

Abstract
Recent studies involving phytochemical polyphenolic compounds have suggested flavones often exert pro-oxidative effect in vitro against wide array of cancer cell lines. The aim of this study was to evaluate the in-vitro pro-oxidative activity of apigenin, a plant based flavone against colorectal cancer cell lines and investigate cumulative effect on long term exposure. In the present study, treatment of colorectal cell lines HT-29 and HCT-15 with apigenin resulted in anti-proliferative and apoptotic effects characterized by biochemical and morphological changes, including loss of mitochondrial membrane potential which aided in reversing the impaired apoptotic machinery leading to negative implications in cancer pathogenesis. Apigenin induces rapid free radical species production and the level of oxidative damage was assessed by qualitative and quantitative estimation of biochemical markers of oxidative stress. Increased level of mitochondrial superoxide suggested dose dependent mitochondrial oxidative damage which was generated by disruption in anti-apoptotic and pro-apoptotic protein balance. Continuous and persistent oxidative stress induced by apigenin at growth suppressive doses over extended treatment time period was observed to induce senescence which is a natural cellular mechanism to attenuate tumor formation. Senescence phenotype inducted by apigenin was attributed to changes in key molecules involved in p16-Rb and p53 independent p21 signaling pathways. Phosphorylation of retinoblastoma was inhibited and significant up-regulation of p21 led to simultaneous suppression of cyclins D1 and E which indicated the onset of senescence. Pro-oxidative stress induced premature senescence mediated by apigenin makes this treatment regimen a potential chemopreventive strategy and an in vitro model for aging research.
Keywords: Apigenin, Oxidative stress, Reactive oxygen species, Pro-oxidation, Senescence
Graphical abstract
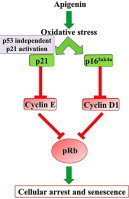
Highlights
-
•
Effect of apigenin on human colorectal cancer cell lines HCT-15 and HT-29 investigated.
-
•
Pro-oxidative stress attributed to reactive oxygen and nitrogen species generation.
-
•
Acute exposure to apigenin mediated apoptosis while chronic exposure caused senescence.
-
•
Chronic exposure affected key proteins in p16-Rb and p53 independent p21 signaling pathways.
-
•
Apigenin treatment as potential chemopreventive strategy and model for aging research.
Introduction
Polyphenols are secondary metabolites of plants with distinctive biological effects [1]. Flavones represent the most studied group of polyphenols with diverse bioactivity [2]. Widespread phytochemical screening efforts have yielded numerous plant based bioactive compounds with interesting therapeutic and pharmaceutical virtue [3]. Recent advances in chemopreventive strategies include optimization and development of diet derived phytochemical therapeutic agents [4].
Apigenin (4′,5,7-trihydroxyflavone) is a plant flavone predominantly found in fruits and vegetables with interesting therapeutic properties which includes chemopreventive, anti-mutagenic and anti-carcinogenic activities [5]. These effects have often been related to apoptosis induction, enhanced free radical production and increased oxidative stress [6,7]. Recent studies involving phytochemical polyphenolic compounds have reported that flavones exert a pro-oxidative effect in vitro and the phenoxyl radicals generated result in mitochondrial membrane potential collapse in a wide array of cancer cell lines [8,9]. The aim of the present study was to evaluate the in-vitro pro-oxidative activity of apigenin against colorectal cancer cell lines and also to investigate the cumulative effect on long term exposure, to utilize it as a potential chemotherapeutic drug.
The present study reports the biochemical changes involving free radicals in vitro when colorectal cancer cells are treated with bioactive flavone apigenin. Primary screening over a wide concentration range yielded loss of viability of the colorectal cell lines chosen at higher doses. The IC50 (median inhibitory concentration) in two different colorectal cell lines was determined (data unreported) and concentration range of apigenin selected for the study included concentrations above and below the respective IC50 molar concentrations of the individual cell lines. The present study reports the ability of apigenin to elicit pro-oxidative damage in both the colorectal cell lines. Dose–response studies yielded increased apoptotic potential of apigenin at higher dosages even in shorter treatment regimens (high dose stress over time periods of 24 or 48 h) while senescence was elicited at low dosages over longer treatment durations (low dose stress over a week treatment regimen). Hence, apigenin mediated acute toxicity in vitro in colorectal cell lines leads to apoptosis while chronic toxicity leads to senescence. The observations reported in this study suggested apigenin treatment to be a potential chemo-preventive strategy and potential cellular aging model.
Materials and methods
Cell lines and cell culture conditions
Human colon carcinoma (CRC) cell lines HCT-15 (p53 mutant) and HT-29 (p53 mutant) obtained from the National Centre for Cell Science (NCCS), Pune, India were grown as adherent cultures in l-glutamine supplemented RPMI-1640 medium with 10% heat-inactivated FBS, 100 units/ml penicillin and 0.1 mg/ml streptomycin at 37 °C in a 5% CO2 and 95% humidified incubator (Heraeus, Hera Cell, Germany) [10]. After the cells reached 80% confluency, they were trypsinized (0.25% Trypsin and 0.1% EDTA), centrifuged (Heraeus Labofuge 400R, Germany), and suspended in RPMI-1640 medium. For subsequent experiments, the cells were seeded in sterile 96-well plates, glass cover slips and 60 mm culture plates respectively.
