

Abstract
The promising clinical results obtained for ocular gene therapy in recent years have paved the way for gene supplementation to treat recessively inherited forms of retinal degeneration. The situation is more complex for dominant mutations, as the toxic mutant gene product must be removed. We used spliceosome-mediated RNA trans-splicing as a strategy for repairing the transcript of the rhodopsin gene, the gene most frequently mutated in autosomal dominant retinitis pigmentosa. We tested 17 different molecules targeting the pre-mRNA intron 1, by transient transfection of HEK-293T cells, with subsequent trans-splicing quantification at the transcript level. We found that the targeting of some parts of the intron promoted trans-splicing more efficiently than the targeting of other areas, and that trans-splicing rate could be increased by modifying the replacement sequence. We then developed cell lines stably expressing the rhodopsin gene, for the assessment of phenotypic criteria relevant to the pathogenesis of retinitis pigmentosa. Using this model, we showed that trans-splicing restored the correct localization of the protein to the plasma membrane. Finally, we tested our best candidate by AAV gene transfer in a mouse model of retinitis pigmentosa that expresses a mutant allele of the human rhodopsin gene, and demonstrated the feasibility of trans-splicing in vivo. This work paves the way for trans-splicing gene therapy to treat retinitis pigmentosa due to rhodopsin gene mutation and, more generally, for the treatment of genetic diseases with dominant transmission.
Introduction
Retinitis pigmentosa (RP), one of the most common visual impairments worldwide, is a group of inherited retinal degenerative diseases for which there is currently no effective treatment. It is characterized by a progressive degeneration of rod photoreceptors, leading to night-blindness and constriction of the visual field, followed by the degeneration of cone photoreceptors, resulting in a total loss of vision. The genetic causes of RP are heterogeneous, and autosomal dominant, autosomal recessive, X-linked and sporadic forms have been identified, involving mutations of more than 60 different genes expressed in either the rod photoreceptors or the retinal pigment epithelium (RetNet, https://sph.uth.edu/retnet/). The rhodopsin gene (RHO) alone accounts for 30–40% of the autosomal dominant forms of RP (adRP). This gene is the most frequently mutated in this disease, with at least 120 mutations identified to date (RetNet). Rhodopsin (RHO) is the only photopigment of rod photoreceptor cells, and is responsible for converting light stimuli into electrochemical signals. Rod photoreceptor cells are required for vision in dim lighting conditions and for peripheral vision, and are essential for the long-term maintenance of a functional retina.1,2 The speed of the degeneration of rod photoreceptor cells induced by RHO mutations is variable, depending on the aggressiveness of the mutation.3 Various studies for preventing or slowing disease progression, before or after the start of degeneration, are currently being investigated. Most involve gene replacement or neuroprotection through neurotrophic4,5,6 and antiapoptotic factors.7 The principal strategy involves the silencing of the mutant gene, by RNA interference, with or without supplementation through the provision of a silencing-insensitive RHO allele.8,9,10 However, this approach requires control over the level of RHO expression, and may be potentially toxic,11 with “off-target effects.” By contrast, neurotrophic factors have been shown to protect photoreceptors effectively in therapeutic trials,12,13,14 but may have adverse effects due to their pleiotropic mode of action,15 and may be toxic at higher doses.16 This approach enlarges the treatment window, by slowing the disease rather than stopping it.
Efforts to develop an optimal approach for preventing retinal degeneration due to RHO mutation must take the following into account: (i) most RHO mutations have gain-of-function or dominant negative effects, making it necessary not only to restore synthesis of the normal protein, but also to remove the mutant protein, (ii) RHO expression level must be kept close to the endogenous level, to prevent deleterious effects on the retina,17,18 and (iii) given the large number of point mutations (more than 120) identified to date in RHO, a generic therapeutic tool is required, to target a wide array of RHO mutations.
