

Abstract
Triggering receptor expressed by myeloid cells 2 (TREM2), a member of the immunoglobulin superfamily, has anti‐inflammatory phagocytic function in myeloid cells. Several studies have shown that TREM 2 gene variant rs75932628‐T increased the risks for Alzheimer's disease (AD), Parkinson's disease, frontotemporal dementia and amyotrophic lateral sclerosis. It has been suggested that the risks could be resulted from the loss of TREM2 function caused by the mutation. Indeed, new evidence showed that several mutations in the immunoglobulin‐like V‐region led to low cell surface expression of TREM2 and reduced phagocytic function. Because of the emerging importance in understanding TREM2 expression and functions in human neurodegenerative diseases, we conducted biochemical and morphological studies of TREM2 expression in human post‐mortem temporal cortical samples from AD and normal cases. Increased expression of TREM2 protein was found to significantly correlate with increases of phosphorylated‐tau and active caspase 3, a marker of apoptosis, and also loss of the presynaptic protein SNAP25. Strong intensities of TREM2 immunoreactivity were observed in the microglia associated with amyloid plaques and in neuritic pathology‐enriched areas. Based on the findings that TREM2 expression correlated with neurodegenerative markers, further investigation on whether there is abnormality of TREM2 functions in AD brains with nonmutated TREM2 is needed.
Keywords: amyloid, brain, human, microglia, tau, TREM2
Introduction
Several mutations of the TREM2 (triggering receptor expressed by myeloid cells 2) gene have been linked to the development of dementia. This was first established in a pre‐senile dementia in patients with polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy (PLOSL), also known as Nasu Hakola disease 34, 35. In this disease, homozygous mutations or deletion of the TREM2 gene or its functional partner protein, the gene encoding TYRO protein tyrosine kinase binding protein (TYROBP; formerly DAP12), caused a loss of function, leading to prominent pathological features of demyelination, axonal spheroids, axonal loss and white matter gliosis along with multiple cysts in bones 2, 21, 22, 24, 34, 49. Recently, whole genome sequencing identified TREM2 gene variant rs75932628‐T to significantly increase the risk of early and late‐onset sporadic Alzheimer's disease (AD), while the mutation carriers without AD between ages of 80–100 had lower cognitive function than the age‐matched noncarrier controls 15, 20. This TREM2 mutation was later reported to also increase the risk of frontotemporal dementia (FTD), Parkinson's disease and amyotrophic lateral sclerosis 3, 7, 11, 14, 40. All of these findings support the hypothesis that defects in TREM2 gene could lead to neurodegeneration.
TREM2 gene was initially cloned by Bouchon and colleagues while investigating leukocyte activating receptors that could associate with the DNAX‐activation protein 12 (DAP12) 5. At the same time as they identified a novel inflammation activating receptor TREM1 that was specifically expressed in neutrophils and granulocytes, they also found TREM2 that was expressed in macrophages and dendritic cells. As expected, TREM2 is expressed by brain microglia, consistent with their myeloid lineage and their roles in brain innate immune functions 12, 18, 22, 33, 39, 41, 42, 44, 47. Although TREM2 expressions in neurons, astrocytes and oligodendrocytes have also been observed, the findings in these cell types are less conclusive 8, 17, 22, 41, 42, 49.
TREM2 is a phagocytic receptor for apoptotic neuronal elements, neuritic debris and bacteria 18, 32, 43, 44, 45. Emerging evidence also showed the capability of TREM2 in phagocytosing amyloid β (Aβ). In the microglial cell line BV2, TREM2 expression levels have been shown to highly correlate with Aβ1‐40 uptake 29. Further confirmation that TREM2 is an Aβ phagocytic receptor came from a recent study in primary microglia isolated from TREM2 knockout mice. In this study, TREM2 gene knockout resulted in significant reduction in phagocytosis of fluorescent dye‐labeled Aβ1‐42 23.
There have been a few studies of TREM2 expression in animal models of AD, and in AD brain tissues. Studies in APP23 transgenic (Tg) mice showed that TREM2 protein‐expressing microglia were localized at the outer zone of amyloid plaques and the increases of TREM2 expression coincided with the progression of amyloid pathology 12, 29. In Tg CRND8 mice, TREM2 immunoreactivities were observed in microglia as well as in neuronal cytoplasm 15. Interestingly, a new study of hemizygous TREM2 APP/PS1 mice and wild‐type TREM2 APP/PS1 mice showed that reducing TREM2 gene expression by 50% did not alter amyloid load in 3‐ and 7‐month‐old mice. Instead, it reduced the number and area of microglia associated with amyloid plaques 48. These findings suggest that TREM2 is important for chemotactic response in microglia and it is possible that TREM2 phagocytosis might not properly function at sites of amyloid accumulation.
The induction of TREM2 functions requires binding of ligand at the cell surface to activate the co‐receptor DAP12‐mediated signaling 36. At present, heat shock protein 60 (HSP60) is the only ligand identified for activating TREM2‐mediated phagocytosis of apoptotic cells 43. TREM2 phagocytic function is not associated with inflammation. Instead, it suppresses inflammation and promotes tissue repair. This has been supported by the evidence from animal models of multiple sclerosis 37, 45. In these studies, blockade of TREM2 binding with ligand by a monoclonal antibody worsened disease, whereas overexpressing TREM2 in microglia improved it. It has also been shown that transduced TREM2 expression in microglia prevented induction of the pro‐inflammatory cytokine tumor necrosis factor‐α and nitric oxide synthase 2 transcripts 44.
To move toward understanding of the roles of TREM2 in AD, we conducted both biochemical and morphological characterization of TREM2 expression in brain tissues of neuropathologically defined AD, possible AD (PossAD) and normal control (NC) subjects. Our results demonstrated that TREM2 expression was predominant in the microglia; the levels of expression were significantly elevated in temporal cortical tissues of AD, and this negatively correlated with the levels of the presynaptic protein SNAP25 and positively correlated with the amount of abnormal tau, suggesting an active involvement of TREM2‐expressing microglia in neurodegenerative processes.
Materials and Methods
Brain tissue from human subjects
Human post‐mortem brain tissues for this study were from subjects who had enrolled in the Brain and Body Donation Program (BBDP) of Banner Sun Health Research Institute (BSHRI). The operations of the BBDP have been approved by the Banner Health Institutional Review Board. Most subjects had received annual standardized batteries of neurological and neuropsychological assessments prior to death. For those subjects lacking standardized ante‐mortem evaluations, information was obtained from a post‐mortem telephone interview with a contact, including an adaptation of the Clinical Dementia Rating (CDR) Scale. An overview of the program has been published 1. Neuropathological diagnosis was made from brain tissue fixed with 4% neutral‐buffered formaldehyde and cut from both paraffin embedded and cryoprotected blocks of various brain regions. Tissue sections were processed following our standard histological procedures. Histopathological grading of AD followed Braak's neurofibrillary tangle staging and Consortium to Establish a Registry for Alzheimer's Disease (CERAD) templates 6, 30. Cases were classified as AD if they had the intermediate or high amyloid plaques and neurofibrillary tangles according to the recommendation of NIA‐Reagan criteria 53. The PossAD cases were noncognitive impaired subjects with moderate presence of amyloid plaques and neurofibrillary tangles, whereas NCs were subjects who were clinically nondemented and had sparse AD pathology. A collection of autopsy cases meeting these criteria was used for the biochemical and morphological studies. The demographic and AD pathological features of the subjects in this study are summarized in Table 1.
Table 1.
