

Abstract
We used a VSV-cDNA library to treat recurrent melanoma, identifying immunogenic antigens, allowing us to target recurrences with immunotherapy or chemotherapy. Primary B16 melanoma tumors were induced to regress by frontline therapy. Mice with recurrent tumors were treated with VSV-cDNA immunotherapy. A Th17 recall response was used to screen the VSV-cDNA library for individual viruses encoding rejection antigens, subsequently targeted using immunotherapy or chemotherapy. Recurrent tumors were effectively treated with a VSV-cDNA library using cDNA from recurrent B16 tumors. Recurrence-associated rejection antigens identified included Topoisomerase-IIα, YB-1, cdc7 kinase, and BRAF. Fourteen out of 16 recurrent tumors carried BRAF mutations (595–605 region) following frontline therapy, even though the parental B16 tumors were BRAF wild type. The emergence of mutated BRAF-containing recurrences served as an excellent target for BRAF-specific immune-(VSV-BRAF), or chemo-(PLX-4720) therapies. Successful PLX-4720 therapy of recurrent tumors was associated with the development of a broad spectrum of T-cell responses. VSV-cDNA technology can be used to identify recurrence specific antigens. Emergence of mutated BRAF may be a major effector of melanoma recurrence which could serve as a target for chemo or immune therapy. This study suggests a rationale for offering patients with initially wild-type BRAF melanomas an additional biopsy to screen for mutant BRAF upon recurrence.
Introduction
Aggressive recurrent tumors following frontline therapy represent a major clinical problem. To address this in a preclinical context, we recently established several models in which frontline treatment of established subcutaneous (s.c.) tumors induced complete macroscopic regressions, tumor dormancy/minimal residual disease (MRD), and subsequent aggressive tumor recurrences.1,2 In those studies, we demonstrated that recurrent tumors growing in immune-competent mice evade an innate immune response directed against them at the time when recurrent tumors transit from a state of MRD into an actively proliferating recurrent tumor.2 In addition, we identified Topisomerase-IIα (TOPO-IIα) as a potential recurrence-specific tumor antigen, which is expressed in recurrences of both TC2 prostate and B16 melanomas and which could be targeted by chemotherapy to prevent, or treat, recurrences.
In the current study, we sought to identify additional antigens which are associated with recurrent melanoma in order to identify therapeutic targets to treat this disease. In this respect, malignant melanoma incidence rates are rising by 1.8% each year,3 and patients with unresectable or metastatic disease currently have few therapies offering long-term remission. Identification of BRAF mutations, which occur in 50% of patients with malignant melanoma, plays a major role in treatment selection. Up to 90% of these mutations consist of a glutamic acid to valine substitution at amino acid 600 (BRAFV600E). Targeting oncogenic BRAF represents a major advance in the clinical setting through the development of BRAF kinase inhibitors. Vemurafenib was the first BRAF kinase inhibitor approved by the US Food and Drug Administration in 2011, for patients with unresectable or metastatic melanoma who have the BRAFV600E mutation. The BRIM-3 phase 3 clinical trial demonstrated a survival benefit in patients who received frontline vemurafenib.4 Recently, newer agents have been approved for clinical use including BRAF (dabrafenib) and MEK (trametinib) inhibitors.5,6 Patients without a BRAFV600E mutation in the primary tumor are treated using the anti-CTLA-4 antibody ipilumumab. Recently, therapies targeting the PD-1/L1 have demonstrated promising results in early-phase clinical trials. Importantly, however, patients whose melanoma is initially BRAF wild type are not routinely biopsied upon recurrence of the disease to investigate whether BRAF status of the recurrence is different from that of the primary tumor.
Here, we used our previously described model of B16 cells expressing the HSVtk suicide gene (B16tk), to establish primary tumors which could be apparently cured by treatment with ganciclovir (GCV). However, following a period of MRD, a proportion of these tumors recur aggressively.2 We demonstrate that B16tk tumor recurrences (B16tk REC) following suboptimal frontline GCV therapy were effectively treated with a vesicular stomatitis virus (VSV) cDNA library constructed using cDNA from recurrent tumors (the Ganciclovir Escape Epitope Library (GEEL)), but not from cDNA derived from primary tumors. Consistent with our previous results,1 screening of the GEEL identified TOPO-IIα as an immunogen in the library responsible for rejection of emerging B16tk REC recurrences. In addition, the same screen identified the murine BRAF gene as a potential immunogen of B16tk recurrence. Further analysis of B16tk REC recurrent tumors following different frontline therapies (chemo-, viro-, or immunotherapy) revealed that 14/16 B16tk REC recurrences had acquired a mutated BRAF, including BRAFV600E, even though the parental B16tk cells were BRAF wild type. We exploited BRAF as a major immunogenic target of B16tk recurrences by showing that VSV-mediated expression of BRAF prevented recurrences in this model following frontline GCV therapy. We also targeted these recurrences using a BRAFV600E inhibitor which concomitantly generated T-cell responses against known melanoma-associated antigens, BRAF itself, and the recently identified tumor recurrence antigen TOPO-IIα.1 These studies suggest that the emergence of mutated BRAF may be a major effector of melanoma recurrence and highlights a clinical rationale for offering patients with initially wild-type BRAF tumor recurrences a biopsy to screen for mutant BRAF.
Results
Antigens of melanoma recurrence
We have previously described a model in which 2 weeks of GCV treatment of s.c. B16 melanomas expressing the Herpes Simplex Virus thymidine kinase gene (B16tk) in C57BL/6 mice, typically induced complete macroscopic regressions, a period of tumor dormancy/MRD (no palpable tumor) followed, in many mice, by aggressive tumor recurrences (Figure 1a).1,2 B16tk recurrences (B16tk REC), which emerged in vivo following frontline GCV therapy, were effectively treated by a VSV-cDNA GEEL derived from the pooled cDNA of three different B16 REC tumors, provided mice in a state of MRD were treated before tumor recurrence became apparent or as recurrences became palpable. In contrast, treatment with a VSV-cDNA library constructed from cDNA of normal human prostate was completely ineffective at treating B16tk REC tumor recurrence (Figure 1b).
Figure 1.