Chemicals and reagents
Apigenin, Senescence Cells Histochemical Staining Kit, Griess reagent were purchased from Sigma Chemicals Co., USA. Dulbecco's modified Eagle's medium (DMEM) and Roswell Park Memorial Institute 1640 medium (RPMI-1640) supplemented with l-glutamine, fetal bovine serum (FBS), penicillin, streptomycin, Dulbecco's phosphate-buffered saline (D-PBS) and Hank's balanced salt solution (HBSS) were all procured from Gibco (Invitrogen), USA. JC-1 fluorescent dye was obtained from Life Technologies (Thermo Fisher Scientific Corporation, Carlsbad, CA, USA). Phospho-Rb (Ser780) Antibody, Bax Antibody, Bcl-2 Antibody, Anti-mouse IgG and Anti-rabbit IgG were procured from Cell Signaling Technology®, USA while Anti-p21WAF1 (Ab-1) was obtained from Calbiochem®, Darmstadt, Germany. Cyclin D1, Cyclin E, p53, p16 antibodies were procured from Santa Cruz Biotechnology, Inc., Dallas, USA while β-actin antibody was obtained from Sigma Chemicals Co., USA. JC-1 fluorescent dye was obtained from Life Technologies (Thermo Fisher Scientific Corporation, Carlsbad, CA, USA). All other chemicals used were of the highest analytical grade available. The chemicals were used as obtained without further purification. Milli-Q water obtained from Milli-Q Integral 3 system (Merck Millipore, Germany) was used for all experiments.
Qualitative and quantitative assessment of reactive oxygen species (ROS)/reactive nitrogen species (RNS) generation
ROS/RNS generation was detected by using oxidant-sensitive probe 2′,7′-dichlorodihydrofluorescein diacetate (H2DCFDA) as described previously [11] with slight modifications. Briefly, both HCT-15 and HT-29 cells were seeded at a density of 2×104 cells on sterile poly-l-lysine-coated glass cover slips or 60 mm petri dishes. The adhered cells were treated with apigenin over a concentration range of 1.5625–100 µM for 24 h respectively. ROS was qualitatively and quantitatively assessed using 5 µM of oxidant-sensitive probe 2′,7′-dichlorofluorescein diacetate (H2DCFDA dissolved in DMSO). Positive control consisted of tert-butyl hydroperoxide (100 µM) treated cells while negative control consisted of untreated cells. Treated and untreated sets of the respective cell lines were stained with H2DCFDA for 30 min at 37 °C. For qualitative assessment the cells seeded on glass cover slips were observed under a Zeiss Observer. Z1 microscope with appropriate filters (Ex/Em: ~492–495/517–527 nm). The generated reactive oxygen and nitrogen species converted the non-fluorescent H2DCFDA to fluorescent 2′,7′-dichlorofluorescein (DCF) which and was estimated with a fluro-spectrophotometer (Hitachi F-7000).
Quantitative assessment of reactive nitrogen species (RNS) generation
Both HCT-15 and HT-29 cells were seeded at a density of 2×104 cells in sterile 96 well tissue culture plates. The adhered cells were treated with apigenin over a concentration range of 1.5625–100 µM for 24 and 48 h respectively. The RNS generated was quantitatively assessed using Griess reagent [1% (w/v) sulfanilamide in 0.1 mol/l HCl and 0.1% N-(1-naphthyl)ethylenediamine dihydrochloride] (Sigma Chemicals Co., USA) as described by the manufacturer [12]. 100 µl Griess reagent was added to equivalent amount of culture media and incubated at room temperature for 15 min in dark. The absorbance was then measured spectrophotometrically at 540 nm using a benchmark microplate reader (BioRad, USA).
Qualitative and quantitative analysis of mitochondrial transmembrane potential changes (ψm) based on epi-fluorescent microscopy and cytometry
1×106 HCT-15 and HT-29 cells were seeded in 60 mm tissue culture plates and treated with apigenin at different concentrations for 24 h. Both adhered and floating cells were collected, washed three times with PBS (1×), incubated with 10 µg/ml of cyanine dye JC-1 (5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazolylcarbocyanine iodide) for 30 min at 37 °C. Mitochondrial membrane potential analysis was then performed with FACS Aria (BD Bioscience, USA) flow cytometer with 10,000 events acquired for each sample and data obtained was processed with BD FACS Diva software package bundled with the instrument. For qualitative assessment cells were seeded on cover slips, treated with different concentration of apigenin for 24 h, washed thrice with PBS (1×) and incubated with 10 µg/ml of JC-1 for 30 min at 37 °C. The cover slips were then observed under a Zeiss Observer. Z1 microscope after 24 h.
Mitochondrial superoxide detection
Mitochondrial superoxide was detected using MitoSOX™ Red, a novel fluorogenic dye highly selective for superoxide in the mitochondria of live cells. Both HCT-15 and HT-29 cells were seeded at a density of 2×104 cells on sterile poly-l-lysine-coated glass cover slips. The adhered cells were untreated or treated with apigenin over a concentration range of 1.5625–100 µM for 24 h. Treated and untreated sets of the respective cell lines were stained with 5 µM MitoSOX™ for 10 min at 37 °C. The cells were washed gently three times with PBS at room temperature and observed under a Zeiss Observer. Z1 microscope with appropriate filter (Ex/Em: 510/580 nm).
In situ cell death detection
The apoptotic cells were qualitatively visualized by using the in situ cell death detection kit (Roche, USA) as described by the manufacturer. Both HCT-15 and HT-29 cells were seeded at a density of 5×103 cells on sterile glass cover slips. The adhered cells were untreated or treated with apigenin over a concentration range of 1.5625–100 µM for 24 h. The cells were washed three times with PBS (1×) and then fixed with freshly prepared 4% paraformaldehyde (in PBS, pH 7.4) for 1 h at room temperature. Cells were permeabilized with 0.1% Triton-X solution for 2 min. Appropriate amounts of enzyme and labeling solutions were mixed based on manufacturer’s instructions to make the TUNEL reaction mixture. 50 µl of TUNEL reaction mixture was added each sample and incubated at 37 °C for 60 min in dark. The samples were washed thrice with PBS, mounted in fluorescent mounting medium and viewed under Zeiss Observer. Z1 microscope.
SA-β-Gal activity assay (senescence estimation)
The SA-β-gal activity assay was performed by using the Senescence Cells Histochemical Staining Kit (Sigma Chemicals Co., USA) as described by the manufacturer. HCT-15 cells were seeded at a density of 2×104 cells on sterile 60 mm tissue culture plates. The cells were treated at a concentration range from 1.5625 to 25 µM over an extended time period of 6 days as described previously [13]. The cells in sub-confluent cultures were washed gently with PBS. The cells were subsequently fixed in 2% (v/v) formaldehyde in PBS for 10 min at room temperature and then incubated with a staining mixture containing 5-bromo-4-chloro-3-indolyl β-d-galactoside, potassium ferricyanide, potassium ferrocyanide in citric acid–sodium phosphate buffer (pH 6.0). The assay is based on a histochemical stain for β-galactosidase activity (pH 6.0), resulted in the senescence positive cells being stained blue/green.