Spliceosome-mediated RNA trans-splicing (SMaRT) technology satisfies all three of these criteria.19 Trans-splicing is a natural splicing mechanism that was first discovered in trypanosomes20,21 but has since been found in rats22 and humans.23,24,25,26 Poorly represented compared to cis-splicing, trans-splicing occurs between two different pre-mRNAs and results in a final mRNA consisting of the 5′ part of the first pre-mRNA and the 3′ part of the second pre-mRNA. SMaRT technology makes use of this natural phenomenon to develop a powerful tool for RNA repair strategies based on the engineering of molecules able to trans-splice a specific endogenous pre-mRNA (target mRNA). The sequence located upstream (5′-replacement) or downstream (3′-replacement) from the trans-splicing site is thus replaced. Unlike ribozyme trans-splicing, SMaRT makes use of the endogenous cell spliceosome machinery and requires only the introduction of an exogenous RNA, the PTM or pre-mRNA trans-splicing molecule. This PTM consists of (i) a binding domain for specific targeting of the endogenous pre-mRNA, (ii) an artificial intron sequence containing all the elements required for splicing, and (iii) the cDNA replacement sequence. Approaches based on 3′-replacement trans-splicing have already been used, both in vitro and in vivo, to correct several genetic diseases, including cystic fibrosis,27,28,29 spinal muscular atrophy,30,31,32 hemophilia A,33 tauopathies,34,35 epidermolysis bullosa simplex,36 X-linked immunodeficiency with hyper-IgM,37 severe combined immune deficiency,38 myotonic dystrophy type 1,39 frontotemporal dementia with parkinsonism linked to chromosome 17,34 Duchenne muscular dystrophy,40 dysferlinopathy.41 Trans-splicing has also recently been considered as a possible basis of new strategies for treating cancer,42,43 producing therapeutic proteins44 and for molecular imaging.45
SMaRT technology has several advantages, including the preservation of endogenous target mRNA regulation in terms of time, space and amount of protein generated. Indeed, as the PTM lacks exon 1 and thus has no AUG initiation codon, it should be inert in cells in the absence of its target pre-mRNA. However, one recent study showed that translation could be initiated from secondary AUG codons.41 Moreover, the replacement of the mutated sequence makes it possible to decrease mutant protein synthesis and to promote synthesis of the normal protein in a single step. This is particularly important in cases of dominantly inherited disease. As the PTM includes only part of the gene sequence to be repaired, SMaRT technology has another key advantage in the smaller size of the restorative molecule than for other gene therapy systems. Finally, by targeting the first intron of rhodopsin pre-mRNA, thereby replacing exons 2 through 5, we should be able to repair about 70–80% of all known RHO mutations with a single PTM.
We describe here the use of SMaRT technology as a therapeutic tool for correcting RHO mutations, and we provide the first evidence of a level of efficacy, which opens the door to its possible use for the treatment of a genetic disease with dominant inheritance. We used an in vitro cellular assay to screen 14 PTMs differing only by the target of their binding domains on rhodopsin pre-mRNA intron 1. We quantified the trans-splicing rate at the RNA level; the highest rate recorded was 25%. We increased efficiency to 41% by adding additional intron sequence to the partial replacement cDNA of the PTM. Furthermore, after developing cell lines derived from HEK293T cells with stable mutant RHO gene expression, we demonstrated that the trans-splicing repair of rhodopsin mRNA led to synthesis of the normal protein, which was correctly targeted to the plasma membrane, as observed by flow cytometry imaging. Finally, we brought the evidence that trans-splicing occurred in vivo, in a humanized mouse model of RP due to a RHO mutation.
Results
PTM engineering and screening: influence of binding domain and replacement sequence
The PTM sequence encodes (i) a binding domain, complementary to 150 nucleotides of the rhodopsin pre-mRNA first intron, (ii) an intron sequence containing all the elements required for spliceosome recruitment: a branch point, a polypyrimidine tract and a 3′ acceptor splice site, and (iii) replacement exons 2 to 5 of the rhodopsin cDNA, devoid of mutation (Figure 1b). The key challenge in the use of this technology is getting the PTM to bind to the rhodopsin pre-mRNA, thereby promoting splicing in trans rather than cis, leading to the repair of any mutations present in exons 2 to 5 (Figure 1a).
Figure 1.

Pre-trans-splicing molecule construct and trans-splicing mechanism. (a) After transcription of the rhodopsin gene, the PTM binding domain recognizes a sequence in the first intron of the rhodopsin pre-mRNA, promoting splicing in trans rather than in cis, and leading to the production of a chimeric mRNA with no mutation in exons 2 to 5. *Potential mutation. (b) The PTM expression plasmid, in which the coding sequence of the PTM is under the control of a cytomegalovirus promoter (CMV prom), and consists, from 5′ to 3′, of a binding domain (BD), an intron sequence (IS), the rhodopsin replacement cDNA (ex2 to 5) and the untranslated terminal region (UTR) of the bovine growth hormone gene containing the polyadenylation signal.