VSV-expressed antigens treat recurrence. (a) Kinetics of the in vivo model of tumor treatment, regression, minimal residual disease, and recurrence. Representative data are shown from a C57BL/6 mouse seeded with a B16tk tumor at day 0. By day 5, tumor was established (longest tumor diameter 0.4 cm). GCV was then given i.p. at 50 mg/kg for 5 consecutive days (days 6–10), followed by 2 days rest, during which time tumor regressed (0.8 cm, day 8 and 0.2 cm at day 11). Five further consecutive injections of GCV were then given on days 13–17. With this regimen, most tumors regressed macroscopically by about days 15–20 after tumor seeding (day 15 undetectable in this mouse). Tumor regression was followed by later local recurrence in ~50–80% of the mice at different time points (typically between days 30–40) after the last injection of GCV. Once recurrence was palpable, the secondary tumor always grew very rapidly and much more aggressively than the primary tumor (from palpable (day 32) to 1.0 cm (day 37) in 5 days in this example). (b) C57BL/6 mice seeded with B16tk tumors 5 days previously were treated with GCV i.p. at 50 mg/kg for 5 consecutive days, followed by 2 days rest, followed by five further consecutive injections of GCV (days 6–10 and 13–17) as in a. Mice were injected i.v. with PBS or VSV-cDNA libraries (GEEL or ASEL, 107 pfu/injection) starting on day 20, by which time primary tumors had regressed (as in a), Subsequent i.v. injections were given on days 22, 24, 27, 29, and 31. Survival of mice (n = 7/8 per group) treated sequentially with GCV, then different combinations of VSV-cDNA as shown. Representative of two separate experiments. (c) C57BL/6 mice seeded with B16tk tumors 5 days previously were treated i.v. with PBS, VSV-cDNA libraries (ASMEL or GEEL), or with VSV-GFP (107 pfu/injection) on days 6, 8, and 10; 13, 15, and 17; 20, 22, and 24. Cumulative number of mice surviving tumor free at day 60 in two separate experiments is shown. (d) Splenocyte/LN cultures from tumor cured, GEEL-vaccinated mice were screened for IL-17 secretion following no infection/restimulation; following restimulation with freeze thaw cell lysates of B16tk, B16tk REC1, B16tk REC2, and B16tk REC3 cells; or following restimulation by infection (multiplicity of infection 10) with VSV-GFP; VSV-TOPO-IIα; VSV-cdc7k; VSV-YB-1; or VSV-BRAF; with a combination of VSV-TOPO-IIα + VSV-YB-1; or VSV-TOPO-IIα + VSV-BRAF; or with a combination of VSV-TOPO-IIα + VSV-YB-1 + VSV-BRAF. (e) Functional cloning of viruses encoding tumor rejection antigens for recurrent B16tk melanomas. LN/splenocyte cultures (104/well) from mice cured of B16tk recurrent tumors by a combination of GCV and GEEL (as in b) were screened for secretion of IL-17 induced by infection with aliquots of ~104 pfu of the parental GEEL virus stock in the presence of recombinant hsp70 aliquots which contained virus competent for inducing the IL-17 recall response were pooled and expanded in BHK cells (24–36 hours).7 New LN/splenocyte cultures from GEEL-vaccinated mice were infected with serial dilutions of this expanded stock in the presence of recombinant hsp70 and assayed for IL-17 production. The highest dilution of the virus stock which induced IL-17 at levels significantly above background was amplified by passaging through BHK cells for 24–36 hours. Serial dilutions of this expanded stock were screened for their ability to induce IL-17. Ten microliters of aliquots of the highest dilution of the virus which induced IL-17 were used as the starting point for limiting dilution cloning on BHK cells to identify the dilution at which a single virus particle generated cytopathic effect (+).
As reported previously, mice treated with the ASMEL VSV-cDNA library, constructed from cDNA of two different human melanoma cell lines, rejected about 70% of 5-day established B16tk primary tumors (Figure 1c). However, treatment of primary B16tk tumors with the GEEL gave no significant treatment compared to treatment with VSV-GFP or PBS (Figure 1c). Consistent with this profile of in vivo treatment, splenocytes/LN from mice cured of B16tk REC by the GEEL secreted IL-17 following re-stimulation with B16 REC lysates, but not B16tk lysates (Figure 1d). Therefore, the antigenic repertoire which protected against B16tk REC recurrences (in the GEEL) differed significantly from that which protected against primary B16tk tumors (in the ASMEL).
We used an in vitro assay to screen for individual viruses within the GEEL which induced B16tk REC-specific IL-17 recall responses (Figure 1e).1,7 Of multiple viruses recovered from this screen, three encoded 5′ sequences of the murine TOPO-IIα gene, seven encoded the 5′ end of the murine YB-1 gene, and four encoded the 5′ end of the murine BRAF gene. VSV-GFP was unable to induce IL-17 following re-stimulation of splenocyte/LN cultures from GEEL-vaccinated mice, even at high multiplicity of infection (Figure 1d). Restimulation of splenocytes/LN from GEEL-treated mice with either VSV-TOPO-IIα, VSV-cdc7k, VSV-YB-1, or VSV-BRAF alone induced either very low levels of levels of IL-17 or levels not significantly above background (Figure 1d). However, a combination of VSV-TOPO-IIα + VSV-YB-1 + VSV-BRAF induced significantly increased levels of IL-17 compared with any of the viruses alone (but always lower than levels induced by restimulation with B16tk REC cell lysates). These data suggested that the combination of TOPO-IIα, YB-1, and BRAF might serve as potential tumor antigens in B16tk REC tumors. The ability of VSV-cDNA viruses to induce IL-17 in combination depended on the nature of the cDNA insert because combining either alone with VSV-GFP did not induce IL-17.
Melanoma recurrences acquire mutated BRAF
The VSV-BRAF virus which contained the longest insert of BRAF cDNA isolated from the screening of the GEEL (Figure 1e) contained a G-R mutation compared to the wild-type BRAF sequence at position 596 (numbering according to the human BRAF sequence) (Figure 2a, i and ii). The three separate B16tk REC tumor explants used to generate the cDNA cloned into the GEEL also contained mutated BRAF sequences, two with the G-R (596) mutation and one with a L-V mutation at position 597 (Figure 2a iii and iv). However, repeated sequencing of the same region of BRAF from the parental, cultured B16tk cells (used to generate B16tk primary, B16tk REC recurrent tumors) consistently demonstrated wild-type BRAF (Figure 2a). Libraries were sequenced on an Illumina HiSeq2000, with a 50× coverage (50 nucleotide reads per kilo base of transcript per million mapped reads). No mutations were found in Braf or N-ras.
Figure 2.

Melanoma recurrences acquire mutant BRAF. (a) Sequence comparisons between the wild-type murine BRAF sequence around positions 595–605 (i) with the cDNA insert in the VSV-cDNA single pfu clone #3 isolated from the screen of Figure 1e (ii); two different B16tk REC recurrence explants which relapsed following GCV therapy with a G596R mutation (Figure 1a) (iii); a further B16tk REC recurrence explant with a L597V mutation (iv); and the BRAF sequence from parental B16tk cells which are maintained in culture and from which all primary and recurrent B16tk tumors in vivo derived (v). (b) BRAF sequence status of primary tumors treated with PBS or of early (0.2–0.3 cm diameter) explant recurrent tumors derived from primary B16tk, or B16ova, tumors in C57BL/6 mice induced to regress, enter a state of MRD and then recur, following Paclitaxel/reovirus,26 VSV-cDNA immune,7 GCV chemo1 or (B16ova) OT-I Adoptive T cell,8,9 frontline therapies (as described in Materials and Methods). (c) BRAF sequence status of primary tumors treated with PBS or of early (0.2–0.3 cm diameter) explant recurrent tumors derived from primary TC2 tumors in C57BL/6 mice induced to regress, enter a state of MRD and then recur, following Paclitaxel/reovirus or VSV-cDNA immune-, frontline therapies.
These data suggested that de novo acquisition of a mutated BRAF sequence may be a hallmark of recurrent B16tk tumors following in vivo treatment, selection, and/or evolution of a tumor reduced to a state of MRD which was subsequently able to emerge as an actively growing recurrence.