Pathological features of the autopsy cases used in the biochemical study
| Disease category (number of the cases) | Normal controls (n = 11) | Possible Alzheimer's (n = 11) | Alzheimer's disease (n = 11) |
|---|---|---|---|
| Age, years (mean ± SE) | 85.4 ± 2.1 | 86.5 ± 1.7 | 82.4 ± 2.6 |
| Gender (male : female) | 7:4 | 3:8 | 6:5 |
| Apolipoprotein Eε4 (carriers : noncarriers) | 1:10 | 2:9 | 5:6 |
| Braak's stages (mean ± SE) | 2.8 ± 0.2 | 2.9 ± 0.2 | 5.2 ± 0.2 |
| Temporal tangle scores (mean ± SE) | 0.4 ± 0.11 | 0.4 ± 0.22 | 2.8 ± 0.21,2 |
| Temporal plaque scores (mean ± SE) | 1.2 ± 0.21,2 | 2.4 ± 0.21,3 | 3.0 ± 01,2 |
| Total tangle scores (mean ± SE) | 4.2 + 0.61 | 4.1 + 0.72 | 14.2 ± 0.41,2 |
| Total plaque scores (mean ± SE) | 3.7 ± 0.61,2 | 10.3 ± 0.92,3 | 13.8 ± 0.41,3 |
Same superscript numbers denote significant differences between two values (P < 0.05).
TREM2 rs75932628‐T polymorphism genotyping
To determine that the autopsy cases used in this study did not carry the TREM2 gene variant rs75932628‐T, we used a polymerase chain reaction (PCR) method developed in‐house for screening a large series of DNA samples derived from human post‐mortem cases. Approximately 0.5 μg of DNA derived from cerebellum was amplified using the following primers (5P) GAAGGACAGCAGCCACAAG and (3P) GAGCCCACAACACCACAG to produce a fragment of 172 base pairs (bp). PCR was carried out using Promega Hot Start DNA polymerase in a reaction mixture of 1x Promega Green Buffer (Madison, WI, USA), primers (0.5 μM), deoxynucleotides (0.2 μM), MgCl2 (1.5 mM), DNA polymerase (0.125 units/reaction) and 5% dimethyl sulfoxide (DMSO). PCR amplification involved 35 cycles of 94°C for 30 s, 58°C for 30 s and 72°C for 1 minute after a 2‐minute step at 94°C to activate the enzyme. The DNA product was digested with the restriction enzyme HhaI (New England Biolabs, Ipswich, MA, USA). The change from C to T in this polymorphism resulted in loss of the HhaI restriction enzyme site. Digested DNA fragments were separated through 8% polyacrylamide gel and imaged after staining using Gel Red stain (Biotium, Hayward, CA, USA). A representative gel showing the patterns of the different genotypes of rs75932628‐T is shown in Figure 1.
Figure 1.

Identification of TREM2 (triggering receptor expressed by myeloid cells 2) rs75932628‐T. DNA samples from cerebellum were amplified by polymerase chain reaction (PCR) followed by restriction enzyme digestion using primer pairs that allow the identification of rs75932628 genotypes. The gel image of amplified PCR products is shown here. Lane 1 is the DNA ladder. In lane 2, the band at 172 bp indicated the presence of rs75932628‐T in this case caused by the loss of a HhaI restriction enzyme site. The other lanes were from DNA samples without this polymorphism. For the purpose of this study, we only used autopsy cases that did not have rs75932628‐T genotype.
Immunohistochemistry of free‐floating brain tissues
Twenty‐micron (μm)‐thick, 4% formaldehyde‐fixed post‐mortem brain tissue sections of various regions were stained for antigen localization following our published free‐floating immunohistochemical procedures 26. The primary and secondary antibodies used in the morphological and biochemical studies are summarized in Table 2.
Table 2.
Information of the primary antibodies used in this study.Abbreviations: GFAP = glial fibrillary acidic protein; IHC = immunohistochemistry; WB = Western blot
| Antibody | Supplier (Catalog No.) | Host | Dilution | Applications |
|---|---|---|---|---|
| TREM2 | R&D Systems (AF1828) | Goat | 1:500 1:750 | WB, IHC |
| TREM2 | Fitzgerald (10R‐1115) | Mouse | 1:1000 | WB |
| TREM2 | Novus Biologicals (NBP1‐06095) | Goat | 1:1000 | IHC |
| p‐tau (AT8) | Thermo Scientific (MN1020) | Mouse | 1:1000 | WB, IHC |
| MHCII (LN3) | MP Biomedicals (0869303) | Mouse | 1:750 | IHC |
| 6E10 | Covance (SIG‐39320) | Mouse | 1:2500 | WB, IHC |
| Iba1 | Wako (016–20001) | Rabbit | 1:1000 | WB |
| DAP12 | Abcam (ab124834) | Rabbit | 1:500 | WB, IHC |
| SNAP25 | Abcam (ab108990) | Rabbit | 1:2000 | WB |
| PSD95 | Abcam (ab76115) | Rabbit | 1:2000 | WB |
| Active caspase‐3 | Abcam (ab32042) | Rabbit | 1:1000 | WB |
| β‐Actin | Sigma | Mouse | 1:5000 | WB |
| GFAP | Dako (Z0334) | Rabbit | 1:2000 | IHC |
| Oligodendrocyte | Abcam (ab7474) | Rabbit | 1:500 | IHC |
Double immunohistochemistry was carried out to determine the relationship of TREM2 expression with various cell types and AD pathology. The markers of cell types used were major histocompatibility complex II (MHCII) for activated microglia, glial fibrillary acidic protein (GFAP) for astrocytes, oligodendrocyte‐specific protein for oligodendrocytes, whereas for pathology, the markers were Aβ for amyloid plaques and phosphorylated tau (p‐tau) for neurofibrillary tangles. The duration of primary antibody incubation was 24 h at room temperature except the goat anti‐TREM2 antibody, which was incubated with tissues for 72 h at 4°C. Tissues were then mounted, dehydrated and coverslipped with Permount (Thermo Fisher Scientific, Waltham, MA, USA). Some of the immunoreacted tissue sections were counterstained before dehydration with a 1% neutral red solution to show the presence of nucleus of the cells. In order to obtain an overall assessment of the immunolabeling in line with our purpose, we first surveyed the entire sections to assess the relationship of TREM2 with cell types and AD pathologies using 4× and 10× objectives. Once TREM2 immunoreactive profiles were identified, we carried out detailed observation in the morphology of the immunoreactive cells and their relationship with AD pathology profiles. The digital images of the staining patterns were captured with 40× and 100× objectives. The results reported here were obtained from multiple tissue sections from each autopsy case and five autopsy cases from each disease category.
Semiquantitative analysis of TREM2 expression pattern
To evaluate the staining pattern of TREM2 protein in microglia, a semiquantitative ranking system was used. To identify TREM2‐positive microglia, mounted sections were viewed with a 20× objective with a 1‐mm2 grid in the lens of one eyepiece. The intensity of TREM2 immunoreactivity was ranked in three categories—low, medium or high—in four 1‐mm‐wide cortical columns in different gyrus on each brain section. The cell number assessed in each column was recorded along with the intensity rank of TREM2 immunoreactivity. The percentages of the cells in each intensity category were calculated from each case according to the formula: 100% × (cell number for each intensity category/total number of the immunoreactive cells assessed). Finally, the mean values of the percentages for each intensity category in each disease group were obtained.
