both brain regions; 0.60 ± 0.13 (mean ± SE) in cingulate cortex and 0.33 ± 0.13 in temporal cortex, demonstrating a limited presence of LTS histopathology in these two brain regions at this stage of LB disorders. In fact, 10 of 15 stage IIb cases had no LTS pathology in temporal cortex, whereas six cases had no LTS pathology in cingulate cortex. The LTS scores remained no greater than 1 in temporal cortex as the disease progressed to Unified stage III. Conversely, the majority of stage III cases had LTS density scores greater than 1 in cingulate cortex. These data show that LTS pathological development in temporal cortex is behind cingulate cortex, which is consistent with our previously published observations and classification system that showed appearance of LTS pathology in cingulate cortex preceding neocortical LTS in a large studied population (7).
Levels of pαsyn and tαsyn, and their ratios in cingulate and temporal cortices
The main goal of this study was to determine the temporal relationship between the spread of LTS histopathology and changes in the levels of pαsyn and tαsyn in RIPA buffer brain extracts. These αsyn proteins were measured by Western blotting with a serine 129 phosphorylation‐specific antibody and an antibody‐detecting epitopes before serine 129, respectively; then normalized by the levels of β actin in each sample. A panel of Western blot images of pαsyn, tαsyn and β actin from representative cases across all Unified LTS stages is shown in Figure 2A. This figure shows the arrangement of cingulate (“C” in the graph) and temporal cortical (“T” in the graph) samples from the same autopsy case in adjacent lanes on the same gel. Strong immunoreactivity for pαsyn protein was observed in the samples from most of stage IV and some stage III cases.
Figure 2.
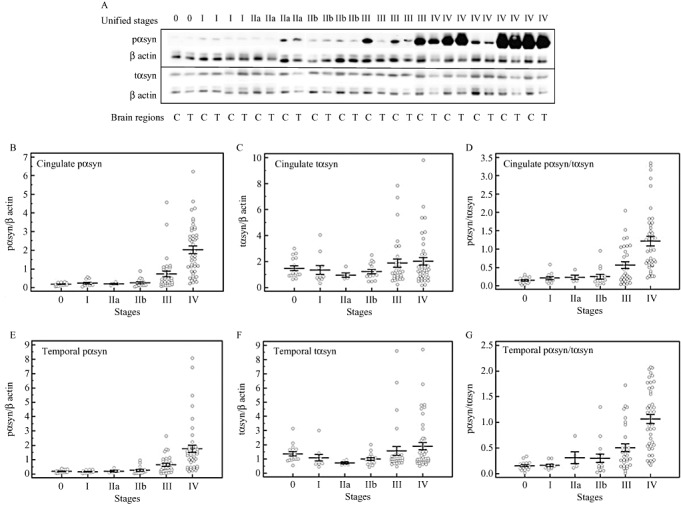
Levels of pαsyn and total αsyn (tαsyn), and their ratios according to Unified stages. A. Immunoblot images of pαsyn and tαsyn from cingulate (C) and temporal (T) cortical samples across Unified stages (0, I, IIa, IIb, III and 4), the bars in the graphs represent mean values and standard errors of pαsyn in cingulate and temporal cortices (B and E) tαsyn in cingulate and temporal cortices (C and F); and the ratios of pαsyn to tαsyn in cingulate and temporal cortex (D and G).
The intensities of full‐length pαsyn‐ and tαsyn‐immunoreactive bands at 18 kD, based on the molecular weight marker run simultaneously, were quantified. The pαsyn and tαsyn proteins were detected on separate membranes to avoid overlapping of the band images caused by minor differences in molecular weights between pαsyn and tαsyn proteins. To assess if there would be a gel‐to‐gel discrepancy occurred at different steps of the procedure, same samples were repeatedly run on different gels. Our results showed no evidence of gel‐to‐gel differences.
We first used one‐way ANOVA to determine if the levels of pαsyn and tαsyn were statistically different between samples from different Unified stages. The results were shown in Figure 2B–G. There were no significant changes detected between stages zero, I, IIa, IIb and III. A drastic increase in the levels of pαsyn and the ratio of pαsyn to tαsyn was detected between stage IV samples and the samples of other stages (P < 0.05). From stage III to stage IV, there were approximately 1.9‐fold and 2.1‐fold increases in pαsyn, and 1.4‐fold and 1.1‐fold increases in the ratios in cingulate and temporal cortices, respectively. Because we were interested in determining the changes in pαsyn levels at earlier disease stages, we reanalyzed the data by excluding stage IV cases. This time, the analysis showed significant twofold and 1.5‐fold increases in cingulate and temporal cortices in stage III, respectively, from the earlier stages.
Because our stage IV subjects consisted of more DLB cases than other disease categories (27 DLB, nine PDAD and seven pure PDD), we determined whether there were disease‐associated differences in the levels of pαsyn in this particular stage. One‐way ANOVA results showed that the levels in DLB cases were significantly greater than the levels in other two disease groups, and this observation was found in cingulate (DLB: 2.33 ± 0.21; PDAD: 1.08 ± 0.33, pure PDD: 1.12 ± 0.27; P < 0.05) and temporal cortices (DLB: 1.88 ± 0.24; PDAD: 0.908 ± 0.20; Pure PDD: 0.767 ± 0.22, P < 0.05). We then examined whether this would also occur in stage III subjects with DLB. There were eight DLB cases in stage III group, in addition to 12 pure PDD, eight ADPD and five PD cases. There were no disease group‐associated differences detected in both brain regions in stage III cases. To determine whether DLB cases were responsible for the statistical significances in the pαsyn levels analyzed by Unified stages, we analyzed the data again by excluding all of the DLB cases from stages III and IV. The results showed that the significant increases in pαsyn in these stages remained.