To test this hypothesis, very early (~0.2–0.3 cm diameter) recurrent B16 tumors were explanted from mice following initial tumor regression, a period of MRD and subsequent relapse, induced by four different frontline therapies. Consistent with the data of Figure 2a, no mutated BRAF sequences were detected in 8/8 B16 primary tumors, from mice treated with PBS (Figure 2b). In contrast, when established primary B16tk tumors were induced to regress, enter a state of MRD, and then recur following either chemo/virotherapy,1 VSV-cDNA immunotherapy with the ASMEL GCV chemotherapy1,7 or adoptive T-cell therapy (using the B16ova/OT-I model),8,9 14/16 recurrences contained a mutated BRAF sequence within the 595–605 region (Figure 2b). These mutations not only included both the G596R and L597V mutations detected in the B16tk REC cell lines used to construct the GEEL (Figure 2a), but also included explants with a V-E mutation at position 600 induced following frontline therapy with either chemo/viro-, VSV-cDNA, or GCV chemotherapies (Figure 2b). Similar recurrences derived from treatment of s.c. prostate TC2 primary tumors by either chemo/viro- or VSV-cDNA immunotherapies did not contain any detectable mutant BRAF sequences (Figure 2c). Taken together, these data suggested that acquisition of a mutated BRAF sequence represents a melanoma-specific effector of tumor recurrence in vivo.
Melanoma recurrences overexpress G596R and L597V mutant BRAF
In B16 REC recurrences which contained either the G596R or the L597V mutations, irrespective of the frontline therapy, BRAF was consistently overexpressed compared to primary B16 tumors from PBS-treated mice in the same experiments (Figure 3a–d). In contrast, B16 REC recurrences in which the V600E mutation was present did not generally overexpress BRAF relative to corresponding primary tumors treated with PBS (Figure 3a–d). No overexpression, or mutation, of BRAF was detected in TC2 REC recurrences following frontline immunotherapy with a VSV-cDNA library (Figure 3e).
Figure 3.
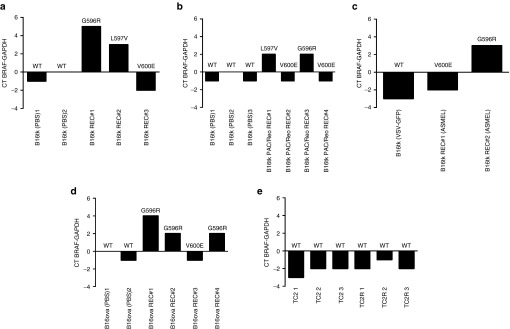
G596R and L597V, but not V600E, mutant BRAF are overexpressed in melanoma recurrences. cDNA was analyzed by qrtPCR for expression of BRAF from primary tumors treated with PBS, or early (0.2–0.3 cm diameter) explant recurrent tumors derived from primary B16tk (a–c) or B16ova (d), tumors in C57BL/6 mice induced to regress, enter a state of MRD and then recur, following (a) GCV chemo,1 (b) paclitaxel/reovirus,26 (c) VSV-cDNA ASMEL immune,7 or (d) (B16ova) OT-I adoptive T cell8,9 frontline therapies (as described in Materials and Methods). The threshold cycle (Ct) at which amplification of the target BRAF sequence was detected was used to compare the relative levels of mRNA between samples. CT BRAF-GAPDH: relative quantities of BRAF mRNA were normalized with Ct of GAPDH amplification. (e) cDNA was analyzed by qrtPCR for expression of BRAF from primary TC2 tumors treated with PBS, or early (0.2–0.3 cm diameter) explant recurrent tumors derived from primary TC2 tumors in C57BL/6 mice induced to regress, enter a state of MRD and then recur, following VSV-cDNA ASEL immune frontline therapy (as described in Materials and Methods).24
BRAF mutant B16 cells dominate in vitro cultures
We were unable to detect any mutated BRAF sequences from cultured B16tk, or B16ova, parental cells (Figure 2a), or from B16 primary tumors grown in C57BL/6 mice treated with PBS (Figure 2b). These data suggest that an acquired mutation in the BRAF gene that occurs in vivo represents a powerful effector mechanism by which dormant B16 cells can actively progress as recurrences in vivo. Alternatively, it is possible that a very low level subpopulation of B16 cells exists in the parental population and that this already carries G596R, L597V, or V600E mutations. In this scenario, these minority populations would be strongly selected during the process of tumor recurrence, but their selective advantage would not be evident in the parental cell culture population (otherwise they would predominate). In this respect, three different B16tk REC explants, recovered following in vivo GCV-treatment and each with a different BRAF mutation (B16tk REC#1 (G596R); B16tk REC#2 (L597V); and B16tk REC#3 (V600E); Figure 2b), were able to outcompete the parental B16tk cell population within 72–96 hours in vitro (Figure 4). Thus, GFP-labeled B16tk cells cocultured with a B16tk primary tumor explanted from a mouse treated with PBS (wild-type BRAF; Figure 2b), were present in the same proportion as initially plated 7 days following mixing (Figure 4a). In contrast, parental B16tk cells were almost completely outcompeted 7 days after mixing in a 1:1 ratio by B16tk REC#1 (G596R) cells (Figure 4b). This in vitro competition was even more pronounced on mixing B16tk parental and B16tk REC#3 (V600E) cells in culture, with as few as 1% of B16tk REC#3 (V600E) cells dominating the culture 7 days later (Figure 4c). We do not yet know whether these B16tk REC explants have further genetic/epigenetic changes, in addition to their different BRAF mutations, which could contribute to their potent outgrowth of B16tk parental cells in culture. Nonetheless, these data suggest that BRAF mutation would confer a selective advantage to even a very minor population present in the parental population of B16tk cells in culture. This supports the hypothesis that BRAF mutation is acquired de novo within the small population of B16tk cells which survive frontline therapy as MRD and is a direct result of genetic plasticity of at least a proportion of these cells which can respond to applied selective pressures and acquire a phenotype which allows re-emergence as a progressively growing recurrence.
Figure 4.
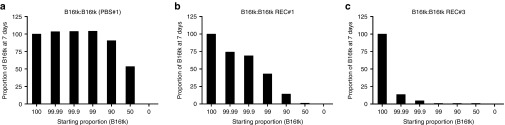
BRAF mutant cells outcompete parental B16tk in culture. Parental B16tk cells, infected with a lentiviral vector expressing GFP, were cocultured with (a) an explant of a B16tk primary tumor from a mouse treated with PBS; (b) with B16tk REC#1 (BRAFG596R) cells; or (c) with B16tk REC#3 (BRAFV600E) cells at varying ratios as shown starting at 100% B16tk:0% partner cell type (105 cells total). Cultures were split 1:5 every 48 hours and 7 days later the % of B16tk parental cells (GFP +ve) was analyzed by FACS. Results representative of three separate experiments.
VSV-BRAF is a component of vaccination against recurrence
The data so far suggest that BRAF can serve as a major immunogenic target of immune responses against B16tk recurrences (Figure 1d), either through overexpression or mutation in recurrent melanomas (Figures 2 and 3). Consistent with this, VSV-mediated expression of BRAF clone #3 (Figure 2a, ii), in combination with VSV-YB-1 and VSV-TOPO-IIα, prevented recurrence of B16tk REC recurrences following frontline GCV therapy in 100% of mice (Figure 5a,b). Therapy was only successful when VSV-cDNA was initiated during the state of MRD before recurrences became palpable (Figure 5a). Pooled spleens and lymph node cells from surviving, recurrence-free mice euthanized from this experiment had significant IL-17 memory recall responses against freeze thaw lysates of B16tk REC #1, #2, and #3 recurrences, but not against freeze thaw lysates of either cultured B16tk cells or B16tk primary tumors explanted from mice treated with PBS (Figure 5c,d). Spleen/LN from a mouse which developed a progressively growing B16tk REC tumor (treated with GCV/PBS/PBS) had no significant IL-17 memory recall response against B16tk primary or REC tumors (Figure 5e). No Th1 IFN-γ recall responses were observed against either B16 primary, or recurrent, tumors in splenocyte/LN cells from either survivors (Figure 5c,d) or nonsurvivor (Figure 5e) mice.