Various strategies can be used to tip the balance in favor of trans-splicing. For example, previous studies have shown that the length and position of the binding domain are crucial for the engineering of efficient PTMs.37,38,39,46 We began by designing and testing 14 different PTMs targeting RHO intron 1, differing only in their binding domains; all these binding domains were 150 bp long, but they bound to different sites within the intron (Supplementary Figure S1). We evaluated trans-splicing efficiency, by cotransfecting HEK-293T (293T) cells with the PTM-expressing constructs and a plasmid carrying the whole rhodopsin gene. We then used primers binding to exons 1 and 5 of RHO, to amplify both cis- and trans-spliced RHO mRNA by PCR (Supplementary Figure S2). The use of a primer binding to exon 1, which is not present in the PTM, ruled out amplification of the PTM that had not reacted with the endogenous rhodopsin pre-mRNA (Figure 2a). We also eliminated other potential artifacts in trans-splicing quantification by checking that amplification of the PCR product was not due to trans-connection between RNAs or cDNAs during RT or PCR steps (Supplementary Figure S3). We then quantified the trans-splicing rate, by using a specific restriction cutting to distinguish between the cis- and trans-spliced transcripts on the basis of a silent mutation introduced by site-directed mutagenesis into the replacement cDNA of the PTM construct (Supplementary Figure S2). The fragments were visualized by gel electrophoresis (Figure 2b) and the trans-splicing rate was precisely determined by quantifying the peak areas on electrophoregrams obtained on a Bioanalyzer (Figure 2c). This method was used for the quantification of trans-splicing efficiency at the RNA level throughout this study.
Figure 2.

Method for the precise quantification of trans-splicing rate by end-point PCR. (a) PCR products obtained from cis- and trans-spliced rhodopsin mRNAs, subjected to electrophoresis in a 2% agarose gel. Expected size: 771 bp. (b) Electrophoresis, in a 2% agarose gel, of the restriction digestions of the PCR products with NheI and BglI. Expected fragment sizes: 165 bp + 606 bp. (c) Schematic diagram of trans-spliced and cis-spliced mature mRNA, with the localization of BglI and NheI restriction sites. Electrophoregrams of digestion products on capillary electrophoresis (corresponding to lanes 4 and 5 of b). Each peak represents a DNA product, the amount of which can be accurately determined by calculating peak area. The two most extreme peaks of each electrophoregram are internal controls and are not included in the calculation of peak area.
The first three PTMs (1-2-3) that we tested targeted the 5′-, central, and 3′-parts of intron 1, respectively (Figure 3a and Supplementary Figure S1). Our results demonstrate the critical importance of the binding sequence for trans-splicing events, with efficiencies of between almost zero (PTM0: negative control with no binding domain, PTM2, PTM3) to 25% (PTM1, P < 0.0001, with respect to PTM0 in Dunn's multiple comparisons test; Figure 3b) recorded. We then tried to increase trans-splicing efficiency further, by engineering new PTMs targeting specific sequences of the rhodopsin pre-mRNA, surrounding the PTM1 target site (PTM4-5-6-7-8-9-10, Figure 3a and Supplementary Figure S1) or directed against the 3′ acceptor splice site of RHO exon 2 (PTM13 and 14, Figure 3a and Supplementary Figure S1). Trans-splicing quantification showed that PTM1 was still the most efficient PTM (Figure 3b). However, PTM7 and 8, which had binding domain sequences overlapping that of PTM1, also displayed significant levels of trans-splicing (P < 0.05 and 0.01, respectively, in Dunn's multiple comparisons tests against PTM0; Figure 3b). These findings suggest that there is at least one region more favorable for trans-splicing in intron 1 of RHO.
Figure 3.