The levels of tαsyn did not change with stages in both brain regions. Thus, the changes in the levels of pαsyn are accountable for the changes in the ratios. When two‐way ANOVA was used to assess whether there were effects of LTS histopathological stages and brain regions on the levels of pαsyn, the results showed significant effects caused by the LB stages, but not by brain regions. No interaction between the effects of LB stages and brain regions were detected.
Relationship between the biochemical measures in cingulate and temporal cortices
We then determined if the levels of pαsyn and tαsyn, and the ratios of pαsyn to tαsyn were correlated between two cortical regions using a linear regression model. Highly significant (P < 0.001) positive linear relationships were detected between these two brain regions in all of the three measures (R2 = 0.4643 for pαsyn, R2 = 0.7497 for total αsyn and R2 = 0.4295 for the ratios).
To determine how well the pαsyn levels in cingulate and temporal cortices correlated with LTS density scores, we performed Spearman's rank correlation analysis. The correlation coefficients Rho showed that across four Unified stages, cingulate and temporal pαsyn levels correlated very well with LTS scores not only within the same brain regions, but also in other brain regions that are affected at earlier stages, including the olfactory bulb, substantia nigra and amygdala (Table 3). The correlation coefficients between expression levels of pαsyn and LTS scores are 0.732 (P < 0.0001) in cingulate cortex and 0.711 (P < 0.0001) in temporal cortex.
Table 3.
Spearman's rank correlation coefficients between Unified staging LTS density scores and the levels of phosphorylated α synuclein in cingulate and temporal cortices.
| Brain regions | Phospho‐α synuclein levels in cingulate cortex (Rho, significance level) | Phospho‐α synuclein levels in temporal cortex (Rho, significance level) |
|---|---|---|
| Olfactory bulb | ||
| LTS density scores | 0.412, P < 0.0001 | 0.411, P < 0.0001 |
| Substantia nigra | ||
| LTS density scores | 0.623, P < 0.0001 | 0.589, P < 0.0001 |
| Amygdala | ||
| LTS density | 0.476, P < 0.0001 | 0.477, P < 0.0001 |
| Trans‐entorhinal | ||
| Cortex LTS density scores | 0.548, P < 0.0001 | 0.560, P < 0.0001 |
| Cingulate | ||
| Cortex LTS density scores | 0.732, P < 0.0001 | 0.660, P < 0.0001 |
| Temporal | ||
| Cortex LTS density scores | 0.745, P < 0.0001 | 0.711, P < 0.0001 |
| Parietal | ||
| Cortex LTS density scores | 0.722, P < 0.0001 | 0.677, P < 0.0001 |
| Frontal | ||
| Cortex LTS density scores | 0.701, P < 0.0001 | 0.660, P < 0.0001 |
We have already shown that pαsyn levels in cingulate and temporal cortical regions significantly elevated at more advanced Unified stages. We then analyzed the levels of pαsyn by LTS histopathology density scores (0 to 4) in cingulate and temporal cortices. The results were shown in Figure 3A (cingulate cortex) and 3B (temporal cortex). One‐way ANOVA showed that in the subjects with cingulate LTS density scores = 3 and 4, the levels of pαsyn within the same brain region were significantly higher than lower LTS density scores (P < 0.05), while there were no difference in levels of pαsyn between the samples with LTS scores = 3 and 4. There was also no difference in pαsyn levels between LTS scores = 0 and 1. However, when LTS score = 2, the levels of pαsyn were not different from the samples with LTS = 0 and 1, but significantly lower than the samples with LTS = 3 and 4. In temporal cortex, the levels of pαsyn were significantly elevated in the samples with LTS scores > 0 than in the samples with LTS scores = 0 (P < 0.05).
Figure 3.
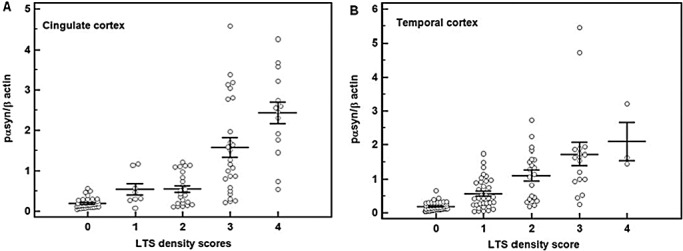
Increases in the levels of pαsyn with LTS histopathological scores. The biochemical levels of pαsyn are grouped by LTS histopathological scores 1 to 4. Horizontal lines indicated means and the vertical bars represent standard errors. Each circle in the graph represents the data from one individual. One‐way ANOVA showed that in the cases that cingulate LTS density scores = 3 and 4, the levels of pαsyn within the same brain region were significantly higher than lower LTS density scores (P < 0.05), while there were no difference in levels of pαsyn between the samples with LTS scores = 3 and 4. There was also no difference in pαsyn levels between LTS scores = 0 and 1. When LTS score = 2, the levels of pαsyn were not different from the samples with LTS = 0 and 1, but significantly lower than the samples with LTS = 3 and 4. In temporal cortex, the levels of pαsyn were significantly elevated in the samples with LTS scores > 0 than the samples with LTS scores = 0 (P < 0.05).