Figure 5.
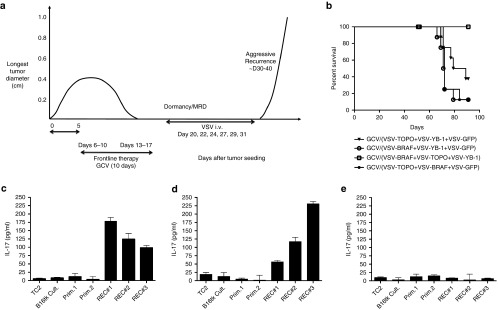
(a) C57BL/6 mice seeded with B16tk tumors 5 days previously were treated with GCV i.p. at 50 mg/kg for 5 consecutive days, followed by 2 days rest, followed by five further consecutive injections of GCV (days 6–10 and 13–17). Mice were injected i.v. with combinations of VSV expressing different cDNAs as recovered from the screening of the GEEL library (Figure 1d,e) starting on day 20, by which time primary tumors had regressed. Subsequent i.v. injections were given on days 22, 24, 27, 29, and 31. (b) Survival of mice (n = 7/8 per group) treated sequentially with GCV, then with combinations of VSV-cDNA as per the protocol of a, with i.v. injections of VSV-BRAF+VSV-YB-1+VSV-GFP; VSV-TOPOIIα+VSV-BRAF+VSV-GFP; VSV-TOPOIIα+VSV-YB-1+VSV-GFP; or VSV-BRAF+VSV-TOPOIIα+VSV-YB-1 (3 × 106 pfu/virus/injection). (c–e) Splenocyte/LN cultures from two different survivor mice (c,d), cured of B16tk recurrences by the GCV/VSV-BRAF+VSV-TOPOIIα+VSV-YB-1 combination therapy of (b), or from a nonsurvivor mouse from the group treated with the GCV/ VSV-BRAF+VSV-YB-1+VSV-GFP combination (e) were screened for IL-17 secretion following restimulation with freeze thaw cell lysates of TC2 cells, parental B16tk cells, cells from two different B16tk primary tumor explants from mice treated with PBS (Prim.1 & 2), or with freeze thaw lysates of B16tk REC#1, B16tk REC#2, and B16tk REC#3 cells as described in Materials and Methods.
BRAF mutant melanoma recurrences can be targeted by drug therapy
Both the B16tk REC#3 (BRAFV600E) and B16tk REC#1 (BRAFG596R) recurrence-derived cell lines proliferated significantly faster in vitro than parental B16tk (Figure 6a) or B16tk REC#2 (BRAFL597V) (data not shown) cells (P < 0.001) (no significant difference between B16tk parental cells and B16tk REC#2 (BRAFL597V)). Consistent both with the data of Figure 4, and the fact that the V600E mutation is associated with the most potent transforming activity of known BRAF mutations, B16tk REC#3 (BRAFV600E) cells proliferated significantly faster than the B16tk REC#1 (BRAFG596R) cells (P < 0.01; Figure 6a). Similarly, B16tkREC#3 (BRAFV600E) were highly sensitive to PLX-4720, a potent inhibitor of BRAFV600E, compared to both B16tk parental and B16tk REC#1 (BRAFG596R) cells (Figure 6a). B16tk REC#1 (BRAFG596R) cells were also significantly more sensitive than parental B16tk cells (P = 0.01; Figure 6a). However, in only one of two different experiments, growth of B16tk REC#2 (BRAFL597V) cells was significantly inhibited by PLX-4720 compared to B16tk parental cells (P = 0.05). In vivo, PLX-4720 exerted a significant growth inhibitory effect on all three BRAF mutant recurrent B16tk REC cell lines (Figure 6b).
Figure 6.

BRAF mutant melanoma recurrences can be treated by chemotherapy. (a) Parental B16tk, B16tk REC#1 (BRAFG596R), B16tk REC#3 (BRAFV600E) cells were plated (104 cells/well in triplicates) in diluent or medium containing PLX-4720 at 1 µmol/l. Seventy-two hours later, cells were washed in PBS, 3×, trypsinised and counted. (b) C57Bl/6 mice were seeded with B16tk or B16tkREC #1, #2, or #3 tumors subcutaneously on day 1. On days 6–10, 13–17, and 20–24, tumor bearing mice were treated with PBS or with PLX-4720 (30 mg/kg/day). Survival of mice in each treatment group (n = 5) in which euthanasia was necessary due to tumor reaching 1.0 cm diameter by day 45 is shown. At day 45, surviving mice bearing B16tkREC #1, #2, or #3 tumors, and treated with PLX-4720, had tumors of longest diameter 0, 0, 0.2 cm (REC #1); 0, 0.2, 0.4, 0.6, and 0.6 cm (REC #2); and 0, 0, 0, 0, and 0 cm (REC #3) respectively. (c) C57BL/6 mice seeded with B16tk tumors 5 days previously were treated with PBS, PLX-420 at 30 mg/kg/day or with 100 µl GCV i.p. at 50 mg/kg for 5 consecutive days, followed by 2 days rest, followed by five further consecutive injections of PBS, PLX-4720, or GCV (d6-10 and 13–17) (first cycle). Mice were then injected daily i.p. with PBS or with PLX-4720 on days 20–24 (second cycle). Finally, mice were injected daily i.p. with GCV or PLX-4720 on days 27–31 and 34–38 (third cycle). (d) Survival of mice (n = 7/8 per group) treated sequentially with first, second, and third cycles of treatment as shown and as described in c. (e) 104 cultured B16 cells (not transduced with the HSVtk gene) or cultured B16tk cells were grown for 96 hours in GCV (5 µg/ml) in triplicate cultures. In addition, the same number of cells from an explanted B16tk primary tumor treated in vivo with PBS; or 3 B16tk REC tumors, REC A, REC B, and REC C, explanted from mice which had failed second line GCV therapy (following GCV-induced initial regressions and MRD) in c, were similarly treated in vitro with GCV. The number of surviving cells is shown. (f) Western blot for the expression of the HSVtk protein in cultured B16 or cultured B16tk cells or in explanted B16tk primary tumor cells from a mouse treated in vivo with PBS; or from the three B16tk REC tumors REC A, B, and C, explanted from mice which had failed second line GCV therapy (following GCV-induced initial regressions and MRD) in c. (g–j) Splenocyte/LN cultures from (g) three different mice treated with PBS (first cycle) and PBS (second cycle) (no survivors for third cycle); (h) three mice treated with PLX-4720/PLX-4720 (first and second cycles, no survivors for third cycle); (i) four mice treated with GCV/PBS/GCV (first, second, and third cycles); or (j) from six mice treated with GCV/PBS/PLX-4720 (first, second, and third cycles); were screened for IFN-γ secretion following re-stimulation with freeze thaw cell lysates of parental B16 cells; B16tk REC#3 (BRAFV600E) cells; TC2 cells; or with peptide epitopes from wild-type BRAF, mutated BRAFV600E, murine TRP-2, Ovalbumin SIINFEKL, hgp-100 or murine TOPO-IIα as described in Materials and Methods.