Lectin‐binding histochemistry for assessing microglia cell density
Temporal brain sections were blocked with 1% hydrogen peroxide and protein‐free blocking buffer (Catalog No. 37572, Pierce, Thermo Fisher Scientific, Waltham, MA, USA), respectively, for 30 minutes and washed three times with PBST (phosphate buffered saline with 0.3% triton X‐100) prior to incubation with biotinylated tomato lectin (Catalog No. B‐1175, Vector Laboratories, Burlingame, CA, USA) at 2 μg/mL for 1 hour. The rest of the procedure followed our standard immunohistochemistry described earlier. Lectin‐positive microglia were counted in four 1‐mm‐wide gray matter columns starting from the surface of the temporal cortex to the border with white matter, guided by a 1‐mm2 grid on the lens of eyepiece. The counting was performed in mid‐temporal cortical tissues available from seven autopsy cases per disease group. The counting procedure was described in our previous publication 31. The total cell number obtained from each cortical column was divided by the area of the column (cell number/mm2). Results were statistically analyzed according to disease groups.
Double immunofluorescence
We also conducted double immunofluorescence to determine the relationship between TREM2 and DAP12 expression in the brain tissues. A sequential labeling procedure was applied. We first processed the immunolabeling of TREM2 with primary antibody as described earlier, followed by biotinylated anti‐goat immunoglobulin G (IgG) and streptavidin‐conjugated Alexa 488 (Molecular Probe, Life Technologies, Carlsbad, CA, USA). After completion of the first round of immunolabeling, we applied the primary antibody of DAP12 overnight, followed by 2‐hour incubation with anti‐rabbit IgG (H + L) conjugated with Alexa 568. Both fluorescence‐dye‐conjugated antibodies were used at 1:2000 from the stock provided by the manufacturer. To reduce nonspecific autofluorescence background from human brain tissues, mounted tissue sections were immersed in 1% Sudan Black solution (Sigma‐Aldrich, St. Louis, MO, USA) in 70% alcohol for 5 minutes. The excessive Sudan Black was removed by brief immersion of the slides in 70% alcohol and distilled water before coverslipping 26. Slides were coverslipped with Vectashield (Vector Laboratories, Burlingame, CA, USA). The images were captured with a charge‐coupled device (CCD) camera attached to the Olympus IX51 inverted fluorescence microscope (Olympus, Center Valley, PA, USA). Image overlay was composed by the Olympus software DP Controller version 3.2.1.276.
Western blotting
Gray matter tissues derived from middle temporal cortices (MTG) of cases described in Table 1 were extracted in RIPA (radioimmunoprecipitation assay) buffer (Thermo Scientific, Thermo Fisher Scientific) following the procedure described in our previous work 27. RIPA‐buffer brain extracts were mixed with 4x lithium dodecyl sulfate (LDS) sample buffer (Life Technologies), water and the reducing reagent dithiothreitol to make Western blot samples that contained 1 μg/μL of the total protein. Samples were heated at 70°C immediately before loading on NuPage 4%–12% Bis‐Tris Mini gels (Invitrogen, Life Technologies). Ten micrograms of protein from each individual sample was loaded in each gel lane. The order of the samples was arranged to ensure that different diseases were included on one gel along with the molecular weight markers (Spectrum, Thermo Fisher Scientific). We also included the same samples as internal controls on different gels to assess the extent of discrepancy in processing and detection of the samples between the gels.
Western blot samples were electrophoresed on NuPage Bis‐Tris gels at 200 V for 36 minutes using 2‐(N‐morpholino) ethanesulfonic acid (MES) buffer (Invitrogen, Life Technologies) and then transferred to nitrocellulose membranes at 30 V for 60 minutes. Blots were air‐dried before blocking with 25 mM Tris–HCl (pH7.4) buffered saline containing 0.05% Tween 20 (TBST) and 5% nonfat dry milk. Primary antibodies (TREM2, DAP12, IBA1, p‐tau, 6E10, SNAP25, PSD95, active caspase 3 and β‐actin) at predetermined dilutions in TBST containing 2% nonfat milk with 0.002% sodium azide were incubated with membranes for 18 hours at room temperature. Western Extended Dura Chemiluminescence substrate for horseradish peroxidase was used for detection (Pierce, Thermo Scientific). Images of immunoreactive bands were captured using a FluorChem Q (Protein Simple, San Jose, CA, USA) Chemi‐Imager. The intensities of the immunoreactive bands were obtained from each lane at the expected molecular weight using multiplex band analysis function of the FluorChem Q SA software version 3.2.2. The levels of β‐actin in each sample were used for normalization purposes.
Characterization of TREM2 and DAP12 antibodies
To determine which of the TREM2 antibodies were specific and suitable for use in the study, we transfected human embryonic kidney (HEK) 293 cells with TREM2 or DAP12 plasmids. Expression‐validated and sequence‐verified cDNA clones of human TREM2 and DAP12 in pCMV6 vectors (RC521132 and RC522502, OriGene Technologies, Rockville, MD, USA) were used. One microgram of TREM2 plasmid or/and DAP12 plasmid was used for transient transfection of HEK cells for 72 hours using the Neon transfection system (MPK5000, Life Technologies). Antibody characterization results with these materials are shown in Figure 2.
Figure 2.

Antibody characterization in the HEK cells and human brain homogenates. A. Lysate samples of HEK cells transfected with human TREM2 (triggering receptor expressed by myeloid cells 2) and DAP12 (DNAX‐activation protein 12) plasmids were used to characterize TREM2 antibodies by immunoblotting. TREM2 protein bands were detected in the range of molecular weights of 35–40 kDa with three antibodies, only in the samples with TREM2 transfection. Among these antibodies, the goat polyclonal antibody from R&D System performed more robustly in TREM2‐transfected cells. DAP12 protein bands were detected in HEK cells transfected with human DAP12 plasmids at 12 kDa, but not in the TREM2‐transfected samples.
Statistical analysis
Statistical analysis was performed with MedCalc software version 9.8 (Oostende, Belgium). The significance level for all comparisons was set at P < 0.05. One‐way analysis of variance (ANOVA) was used to determine the statistical differences in the expression levels of the TREM2 and DAP12 proteins between the groups classified by diseases, and by the Braak's stages in MTG followed by Student–Newman–Keuls pairwise comparisons. Microglia cell density detected with lectin‐binding histochemistry was also analyzed according to disease groups. The Spearman's rank correlation was used to correlate between the results from biochemical analysis of TREM2 with other parameters including DAP12, microglial markers IBA1, apoptotic marker active caspase 3 and synaptic proteins SNAP25 and PSD95, phosphorylated‐tau protein (p‐tau), as well as with neuropathological parameters (regional plaque and neurofibrillary tangle scores).
Results
Characteristics of the autopsy cases used in this study
Demographic and neuropathological features, including age at death, post‐mortem interval, apolipoprotein Eε4 (ApoEε4) genotype, amyloid plaque density and Braak stage, of the cases used in this study are summarized in Table 1. These are classified into three disease groups: nondemented controls (ND), possible AD (PossAD) and AD. As expected, the AD cases have significantly higher mean values of temporal cortex plaque and tangle scores, and Braak stage. The PossAD group was characterized by having intermediate mean value of plaque scores and similar Braak's stage and temporal tangle scores compared with the ND cases. AD temporal tissues also contained significantly higher density of microglia (P = 0.02) compared with ND and PossAD. The mean and standard errors of microglia density (cell number/mm2) were 4.011 ± 0.356 for ND, 4.279 ± 0.333 for PossAD and 5.304 ± 0.371 for AD. Microglia density was not significantly different between ND and PossAD, or between PossAD and AD.
TREM2 rs75932628 polymorphism genotype
In screening for TREM2 rs75932628‐T polymorphism in 206 samples of DNA, we identified only one heterozygous positive case (Figure 1). We excluded this case from our study. All of the cases included in this study carried the normal rs75932628 TREM2 genotype.