Cell signaling studies
1×106 HCT-15 and HT-29 cells were seeded in 100 mm sterile tissue culture plates and allowed to reach 75–90% confluence. Both the cells were treated with apigenin at their respective apoptotic-inducting and senescence-inducing dosage and were harvested at indicated time point using NP-40 Cell Lysis Buffer supplemented with 1 mM phenylmethylsulfonyl fluoride (Sigma Chemicals Co., USA) and protease and phosphatase inhibitors (Roche, USA). Proteins were quantitated using the Bicinchoninic Acid (BCA) Protein Assay kit (GeNei, Merck Millipore, Germany) in a benchtop plate reader. Equivalent concentration of proteins were then carefully loaded in each lane of a 12% polyacrylamide gel and electrophoresed appropriately. The proteins were meticulously transferred onto a nitrocellulose membrane (GE Healthcare Life Sciences, United Kingdom), and the blots were probed with appropriate primary antibodies followed by secondary antibodies at supplier's recommended dilutions. Immunoblots were then developed using chemiluminescence (ECL) detection system (Sigma Chemicals Co., USA).
Statistical analysis
Graphpad Prism 5 software (GraphPad Software, Inc., La Jolla, USA) was used for statistical analyses reported in this manuscript. Data are presented as mean±S.D. (n=3). The statistical significance or variance was determined by using paired or unpaired Student's t-test in experiments where control set values were reported and one-way analysis of variance (ANOVA) in case of tests where variance between experimental (test) sets were accessed. *p<0.05 was considered statistically significant while ***p<0.001 was considered statistically highly significant.
Results and discussion
Apigenin affects reactive oxygen species (ROS)/reactive nitrogen species (RNS) generation
The increased green fluorescent intensity observed in the epi-fluorescent images demonstrated the gradual surge in ROS/RNS production with increasing dosage of apigenin in both HT-29 and HCT-15 cell lines treated over 24 as compared with untreated cells [Fig. 1g and h]. While the initial burst of ROS/RNS production in treated sets of HCT-15 [Fig. 1a] was less than HT-29 [Fig. 1c] after 24 h, the trend was reversed after 48 h [Fig. 1b and d]. Treatment with increasing concentrations of apigenin for 24 and 48 h resulted in a significantly increased ROS generation in both cell lines respectively, as compared with untreated cells [Fig. 1e and f]. Since ROS has been previously reported as a possible mediator of apoptosis, the observed resulted indicated the potential of apigenin to induce apoptosis in treated cells [14].
Fig. 1.
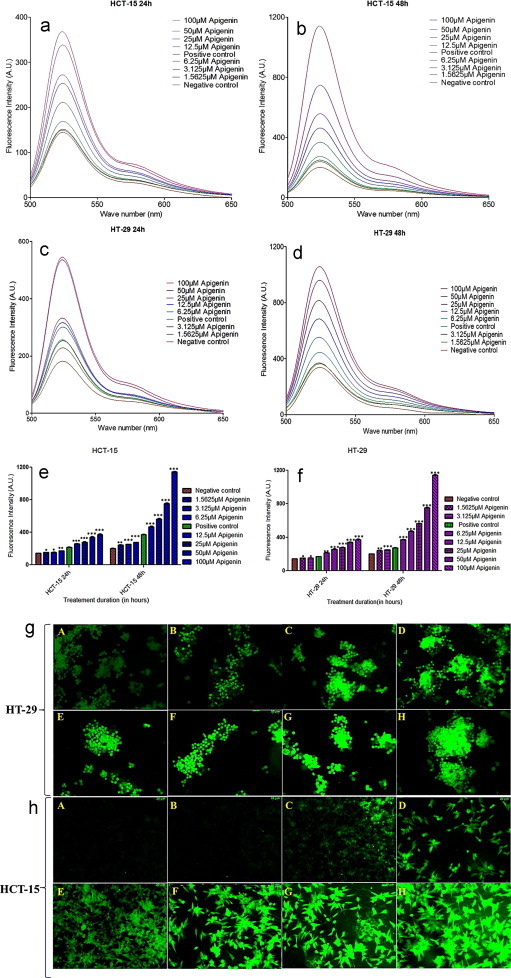
The effect of apigenin on the reactive oxygen species/reactive nitrogen species generation in HT-29, HCT-15 cells assessed using H2DCFDA over a treatment time period of 24 and 48 h respectively. (a and b) Fluorescence emission spectra of HCT-15 treated and untreated sets at 24 and 48 h respectively. (c and d) Fluorescence emission spectra of HT-29 treated and untreated sets at 24 and 48 h respectively. (e and f) Comparative assessment of the fluorescence maxima representing the quantitative generation of reactive oxygen species/reactive nitrogen species in treated/untreated sets of both cell lines at 24 and 48 h. (g and h) Epi-Fluorescent images of HCT-15 and HT-29 cells (A) untreated control (B–H) apigenin treated cells with dosage starting from 1.5625 µM and ranging to 100 µM. Comparative assessment of the fluorescence maxima representing the quantitative generation of reactive oxygen species/reactive nitrogen species in treated/untreated sets of both cell lines at 24 and 48 h. Data represent the means±S.D. of three independent experiments. Significant differences are indicated as ***p<0.01, **p<0.01 or *p<0.05.