In vitro sensitivity to PLX-4720 translated into in vivo control of B16tk tumor recurrence following frontline GCV chemotherapy (Figure 6c,d). Thus, treatment of primary B16tk tumors with PLX-4720 was no more effective than PBS, and significantly less effective than GCV (Figure 6d). However, once B16tk recurrences started to grow again in vivo, although they were insensitive to further GCV, they could be completely eradicated by treatment with PLX-4720 (Figure 6d). Three separate B16tkREC tumors (REC A, B, and C), which grew as recurrences following GCV-induced regression and retreatment with GCV in vivo upon recurrence, were explanted. Consistent with their regrowth in vivo despite multiple rounds of GCV therapy, all three B16tkREC tumors were insensitive to GCV in vitro, compared to the parental B16tk cell line from which the initial tumors had been seeded (Figure 6e). All three of these recurrences had lost detectable expression of the HSVtk protein by western blot (Figure 6f). These data are consistent with the acquisition of resistance to GCV therapy through mechanisms which silence the expression of the protein responsible for efficacy of the frontline therapy (GCV). We are currently investigating whether this occurred through the selection in vivo of a subclone of B16tk, which never expressed the HSVtk protein, or by mechanisms in which initially HSVtk-expressing tumor cells lost expression through the action of potent selective pressures in vivo.
Splenocytes from mice in which primary B16tk tumors grew progressively with either PBS (Figure 6g) or PLX-4720 (Figure 6h) treatments had no T cell recall responses to B16tk, B16tk REC, or any of the specific antigens tested. Frontline therapy with GCV, which caused initial tumor regressions, generated weak, but significant, Th1 T cell memory responses against both B16tk parental and B16tk REC cell freeze thaw lysates, as well as against the melanoma associated antigens TRP-2 and, very weakly, gp-100, but not against the BRAF-derived peptides or the negative control SIINFEKL peptide from the OVA protein (Figure 6i). In contrast, all the mice tested, which were cured of both primary and recurrent tumors (Figure 6d), developed potent Th1 T cell responses against both primary B16tk and recurrent B16tk REC cells and against melanoma-associated antigens (TRP-2 and gp-100; Figure 6j). Most significantly, these cured mice also developed T cell memory recall responses against BRAF peptides, although there was no significant difference between the T cell response to wild-type or V600E-mutated epitopes (Figure 6j). In addition, these mice cured of recurrence by PLX-4720, also developed T cell responses to a peptide epitope from TOPO-IIα, which was identified from the immunogenic screen of Figure 1, and the therapy experiments of Figure 5, as a potential antigen of B16tk recurrence (Figure 6j). Interestingly, and in contrast to the immunity generated by treatment with VSV-BRAF in Figure 5, no Th17 T cell responses could be detected in any of these recurrence-cured mice against any of the targets tested.
Taken together, these data show that the acquisition of a mutated BRAF as an effector of B16tk melanoma recurrence can be targeted by either immunotherapeutic strategies (Figure 5) or by chemotherapy (Figure 6). Moreover, they also suggest that the PLX-4720-mediated killing of BRAF-mutant recurrences is itself potently immunogenic and that, therefore, the therapeutic effects of chemotherapy against melanoma recurrence may be mediated in part by T cell responses against the emerging relapsing tumor.
Discussion
Tumor recurrence following primary therapy in malignant melanoma represents a major clinical challenge and is usually incurable. Here, we used our previously described model of tumor dormancy where frontline therapy leads to a macroscopic resolution of tumor bulk.1,2 The initial response is followed by a period of dormancy/MRD visualized microscopically, recurring with a more aggressive phenotype.1 We show here that B16tk s.c. tumors treated with frontline GCV, which would otherwise have recurred aggressively, were successfully treated with systemic injections of the VSV-cDNA GEEL (Figure 1b). Significantly, the GEEL did not treat established primary B16tk tumors (Figure 1c). Similarly, although the VSV-cDNA ASMEL treated primary B16tk tumors, it was ineffective against B16tk REC tumors.7 These data show that the antigenic repertoire which is relevant for protection against recurrent melanomas is significantly different from that protecting against primary tumors, consistent with our observations in the prostate model.1 Also, consistent with our previous findings in the prostate model, immunological screening of the GEEL (Figure 1e) identified VSV-expressed TOPO-IIα as a major component of the immunogenic repertoire of recurrent melanomas (Figure 1d,e). In addition, however, the current screen also identified VSV-expressed BRAF as an essential contributor, in combination with VSV-TOPO IIα and VSV-YB-1, to the Th17 recall response induced by the GEEL which led to rejection of B16tk REC tumors (Figure 1d).
Sequencing of one of the VSV-BRAF clones isolated from the screen of Figure 1e showed that it contained a BRAF cDNA with the G596R mutation (Figure 2a). Further analysis of two B16tk REC recurrent tumors showed that they also contained mutated BRAF cDNA (G596R and L597V) (Figure 2a). In contrast, B16tk parental cells did not have detectable mutations between positions 595 and 605 (BRAFWT). To investigate whether this acquired BRAF mutant status was specific to frontline therapy with GCV, B16 primary tumors were treated with several other chemo-, viro-, or immunotherapies (Figure 2b). Fourteen out of 16 B16 REC tumors contained a predominant species of mutated BRAF (at positions G596R, L597V, or V600E), irrespective of the frontline therapy (Figure 2b). No BRAF mutations were observed in recurrent tumors derived from TC2 prostate model (Figure 2c). Interestingly, although recurrent tumors which carried either the G596R or L597V mutations were markedly overexpressed in recurrences compared to primary B16 tumors (Figure 3), BRAF with the V600E mutation was expressed at wild-type levels. Kinase assays on these BRAF mutant lines are underway to test how the nature of the BRAF mutation functionally affects the ability of B16 REC to recur in vivo. In this respect, although patients are selected for BRAF kinase inhibitors on the basis of the presence of a V600E/K mutation, recent studies demonstrated that the L597R mutation also confers sensitivity to MEK inhibition.10
Although the parental B16tk tumor cells were consistently wild-type for BRAF (Figure 2a,b), it was possible that the emergence of B16tk REC recurrences mutant in BRAF resulted from the in vivo selection for a very low frequency population of BRAF mutant cells in the original B16tk population. In vitro coculture experiments suggested that this was not the case (Figure 4). Thus, even very low proportions of the BRAF mutant recurrent cells readily outcompeted the BRAF wild-type parental B16tk cultures in 7d cultures, although the extent of this competition was dependent on the nature of the BRAF mutation. It remains a formal possibility that BRAF mutant cells are present in our parental B16 stocks at levels below the threshold of detection by our sequencing protocols and become heavily selected for during the evolution of recurrent tumors.