TREM2 antibody characterization
Three commercially available TREM2 antibodies were compared in our antibody characterization study: a goat polyclonal antibody from R&D Systems (AF1828, Minneapolis, MN, USA), raised against recombinant TREM2 molecule spanning amino acids 19–174; a goat polyclonal antibody from Novus Biologicals (NBP1‐06095, Littleton, CO, USA), raised against the amino acids 199–212; and a mouse monoclonal antibody from Fitzgerald (10R‐1115, Acton, MA, USA) raised against amino acids 19–161. The transmembrane domain of human TREM2 protein is amino acid positions 168–200. All of these three TREM2 antibodies detected two major bands with molecular weights of 35 and 40 kilodalton (kDa) (Figure 2). The AF1828 TREM2 antibody also detected a diffuse immunoreactivity pattern between the two major bands, indicative of post‐translational glycosylation of the expressed protein. The specificity of each TREM2 antibody was confirmed by the lack of reactivity with HEK cells that had been transfected with the plasmid for DAP12.
Levels of TREM2 protein and AD degenerative markers in studied samples
The primary goal of this study was to determine using biochemical measurements if TREM2 protein expression was linked to neuropathological features of AD. In addition to measuring the protein levels of TREM2, its co‐receptor DAP12 and a microglia marker IBA1, we also measured the expression levels of the presynaptic protein (SNAP25), the post‐synaptic proteins (PSD95), p‐tau, an apoptotic marker detected by cleaved caspase 3 (the p17 subunit) and 4‐kDa Aβ.
Our results showed that in MTG from AD cases, there was higher level of TREM2 protein than in the PossAD and ND (P < 0.05) cases, whereas there was no difference between the PossAD and ND disease cases (Figure 3A). Likewise, DAP12 was significantly increased in the AD cases compared with the PossAD and ND cases (Figure 3B). The expression levels of the microglia‐specific protein IBA1 were only significantly elevated in AD cases (Figure 3C). Of the degenerative markers, we detected a significant increase in the levels of active caspase 3, and decreased levels of the synaptic proteins SNAP25 and PSD95 in the AD cases (Figure 3D–F) compared with the PossAD and ND groups. The representative immunoblot images of these proteins are shown in Figure 3G.
Figure 3.
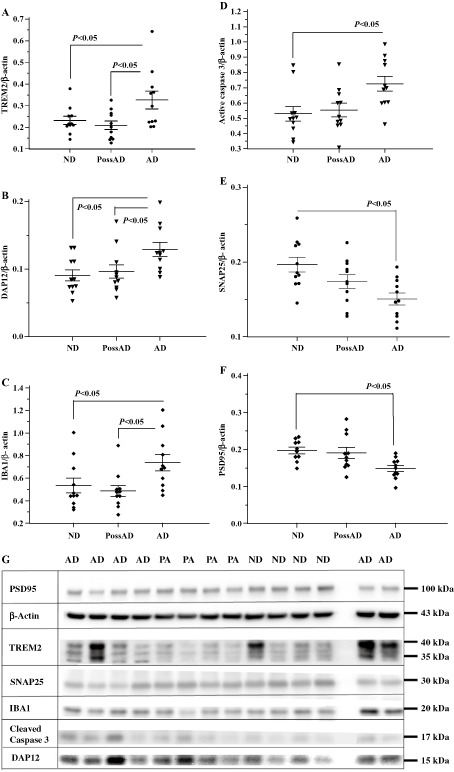
The expression levels of TREM2 (triggering receptor expressed by myeloid cells 2), DAP12 (DNAX‐activation protein 12), IBA1, cleaved caspase 3, SNAP25 and PSD95 by disease groups. The expression levels of microglial proteins TREM2, DAP12 and IBA1, apoptotic marker active caspase 3, and synaptic proteins SANP25 and PSD95 were analyzed in the brain samples using protein‐specific antibodies by our standard immunoblotting procedure. The intensities of the specific band in each sample were normalized by β‐actin followed by statistical analysis by disease groups: nondemented controls (ND); possible AD (PossAD); and Alzheimer's disease (AD). Significant changes (P < 0.05) were detected between AD and ND groups in TREM2 (A), DAP12 (B), IBA1 (C), and active caspase 3 (D), SNAP25 (E) and PSD 95 (F). However, the significant differences between AD and PossAD groups were only detected in TREM2, DAP12 and IBA1. Expression levels of the apoptotic marker active caspase 3, presynaptic protein SNAP25 and post‐synaptic protein PSD95 were not significantly different between PossAD and ND groups. There were no significant differences between ND and PossAD in all of the markers. The horizontal lines in the dot plots represent the values of mean and standard errors. Representative immunoblot images for the proteins TREM2, DAP12, IBA1, PSD95, SNAP25 and cleaved caspase 3 were compiled in G. The disease groups for the samples in each lane were shown above gel images: AD for Alzheimer's disease; PA for possible Alzheimer's disease, ND for nondemented normal controls. The blank lane was the lane that molecular weight ladders were loaded. The molecular weights corresponding to each protein were indicated at the right side of the gel images. The brain TREM2 has molecular weights of 35–40 kDa.
Relationship between TREM2 levels and AD pathological parameters
As both TREM2 and DAP12 protein levels were elevated in the AD group, we performed correlation analysis to determine their relationship, and with IBA1 to determine if their expression levels correlate with this marker of microglia. We found that there was a significant positive association between TREM2 and IBA1 (ρ = 0.490, P = 0.0038), but no such relationship between DAP12 and IBA1 (ρ = 0.102, P = 0.574). There was also a lack of association between TREM2 and its co‐receptor DAP12 (ρ = 0.098, P = 0.586). When a linear regression analysis was performed based on disease groups, we detected a linear relationship between the expression levels of TREM2 and IBA1 in AD subjects (R 2 = 0.4742, F ratio = 8.115, P = 0.0191). Although the linear relationship was not present within the PossAD or ND group, the coefficients of determination (R 2) were increased in the PossAD group, 0.341 (F ratio = 4.662, P = 0.0592) in contrast to 0.0168 (F ratio = 0.154, P = 0.704) in the ND group. No such relationship was detected between DAP12 and TREM2, or between DAP12 and IBA1.
We further determined if there were relationships between microglial proteins TREM2, DAP12 and IBA1, and neuropathological markers. Spearman's correlation coefficients for these measures are summarized in Table 3. The expression levels of TREM2 in MTG had significant correlations with tangle scores, PHF‐tau and active caspase 3, and significant negative association with the levels of SNAP25 protein, but not with the post‐synaptic protein PSD95 and plaque scores. The protein levels of DAP12 had significant positive correlations with plaque scores, tangle scores, 4‐kDa Aβ and active caspase 3, significant negative correlation with PSD95, but not with presynaptic protein SNAP25. IBA1 protein levels significantly correlated with tangle scores, but not with synaptic proteins, Aβ, p‐tau or caspase 3.
Table 3.