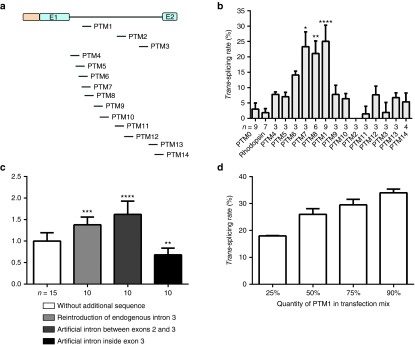
Effect of the binding domain and intron sequence on trans-splicing efficiency. (a) Schematic diagram of the sites of binding domains in intron 1. Each binding domain is represented by a dash, indicating its location, following a vertical line above the intron diagram. (b) Rate of trans-splicing of PTM0 to 14. PTM0 with no binding domain served as a negative control. Each experiment was performed with a minimum of three culture wells and was replicated several times. The number of replicates is indicated under the x-axis for each PTM. Statistical analysis was performed by comparing PTM0 with all other groups in Dunn's test. (c) Effect of introducing an intron into the replacement cDNA sequence. Trans-splicing rates were calculated after modifying PTM1 and 8 by adding various intron sequences (see Supplementary Figure S5). The efficiency of each intron addition was normalized against the trans-splicing rate obtained for PTM1 and 8 before the addition of the intron sequence. Statistical analysis was performed by comparing PTM1 and 8 with all other groups in Dunnett's test. (d) Variations of PTM1 trans-splicing rate as a function of the proportion of PTM1 in the transfection mix. The cells were transfected with the same amount of total DNA. The proportion of the total DNA accounted for by PTM-encoding plasmids is indicated under the x-axis. In all these experiments, trans-splicing rates were determined at the RNA level, after the transient cotransfection of HEK-293T cells with the PTM and RHO constructs. All values are presented as medians ± SD.
We then checked that both cis-splicing and trans-splicing reactions led to the correct mRNA sequence, by reverse transcribing the mRNA and sequencing the PCR product. We demonstrated that the sequence at the trans-splicing site, i.e., at the junction between exons 1 and 2, was correct. The electrophoregram of the Sanger sequencing reaction also demonstrated the production of a mixture of cis- and trans-spliced transcripts based on the site at which the silent mutation was inserted (Supplementary Figure S4).
Another group reported that modifications of the PTM cDNA replacement sequence could have a considerable effect on efficiency of trans-splicing.47 We thus tried to improve the trans-splicing rates obtained with the two most efficient molecules (PTM1 and 8), by adding natural or artificial intron sequences to this part of the molecule. We reintroduced RHO intron 3 in its original position, to create PTM19 and 22, respectively, or added an artificial intron sequence between RHO exons 2 and 3 (PTM20 and PTM23) or within exon 3 (PTM21 and PTM24) (Supplementary Figure S5a). Trans-splicing quantification showed that the efficiency of PTM1 and 8 could be increased or decreased, depending on the sequence and location of the added intron (endogenous intron 3, significant increase P < 0.001; artificial intron between exons 2 and 3, significant increase P < 0.0001; artificial intron within exon 3, significant decrease P < 0.01, in Dunnett's multiple comparisons with PTM1 and 8 without additional sequence; Figure 3c and Supplementary Figure S5b). Thus, trans-splicing efficiency is also determined by the cDNA replacement sequence of the PTM. The most efficient PTM obtained to date was PTM20, which has an artificial intron between RHO exons 2 and 3, with a trans-splicing efficiency greater than 40%, as assessed by the transient cotransfection of 293T cells with two plasmids carrying the RHO gene and the PTM. Finally, we investigated the correlation between trans-splicing efficiency and PTM expression level, by transfecting 293T cells with different ratios of the RHO-encoding and PTM1-encoding plasmids. As expected, this experiment demonstrated that trans-splicing rate was directly linked to PTM expression level (Figure 3d).
Development of cell lines stably expressing the rhodopsin gene: trans-splicing in physiological conditions and quantification at the protein level
The experiments described above were carried out in conditions of transient cotransfection, to facilitate and accelerate the first PTM screen. However, this methodology might not be entirely representative of the situation in vivo. For example, cotransfection may bring the PTM RNA and RHO RNA into closer proximity, thereby promoting trans-splicing events. We therefore created cell lines derived from 293T cells and stably expressing wild-type (WT) or mutant alleles of RHO, using lentiviral vectors encoding the full-length gene. This small gene (5 kb including the 5′UTR) is of a size compatible with the packaging capacity of lentiviral vectors, which integrate into the genome of the host cells, resulting in the stable expression of the transgene.