Irrespective of the source of the BRAF mutation in B16 REC cells (probably acquired de novo rather than preexisting in the population), these data show that BRAF mutation represents a major, melanoma-specific, effector of tumor recurrence in vivo. Although BRAFV600E mutations account for 50% of primary human tumors, we observed a higher frequency of BRAF mutations that were not BRAFV600E in this group of tumor recurrences. We are currently investigating exactly what phenotypic properties are conferred upon B16 REC recurrences as a result of the expression of mutant G596R or L597V BRAF or by expression of the V600E mutant BRAF. In addition to the strong growth enhancing phenotype conferred by the mutant BRAF status observed in Figure 4, we are particularly investigating how mutant BRAF also affects the potency of the proangiogenic and/or anti-immunogenic signals that are critical to mediate recurrence in this in vivo model system.2 These studies will be important in understanding how melanomas which have been shaped by frontline therapy can recur in the face of selective immune pressure.
We also sought to exploit these findings by targeting the apparent dependence of B16 recurrences, irrespective of the frontline therapy, on emergence of mutant BRAF tumors. VSV-expressed BRAF was identified as a major component of the immunogenicity of the GEEL against B16tk REC tumors (Figure 1). Consistent with this, systemic VSV-BRAF, in combination with VSV-TOPO IIα and VSV-YB-1, cured mice of recurrences (Figure 5a,b). Mice cured of recurrence by this VSV-cDNA combination immunotherapy exhibited potent Th17 recall responses to recurrent B16 REC cell lysates but not to primary B16 tumors or cultured B16 cells (Figure 5c,d,e). An absence of this Th17 recall response correlated with the efficacy of the treatment, as nonsurvivors did not mount this recurrence-specific Th17 recall response (Figure 5e). These data confirmed, therefore, that the immunogenic targets of recurrent melanomas are significantly different to those of primary tumors and that effective identification of such antigens can be used to treat recurrence.
In addition to targeting the BRAF status of recurrences with immunotherapy, we hypothesized that B16 Rec tumors would be vulnerable to chemotherapy with BRAF kinase inhibitors. Cell viability was significantly reduced (almost 3 log) in vitro in B16tk REC tumors expressing the V600E mutation treated with the BRAF inhibitor PLX-4720 (Figure 6a). Significant, but smaller, reductions in cell numbers were achieved against the B16tk REC line carrying the G596R mutation (Figure 6a). However, PLX-4720 did not consistently reduce cell proliferation in the L597V mutated cells. Consistent with these data, preclinical and clinical studies support the use of vemurafenib if the BRAF mutation is confirmed as V600E or V600K.11,12 However, although primary B16tk tumors were completely unaffected by treatment with PLX-4720 (Figure 6d), once B16tk recurrences started to recur in vivo, although they were insensitive to further GCV, they were completely eradicated by treatment with PLX-4720 (Figure 6d). This result was unexpected since our analysis of B16tk REC suggested that only a proportion evolved the V600E mutation (Figure 2b) which would be optimally sensitive to the BRAF inhibitor (Figure 6c). However, immunological analysis of the mice cured of recurrences by PLX-4720 indicated that treatment with frontline GCV, followed by BRAF inhibitor, induced very potent T cell responses against both primary B16 cells and against recurrent B16tk REC cells, as well as against known melanoma-associated antigens (TRP-2, gp100) and the recurrence-associated TOPO-IIα antigen (Figure 6j). Significantly, these mice also developed T cell responses against BRAF itself, although independent of the mutational status of the BRAF (Figure 6j; perhaps consistent with the existence of a predicted Kd/Kb epitope of BRAF, SGSHQFEQL, from positions 605–613, situated just downstream of the V600E mutation). GCV treatment alone induced less potent antimelanoma T cell reactivities (as reported previously),13 but not T cell responses to BRAF, or the other recurrence-associated antigen TOPO-IIα (Figure 6i). Therefore, we believe that the efficiency with which PLX-4720 was able to treat all B16tk REC recurrences, even though only a proportion were likely to have harboured a V600E mutation, is due to the strong immune stimulatory nature of the in vivo death of B16tk REC tumors when treated with PLX-4720. Figure 6d shows that all recurrences following frontline GCV therapy were sensitive to PLX-4720 treatment, even though individual recurrences, with different BRAF mutations, showed variable sensitivities both in vitro (Figure 6a) and in vivo (Figure 6b). Our hypothesis is that the initial HSVtk/GCV-mediated killing of the primary tumor cells primed an antitumor T cell response, consistent with studies showing that HSVtk-mediated GCV tumor killing is immunogenic,13 and with the data Figure 6i which shows that GCV treatment of B16tk tumors primes (weak) anti-B16 T cell responses. Thereafter, despite variable sensitivities of different BRAF mutant recurrences to PLX-4720, there is, nonetheless, sufficient killing of these recurrent tumors (Figures 6a,b) to boost those antitumor T cell responses which were initially primed by HSVtk/GCV-mediated killing of the primary tumors. In addition to these proposed antitumor immunizing effects, it may also be that PLX-4720 treatment has direct effects on the tumor microenvironment to facilitate T cell trafficking and effector activity14,15 as has been reported for other agents which disrupt the tumor endothelial cell barrier.16 In addition, pharmacologic inhibition of the BRAF protein has pleiotropic immune-stimulating effects17,18 which counteract its immunosuppressive activity.19 In vivo treatment of the B16tk REC tumors with PLX-4720 may be operating through some of these pathways.
Our data here are significant in several respects. First, we have shown that VSV-cDNA libraries are an efficient way to identify antigens associated with tumor recurrence and that VSV-expressed antigens can, combinatorially, be highly effective at treating recurrent tumors. Second, our results show that recurrent melanomas adopt an antigenic profile which is immunologically separate from that of the primary tumor. Hence, immunotherapies, such as vaccines, targeting antigens identified from analysis of primary tumors may be ineffective against recurrences which have evolved away from the phenotype initially treated by frontline therapy. Moreover, some of the immunogens of recurrence (such as TOPO-IIα) may be shared between tumor types,1 while others (such as BRAF) will be specific to the histological type of tumor. Third, our in vivo data of Figure 6 suggest that even if BRAF mutant melanomas are not optimally sensitive to BRAF inhibitors in vitro, in vivo cytotoxicity of the drugs may still be effective at raising powerful adjuvant T cell responses against tumors. Therefore, combination of BRAF inhibitors with additional cytotoxic agents, such as MEK inhibitors in BRAFV600E tumors, and/or immune stimulators, such as checkpoint inhibitors, may further add to treatment efficacy.20,21 Finally, we demonstrate that acquisition of a mutated BRAF is a major driver of melanoma recurrence, at least in the B16 model system. It remains to be seen whether similar transitions from wild-type BRAF to mutated BRAF are confirmed from our clinical samples of melanoma recurrence and/or metastases, following, for example, ipilumumab therapy. To our knowledge, no such clinical cases have been reported to date, although patients currently are not routinely offered a repeat tumor biopsy following disease recurrence. Therefore, to validate our findings in the C57Bl/6/B16 model, clinical studies are underway to assess the BRAF status of patient recurrences following frontline immunotherapy which induced tumor regression followed by recurrence.
In the meantime, these preclinical data suggest a clear clinical rationale for offering patients with initially wild-type BRAF melanomas an additional biopsy to screen for mutant BRAF upon recurrence, opening the door for treatment of some melanoma recurrences with BRAF inhibitors.