Spearman's correlation coefficients Rho and significant levels P between microglial markers and neurodegenerative markers in middle temporal cortices
| Rho (significant levels, P) | TREM2 | DAP12 | IBA1 |
|---|---|---|---|
| Temporal cortical plaque scores | 0.203 (P = 0.258) | 0.436 (P = 0.011) | 0.190 (P = 0.289) |
| Temporal cortical tangle scores | 0.359 (P = 0.040) | 0.364 (P = 0.037) | 0.529 (P = 0.002) |
| 4‐kDa Aβ | 0.037 (P = 0.836) | 0.371 (P = 0.03) | 0.118 (P = 0.513) |
| p‐tau | 0.483 (P = 0.004) | 0.302 (P = 0.088) | 0.239 (P = 0.180) |
| Active caspase 3 | 0.385 (P = 0.027) | 0.668 (P < 0.0001) | 0.278 (P = 0.117) |
| SNAP25 | −0.367 (P = 0.036) | 0.098 (P = 0.5864) | −0.151 (P = 0.401) |
| PSD95 | −0.513 (P = 0.832) | −0.533 (P = 0.002) | −0.112 (P = 0.542) |
Immunohistochemical detection of TREM2‐expressing cells
Although the main purpose of this study was to determine the biochemical levels of TREM2 in human temporal cortical tissues, to identify the source of increase in TREM2 expression we investigated the cell type and localization of TREM2 protein in post‐mortem brain tissues. We carried out double immunohistochemistry using one of the TREM2 antibodies (R&D Systems) that we had characterized, along with antibodies detecting Aβ, p‐tau, MHCII, GFAP and oligodendrocyte‐specific protein. In ND cases, TREM2 immunoreactivity was observed in a subset of microglia, whereas in AD cases, the intensity of TREM2 in microglia was increased in the areas enriched with AD pathology. Representative images of the micrographs are presented in Figure 4A–N. TREM2‐immunoreactive microglia not associated with AD pathology were also observed (Figure 4A–D). As MHCII has been frequently used as a marker for microglia with increased inflammatory properties, we determined whether TREM2‐expressing microglia were also MHCII‐positive. In the brain sections that we studied, TREM2‐expressing microglia were either negative or positive for MHCII immunoreactivity. Examples of undetectable (Figure 4A), strong (Figure 4B) and weak (Figure 4C,D) co‐localization of MHCII with TREM2, and TREM2‐expressing microglia positioned closely to a neuronal cell body (Figure 4E) and neuronal processes (Figure 4F) are shown. TREM2‐expressing microglia were frequently observed in close vicinity to neurons in brain regions enriched for p‐tau‐immunoreactive neurites (Figure 4G–I). We also observed TREM2‐expressing microglia at the outer edge (Figure 4J,K) as well as inside the amyloid plaques (Figure 4L–N).
Figure 4.

Immunohistochemical characterization of TREM2 (triggering receptor expressed by myeloid cells 2) expressions in postmortem human brain tissues. Representative images of double immunohistochemistry of TREM2 (dark blue) and MHCII (major histocompatibility complex II) (brown) are shown in A–D; TREM2 immunoreactive microglia in association with neurons in E–F; TREM2 (dark blue) and p‐tau (brown) in G–I; and TREM2 and Aβ in J–N. A–D. TREM2‐immunoreactive microglia are present in MHCII‐negative (arrows) cells and also in MHCII‐positive cells (arrow heads). E–I. TREM2 immunoreactive microglia are in close contact with neuronal cell bodies (neutral red stained; E, F) in the tissues with minimal Alzheimer's disease (AD) pathology. Increased TREM2 immunoreactivity (dark blue, arrows) could be seen in the areas enriched with p‐tau immunoreactive neurites (brown; G–I). J–N. TREM2 immunoreactive microglia (dark blue, arrows; J–N) are observed in the vicinity of amyloid plaques (J–K) and inside amyloid plaques with different compactness of the amyloid aggregates (L–N). All of the micrograph images were taken with 40× objective. The 10‐μm magnification calibration bars were shown in the first picture in each row (A, E, G, J). The brain tissues for the morphological study were ND cases in A, B, E; PossAD cases in C, D, F, J, K; AD cases in G–I and L–N.
To assess the expression pattern of TREM2 immunoreactivity in microglia, a semiquantitative system combining the ranking of staining intensity and frequency was adopted. In this system, the intensity of the TREM2 immunoreactivity was ranked as low, medium or high, and the number of microglia counted for each rank was recorded. The percentages of the cell number in each intensity category were calculated according to the formula described earlier and the results were shown in Table 4. Interestingly, we found that 87.5% of TREM2‐immunoreactive microglia in ND cases exhibited low staining intensity, whereas 12.5% of the TREM2‐positive cells exhibited medium intensity. In contrast to the ND and PossAD cases that had no microglia expressing high intensity of TREM2 immunoreactivity, 37.5% of TREM2‐expressing microglia in AD exhibited high immunoreactive intensity.
Table 4.
Semiquantitative assessment of microglial TREM2 expression pattern
| Intensity ranking of TREM2 immunoreactivity | Nondemented controls (%) | Possible Alzheimer's disease (%) | Alzheimer's disease (%) |
|---|---|---|---|
| Low | 87.5 | 75 | 37.5 |
| Medium | 12.5 | 25 | 25 |
| High | 0 | 0 | 37.5 |
TREM2 immunoreactivities were not detected in the cells marked by the astrocyte marker GFAP or oligodendrocyte‐specific protein (Figure 5), as indicated by black arrows (TREM2‐immunoreactive cells) and white arrows (astrocytes in Figure 5A,B; oligodendrocytes in Figure 5C,D). By two‐color immunofluorescence, DAP12 immunoreactivity was determined in relation to TREM2‐expressing cells (Figure 6). TREM2‐expressing cells are labeled with green (Alexa 488) (Figure 6A,D), whereas DAP12 protein immunoreactivities are labeled with red (Alexa 568) (Figure 6B,E), and the co‐localization of TREM2 and DAP12 immunoreactivities is shown in the image overlays (Figure 6C,F). In these figures, there are also DAP12 immunoreactive cells that do not co‐localize with TREM2 immunoreactivity white arrows to be put between parenthesis.
Figure 5.
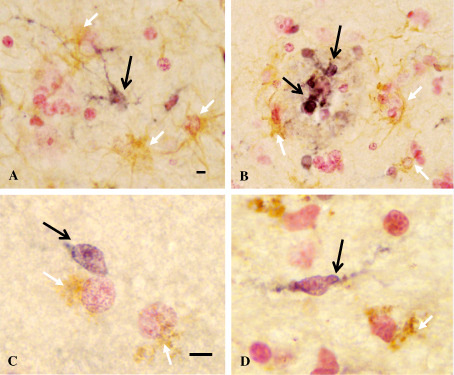
Double immunohistochemistry of TREM2 (triggering receptor expressed by myeloid cells 2) with astrocyte and oligodendrocyte markers. Double immunohistochemistry of TREM2 with astrocyte marker [glial acidic filament protein (GFAP)] or oligodendrocyte‐specific protein shown in this figure were performed in the brain tissues of nondemented controls (ND) (A, C), possible AD (PossAD) (D) and Alzheimer's disease (AD) (B) cases. TREM2 was demonstrated with nickel enhanced DAB (DNAX‐activation protein) substrate (dark blue, indicated by black arrows), whereas GFAP and oligodendrocyte‐specific protein were demonstrated with DAB without enhancement (light brown, indicated by white arrows). TREM2 immunoreactivities were not observed in the same cells marked by GFAP or oligodendrocytes‐specific protein. Images were taken at 40× objective in A, B, and at 100× with oil immersion lens in C, D. The calibration bars represent 10‐μm length.
Figure 6.