The expression levels of biochemically measurable and the histologically detectable pαsyn do not always coincide in the studied subjects. For examples, 25%–30% of stage III cases and 5% of stage IV cases had pαsyn levels lower than the mean values of pαsyn in cingulate and/or temporal cortices in stage II subjects. We showed the LTS histopathological images from some of the representative cases, which had discrepancy between biochemical and histological abundance in Figure 4. LTS histopathological profiles detected with pαsyn antibody were photographed from 5‐µm paraffin sections cut from cingulate and temporal cortices from the representative cases that showed agreement or discrepancy in two types of measures of pαsyn (Figure 4A–H). The pαsyn immunoreactivities appeared in dark blue color in the images. The LTS scores and pαsyn level were written side by side with each histological image. Case 1 had LTS histopathology scores = 4 in cingulate cortex and LTS scores = 3 in temporal cortex, case 2 had LTS score = 4 in cingulate cortex and LTS score = 3 in temporal cortex, whereas case 3 and case 4 all had LTS score = 1 in both cingulate and temporal cortices. Although case 1 had higher biochemically measurable pαsyn level than case 2, there is hardly any difference in the abundance of LTS pathology (Figure 4A–D). Cases 3 and 4 had the same LTS scores in both cingulate and temporal cortices, but case 3 had higher biochemically measurable pαsyn than case 4. Nevertheless, there is no observable difference in the presence of LTS pathology between the cases 3 and 4 (Figure 4E–H).
Figure 4.
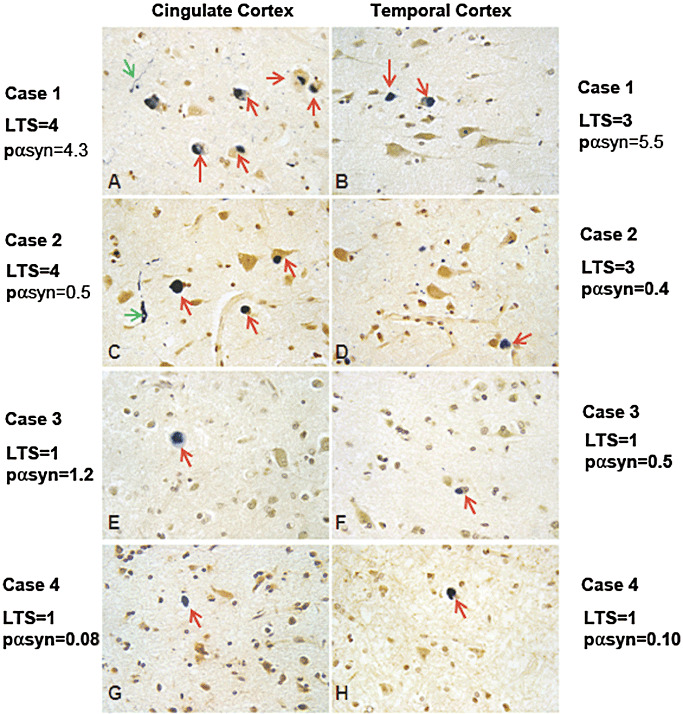
Representative images of pαsyn immunoreactivities in cingulate and temporal cortices. Four representative cases were documented for the abundance of pαsyn immunoreactivities shown in dark blue color. Images from 5 µm paraffin sections from cingulate and temporal cortices were taken with CCD camera and a 40X objective on a light microscope (Olympus). The LTS scores and pαsyn level were written at the sides of each histological image. Case 1 had LTS histopathology scores = 4 in cingulate cortex and LTS scores = 3 in temporal cortex, case 2 had LTS score = 4 in cingulate cortex and LTS score = 3 in temporal cortex, case 3 and case 4 all had LTS score = 1 in both cingulate and temporal cortices. Although case 1 had higher biochemically measurable pαsyn level than case 2, there is hardly any difference in the abundance of LTS pathology in the tissues as shown in Figures A–D. Cases 3 and 4 had the same LTS scores in both cingulate and temporal cortices, but case 3 had higher biochemically measurable pαsyn than case 4. Nevertheless, there is no observable difference in the presence of LTS pathology between the cases 3 and 4 (Figures E–H).
We then determined whether the levels of pαsyn in cingulate and temporal cortices is associated with the presence of AD pathology especially in the cases that LTS pathology is absent. The AD pathology was assessed by total tangle scores, Braak's stages and plaque density scores obtained according to our standard histological procedure. We performed Spearman's rank correlation analysis in the subset of the cases that has no LTS pathology. We found that there was no association between the levels of pαsyn in either cingulate or temporal cortices and the AD pathologies in these cases. Thus, it is likely that the expression of pαsyn in these cases were not affected by AD neuropathology.