Materials and Methods
Cells and viruses. B16tk cells are B16 murine melanoma cells (H-2kb) stably transfected with the HSVtk gene which confers sensitivity to the prodrug GCV at 5 µg/ml.13 The B16ova cell line was derived from a B16.F1 clone transfected with a pcDNA3.1ova plasmid.8 B16ova cells were grown in DMEM (HyClone, Logan, UT) + 10% FBS (Life Technologies, Carlsbad, CA) + 5 mg/ml G418 (Mediatech, Manassas, VA) until challenge. TRAMP-C2 (TC2) cells are derived from a prostate tumor that arose in a TRAMP mouse (H-2kb) and were characterized by Dr Esteban Celis. TC2 cells grow in an androgen-independent manner and are routinely grown as tumors in C57BL/6 male mice.22 The OT-I mouse strain is on a C57BL/6 background (H2-Kb) and expresses a transgenic T cell receptor Vα2/Vβ5 specific for the SIINFEKL peptide of ovalbumin in the context of MHC class I, H-2Kb, as previously described and were bred at the Mayo Clinic.23
The GEEL VSV-cDNA library was constructed as described,24 containing cDNA from three pooled B16tk recurrent tumors explanted early (~0.2–0.3 cm) at recurrence following GCV therapy. VSV-cDNA libraries were generated from BHK cells by cotransfection of pVSV-XN2-cDNA library DNA along with plasmids encoding viral genes. Virus was expanded by a single round of infection of BHK cells and purified by sucrose gradient centrifugation. VSV-GFP was generated by cloning GFP cDNA into the plasmid pVSV-XN2.25 Titers were measured by standard plaque assays on BHK-21 cells.
Animal studies. All procedures were approved by the Mayo Foundation Institutional Animal Care and Use Committee. C57BL/6 mice were purchased from Jackson Laboratories at 6–8 weeks of age. To establish s.c. tumors, 5 × 105 B16tk cells in 100 µl of PBS were injected into the flank of mice. Intravenous injections of virus were administered in 100 µl volumes. Freshly explanted tumors were kept in culture for a single passage from explant before in vitro experiments. For survival studies, tumor diameter in two dimensions was measured three times weekly using calipers, and mice were killed when tumor size was ~1.0 × 1.0 cm in two perpendicular directions.
For apparently curative adoptive therapy experiments, 1 × 107 in vitro-activated OT-I T cells were injected i.v. in 100 μl PBS on days 5 and 7 following tumor seeding. This regimen typically led to tumor regression and tumor-free “cure” of ~50% of mice for over 60 days.8,9 For suboptimal adoptive T cell therapy, in which more than 50% of treated mice would undergo complete macroscopic regression followed by local recurrence, mice seeded s.c. with B16ova tumors 5 days previously were treated with PBS or i.v. with 106 4-day activated OT-I T cells on days 6 and 7 (refs. 8,9).
For GCV chemotherapy experiments, C57BL/6 mice seeded with B16tk tumors 5 days previously were treated with GCV i.p. at 50 mg/kg on days 6–10 and then 13–17. With this regimen, 100% of tumors regressed macroscopically followed by ~50–80% of the mice undergoing later local recurrence (Figure 1a).
For VSV-cDNA library-based immunotherapy experiments, C57BL/6 mice seeded with either B16 melanoma or TC2 prostate tumors 5 days previously were treated with three i.v. injections on days 6, 8, and 10 of the ASMEL or the ASEL, VSV-cDNA libraries previously shown to cure B16 s.c. tumors7 or TC2 tumors, respectively, using 9 i.v. injections.24
For suboptimal systemic oncolytic reovirus virotherapy experiments, C57BL/6 mice with 5-day established B16tk tumors were treated i.p. with PBS or paclitaxel at 10 mg/kg/injection (Mayo Clinic Pharmacy, Rochester, MN) for 3 days followed by i.v. reovirus (2 × 107 TCID50) or PBS for 2 days. This cycle (PAC/Reo, five injections, 2-day rest), modified from a more effective therapy described, was repeated once.26
Cell viability assay. For trypan blue staining, 0.1 ml of cells were incubated for 4 minutes at room temperature with an equal volume of 0.4% (w/v) trypan blue solution. Cells were counted using a hemocytometer and a light microscope. Viable cells were those that excluded the stain.
Splenocyte/LN activation assays. Spleens from either control C57BL/6 mice or C57BL/6 mice previously treated with GCV and GEEL or PLX-4720 were immediately excised from euthanized mice and dissociated in vitro to achieve single-cell suspensions. Red blood cells were lysed with ACK lysis buffer for 2 minutes. Cells were resuspended at 1 × 106 cells/ml in Iscove's Modified Dulbecco's Medium (Gibco, Grand Island, NY) + 5% FBS + 1% Pen-Strep + 40 μmol/l 2-ME. Depending on the experiment, cells were restimulated in vitro by infection with VSV viruses (multiplicity of infection ~10) or at a lower multiplicity of infection (1.0) with added recombinant hsp70 (10 µg/ml); or with freeze thaw lysates from tumor cells (equivalent of 107 cells per stimulation); or with (2.5 μg/ml) the synthetic peptides hgp10025-33: KVPRNQDWL; ova257-264: SIINFEKL; TRP-2180–188: SVYDFFVWL; wild-type BRAF595-613: FGLATVKSRWSGSHQFEQL; V600E mutant BRAF595-613: FGLATEKSRWSGSHQFEQL; TOPO-IIα: NSMVLFDHV; (synthesized at the Mayo Foundation Core Facility, Rochester, MN); or with medium for 48 hours. Restimulations were performed in triplicate, every 24 hours for 3 days. Forty-eight hours later, cell-free supernatants were collected and tested by ELISA for IL-17 (R&D Systems, Minneapolis, MN) or IFN-γ (BD Biosciences, San Jose, CA).
Quantitative rtPCR. RNA was prepared with the QIAGEN-RNeasy-MiniKit. One microgram total RNA was reverse transcribed in a 20 µl volume using oligo-(dT) primers. A cDNA equivalent of 1 ng RNA was amplified by PCR with gene-specific primers using GAPDH as loading control. Expression of the murine BRAF gene was detected using the forward 5′-ACTCGACATGTGAATATCCT-3′ and reverse 5′-AGGTATCCTCGTCCCACCAT-3′ primers.
qrtPCR was carried out using a LightCycler480 SYBRGreenI Master kit and a LightCycler480 instrument (Roche, Indianapolis, IN) according to the manufacturer's instructions. Typically, RNA was prepared from equal numbers of cells from each sample (usually 5,000 cells) and reverse transcribed as described above. PCR (primers at 0.5 µmo/l, annealing = 58 °C) was run with diluted cDNA samples (neat, 1:10, 1:100, 1:1,000). GAPDH amplification was used as a control for equal loading of target cDNAs. The threshold cycle (Ct) at which amplification of the target sequence was detected was used to compare the relative levels of mRNA between samples. Relative quantities of BRAF mRNA were normalized with Ct of GAPDH amplification.
Western blot analysis. Tumors were excised from euthanized mice and homogenized in Lammli's lysis buffer (distilled H20 containing 125 mmol/l Tris, 2% SDS, 10% glycerol, pH 6.8). Proteins (50 mg total) were separated by SDS-PAGE (12% Mini-PROTEAN TGX Gels; Bio-Rad, Hercules, CA) and transferred to 0.2 mm nitrocellulose membranes (Bio-Rad). Membranes were incubated with a polyclonal rabbit anti-HSVtk primary antibody (purchased from Yale University) overnight at 4 °C. Washed membranes were then incubated with horseradish peroxidase–conjugated antirabbit (1:5,000) IgG (Abcam, Cambridge, UK) for 1 hour. Finally, the membranes were coated with Pierce ECL Western Blotting Substrate (Pierce, Rockford, IL) and exposed.