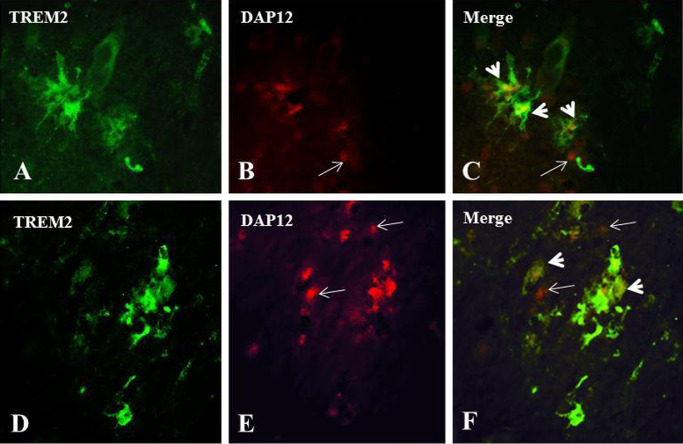
Double immunofluorescence labeling of TREM2 (triggering receptor expressed by myeloid cells 2) and DAP12 (DNAX‐activation protein 12) in human brain tissues. To illustrate the relationship of TREM2 and DAP12 expressions in brain cells, results of double immunofluorescence labeling are shown here: A, D: TREM2 expression in green; B, E: DAP12 expression in red; C, F: overlay of TREM2 and DAP12 images. In both overlay images, the co‐localization of TREM2 and DAP12 immunoreactivities were shown (arrow heads). However, some of the DAP12 immunoreactive profiles are not associated with TREM2 immunoreactivity (indicated by arrows). Magnification: calibration bars: 10 μm.
Discussion
We used both biochemical and immunohistochemical approaches to characterize AD‐associated changes of TREM2 protein expression in post‐mortem temporal cortical tissues. The major findings of our study are that TREM2 protein levels were significantly elevated in AD compared with ND and PossAD cases. This increase correlated with the abundance of neurofibrillary pathology, the biochemical levels of p‐tau and an apoptotic marker, the loss of presynaptic protein SNAP25, and the increases in microglial activation marker IBA1. This is the first time that TREM2 protein expression has been demonstrated in significant association with neuritic pathological changes and microglial activation in AD. These findings suggest the importance of assessing microglial TREM2 function in the context of interaction with neurons. This will include microglial responses to signals sent by neurons undergoing synaptic loss, oxidative stress, apoptosis and progressive modification of the microtubule associated protein tau.
By double immunohistochemistry with TREM2 antibody and antibodies for cell‐type‐specific antigens, we demonstrated that TREM2 was only present in microglia, not in other glial cells in the MTG samples analyzed. Although there are several contradictory reports about the cell types that express TREM2 in the brain, the absence of TREM2 RNA and protein expression in astrocytes, oligodendrocytes and neurons has been reported previously in purified primary cell cultures 44. Our findings confirmed that TREM2 is expressed by microglia, but is undetectable in other glial cells in human elderly brains.
In this study, biochemically‐measured TREM2 levels did not show a correlation with amyloid plaque counts or amounts of the monomeric 4‐kDa Aβ in the RIPA‐extracted brain samples. Interestingly, in transgenic mice modeling AD amyloid accumulation, such as APP23 and TgCRND8 mice, TREM2 expression and amyloid load increased in parallel in the plaque‐laden brain regions 12, 15, 29. In these studies, TREM2 expression were detected by immunoblotting and/or in situ hybridization, and their relationship with Aβ was assessed by amyloid plaque staining. The reasons for the absence of correlation between biochemical expression levels of TREM2 with plaque counts in our study is unclear. We speculate that this could be caused by the transgenic models of AD, such as APP23 and TgCRND8, do not mimic the complete magnitude and all aspects of AD pathology in humans. As one of the constitutive functions of TREM2 is to maintain the healthy environment for neurons by phagocytosis of cellular debris and apoptotic cells, it is possible that TREM2 expression levels are also regulated by these factors.
Moreover, in human AD brain tissues, we observed that the localization of TREM2‐expressing microglia was not limited to certain regions in amyloid plaques (shown in Figure 4J–N). In contrast, TREM2‐expressing microglia were limited to the outer zone of amyloid plaques in APP23 and TgCRND8 mice 15, 28. TREM2 mediates chemotactic responses to amyloid plaques. This is supported by the evidence from a study of the hemizygous APP/PS1‐21 mice generated by crossing APP/PS1‐21 mice with TREM2 knockout mice 48. Amyloid load was not altered at 3 and 7 months because of reduced TREM2 expression but the number of microglia accumulated at the amyloid plaques was markedly reduced at 3 months of age. This study led the authors to suggest that TREM2 mediates chemotactic responses of microglia toward amyloid plaques.
Elevated TREM2 expression might not result in more activation of phagocytosis signaling, as this is dependent on coupling with DAP12 44. In the study of APP23 mice, the expression of the TREM2 co‐receptor DAP12 increased in a similar trend as TREM2 29. We have found that although both DAP12 and TREM2 expression were elevated in the MTG of the AD cases, there was a lack of correlation between them. The levels of DAP12 correlated positively with amyloid plaque scores and Aβ, while TREM2 levels did not correlate with these features. We also showed that TREM2 immunoreactive cells did not consistently contain detectable DAP12 levels. It is unclear whether the lack of association between TREM2 and DAP12 could be caused by differences in their regulation. Although DAP12 is the only signaling receptor identified for TREM2, DAP12 partners with other receptors, including Fcγ receptors, complement receptors, and one of other members of the TREM family, TREM1 16, 19, 25, 46, 50, 51. In contrast to TREM2, the interaction of TREM1 with DAP12 leads to activation of NFκB pathway with pro‐inflammatory consequence 4. Thus, depending on the local immunological environment and what types of molecular complexes are formed, activation of DAP12 signaling could result in different outcomes. In addition, it has been shown that FTD‐associated mutations in TREM2 affected further enzymatic cleavage of the TREM2 C‐terminal fragment (CTF) following ectodomain shedding 23. Whether the accumulation of CTF in the plasma membrane could affect the expression of DAP12 caused by the lack of pairing with full‐length TREM2 is not known. How disease condition might alter DAP12 expression and its coupling with TREM2 in microglia is an important area for further research.
Microglia have heterogeneous phenotypes that represent their activation states 13. Our results showed varied intensity of TREM2 immunoreactivities in microglia. Higher TREM2 immunoreactivities were observed in microglia clustered in AD pathology‐enriched areas and semiquantitative analysis showed that more AD microglia exhibited higher intensity of TREM2 immunoreactivity than ND and PossAD microglia. We also observed high MHCII immunoreactivities in cells expressing higher level of TREM2. A study using BV2 microglial cells demonstrated that higher levels of TREM2 corresponded with greater antigen‐presenting function and neuroprotection, as these cells interacted with activated CD4+ T‐cells without inducing the production of interferon‐γ 29.
As a member of the immunoglobulin superfamily, TREM2 consists of a single extracellular globular domain anchored by a transmembrane domain followed by a cytoplasmic tail. Similar to other members of this family, full‐length TREM2 is subject to ectodomain shedding. Recently, it has been shown that soluble TREM2 fragments are produced through sequential cleavage in the lower part of the extracellular domain by an unidentified sheddases, followed by γ‐secretase cleavage inside the transmembrane domain to generate a soluble secreted CTF 52. Soluble TREM2 has been detected recently in human cerebrospinal fluid (CSF), synovial fluid and serum 9, 10, 23, 38, 43. Possession of the TREM2 single nucleotide polymorphism (SNP) (rs6922617) has been associated with increased tau and phosphorylated tau levels in CSF 10. Nevertheless, a recent analysis of sTREM2 in CSF samples showed significant decreases in the levels of sTREM2 in AD and FTD compared with the levels from ND cases 23.
A new study that compared the missense mutations T66M, Y38C and R47H with wild‐type TREM2 shed light on the functional and expressional abnormalities that these mutations could cause 23. The mutations could lead to defective maturation and transport of TREM2 proteins, resulting in low expression at the cellular surface, thereby leading to deficiency of phagocytic function. Although all of the three mutations are located within the V‐domain of immunoglobulin‐like structure, the magnitude of the above‐mentioned abnormalities varied. The two mutations (T66M and Y38C) associated with risks of FTD and FTD‐like diseases resulted in a higher degree of negative effects than the R47H mutation linked to the risk of AD. This raises the question whether additional mechanisms could be involved in increasing the risk for AD neurodegeneration in cases with the R47H mutation.