The WT RHO protein is located in the plasma membrane in HEK-293S cells and COS-7 cells, whereas the mutant RHO protein may be retained within the cell in various types of aggresomes, depending on the mutation.48,49,50
We generated four different cell lines producing the WT, P23H, R135W, or P347L mutated RHO proteins. The corresponding alleles were selected as follows. The P23H mutation is the most widely studied and most representative mutation in North American patients,51,52,53 and is known to cause the retention of the protein in the cell. However, due to its location in exon 1, it cannot be repaired by our PTM. We therefore used this mutation as a control mutation resulting in a strong phenotype. The P347L mutation is the second most prevalent mutation worldwide,51,52 and the leading mutation in European patients.54 However, this mutation does not result in the mislocalization of RHO in 293T cells. The third mutation studied, R135W, is also a frequent cause of RP in European patients.54 It is located in RHO exon 2 and is thus amenable to our 3′ replacement strategy. It slightly modifies the export of RHO, which is described to be localized in both the plasma membrane and the cell.
Following the transduction of 293T cells with RHO-encoding lentiviral vector, more than 95% of the cells produced RHO protein (data not shown). We investigated the subcellular distribution of this protein in immunocytofluorescence studies. As expected, WT and P347L RHO were present at the plasma membrane, whereas P23H RHO was retained within the cell. Unexpectedly, we also found that R135W RHO was clearly retained within the cell, and this protein was not detected at the plasma membrane (Figure 4a). We also carried out additional immunocytofluorescence labeling before and after cell permeabilization, to establish clearly the subcellular distribution of RHO. These findings confirmed that both the P23H and R135W mutations led to the retention of the protein in the cytoplasm (Figure 4b).
Figure 4.

Cellular distribution of WT, P23H, R135W and P347L human RHO stably expressed in HEK-293T cells. HEK-293T cells were transduced with lentiviral vectors to create cell lines stably expressing normal or mutated RHO. (a) Confocal microscopy slices of confluent HEK-293T cells following the immunofluorescence labeling of rhodopsin (AF488, green) and staining of the nucleus (DAPI; blue). No RHO is detected in nontransduced cells (NT). In cell lines expressing WT or P347L RHO, the staining is localized at the periphery of the cells and the cytoplasm appears to be almost devoid of staining. By contrast, with the P23H and R135W mutations, staining is concentrated in the perinuclear space. (b) Immunolabeling of a representative transduced cell for each cell line, before and after permeabilization. 1st line: DAPI staining; 2nd line: rhodopsin staining before permeabilization; 3rd line: rhodopsin staining after permeabilization; 4th line: merged views. Scale bar: 10 μm.
We quantified the RHO phenotype objectively, using a flow imaging technique based on the ImageStreamX set-up, for the measurement of various cytological parameters on a large population of cells. Digital masks were applied to the cells, to discriminate between RHO present in the membrane and in the cytoplasm and for the automatic calculation of an internalization score by the ImageStreamX device (Figure 5a). WT and P347L RHO had internalization scores of 0.41 and 0.34, respectively, whereas the internalization scores of P23H and R135W RHO were greater than 1, indicating that these proteins were mostly intracellular (scores: 2.26 and 2.25, respectively; P < 0.0001 in Dunnett's multiple comparisons tests against WT RHO; Figure 5b,c, Supplementary Figure S6). We quantified the effect of trans-splicing on the subcellular distribution of RHO in these cell lines, by obtaining an internalization score in cells cotransfected with the PTM1- or PTM20-encoding plasmid and a cerulean fluorescent protein-encoding plasmid, making it possible to select only transfected cells. In the WT RHO cell line, the internalization score was unaffected by the PTM with which the cells were transfected, whereas, in the R135W RHO cell line, the internalization score tended to be lowered by transfection with PTM1, and this effect was even more pronounced with PTM20 (Figure 5d). The difference in internalization scores was expected to be maximal between R135W RHO and WT RHO cells. We observed that PTM20 gave an internalization score similar to that for PTM0 in WT RHO cells (2.18% decrease), whereas it led to 22.32% decrease in R135W RHO cells (P < 0.05 in t-tests; Figure 5e). Thus, trans-splicing also occurred when the cells expressed the target pre-mRNA stably. Moreover, trans-splicing of the mRNA was accompanied by synthesis of the repaired protein, which was correctly localized within the cell.