Statistics. Survival data from the animal studies were analyzed using the log-rank test, and the Mann–Whitney U-test was applied for in vitro assays. Statistical significance was determined at the level of P < 0.05. In vivo data were analyzed using GraphPad Prism 4 Software (GraphPad Software, La Jolla, CA).
Acknowledgments
This work was funded by The Richard M. Schulze Family Foundation, the Mayo Foundation, Cancer Research UK, the European Research Council, the National Institute of Health (R01CA107082, R01CA130878, R01CA132734, and R01CA175386-01A1), and a grant from Terry and Judith Paul. We thank Toni Higgins for secretarial assistance. No potential conflicts of interest were disclosed.
References
- Boisgerault N, Kottke T, Pulido J, Thompson J, Diaz RM, Rommelfanger-Konkol D.et al. (2013Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse Mol Ther 211507–1516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Boisgerault N, Diaz RM, Donnelly O, Rommelfanger-Konkol D, Pulido J.et al. (2013Detecting and targeting tumor relapse by its resistance to innate effectors at early recurrence Nat Med 191625–1631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin. 2012;62:10–29. doi: 10.3322/caac.20138. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J.et al.; BRIM-3 Study Group 2011Improved survival with vemurafenib in melanoma with BRAF V600E mutation N Engl J Med 3642507–2516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FDA approved drug products DABRAFENIB 2013 < http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=202806&DrugName=TAFINLAR&ActiveIngred=DABRAFENIB%20MESYLATE&SponsorApplicant=GLAXOSMITHKLINE&ProductMktStatus=1&goto=Search.DrugDetails >
- FDA approved drug products TRAMETINIB 2013 < http://www.accessdata.fda.gov/scripts/cder/drugsatfda/index.cfm?fuseaction=Search.Set_Current_Drug&ApplNo=204114&DrugName=MEKINIST&ActiveIngred=TRAMETINIB%20DIMETHYL%20SULFOXIDE&SponsorApplicant=GLAXOSMITHKLINE%20LLC&ProductMktStatus=1&goto=Search.DrugDetails >.
- Pulido J, Kottke T, Thompson J, Galivo F, Wongthida P, Diaz RM.et al. (2012Using virally expressed melanoma cDNA libraries to identify tumor-associated antigens that cure melanoma Nat Biotechnol 30337–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza KM, Thompson JM, Kottke TJ, Flynn Gilmer HC, Knutson DL, Vile RG. Adoptive T cell therapy promotes the emergence of genomically altered tumor escape variants. Int J Cancer. 2012;131:844–854. doi: 10.1002/ijc.26447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rommelfanger DM, Wongthida P, Diaz RM, Kaluza KM, Thompson JM, Kottke TJ.et al. (2012Systemic combination virotherapy for melanoma with tumor antigen-expressing vesicular stomatitis virus and adoptive T-cell transfer Cancer Res 724753–4764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dahlman KB, Xia J, Hutchinson K, Ng C, Hucks D, Jia P.et al. (2012BRAF(L597) mutations in melanoma are associated with sensitivity to MEK inhibitors Cancer Discov 2791–797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Puzanov I, Kim KB, Ribas A, McArthur GA, Sosman JA.et al. (2010Inhibition of mutated, activated BRAF in metastatic melanoma N Engl J Med 363809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Søndergaard JN, Nazarian R, Wang Q, Guo D, Hsueh T, Mok S.et al. (2010Differential sensitivity of melanoma cell lines with BRAFV600E mutation to the specific Raf inhibitor PLX4032 J Transl Med 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vile RG, Castleden S, Marshall J, Camplejohn R, Upton C, Chong H. Generation of an anti-tumour immune response in a non-immunogenic tumour: HSVtk killing in vivo stimulates a mononuclear cell infiltrate and a Th1-like profile of intratumoural cytokine expression. Int J Cancer. 1997;71:267–274. doi: 10.1002/(sici)1097-0215(19970410)71:2<267::aid-ijc23>3.0.co;2-d. [DOI] [PubMed] [Google Scholar]
- Liu C, Peng W, Xu C, Lou Y, Zhang M, Wargo JA.et al. (2013BRAF inhibition increases tumor infiltration by T cells and enhances the antitumor activity of adoptive immunotherapy in mice Clin Cancer Res 19393–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilmott JS, Long GV, Howle JR, Haydu LE, Sharma RN, Thompson JF.et al. (2012Selective BRAF inhibitors induce marked T-cell infiltration into human metastatic melanoma Clin Cancer Res 181386–1394. [DOI] [PubMed] [Google Scholar]
- Buckanovich RJ, Facciabene A, Kim S, Benencia F, Sasaroli D, Balint K.et al. (2008Endothelin B receptor mediates the endothelial barrier to T cell homing to tumors and disables immune therapy Nat Med 1428–36. [DOI] [PubMed] [Google Scholar]
- Hu-Lieskovan S, Robert L, Homet Moreno B, Ribas A. Combining targeted therapy with immunotherapy in BRAF-mutant melanoma: promise and challenges. J Clin Oncol. 2014;32:2248–2254. doi: 10.1200/JCO.2013.52.1377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ribas A, Wolchok JD. Combining cancer immunotherapy and targeted therapy. Curr Opin Immunol. 2013;25:291–296. doi: 10.1016/j.coi.2013.02.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sumimoto H, Imabayashi F, Iwata T, Kawakami Y. The BRAF-MAPK signaling pathway is essential for cancer-immune evasion in human melanoma cells. J Exp Med. 2006;203:1651–1656. doi: 10.1084/jem.20051848. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J.et al. (2012Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations N Engl J Med 3671694–1703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim KB, Kefford R, Pavlick AC, Infante JR, Ribas A, Sosman JA.et al. (2013Phase II study of the MEK1/MEK2 inhibitor Trametinib in patients with metastatic BRAF-mutant cutaneous melanoma previously treated with or without a BRAF inhibitor J Clin Oncol 31482–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Sanchez-Perez L, Diaz RM, Thompson J, Chong H, Harrington K.et al. (2007Induction of hsp70-mediated Th17 autoimmunity can be exploited as immunotherapy for metastatic prostate cancer Cancer Res 6711970–11979. [DOI] [PubMed] [Google Scholar]
- Hogquist KA, Jameson SC, Heath WR, Howard JL, Bevan MJ, Carbone FR. T cell receptor antagonist peptides induce positive selection. Cell. 1994;76:17–27. doi: 10.1016/0092-8674(94)90169-4. [DOI] [PubMed] [Google Scholar]
- Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P.et al. (2011Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors Nat Med 17854–859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez M, Porosnicu M, Markovic D, Barber GN. Genetically engineered vesicular stomatitis virus in gene therapy: application for treatment of malignant disease. J Virol. 2002;76:895–904. doi: 10.1128/JVI.76.2.895-904.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Chester J, Ilett E, Thompson J, Diaz R, Coffey M.et al. (2011Precise scheduling of chemotherapy primes VEGF-producing tumors for successful systemic oncolytic virotherapy Mol Ther 191802–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]