Increases in the levels of pαsyn occur at early stages of Lewy pathology formation
To further investigate how early the levels of pαsyn in cingulate and temporal cortices are altered during disease progression, we focused on the relationship of pαsyn with the LTS density scores in brain regions and stages that are selectively affected early in the disorders, namely, olfactory bulb and substantia nigra. We compared the pαsyn levels from subjects with LTS density scores graded as 0 or 1 in olfactory bulb and substantia nigra, and 0 in cingulate and temporal cortices.
A nonparametric test was performed to compare the samples derived from subjects who were at Unified LB stages 0–II. These subjects were grouped by LTS density scores, 0 or 1 in olfactory bulb or substantia nigra, and 0 and 1–2 in amygdala. As shown in Figure 5A–F, the levels of pαsyn were significantly elevated in cingulate and temporal cortices when LTS density scores increased from 0 to 1 in olfactory bulb and substantia nigra. The change in pαsyn levels between the cases with amygdala LB score 0 and greater than 1 were significantly different in the temporal cortex samples, but not in the cingulate samples. This trend continues as the LTS scores in olfactory bulb and substantia nigra increased to 2; there were more than twofold increases observed in pαsyn levels in both brain regions (data not shown).
Figure 5.
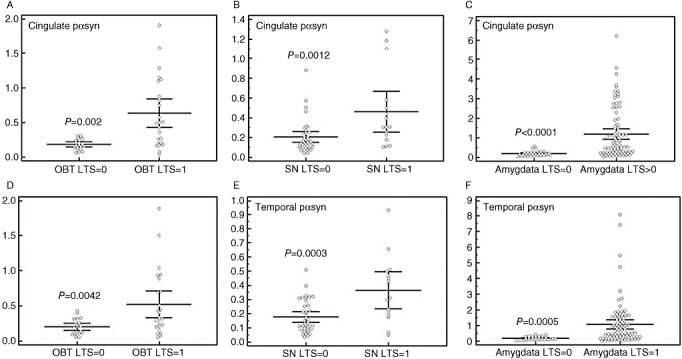
Levels of pαsyn in cingulate and temporal cortices according to Lewy‐type synucleinopathy (LTS) density scores in olfactory bulb, substantia nigra and amygdala. Bar graphs represents mean values and standard errors of pαsyn in cingulate (A, B, C) and temporal (D, E, F) cortices according to LTS density scores in olfactory bulb (OBT) (A and D), substantia nigra (SN) (B and E) and amygdala (C and F).
DISCUSSION
Previously, the distribution patterns and abundance of histological deposits of immunoreactive αsyn in olfactory bulb, brain stem, limbic and neocortex have been used to describe the progression of PD and DLB, two major diseases caused by Lewy‐type α‐synucleinopathy However, a new Unified Staging System for Lewy Body Disorders, based on pαsyn immunohistochemistry, has demonstrated an improved efficacy for the classification of LB disorders. In this system, the grading of pαsyn‐containing LTS histopathology was conducted in 10 brain regions. The LTS histopathology begins in olfactory bulb at Unified stage I, followed by either brain stem (stage IIa) or amygdala (stage IIb) and by stage III, the LTS histopathology coexists in both brain stem and limbic regions. The LTS histopathology spread to neocortical regions in stage IV. This new system has demonstrated that the deposition of αsyn phosphorylated at serine 129 is a valid and sensitive marker of LTS pathological progression in LB disorders (7).
The present study analyzed RIPA buffer‐soluble αsyn and pαsyn in cingulate and temporal cortices from subjects across Unified LTS stages to determine whether the levels of pαsyn are altered prior to its presence in LTS histopathology in LB disorders. Biochemical methodology is effective for detection of pαsyn protein, which is not in LB pathologies 2, 43. The increase in pαsyn expression could be independent of the presence of LTS histopathology as nonaggregated pαsyn could be localized in synapse‐enriched fraction (43).
We chose to study cingulate (belong to limbic system) and temporal cortices because these two brain regions have absent or rare LB pathology at early disease stages such as stages I and II, thus they are suitable for inferring a possible temporal relationship between the biochemical and histological changes of pαsyn at early stages of diseases. Our results showed that pαsyn could be detected in cingulate and temporal cortices in normal controls and across four Unified stages. Our results also showed that pαsyn expression levels in both cingulate and temporal cortices are significantly elevated as Unified stages advance from II to III, and also as early as the LTS histopathology density score increased from 0 to 1 in temporal cortex as well as from 2 to 3 in cingulate cortex. Approximately 70% of our stage III cases have cingulate LTS density scores 2 and 3, while their temporal LTS scores are 1. These results suggested that biochemically measurable pαsyn in temporal cortex could be more sensitive than cingulate cortex for reflecting the increases in LTS histopathology within same brain region during these stages of LTS development.