Reactive nitrogen species (RNS) generation varies with time and dose of apigenin treatment
The data reported in Section 3.1 suggested towards increased reactive nitrogen species at both time points (24 and 48 h). However, specific analysis of RNS generation using Griess reagent revealed an interesting phenomenon. While the RNS levels were significantly reduced in the treated groups of both cell lines with respect to the control untreated set after 24 h, the trend was completely reversed after 48 h. Furthermore the observed decrease in the RNS levels after 24 h in treated sets were found to be concentration dependent, with cells treated with higher doses of apigenin producing reduced RNS generation In both cell lines initially (24 h time point) RNS levels were significantly reduced in the treated groups with respect to the control and the reduction was concomitant with increasing drug treatment regime while this trend was reversed as the treatment time progressed (48 h) [Fig. 2]. these data were further substantiated when a one way ANOVA in both HCT-15 and HT-29 cells for 24 and 48 h treatment periods yielded a significant variance of p<0.0001, representing highly significant changes between the groups. The obtained results indicating the initial reduction in RNS might have thus contributed to the substantial difference between the reactive oxygen/nitrogen species production values (obtained with H2DCFDA) after 24 and 48 h reported in the previous section.
Fig. 2.
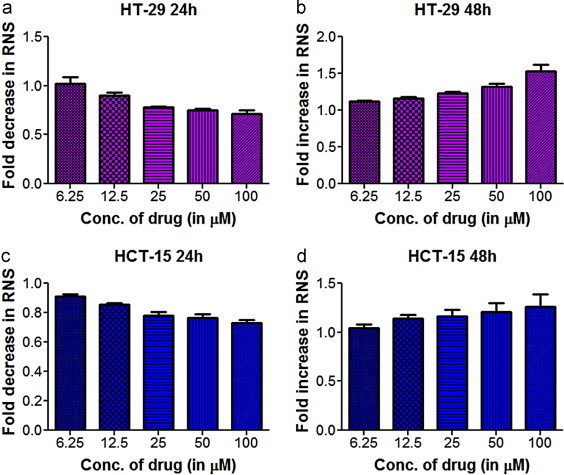
The effect of apigenin on the reactive nitrogen species generation in HT-29, HCT-15 cells over a time period of 24 and 48 h respectively. (a) and (c) Fold decrease in reactive nitrogen species with respect to control in both cell lines at 24 h. (b) and (d) Fold increase in reactive nitrogen species with respect to control in both cell lines at 48 h.
Apigenin induces mitochondrial transmembrane potential (ψm) changes
Previous studies have reported changes in the mitochondria of cells to reveal substantial information in the case of various pathological conditions like cancer and aging since mitochondria is the active site for reactive oxygen, nitrogen and superoxide species generation [15]. The cationic dye JC-1 which at depolarized membrane potentials exist as green-fluorescent monomer transforms into orange-fluorescent JC-1 aggregates at hyperpolarized membrane potentials has been utilized for discerning the changes in mitochondrial membrane potential of HT-29 and HCT-15 cells treated with apigenin. Flow cytometry results revealing higher percentage of cells showing green fluorescence in both cell lines when treated with increasing dose of apigenin suggested drug-induced increased membrane depolarization [Fig. 3b and d] since decreased red/green fluorescence intensity ratio is a measure of mitochondrial depolarization.
Fig. 3.
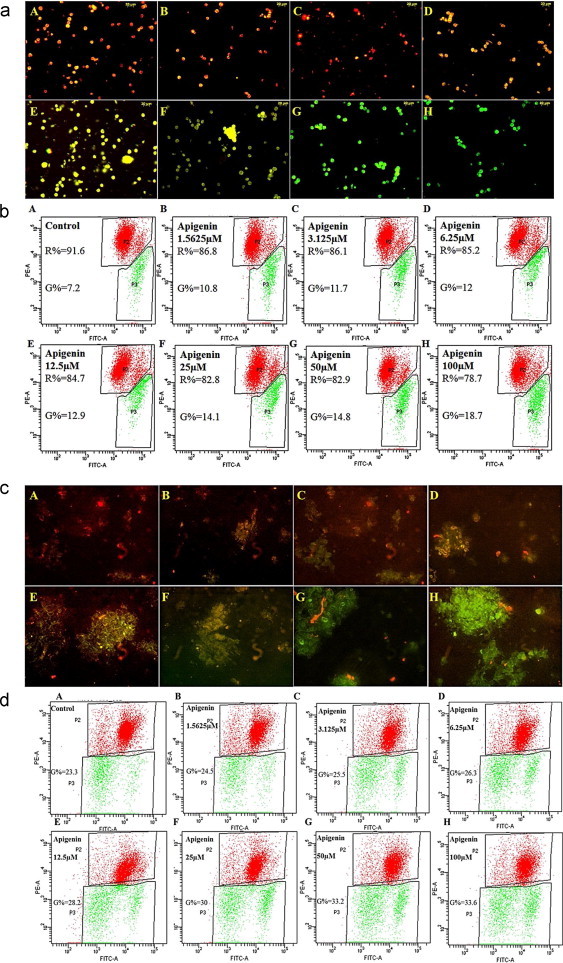
Change in mitochondrial transmembrane potential and early induction of apoptosis estimated by JC-1 in HT-29 and HCT-15 after 24 h treatment duration. (a) HT-29 treated and untreated sets, (c) HCT-15 treated and untreated sets, (b and d) flow cytometry graphs showing the percentage of cells with red or green fluorescence respectively in treated or untreated sets of HCT-15 or HT-29 cells. Apigenin induces mitochondrial transmembrane potential changes in concentration-dependent manner which is an indicator of early apoptosis.
Epi-fluorescence images exhibiting diffused green fluorescence in cells treated with apigenin, unlike the punctate orange-red fluorescent staining of the untreated control cells with healthy polarized mitochondria, indicated the presence of drug-induced depolarized unhealthy mitochondria and corroborated with the flow cytometry results [Fig. 3a and c].