Figure 5.

Quantification of RHO localization by ImageStreamx technology before and after trans-splicing. (a) Representative example of a bright-field image of a cell acquired with ImageStreamX (left). This image was used for the automatic definition of masks corresponding to the cell cytoplasm (middle) and plasma membrane (right). (b) The RHO internalization score for each cell line was calculated from the fluorescence quantified for the two masks. An internalization score above 1 indicates that fluorescence is found mostly within the cell. Data were collected in three independent experiments in each of which, at least 500 cells were analyzed (statistically representative). Statistical analysis was performed by comparing internalization scores between WT RHO and all other groups in Dunnett's test. (c) Representative rhodopsin immunofluorescence staining of the four cell lines visualized on the ImageStreamX. (d) WT and R135W RHO cell lines were cotransfected with a PTM construct and a plasmid encoding the cerulean fluorescent protein (CFP). Internalization scores were determined after the selection of transfected cells on the basis of CFP fluorescence. Data were collected in four independent experiments (represented by the four different symbols), and each point corresponds to the observation of at least 500 cells (statistically representative). (e) Effect of PTM20 in WT RHO and R135W RHO cell lines, expressed as a percentage of the maximal variation obtained with PTM0 in these same cell lines. Statistical analysis was performed by one-tail t-test comparison between the two groups.
In vivo assessment: trans-splicing occurs in the photoreceptors of a humanized mouse model expressing a mutant human allele of RHO
We first investigated whether the expression of the PTM alone led to the translation of a partial protein product in mouse photoreceptors, as observed for other PTMs in different experimental paradigms.41,46 We coinjected an AAV2/8 expressing either PTM0 or PTM20 under the control of a bovine rhodopsin promoter (bRho) with the same vector expressing the GFP reporter gene, in a 5:1 ratio, into the subretinal space of Rho−/− mice on postnatal day 21 (P21). We used Rho−/− mice that are totally devoid of Rhodopsin to rule out the possibility of translation of a truncated protein coming from the PTM. Western blots performed 7 days after injection did not evidence any truncated protein from the PTM in transduced retinas (Figure 6a), while photoreceptors transduction occurred correctly, as shown by detection of GFP in vivo and on western blots (Figure 6a,b).
Figure 6.

Absence of truncated rhodopsin protein translation from the PTM alone. (a) Western blots for RHO and GFP on retinal proteins extracted from noninjected C57Bl6J wild-type mouse (lanes 1 and 5), noninjected Rho−/− mice (lane 2), or 1 week after the subretinal coinjection of AAV2/8-bRho-GFP and AAV2/8-bRho-PTM0 (lane 3) or PTM20 (lane 4) in a 1:5 ratio, and Rho+/−RHOP347S/− mice 1 week after the subretinal coinjection of AAV2/8-bRho-GFP and AAV2/8-bRho-PTM20 (lane 6) in a 1:7 ratio. RHO is detected in the “Rho WT” and the “Rho+/−RHOP347S/− +PTM20+GFP” samples only (predicted molecular weight of 39 kDa, corresponding to the monomeric form). More RHO is observed in WT samples due to the photoreceptor degeneration that occurs in Rho+/−RHOP347S/− mice. GFP is detected in AAV2/8-bRho-GFP transduced retinas samples only, according to in vivo detection of GFP expression in the (b) eye fundus before protein extraction for western blot analyses. The four images were obtained with the same settings of the camera parameters. No truncated protein was thus produced in vivo from PTM20 alone.
We then investigated whether RHO trans-splicing occurred in mouse photoreceptors in vivo. We coinjected AAV2/8-bRho-GFP and AAV2/8-bRho-PTM0 or -PTM20, in a 1:7 ratio, into Rho+/- RHOP347S/- mice on P23 to P30. These mice, which were obtained by crossing the Rho−/− strain and the RHOP347S/P347S transgenic strain, carried one endogenous Rho gene allele and one human P347S RHO gene allele. One week after injection, quantification at the RNA level showed that PTM20 gene transfer had induced significant trans-splicing in vivo (Figure 7b). Moreover, trans-splicing rate reflected GFP fluorescence in the eye fundus (Figure 7a and Supplementary Figure S7a). To validate this observation, we microdissected the transduced retinas, and separated transduced areas from nontransduced areas (Supplementary Figure S7c). We observed that trans-spliced products were present only in PTM20 transduced areas, and trans-splicing rate was higher when estimated on transduced areas compared to total retinas (Figure 7d versus 7b).