Our second major finding was obtained from exploring how early the biochemical levels of pαsyn in cingulate and temporal cortices were altered in relation to LTS pathology developed in the brain regions that are affected at early stages of LB disorders. We compared the pαsyn levels from subjects with LTS density scores graded as 0 or 1 in olfactory bulb and substantia nigra, and 0 in cingulate and temporal cortices. Patients with olfactory bulb LTS density score = 1 were either at Unified stages I or II, whereas patients with substantia nigra LTS score = 1 were usually at stage IIa. Our results showed biochemically measurable pαsyn levels were significantly elevated as LTS density scores in olfactory bulb and substantia nigra increased from 0 to 1, indicating that changes of pαsyn levels in cingulate and temporal cortices coincided with the early appearance of the LB pathology in olfactory bulb and substantia nigra, even though histologically demonstrable LTS was lacking in these two cortical regions. This finding raises the question whether phosphorylation modification is a widespread event across brain regions, even when LTS histopathology has only limited appearance in olfactory bulb, brains stem or amygdala.
Although our data demonstrated phosphorylation of αsyn in cingulate and temporal cortices as an early abnormality in LTS‐associated disorders, we need to point out the existence of the variability in the data obtained in this study. For example, approximately 25%–30% of stage III cases had pαsyn lower than the mean values of stage II samples in one or both of the brain regions. From demographical features and histopathological measures of AD and PD that were available from these cases, we could not identify the specific causes that could have made these cases to express lower levels of pαsyn. Current knowledge on the dynamics between increases in pαsyn compartmentalized in cytoplasm and synapses, and formation of Lewy bodies and neurites are still not clearly understood. The possibility for the coexistence of biochemical and/or pathological modifiers in favor of pathological formation in these cases could not be excluded.
It has been demonstrated that AD pathologies have synergistic effects on the formation of LTS pathologies and it is frequent to find concurrent AD pathology in LB disorders 7, 9, 18, 19, 41. Among the molecules that have been shown to interact with αsyn and pαsyn, amyloid beta (Aβ) peptide and phosphorylated tau are on top of the list. Phosphorylated tau was shown to be increasingly co‐localized with pαsyn in the synaptic‐enriched fractions (43). Aβ peptide increased intraneuronal accumulation of αsyn in cell culture and transgenic mouse model (37). Transgenic mice over‐expressing αsyn increased accumulation of Aβ as well as ApoE (24). We have assessed the cases that did not have LTS pathology in cingulate and temporal cortices and the cases that had higher LTS but low biochemical values of pαsyn for the effects of AD pathology. In general, we did not detect an association of AD hallmark pathologies with the levels of pαsyn in these cases. However, we did not measure the biochemical levels of Aβ and phosphorylated tau in these brain samples, we could not conclude if these proteins had influence on the levels of pαsyn in our studied subjects.
Evidence has indicated that phosphorylation at serine 129 could play an important role in regulation of oligomerization, fibrillogenesis and aggregation of αsyn 17, 25, 27, 33, 44, 45, 49, 52. Increased expression of monomeric pαsyn promotes oligomerization of pαsyn, which causes neurotoxicity and synaptic damages 16, 51. Although the effects of pαsyn on cellular functions have not been completely characterized, a recent study using a molecular pull‐down technique for proteomic analysis showed that pαsyn interacted with a vast range of functional and structural proteins, including cytoskeletal proteins, cell‐signaling proteins and molecules associated with synaptic vesicle trafficking (39).
If phosphorylation of αsyn at serine 129 is indeed a critical step in the pathogenesis of LB disorders, then possible mechanisms leading to this, including increased activity of the specific kinases, reduced dephosphorylation capability and reduced degradation and clearance, could be important processes for therapeutic targeting. To this date, three families of kinases have been shown capable of in vitro phosphorylation of nonaggregated αsyn at serine 129, including casein kinases (CK) 1 and 2, G‐protein‐coupled receptor kinases (GRK) leucin‐rich repeat kinase 2 (LRRK2) and polo‐like kinases (PLK) 1, 2 and 3 38, 48, 54. Among these, PLK1, CK1 and CK2 have also been shown to phosphorylate aggregated αsyn 41, 54, 55. In vitro, phosphorylation of αsyn by PLK2 was shown to compromise the ability of αsyn to regulate the activity of tyrosine hydroxylase (TH), the rate‐limiting enzyme of dopamine synthesis (33). By inhibition of the enzyme that dephosphorylates TH, called phosphor‐protein phosphatase 2A (PP2A), αsyn promotes the activity of the TH (47). The phosphorylation at serine129 of αsyn reduced this function (33). One recent study in a transgenic mouse model over‐expressing αsyn showed that by enhancing phosphatase activity of the PP2A, the phosphorylation and aggregation of αsyn could be reduced (32). As understanding of the mechanisms of phosphorylation and dephosphorylation of αsyn is advancing, to clarify the mechanisms contributing to the accumulation of pαsyn in LB pathologies, it is important also to examine the mechanisms for degradation of pαsyn, which are currently unclear; the ubiquitin‐independent proteasome pathway or a ubiquitin‐dependent pathway after dephosphorylation have both been implicated (34).
Taken together, we have shown significant, Unified stage‐associated increases in RIPA buffer‐soluble pαsyn in cingulate and temporal cortices; and we have also shown significant increases in these measures coincided with the appearance of the LTS histopathology in olfactory bulb and substantia nigra. This study support that the mechanisms driving the increases in phosphorylation of αsyn are at work at early stages of LB disorders and the effects are more widespread than what have known previously, thereby justifies the importance of identifying the causes and mechanisms leading to aberrant phosphorylation of αsyn in these conditions.