Based on our current findings in nonmutated TREM2 protein expressions in AD brain, we speculate that the consequences of TREM2 functional deficiency might be different from mutated TREM2. Non‐mutated TREM2 in AD might be the inaccessibility or defect in the ligand binding domains on TREM2, caused by interacting with amyloid plaques. The ligand binding domain of TREM2 might become inaccessible for HSP60, the only ligand identified important for signaling the clearance of apoptotic cells 43. Nevertheless, this hypothesis will need to be tested in a future study.
Taken together, in this study, we have demonstrated in post‐mortem human temporal cortex a significant upregulation of TREM2 protein expressions in AD cases. We also demonstrated that TREM2 protein expression increased with p‐tau‐containing neuritic pathology, presynaptic loss, apoptotic activation and microglial activation. These findings provide the foundation for further mechanistic investigation. We propose that microglia TREM2 upregulation in AD could be a response to phagocytic demand caused by increased burden of neuritic pathology and apoptotic cells as disease progresses. Whether TREM2 functions are defective during these processes remains to be determined.
Acknowledgments
The post‐mortem human brain tissues were made available through funding from the Arizona Department of Health Services (Contract No. 211002, Arizona Alzheimer's Research Center) and the National Institute on Aging (P30 AG19610, Arizona Alzheimer's Disease Core Center).
References
- 1. Beach TG, Sue LI, Walker DG, Roher AE, Lue L, Vedders L et al (2008) The Sun Health Research Institute Brain Donation Program: description and experience, 1987–2007. Cell Tissue Bank 9:229–245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Bianchin MM, Capella HM, Chaves DL, Steindel M, Grisard EC, Ganev GG et al (2004) Nasu‐Hakola disease (polycystic lipomembranous osteodysplasia with sclerosing leukoencephalopathy—PLOSL): a dementia associated with bone cystic lesions. From clinical to genetic and molecular aspects. Cell Mol Neurobiol 24:1–24. [DOI] [PubMed] [Google Scholar]
- 3. Borroni B, Ferrari F, Galimberti D, Nacmias B, Barone C, Bagnoli S et al (2013) Heterozygous TREM2 mutations in frontotemporal dementia. Neurobiol Aging 35(4):p934.e7–934.e10. [DOI] [PubMed] [Google Scholar]
- 4. Bouchon A, Dietrich J, Colonna M (2000) Cutting edge: inflammatory responses can be triggered by TREM‐1, a novel receptor expressed on neutrophils and monocytes. J Immunol 164:4991–4995. [DOI] [PubMed] [Google Scholar]
- 5. Bouchon A, Hernandez‐Munain C, Cella M, Colonna M (2001) A DAP12‐mediated pathway regulates expression of CC chemokine receptor 7 and maturation of human dendritic cells. J Exp Med 194:1111–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Braak H, Braak E (1997) Diagnostic criteria for neuropathologic assessment of Alzheimer's disease. Neurobiol Aging 18:S85–S88. [DOI] [PubMed] [Google Scholar]
- 7. Cady J, Koval ED, Benitez BA, Zaidman C, Jockel‐Balsarotti J, Allred P et al (2014) TREM2 variant p.R47H as a risk factor for sporadic amyotrophic lateral sclerosis. JAMA Neurol 71:449–453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Colonna M (2003) TREMs in the immune system and beyond. Nat Rev Immunol 3:445–453. [DOI] [PubMed] [Google Scholar]
- 9. Crotti TN, Dharmapatni AA, Alias E, Zannettino AC, Smith MD, Haynes DR (2012) The immunoreceptor tyrosine‐based activation motif (ITAM)‐related factors are increased in synovial tissue and vasculature of rheumatoid arthritic joints. Arthritis Res Ther 14:R245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Cruchaga C, Kauwe JS, Harari O, Jin SC, Cai Y, Karch CM et al (2013) GWAS of cerebrospinal fluid tau levels identifies risk variants for Alzheimer's disease. Neuron 78:256–268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Cuyvers E, Bettens K, Philtjens S, Van LT, Gijselinck I, van der Zee J et al (2014) Investigating the role of rare heterozygous TREM2 variants in Alzheimer's disease and frontotemporal dementia. Neurobiol Aging 35:726.e11–726.e19. [DOI] [PubMed] [Google Scholar]
- 12. Frank S, Burbach GJ, Bonin M, Walter M, Streit W, Bechmann I, Deller T (2008) TREM2 is upregulated in amyloid plaque‐associated microglia in aged APP23 transgenic mice. Glia 56:1438–1447. [DOI] [PubMed] [Google Scholar]
- 13. Gehrmann J (1996) Microglia: a sensor to threats in the nervous system? Res Virol 147:79–88. [DOI] [PubMed] [Google Scholar]
- 14. Giraldo M, Lopera F, Siniard AL, Corneveaux JJ, Schrauwen I, Carvajal J et al (2013) Variants in triggering receptor expressed on myeloid cells 2 are associated with both behavioral variant frontotemporal lobar degeneration and Alzheimer's disease. Neurobiol Aging 34:2077–2078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Guerreiro R, Wojtas A, Bras J, Carrasquillo M, Rogaeva E, Majounie E et al (2013) TREM2 variants in Alzheimer's disease. N Engl J Med 368:117–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Hamerman JA, Ni M, Killebrew JR, Chu CL, Lowell CA (2009) The expanding roles of ITAM adapters FcRgamma and DAP12 in myeloid cells. Immunol Rev 232:42–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Hickman SE, El KJ (2014) TREM2 and the neuroimmunology of Alzheimer's disease. Biochem Pharmacol 88:495–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hsieh CL, Koike M, Spusta SC, Niemi EC, Yenari M, Nakamura MC, Seaman WE (2009) A role for TREM2 ligands in the phagocytosis of apoptotic neuronal cells by microglia. J Neurochem 109:1144–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ivashkiv LB (2009) Cross‐regulation of signaling by ITAM‐associated receptors. Nat Immunol 10:340–347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Jonsson T, Stefansson H, Steinberg S, Jonsdottir I, Jonsson PV, Snaedal J et al (2013) Variant of TREM2 associated with the risk of Alzheimer's disease. N Engl J Med 368:107–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Kaneko M, Sano K, Nakayama J, Amano N (2010) Nasu‐Hakola disease: the first case reported by Nasu and review. Neuropathology 30:463–470. [DOI] [PubMed] [Google Scholar]
- 22. Kiialainen A, Hovanes K, Paloneva J, Kopra O, Peltonen L (2005) Dap12 and Trem2, molecules involved in innate immunity and neurodegeneration, are co‐expressed in the CNS. Neurobiol Dis 18:314–322. [DOI] [PubMed] [Google Scholar]
- 23. Kleinberger G, Yamanishi Y, Suárez‐Calvet M, Czirr E, Lohmann E, Cuyvers E et al (2014) TREM2 mutations implicated in neurodegeneration impair cell surface transport and phagocytosis. Sci Transl Med 6:243ra86. [DOI] [PubMed] [Google Scholar]
- 24. Klunemann HH, Ridha BH, Magy L, Wherrett JR, Hemelsoet DM, Keen RW et al (2005) The genetic causes of basal ganglia calcification, dementia, and bone cysts: DAP12 and TREM2. Neurology 64:1502–1507. [DOI] [PubMed] [Google Scholar]
- 25. Linnartz B, Neumann H (2013) Microglial activatory (immunoreceptor tyrosine‐based activation motif)‐ and inhibitory (immunoreceptor tyrosine‐based inhibition motif)‐signaling receptors for recognition of the neuronal glycocalyx. Glia 61:37–46. [DOI] [PubMed] [Google Scholar]