Figure 7.
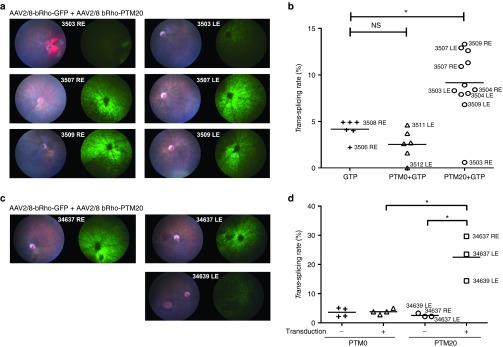
Trans-splicing efficiency in Rho+/− RHOP347S/− mouse photoreceptors following the subretinal delivery of AAV2/8-bRho-PTM20. (a) Color fundus images (left picture in each panel) and green fluorescence (right picture in each panel) showing the general appearance of the retina and GFP expression, respectively, following the subretinal injection of AAV2/8-bRho-GFP associated with AAV2/8-bRho-PTM20 (3503, 3507, and 3509) in a 1:7 ratio. Injection was performed at P23 and retinal imaging was carried out 1 week later. Total vector: 8.1010 vg/eye. The numbers at the middle of each panel correspond to mouse identification numbers and are reported in b. (b) Quantification of trans-splicing rate at the RNA level in the whole retina sampled immediately after retinal imaging. Each sample is represented by a symbol. Eye identification, as in a, is reported for the corresponding symbols. Statistical analysis: Dunn's test. (c) Color fundus images (left picture in each panel) and green fluorescence (right picture in each panel) showing the general appearance of the retina and GFP expression, respectively, following the subretinal injection of AAV2/8-bRho-GFP associated with AAV2/8-bRho-PTM20 (34637, 34639) in a 1:7 ratio. Injection was performed at P34 and retinal imaging was carried out 1 week later. Total vector: 8.1010 vg/eye. The numbers at the middle of each panel correspond to mouse identification numbers and are reported in d. (d) Quantification, of trans-splicing rate at the RNA level in microdissected transduced and nontransduced areas, immediately after retinal imaging. Each sample is represented by a symbol. Eye identification, as in c, is reported for the corresponding symbols. Statistical analysis: One-tail Mann–Whitney test. LE, left eye; RE, right eye.
SD-OCT follow-up in a second series of experiments on these mice from P30 to P60 showed that trans-splicing did not stop or slow PR degeneration with respect to that observed in mice receiving injections of PTM0 or GFP alone (Figure 8). The extreme severity of our model, leading to almost total ONL degeneration at P60, might account for the lack of therapeutic efficiency. However, our data clearly show that the trans-splicing of RHO mRNA occurs in vivo in mouse photoreceptors. Moreover, the rate of trans-splicing was clearly much higher in individual transduced cells (mean of 22.5% in transduced areas) than the mean rate of about 9% observed for the whole retina, given that only a subset of the photoreceptors were transduced.
Figure 8.
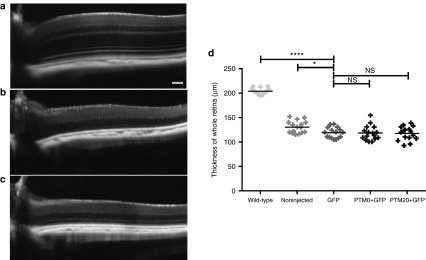
PTM20 is not sufficiently efficient to rescue the degeneration of Rho+/− RHOP347S/− photoreceptors. (a) SD-OCT retinal images at P60 of a noninjected wild-type C57Bl6J mouse, and (b) Rho+/− RHOP347S/− mice injected with either AAV2/8-bRho-GFP alone or (c) associated with AAV-PTM20 in a 1:7 ratio. (d) Graphic representation of the thickness of the entire retina in wild-type mice or noninjected, AAV-bRho-GFP-injected, AAV-bRho-PTM0 + AAV-bRho-GFP-injected, and AAV-bRho-PTM20 + AAV-bRho-GFP-injected Rho+/− RHOP347S/− mice. Statistical analysis: Dunnett's test. *P<0.05; ****P<0.0001.