ACKNOWLEDGMENTS
The Prescott Family Initiative of the Michael J Fox Foundation for Parkinson's Research, the Arizona Biomedical Research Commission (contracts 4001,0011, 05‐901 and 1001 to the Arizona Parkinson's Disease Consortium), the Arizona Department of Health Services (Contract 211002, Arizona Alzheimer's Research Center), the National Institute on Aging (P30 AG19610, Arizona Alzheimer's Disease Core Center).
Conflict of interest: All of the authors declare no conflict of interest.
REFERENCES
- 1. Adler CH, Hentz JG, Joyce JN, Beach T, Caviness JN (2002) Motor impairment in normal aging, clinically possible Parkinson's disease, and clinically probable Parkinson's disease: longitudinal evaluation of a cohort of prospective brain donors. Parkinsonism Relat Disord 9:103–110. [DOI] [PubMed] [Google Scholar]
- 2. Anderson JP, Walker DE, Goldstein JM, de Laat R, Banducci K, Caccavello RJ et al (2006) Phosphorylation of Ser‐129 is the dominant pathological modification of alpha‐synuclein in familial and sporadic Lewy body disease. J Biol Chem 281:29739–29752. [DOI] [PubMed] [Google Scholar]
- 3. Arai T, Ueda K, Ikeda K, Akiyama H, Haga C, Kondo H et al (1999) Argyrophilic glial inclusions in the midbrain of patients with Parkinson's disease and diffuse Lewy body disease are immunopositive for NACP/alpha‐synuclein. Neurosci Lett 259:83–86. [DOI] [PubMed] [Google Scholar]
- 4. Arima K, Ueda K, Sunohara N, Hirai S, Izumiyama Y, Tonozuka‐Uehara H, Kawai M (1998) Immunoelectron‐microscopic demonstration of NACP/alpha‐synuclein‐epitopes on the filamentous component of Lewy bodies in Parkinson's disease and in dementia with Lewy bodies. Brain Res 808:93–100. [DOI] [PubMed] [Google Scholar]
- 5. Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L et al (2008) The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank 9:229–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Beach TG, White CL, Hamilton RL, Duda JE, Iwatsubo T, Dickson DW et al (2008) Evaluation of alpha‐synuclein immunohistochemical methods used by invited experts. Acta Neuropathol 116:277–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Beach TG, Adler CH, Lue L, Sue LI, Bachalakuri J, Henry‐Watson J et al (2009) Unified Staging System for Lewy body disorders: correlation with nigrostriatal degeneration, cognitive impairment and motor dysfunction. Acta Neuropathol 117:613–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Beach TG, White CL III, Hladik CL, Sabbagh MN, Connor DJ, Shill HA et al (2009) Olfactory bulb alpha‐synucleinopathy has high specificity and sensitivity for Lewy body disorders. Acta Neuropathol 117:169–174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Braak H, Del TK (2009) Neuroanatomy and pathology of sporadic Parkinson's disease. Adv Anat Embryol Cell Biol 201:1–119. [PubMed] [Google Scholar]
- 10. Braak H, Del TK, Rub U, de Vos RA, Jansen Steur EN, Braak E (2003) Staging of brain pathology related to sporadic Parkinson's disease. Neurobiol Aging 24:197–211. [DOI] [PubMed] [Google Scholar]
- 11. Braak H, Rub U, Jansen Steur EN, Del TK, de Vos RA (2005) Cognitive status correlates with neuropathologic stage in Parkinson disease. Neurology 64:1404–1410. [DOI] [PubMed] [Google Scholar]
- 12. Braak H, Bohl JR, Muller CM, Rub U, de Vos RA, Del TK (2006) Stanley Fahn Lecture 2005: the staging procedure for the inclusion body pathology associated with sporadic Parkinson's disease reconsidered. Mov Disord 21:2042–2051. [DOI] [PubMed] [Google Scholar]
- 13. Braak H, Muller CM, Rub U, Ackermann H, Bratzke H, de Vos RA, Del TK (2006) Pathology associated with sporadic Parkinson's disease—where does it end? J Neural Transm Suppl 70:89–97. [DOI] [PubMed] [Google Scholar]
- 14. Burke RE, Dauer WT, Vonsattel JP (2008) A critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 64:485–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Caviness JN, Driver‐Dunckley E, Connor DJ, Sabbagh MN, Hentz JG, Noble B et al (2007) Defining mild cognitive impairment in Parkinson's disease. Mov Disord 22:1272–1277. [DOI] [PubMed] [Google Scholar]
- 16. Chen L, Feany MB (2005) Alpha‐synuclein phosphorylation controls neurotoxicity and inclusion formation in a Drosophila model of Parkinson disease. Nat Neurosci 8:657–663. [DOI] [PubMed] [Google Scholar]