- 26. Lue LF, Walker DG, Brachova L, Beach TG, Rogers J, Schmidt AM et al (2001) Involvement of microglial receptor for advanced glycation endproducts (RAGE) in Alzheimer's disease: identification of a cellular activation mechanism. Exp Neurol 171:29–45. [DOI] [PubMed] [Google Scholar]
- 27. Lue LF, Walker DG, Adler CH, Shill H, Tran H, Akiyama H et al (2012) Biochemical increase in phosphorylated alpha‐synuclein precedes histopathology of Lewy‐type synucleinopathies. Brain Pathol 22:745–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Melchior B, Puntambekar SS, Carson MJ (2006) Microglia and the control of autoreactive T cell responses. Neurochem Int 49:145–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Melchior B, Garcia AE, Hsiung BK, Lo KM, Doose JM, Thrash JC et al (2010) Dual induction of TREM2 and tolerance‐related transcript, Tmem176b, in amyloid transgenic mice: implications for vaccine‐based therapies for Alzheimer's disease. ASN Neuro 2:e00037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Mirra SS, Heyman A, McKeel D, Sumi SM, Crain BJ, Brownlee LM et al (1991) The Consortium to Establish a Registry for Alzheimer's Disease (CERAD). Part II. Standardization of the neuropathologic assessment of Alzheimer's disease. Neurology 41:479–486. [DOI] [PubMed] [Google Scholar]
- 31. Moses GS, Jensen MD, Lue LF, Walker DG, Sun AY, Simonyi A, Sun GY (2006) Secretory PLA2‐IIA: a new inflammatory factor for Alzheimer's disease. J Neuroinflammation 3:28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. N'Diaye EN, Branda CS, Branda SS, Nevarez L, Colonna M, Lowell C et al (2009) TREM‐2 (triggering receptor expressed on myeloid cells 2) is a phagocytic receptor for bacteria. J Cell Biol 184:215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Neumann H, Takahashi K (2007) Essential role of the microglial triggering receptor expressed on myeloid cells‐2 (TREM2) for central nervous tissue immune homeostasis. J Neuroimmunol 184:92–99. [DOI] [PubMed] [Google Scholar]
- 34. Paloneva J, Kestila M, Wu J, Salminen A, Bohling T, Ruotsalainen V et al (2000) Loss‐of‐function mutations in TYROBP (DAP12) result in a presenile dementia with bone cysts. Nat Genet 25:357–361. [DOI] [PubMed] [Google Scholar]
- 35. Paloneva J, Manninen T, Christman G, Hovanes K, Mandelin J, Adolfsson R et al (2002) Mutations in two genes encoding different subunits of a receptor signaling complex result in an identical disease phenotype. Am J Hum Genet 71:656–662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paradowska‐Gorycka A, Jurkowska M (2013) Structure, expression pattern and biological activity of molecular complex TREM‐2/DAP12. Hum Immunol 74:730–737. [DOI] [PubMed] [Google Scholar]
- 37. Piccio L, Buonsanti C, Mariani M, Cella M, Gilfillan S, Cross AH et al (2007) Blockade of TREM‐2 exacerbates experimental autoimmune encephalomyelitis. Eur J Immunol 37:1290–1301. [DOI] [PubMed] [Google Scholar]
- 38. Piccio L, Buonsanti C, Cella M, Tassi I, Schmidt RE, Fenoglio C et al (2008) Identification of soluble TREM‐2 in the cerebrospinal fluid and its association with multiple sclerosis and CNS inflammation. Brain 131:3081–3091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Prada I, Ongania GN, Buonsanti C, Panina‐Bordignon P, Meldolesi J (2006) Triggering receptor expressed in myeloid cells 2 (TREM2) trafficking in microglial cells: continuous shuttling to and from the plasma membrane regulated by cell stimulation. Neuroscience 140:1139–1148. [DOI] [PubMed] [Google Scholar]
- 40. Rayaprolu S, Mullen B, Baker M, Lynch T, Finger E, Seeley WW et al (2013) TREM2 in neurodegeneration: evidence for association of the p.R47H variant with frontotemporal dementia and Parkinson's disease. Mol Neurodegener 8:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Schmid CD, Sautkulis LN, Danielson PE, Cooper J, Hasel KW, Hilbush BS et al (2002) Heterogeneous expression of the triggering receptor expressed on myeloid cells‐2 on adult murine microglia. J Neurochem 83:1309–1320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Sessa G, Podini P, Mariani M, Meroni A, Spreafico R, Sinigaglia F et al (2004) Distribution and signaling of TREM2/DAP12, the receptor system mutated in human polycystic lipomembraneous osteodysplasia with sclerosing leukoencephalopathy dementia. Eur J Neurosci 20:2617–2628. [DOI] [PubMed] [Google Scholar]
- 43. Stefano L, Racchetti G, Bianco F, Passini N, Gupta RS, Panina BP, Meldolesi J (2009) The surface‐exposed chaperone, Hsp60, is an agonist of the microglial TREM2 receptor. J Neurochem 110:284–294. [DOI] [PubMed] [Google Scholar]
- 44. Takahashi K, Rochford CD, Neumann H (2005) Clearance of apoptotic neurons without inflammation by microglial triggering receptor expressed on myeloid cells‐2. J Exp Med 201:647–657. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Takahashi K, Prinz M, Stagi M, Chechneva O, Neumann H (2007) TREM2‐transduced myeloid precursors mediate nervous tissue debris clearance and facilitate recovery in an animal model of multiple sclerosis. PLoS Med 4:e124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Tessarz AS, Cerwenka A (2008) The TREM‐1/DAP12 pathway. Immunol Lett 116:111–116. [DOI] [PubMed] [Google Scholar]
- 47. Thrash JC, Torbett BE, Carson MJ (2009) Developmental regulation of TREM2 and DAP12 expression in the murine CNS: implications for Nasu‐Hakola disease. Neurochem Res 34:38–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Ulrich JD, Finn MB, Wang Y, Shen A, Mahan TE, Jiang H et al (2014) Altered microglial response to Abeta plaques in APPPS1‐21 mice heterozygous for TREM2. Mol Neurodegener 9:20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Verloes A, Maquet P, Sadzot B, Vivario M, Thiry A, Franck G (1997) Nasu‐Hakola syndrome: polycystic lipomembranous osteodysplasia with sclerosing leucoencephalopathy and presenile dementia. J Med Genet 34:753–757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Wakselman S, Bechade C, Roumier A, Bernard D, Triller A, Bessis A (2008) Developmental neuronal death in hippocampus requires the microglial CD11b integrin and DAP12 immunoreceptor. J Neurosci 28:8138–8143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang L, Gordon RA, Huynh L, Su X, Park Min KH, Han J et al (2010) Indirect inhibition of Toll‐like receptor and type I interferon responses by ITAM‐coupled receptors and integrins. Immunity 32:518–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Wunderlich P, Glebov K, Kemmerling N, Tien NT, Neumann H, Walter J (2013) Sequential proteolytic processing of the triggering receptor expressed on myeloid cells‐2 (TREM2) protein by ectodomain shedding and gamma‐secretase‐dependent intramembranous cleavage. J Biol Chem 288:33027–33036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. The National Institute on Aging and Reagan Institute Working group (1997) Consensus recommendations for the postmortem diagnosis of Alzheimer's disease. The National Institute on Aging, and Reagan Institute Working Group on Diagnostic Criteria for the Neuropathological Assessment of Alzheimer's Disease. Neurobiol Aging 18:S1–S2. [PubMed] [Google Scholar]