Apigenin increases mitochondrial superoxide production
Recent advances in mitochondrial research have brought free radical superoxide anion into the limelight as biologically significant molecule [15]. Mitochondrial superoxide production related cellular oxidative damage has been reported to be one of the underlying causes of molecular alteration in many degradative diseases and aging [16]. The fluorogenic dye MitoSOX™ Red targets mitochondria in live cells and is selectively oxidized by mitochondrial superoxide present in live cells to produce red fluorescence but remains unaltered by other ROS and/or RNS-generating systems. The observed increase in red fluorescence intensity in both HT-29 and HCT-15 cells with increasing dose of apigenin indicated increased superoxide production [Fig. 4a and c]. In HT-29 cells, a significant increase (p<0.05) in the fluorescence intensity was observed at 1.5625 µM concentration of apigenin, which further increased at successive higher dosages (p<0.001). However, in the case of HCT-15 cells, a significant increase in the red fluorescence intensity was noticed at the drug concentration of 3.125 µM (p<0.001) but was subsequently found to exhibit highly significant changes above 12.5 µM concentration (p<0.0001) [Fig. 4b and d].
Fig. 4.
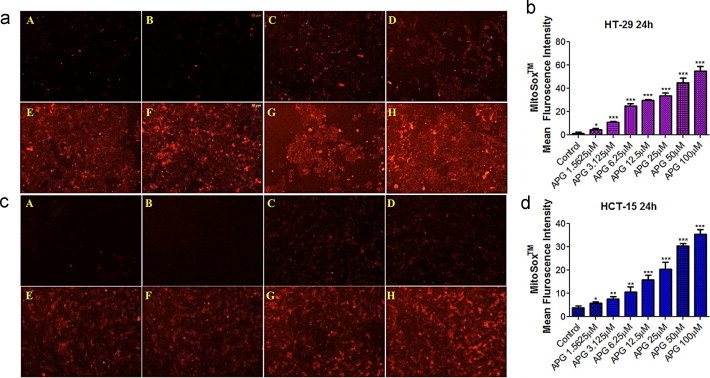
Epi-fluorescent images representing changes in mitochondrial superoxide estimated by MitoSOX™ in HT-29 and HCT-15 after 24 h treatment duration. (a) HT-29 treated and untreated sets, (c) HCT-15 treated and untreated sets. (A) Micrograph of control/untreated group, (B–H) micrograph of treated group with dosage starting from 1.5625 µM and ranging to 50 µM. (b and d) Mean fluorescence intensity of treated and untreated sets in HT-29 and HCT-15 respectively. Data represent the means±S.D. of three independent experiments. Significant differences are indicated as ***p<0.01, **p<0.01 or *p<0.05.
Acute toxicity mediated by apigenin induces apoptosis
Terminal deoxynucleotidyl transferase-mediated dUTP nick end-labeling (TUNEL) assay was performed to detect cells in the late stages of apoptosis which have undergone extensive DNA degradation [17]. Epi-fluorescent microscopy studies revealed dose-dependent increased green fluorescent (TUNEL-positive) apoptotic HT-29 and HCT-15 cells on treatment with apigenin with respect to untreated control cells in the concentration range examined [Fig. 5a and c]. The observed increase in the green fluorescence intensities of the treated cells were analyzed by ImageJ software (Fiji, USA) and were quantified as fold increase with respect to the fluorescent intensity of untreated cells. While a statistically significant increase in the fluorescence intensity was observed from 1.56 µM concentration of apigenin treated HCT-15 cells, it was only observed from 3.125 µM drug dosage in the case of HT-29 cells. Further analysis of the individual sets within the respective test groups yielded highly significant change in the fluorescence intensities amongst the various doses of apigenin treated HCT-15 cells (p<0.0001), unlike HT-29 cells (p 0.0129) [one way ANOVA analysis]. The results thus, suggested greater susceptibility of HCT-15 cells to apigenin in comparison to HT-29 cells [Fig. 5b and d].
Fig. 5.
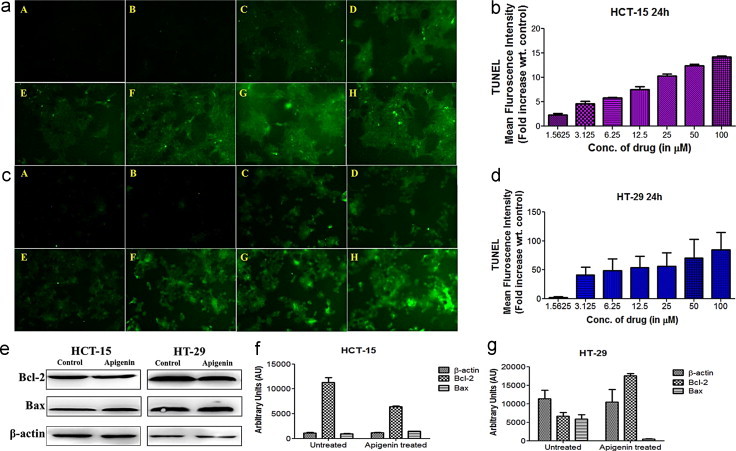
Epi-fluorescent images representing cellular death or apoptosis estimated by in situ cell death detection kit after 24 h treatment duration and apigenin mediated alterations in apoptotic and pro-apoptotic protein profile at high dosage treatment observed in colorectal cell lines HCT-15 and HT-29. (a) HCT-15 treated and untreated sets, (c) HT-29 treated and untreated sets. (A) Micrograph of control/untreated group, (B–H) micrograph of treated group with dosage starting from 1.5625 µM and ranging to 50 µM. (b and d) Mean fluorescence intensity of treated and untreated sets in HT-29 and HCT-15 respectively have been represented as fold increase with respect to control. (e) Western blot analysis of Bcl-2, Bax in colorectal cancer cells untreated/treated with high dose of apigenin. (f and g) Densitometric analysis of the protein bands obtained by western blot in apigenin treated/untreated HCT-15 and HT-29 cells respectively.
Protein profiling of the cells exposed to cytotoxic stimuli elicited by apigenin at higher dose at shorter treatment regimen revealed disruption of the anti-apoptotic and pro-apoptotic protein balance. Simultaneous inhibition of anti-apoptotic protein Bcl-2 and activation of pro-apoptotic effector Bax was observed [Fig. 5e–g] which has been previously attributed to be the underlying cause of the disruption of the mitochondrial outer membrane [18].