- 17. Chen L, Periquet M, Wang X, Negro A, McLean PJ, Hyman BT, Feany MB (2009) Tyrosine and serine phosphorylation of alpha‐synuclein have opposing effects on neurotoxicity and soluble oligomer formation. J Clin Invest 119:3257–3265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Clinton LK, Blurton‐Jones M, Myczek K, Trojanowski JQ, LaFerla FM (2010) Synergistic Interactions between Abeta, tau, and alpha‐synuclein: acceleration of neuropathology and cognitive decline. J Neurosci 30:7281–7289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Deramecourt V, Bombois S, Maurage CA, Ghestem A, Drobecq H, Vanmechelen E et al (2006) Biochemical staging of synucleinopathy and amyloid deposition in dementia with Lewy bodies. J Neuropathol Exp Neurol 65:278–288. [DOI] [PubMed] [Google Scholar]
- 20. Dickson DW, Uchikado H, Fujishiro H, Tsuboi Y (2010) Evidence in favor of Braak staging of Parkinson's disease. Mov Disord 25(Suppl. 1):S78–S82. [DOI] [PubMed] [Google Scholar]
- 21. Ellis CE, Schwartzberg PL, Grider TL, Fink DW, Nussbaum RL (2001) Alpha‐synuclein is phosphorylated by members of the Src family of protein‐tyrosine kinases. J Biol Chem 276:3879–3884. [DOI] [PubMed] [Google Scholar]
- 22. Fujiwara H, Hasegawa M, Dohmae N, Kawashima A, Masliah E, Goldberg MS et al (2002) alpha‐Synuclein is phosphorylated in synucleinopathy lesions. Nat Cell Biol 4:160–164. [DOI] [PubMed] [Google Scholar]
- 23. Fujishiro H, Ferman TJ, Boeve BF, Smith GE, Graff‐Radford NR, Uitti RJ et al (2008) Validation of the neuropathologic criteria of the third consortium for dementia with Lewy bodies for prospectively diagnosed cases. J Neuropathol Exp Neurol 67:649–656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Gallardo G, Schluter OM, Sudhof TC (2008) A molecular pathway of neurodegeneration linking alpha‐synuclein to ApoE and Abeta peptides. Nat Neurosci 11:301–308. [DOI] [PubMed] [Google Scholar]
- 25. Gorbatyuk OS, Li S, Sullivan LF, Chen W, Kondrikova G, Manfredsson FP et al (2008) The phosphorylation state of Ser‐129 in human alpha‐synuclein determines neurodegeneration in a rat model of Parkinson disease. Proc Natl Acad Sci U S A 105:763–768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Irizarry MC, Growdon W, Gomez‐Isla T, Newell K, George JM, Clayton DF, Hyman BT (1998) Nigral and cortical Lewy bodies and dystrophic nigral neurites in Parkinson's disease and cortical Lewy body disease contain alpha‐synuclein immunoreactivity. J Neuropathol Exp Neurol 57:334–337. [DOI] [PubMed] [Google Scholar]
- 27. Iwatsubo T (2003) Aggregation of alpha‐synuclein in the pathogenesis of Parkinson's disease. J Neurol 250(Suppl. 3):III11–III14. [DOI] [PubMed] [Google Scholar]
- 28. Jakes R, Spillantini MG, Goedert M (1994) Identification of two distinct synucleins from human brain. FEBS Lett 345:27–32. [DOI] [PubMed] [Google Scholar]
- 29. Jellinger KA (2010) Critical evaluation of the Braak staging scheme for Parkinson's disease. Ann Neurol 67:550. [DOI] [PubMed] [Google Scholar]
- 30. Kalaitzakis ME, Graeber MB, Gentleman SM, Pearce RK (2008) The dorsal motor nucleus of the vagus is not an obligatory trigger site of Parkinson's disease: a critical analysis of alpha‐synuclein staging. Neuropathol Appl Neurobiol 34:284–295. [DOI] [PubMed] [Google Scholar]
- 31. Kingsbury AE, Bandopadhyay R, Silveira‐Moriyama L, Ayling H, Kallis C, Sterlacci W et al (2010) Brain stem pathology in Parkinson's disease: an evaluation of the Braak staging model. Mov Disord 25:2508–2515. [DOI] [PubMed] [Google Scholar]
- 32. Lee KW, Chen W, Junn E, Im JY, Grosso H, Sonsalla PK et al (2011) Enhanced phosphatase activity attenuates alpha‐Synucleinopathy in a mouse model. J Neurosci 31:6963–6971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lou H, Montoya SE, Alerte TN, Wang J, Wu J, Peng X et al (2010) Serine 129 phosphorylation reduces the ability of alpha‐synuclein to regulate tyrosine hydroxylase and protein phosphatase 2A in vitro and in vivo . J Biol Chem 285:17648–17661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Machiya Y, Hara S, Arawaka S, Fukushima S, Sato H, Sakamoto M et al (2010) Phosphorylated alpha‐synuclein at Ser‐129 is targeted to the proteasome pathway in a ubiquitin‐independent manner. J Biol Chem 285:40732–40744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Marui W, Iseki E, Nakai T, Miura S, Kato M, Ueda K, Kosaka K (2002) Progression and staging of Lewy pathology in brains from patients with dementia with Lewy bodies. J Neurol Sci 195:153–159. [DOI] [PubMed] [Google Scholar]
- 36. Marui W, Iseki E, Kato M, Akatsu H, Kosaka K (2004) Pathological entity of dementia with Lewy bodies and its differentiation from Alzheimer's disease. Acta Neuropathol 108:121–128. [DOI] [PubMed] [Google Scholar]
- 37. Masliah E, Rockenstein E, Veinbergs I, Sagara Y, Mallory M, Hashimoto M, Mucke L (2001) beta‐amyloid peptides enhance alpha‐synuclein accumulation and neuronal deficits in a transgenic mouse model linking Alzheimer's disease and Parkinson's disease. Proc Natl Acad Sci U S A 98:12245–12250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mbefo MK, Paleologou KE, Boucharaba A, Oueslati A, Schell H, Fournier M et al (2010) Phosphorylation of synucleins by members of the Polo‐like kinase family. J Biol Chem 285:2807–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. McFarland MA, Ellis CE, Markey SP, Nussbaum RL (2008) Proteomics analysis identifies phosphorylation‐dependent alpha‐synuclein protein interactions. Mol Cell Proteomics 7:2123–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. McKeith IG, Perry EK, Perry RH (1999) Report of the second dementia with Lewy body international workshop: diagnosis and treatment. Consortium on Dementia with Lewy Bodies. Neurology 53:902–905. [DOI] [PubMed] [Google Scholar]
- 41. McKeith IG, Dickson DW, Lowe J, Emre M, O'Brien JT, Feldman H et al (2005) Diagnosis and management of dementia with Lewy bodies: third report of the DLB Consortium. Neurology 65:1863–1872. [DOI] [PubMed] [Google Scholar]
- 42. Mirra SS, Gearing M, McKeel DW Jr, Crain BJ, Hughes JP, van Belle G, Heyman A (1994) Interlaboratory comparison of neuropathology assessments in Alzheimer's disease: a study of the Consortium to Establish a Registry for Alzheimer's Disease (CERAD). J Neuropathol Exp Neurol 53:303–315. [DOI] [PubMed] [Google Scholar]
- 43. Muntane G, Dalfo E, Martinez A, Ferrer I (2008) Phosphorylation of tau and alpha‐synuclein in synaptic‐enriched fractions of the frontal cortex in Alzheimer's disease, and in Parkinson's disease and related alpha‐synucleinopathies. Neuroscience 152:913–923. [DOI] [PubMed] [Google Scholar]
- 44. Neumann M, Kahle PJ, Giasson BI, Ozmen L, Borroni E, Spooren W et al (2002) Misfolded proteinase K‐resistant hyperphosphorylated alpha‐synuclein in aged transgenic mice with locomotor deterioration and in human alpha‐synucleinopathies. J Clin Invest 110:1429–1439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Okochi M, Walter J, Koyama A, Nakajo S, Baba M, Iwatsubo T et al (2000) Constitutive phosphorylation of the Parkinson's disease associated alpha‐synuclein. J Biol Chem 275:390–397. [DOI] [PubMed] [Google Scholar]
- 46. Parkkinen L, Pirttila T, Alafuzoff I (2008) Applicability of current staging/categorization of alpha‐synuclein pathology and their clinical relevance. Acta Neuropathol 115:399–407. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Peng X, Tehranian R, Dietrich P, Stefanis L, Perez RG (2005) Alpha‐synuclein activation of protein phosphatase 2A reduces tyrosine hydroxylase phosphorylation in dopaminergic cells. J Cell Sci 118:3523–3530. [DOI] [PubMed] [Google Scholar]
- 48. Qing H, Wong W, McGeer EG, McGeer PL (2009) Lrrk2 phosphorylates alpha synuclein at serine 129: Parkinson disease implications. Biochem Biophys Res Commun 387:149–152. [DOI] [PubMed] [Google Scholar]
- 49. Saito Y, Kawashima A, Ruberu NN, Fujiwara H, Koyama S, Sawabe M et al (2003) Accumulation of phosphorylated alpha‐synuclein in aging human brain. J Neuropathol Exp Neurol 62:644–654. [DOI] [PubMed] [Google Scholar]
- 50. Saito Y, Ruberu NN, Sawabe M, Arai T, Kazama H, Hosoi T et al (2004) Lewy body‐related alpha‐synucleinopathy in aging. J Neuropathol Exp Neurol 63:742–749. [DOI] [PubMed] [Google Scholar]
- 51. Sato H, Arawaka S, Hara S, Fukushima S, Koga K, Koyama S, Kato T (2011) Authentically phosphorylated alpha‐synuclein at Ser129 accelerates neurodegeneration in a rat model of familial Parkinson's disease. J Neurosci 31:16884–16894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Smith WW, Margolis RL, Li X, Troncoso JC, Lee MK, Dawson VL et al (2005) Alpha‐synuclein phosphorylation enhances eosinophilic cytoplasmic inclusion formation in SH‐SY5Y cells. J Neurosci 25:5544–5552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Spillantini MG, Crowther RA, Jakes R, Hasegawa M, Goedert M (1998) alpha‐Synuclein in filamentous inclusions of Lewy bodies from Parkinson's disease and dementia with lewy bodies. Proc Natl Acad Sci U S A 95:6469–6473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Waxman EA, Giasson BI (2008) Specificity and regulation of casein kinase‐mediated phosphorylation of alpha‐synuclein. J Neuropathol Exp Neurol 67:402–416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Waxman EA, Giasson BI (2011) Characterization of kinases involved in the phosphorylation of aggregated alpha‐synuclein. J Neurosci Res 89:231–247. [DOI] [PMC free article] [PubMed] [Google Scholar]