Chronic toxicity mediated by apigenin induces aging
Cellular senescence is believed to represent a natural cellular process to suppress tumor formation [19]. Senescent cells have been reported to be characterized by expression of the senescence associated β-galactosidase (SA-β-gal) [20]. The SA-β-gal activity assay exhibited the occurrence of senescence in long term treatment (6 days) with low doses of apigenin in HCT-15 cells [Fig. 6]. Apigenin at concentrations above 25 µM failed to induce senescence since it abrogated cellular viability and yielded very less cells which could advance to growth and were thus not reported here. The results from the present study indicated the role of apigenin to exhibit oxidative stress mediated premature senescence which might represent a natural tumor suppressor mechanism. This finding also corroborates with previous reports of oxidative stress induced senescence in fibroblast cells [21].
Fig. 6.
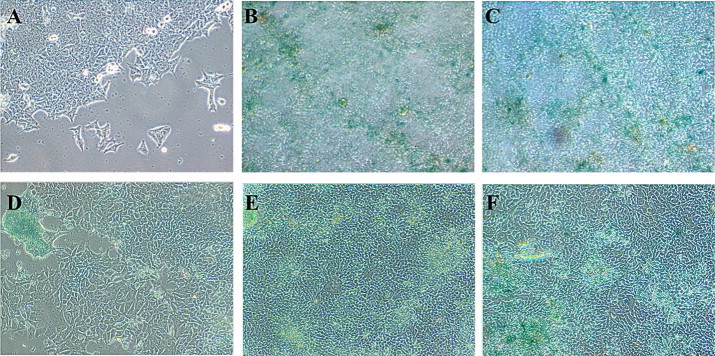
Phase contrast micrographs of (A) untreated/control HCT-15 cells, and (B–H) treated HCT-15 cells with apigenin (conc. range from 1.5625 to 25 µM) stained with senescence Cells Histo-chemical staining kit after 5 day drug treatment regimen. Cells stained as blue/green represent senescent cells.
Apigenin activates signaling pathways driving cellular senescence
Amongst the wide array of extrinsic stresses which activate the cellular senescence program, ROS-dependent oxidative stress is an integral component. In order to assess stress mediated changes in the cellular signaling cascades, the expression and phosphorylation status of various cellular proteins was quantified. Since chronic toxicity of apigenin was observed in earlier sections to elicit cellular senescence in CRC cell lines, the induction of this process was predicted to trigger key signaling pathways important for cellular aging.
Protein profiling of apigenin-induced senescent CRC cells revealed that both p16Ink4a-Rb and p53 independent p21 signaling cascades were activated. Western blotting technique was utilized to validate the involvement of the key downstream proteins of both p16Ink4a-Rb and p53 independent p21 signaling pathways [Fig. 7]. Retinoblastoma protein is a known tumor suppressor protein which remains inactive in its phosphorylated state pRb. While hyperphosphorylation of pRb results in the loss of its capacity to restrict cell cycle progression from G1 to S phase, hypophosphorylation of pRb induces growth-suppression [22]. Apigenin induced ROS generation primarily triggered sustained and robust activation of p16Ink4a. In both HCT-15 and HT-29 which carry mutant p53, apigenin treatment caused negligible change in p53 however, simultaneous increase in p21 levels were observed in both the cell lines. Thus, apigenin mediated increase in p21 was executed through a p53 independent pathway. While the activation of p21 inhibited cyclin E expression, the activation of p16Ink4a down-regulated cyclin D1. This ultimately caused temporal cell-cycle arrest and inhibition of cell-cycle progression. Reminiscent to previous reports the changes in the profiles of cyclins D1 and E observed in our study hindered the Rb phosphorylation which resulted in cell cycle arrest [23,24] corresponding to the onset of senescence. The present study reported the specific role of cyclin D1 in Rb phosphorylation at Ser780 which corroborated with previous in vivo reports [25]. The p53 independent induction of p21 elicited by apigenin is similar to earlier reports of effect of cytotoxic agents as observed in numerous colorectal cell lines bearing p53 mutation [26].
Fig. 7.
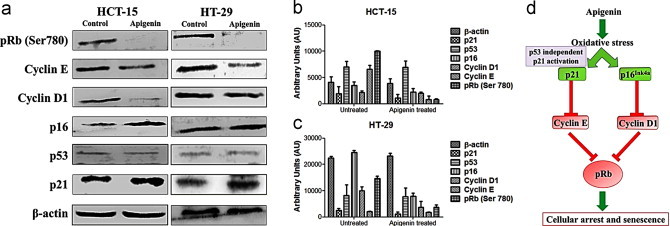
Apigenin mediated alterations in signaling pathways observed in colorectal cell lines HCT-15 and HT-29. (a) Western blot analysis of pRb (Ser780), cyclin E, cyclin D1, p16, p53, p21WAF-1 proteins in colorectal cancer cells untreated/treated with senescence inductive dose of apigenin. (b and c) Densitometric analysis of the protein bands obtained by western blot in apigenin treated/untreated HCT-15 and HT-29 cells respectively. (d) Schematic summary of p16INK4a-Rb and p53–p21 signaling pathway alterations in colorectal cells occurring following apigenin treatment.
The observed alterations in the key signaling molecules regulating the cell cycle suggested that apigenin triggered temporal cell-cycle arrest and inhibited cell-cycle progression. This ultimately induced senescence in the cells which represents a natural cellular process to suppress tumor formation.
Conclusions
The theory of “free radical theory of aging” first proposed by Dr. Denham Harman in the 1950s [27] about the effects of reactive species on aging has been demonstrated in this study, whereby the treatment of colorectal cancer cells in vitro by apigenin mediated free radical stress induced premature senescence. For shorter treatment regime while higher concentrations of apigenin resulted in stress which caused immediate apoptosis, lower concentrations of the drug induced stress that led to marginal increase in ROS levels. Prominent and continuous free radical production induced senescence phenotype at low drug concentrations for longer period. Thus, the present study unearthed the cellular response either of senescence or apoptosis of the colorectal cancer cells to apigenin to be a function of the treatment duration rather than the severity of the stress.
The generation of pro-oxidants (like reactive oxygen species) under normal physiological conditions is balanced by an equivalent rate of its consumption by antioxidants enzymatically and/or non-enzymatically [28]. It has already been reported that a disruption in the pro-oxidant-antioxidant equilibrium leads to “oxidative stress” [28]. In the present study, apigenin was found to induce increased ROS production at growth-suppressive concentrations in both HT-29 and HCT-15 colon cancer cell lines. Further study of the other markers of oxidative stress revealed that apigenin triggered increased pro-oxidative stress by eliciting increased mitochondrial superoxide generation which resulted in enhanced membrane depolarization in colon cancer cell lines. Since increased mitochondrial membrane depolarization is known to be an early marker of apoptosis, the obtained results pointed towards the anti-tumorigenic potential of the flavones. The molecular basis of the apoptotic potential of apigenin is ascribed to the disruption in pro-apoptotic and anti-apoptotic protein balance between Bcl-2 and Bax, which is the underlying cause of the enhanced membrane depolarization in the colon cancer cell lines.
The study also reported that while exposure of cancer cells to higher concentrations of apigenin over shorter time periods induced acute toxicity which led to increased apoptosis, treatment with lower concentrations of the drug for longer time points (6 days) resulted in chronic toxicity through increased reactive nitrogen species generation and senescence induction. Furthermore the present study also revealed that although an increase in the total reactive species production was observed, specific probing of the RNS generation yielded decreased production at shorter time points that was subsequently reversed with treatment time progression. The nitric oxide free radical (NO•) generated has already been reported to be capable to form other reactive intermediates which could trigger nitrosative damage on biomolecules, leading to age-related diseases due to structural alteration of proteins or enzymatic inhibition [29]. Thus, apigenin-induced nitrosative damage over longer time periods may be the underlying basis of the subsequent senescent phenotype which was characterized by the dysregulation in key signaling proteins related to cell cycle progression that included pRb, p16Ink4a and activated the p53 independent p21 signaling cascade. Therapeutic molecules like apigenin which bypass the p53 mediated pathway are increasingly generating interest in the context of clinical application since loss of p53 function is associated with most instances of colorectal cancer. The observed cellular senescence induced by extended low levels of oxidative stress caused by apigenin could be utilized for chemoprevention as well as an in vitro model for aging research in the future.
Author contributions
Kacoli Banerjee: Conception and design of study, acquisition and analysis of data, drafting of article and/or critical revision, final approval of manuscript.
Mahitosh Mandal: Conception and design of study, drafting of article and/or critical revision, final approval of manuscript.
Conflicts of interest
The authors declare that they have no conflict of interest.
Acknowledgements
KB gratefully acknowledges the University Grants Commission, India for awarding her with Senior Research Fellowship [F.No. 2-16/98(SA-I), 04.10.2010]. This work was supported by generous research grants from the Department of Biotechnology (DBT) [Grant no. BT/PR13996/Med/30/309/2010] and Department of Science and Technology (DST) [Grant no. SR/SO/BB-58/2008], Ministry of Science and Technology, Government of India.
References
- 1.Pandey K.B., Rizvi S.I. Plant polyphenols as dietary antioxidants in human health and disease. Oxidative Medicine and Cellular Longevity. 2009;2(5):270–278. doi: 10.4161/oxim.2.5.9498. 20716914 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rice-Evans C. Flavonoids and isoflavones: absorption, metabolism, and bioactivity. Free Radical Biology and Medicine. 2004;36(7):827–828. doi: 10.1016/j.freeradbiomed.2003.12.012. 15019967 [DOI] [PubMed] [Google Scholar]
- 3.Tsuchiya H. Structure-dependent membrane interaction of flavonoids associated with their bioactivity. Food Chemistry. 2010;120(4):1089–1096. [Google Scholar]
- 4.Ferrari N., Tosetti F., De Flora S., Donatelli F., Sogno I., Noonan D.M., Albini A. Diet-derived phytochemicals: from cancer chemoprevention to cardio-oncological prevention. Current Drug Targets. 2011;12(13):1909–1924. doi: 10.2174/138945011798184227. 21158708 [DOI] [PubMed] [Google Scholar]
- 5.Patel D., Shukla S., Gupta S. Apigenin and cancer chemoprevention: progress, potential and promise. International Journal of Oncology. 2007;30(1):233–245. 17143534 [PubMed] [Google Scholar]
- 6.Ruela-de-Sousa R.R., Fuhler G.M., Blom N., Ferreira C.V., Aoyama H., Peppelenbosch M.P. Cytotoxicity of apigenin on leukemia cell lines: implications for prevention and therapy. Cell Death and Disease. 2010;1:e19. doi: 10.1038/cddis.2009.18. 21364620 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shukla S., Gupta S. Apigenin-induced prostate cancer cell death is initiated by reactive oxygen species and p53 activation. Free Radical Biology and Medicine. 2008;44(10):1833–1845. doi: 10.1016/j.freeradbiomed.2008.02.007. 18342637 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Lugli E., Troiano L., Ferraresi R., Roat E., Prada N., Nasi M., Pinti M., Cooper E.L., Cossarizza A. Characterization of cells with different mitochondrial membrane potential during apoptosis. Cytometry Part A. 2005;68(1):28–35. doi: 10.1002/cyto.a.20188. 16184612 [DOI] [PubMed] [Google Scholar]
- 9.Galati G., Sabzevari O., Wilson J.X., O’Brien P.J. Prooxidant activity and cellular effects of the phenoxyl radicals of dietary flavonoids and other polyphenolics. Toxicology. 2002;177(1):91–104. doi: 10.1016/s0300-483x(02)00198-1. 12126798 [DOI] [PubMed] [Google Scholar]
- 10.Banerjee S., Sen K., Pal T.K., Guha S.K. Poly(styrene-co-maleic acid)-based pH-sensitive liposomes mediate cytosolic delivery of drugs for enhanced cancer chemotherapy. International Journal of Pharmaceutics. 2012;436(1–2):786–797. doi: 10.1016/j.ijpharm.2012.07.059. 22884831 [DOI] [PubMed] [Google Scholar]
- 11.Eruslanov E., Kusmartsev S. Identification of ROS using oxidized DCFDA and flow-cytometry. Methods in Molecular Biology. 2010;594:57–72. doi: 10.1007/978-1-60761-411-1_4. 20072909 [DOI] [PubMed] [Google Scholar]
- 12.Chae S.Y., Lee M., Kim S.W., Bae Y.H. Protection of insulin secreting cells from nitric oxide induced cellular damage by crosslinked hemoglobin. Biomaterials. 2004;25(5):843–850. doi: 10.1016/s0142-9612(03)00605-7. 14609673 [DOI] [PubMed] [Google Scholar]
- 13.Ryu S.-W., Woo J.H., Kim Y.-H., Lee Y.-S., Park J.W., Bae Y.-S. Downregulation of protein kinase CKII is associated with cellular senescence. FEBS Letters. 2006;580(3):988–994. doi: 10.1016/j.febslet.2006.01.028. 16442104 [DOI] [PubMed] [Google Scholar]
- 14.Shin G.-C., Kim C., Lee J.-M., Cho W.-S., Lee S.-G., Jeong M., Cho J., Lee K. Apigenin-induced apoptosis is mediated by reactive oxygen species and activation of ERK1/2 in rheumatoid fibroblast-like synoviocytes. Chemico-Biological Interactions. 2009;182(1):29–36. doi: 10.1016/j.cbi.2009.07.016. 19647729 [DOI] [PubMed] [Google Scholar]
- 15.Mukhopadhyay P., Rajesh M., Haskó G., Hawkins B.J., Madesh M., Pacher P. Simultaneous detection of apoptosis and mitochondrial superoxide production in live cells by flow cytometry and confocal microscopy. Nature Protocols. 2007;2(9):2295–2301. doi: 10.1038/nprot.2007.327. 17853886 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cadenas E., Davies K.J.A. Mitochondrial free radical generation, oxidative stress, and aging. Free Radical Biology and Medicine. 2000;29(3–4):222–230. doi: 10.1016/s0891-5849(00)00317-8. 11035250 [DOI] [PubMed] [Google Scholar]
- 17.Kyrylkova K., Kyryachenko S., Leid M., Kioussi C. Detection of apoptosis by TUNEL assay. Methods in Molecular Biology. 2012;887:41–47. doi: 10.1007/978-1-61779-860-3_5. 22566045 [DOI] [PubMed] [Google Scholar]
- 18.Czabotar P.E., Lessene G., Strasser A., Adams J.M. Control of apoptosis by the BCL-2 protein family: implications for physiology and therapy. Nature Reviews Molecular Cell Biology. 2014;15(1):49–63. doi: 10.1038/nrm3722. 24355989 [DOI] [PubMed] [Google Scholar]
- 19.Dasari A., Bartholomew J.N., Volonte D., Galbiati F. Oxidative stress induces premature senescence by stimulating Caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-Rich promoter elements. Cancer Research. 2006;66(22):10805–10814. doi: 10.1158/0008-5472.CAN-06-1236. 17108117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Debacq-Chainiaux F., Erusalimsky J.D., Campisi J., Toussaint O. Protocols to detect senescence-associated beta-galactosidase (SA-βgal) activity, a biomarker of senescent cells in culture and in vivo. Nature Protocols. 2009;4(12):1798–1806. doi: 10.1038/nprot.2009.191. 20010931 [DOI] [PubMed] [Google Scholar]
- 21.Volonte D., Zhang K., Lisanti M.P., Galbiati F. Expression of Caveolin-1 induces premature cellular senescence in primary cultures of murine fibroblasts. Molecular Biology of the Cell. 2002;13(7):2502–2517. doi: 10.1091/mbc.01-11-0529. 12134086 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hu B. Liver yin deficiency tonifying herbal extract induces apoptosis and cell senescence in Bel-7402 human hepatocarcinoma cells. Experimental and Therapeutic Medicine. 2011 doi: 10.3892/etm.2011.364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Narasimha A.M., Kaulich M., Shapiro G.S., Choi Y.J., Sicinski P., Dowdy S.F. Cyclin D activates the Rb tumor suppressor by mono-phosphorylation. eLife. 2014;3 doi: 10.7554/eLife.02872. 24876129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Hwang H.C., Clurman B.E. Cyclin E in normal and neoplastic cell cycles. Oncogene. 2005;24(17):2776–2786. doi: 10.1038/sj.onc.1208613. 15838514 [DOI] [PubMed] [Google Scholar]
- 25.Geng Y., Yu Q., Sicinska E., Das M., Bronson R.T., Sicinski P. Deletion of the p27Kip1 gene restores normal development in cyclin D1-deficient mice. Proceedings of the National Academy of Sciences. 2001;98(1):194–199. doi: 10.1073/pnas.011522998. 11134518 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ravizza R., Gariboldi M.B., Passarelli L., Monti E. Role of the p53/p21 system in the response of human colon carcinoma cells to doxorubicin. BMC Cancer. 2004;4:92. doi: 10.1186/1471-2407-4-92. 15601469 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Harman D. Aging: a theory based on free radical and radiation chemistry. Journal of Gerontology. 1956;11(3):298–300. doi: 10.1093/geronj/11.3.298. 13332224 [DOI] [PubMed] [Google Scholar]
- 28.Sies H. Oxidative stress: from basic research to clinical application. The American Journal of Medicine. 1991;91(3C):31S–38S. doi: 10.1016/0002-9343(91)90281-2. 1928209 [DOI] [PubMed] [Google Scholar]
- 29.Drew B., Leeuwenburgh C. Aging and the role of reactive nitrogen species. Annals of the New York Academy of Sciences. 2002;959:66–81. doi: 10.1111/j.1749-6632.2002.tb02084.x. 11976187 [DOI] [PubMed] [Google Scholar]