Discussion
In this study, we demonstrated the repair of dominant mutations of RHO by RNA trans-splicing. Using two different cellular models of RHO expression, we showed that a significant proportion—up to 40%—of the mRNA could be repaired, leading to an improvement of the phenotype, characterized by correction of the subcellular distribution of RHO.
We first engineered various PTMs, which we screened by using them for the transient cotransfection of 293T cells. A significant advantage of our system is that the size of the RHO expression cassette (5,375 bp from the transcription start site to the end of the 3′ untranslated region) allows the production of an expression plasmid containing the entire gene. We were thus able to evaluate the efficiency of the PTM by cotransfection with a construct expressing the full RHO, rather than with an artificial mini-gene, as generally used in trans-splicing studies. This procedure made it possible to assess the efficacy of PTMs against their real target, the entire rhodospin pre-mRNA, maximizing the likelihood of the results of the screen being replicated in vivo, first in the animal model of the disease and then in patients. The screen of 14 different binding domains showed that this part of the PTM determined trans-splicing efficiency. Indeed, the location of the binding domain was found to be crucial and our results revealed the presence of a hotspot sequence for trans-splicing efficiency in the 5′ part of the target intron. Interestingly, another team has already demonstrated that the location of the binding domain in the 5′ part of the target intron increases trans-splicing efficiency.41 We looked at splicing enhancer proteins recruitment motifs by analyzing the first RHO intron using human splicing finder online tool (www.umd.be/HSF/). Several motifs were found all along the intronic sequence, with no particular preference for PTM1 binding site. This hotspot may be directly linked to the sequence of the binding domain, or to the secondary structure of the PTM, but also to that of the target pre-mRNA, which may be more accessible at this site. PTM binding in the intron 5′ region may also promote trans-splicing by ensuring that the PTM 3′ acceptor splice site is available before the endogenous one. Indeed, if we consider transcription and splicing to be coupled mechanisms,55 PTM binding may occur before transcription of the endogenous 3′ acceptor splice site. PTM13 and 14, which target the endogenous 3′ acceptor splice site, were not efficient, consistent with the findings of a previous study,56 but contrary to the results obtained by other groups using an equivalent strategy.46,57 The small size of the overlap in sequence between the PTM13 and 14 binding domains and exon 2 (only 5 bp) may account for our findings. Nevertheless, even if the masking of the endogenous 3′ acceptor splice site by PTM binding favors trans-splicing, it should be borne in mind that it could also induce exon skipping, by promoting the reaction of the exon 1 donor splice site with the acceptor splice site of exon 3 or followings. The binding domain is not the only factor affecting PTM efficiency. Indeed, we have shown that it is possible to modify PTM efficiency by modifying the replacement cDNA, by introducing sequences promoting splicing, such as introns. Depending on the sequence and location of this intron, it can promote or reduce the trans-splicing efficiency (Supplementary Figure S5). Splicing is increasingly thought to occur sequentially, from the first to the last intron, because of its coupling with transcription,58 but studies of in vivo splicing products have shown that the first intron is not necessarily the first to be removed.59 We thus hypothesized that adding a supplementary intron to the PTM sequence would lead to spliceosome recruitment, favoring the binding of this structure to the PTM 3′ acceptor splice site over binding to the endogenous pre-mRNA site. Changes in binding domain location and the addition of supplementary introns are two examples of PTM modifications that can significantly increase trans-splicing efficiency, but other strategies could also be used. For example, the length of the binding domain could be modified, or an intronic splicing enhancer (ISE) could be introduced just after the binding domain. In the face of this complexity, various systems have been developed to facilitate screening,36,57,60 but it is still difficult to predict the effect of a PTM in vivo precisely, above all because these screening systems have been tested on artificial minigenes that may not faithfully reflect the physiological context.
Another key factor governing trans-splicing efficiency is the ratio between the amounts of PTM and its target pre-mRNA. Intuitively, we might expect higher proportions of PTM to be associated with higher rates of trans-splicing. We tested this hypothesis, by transfecting 293T cells with different ratios of the PTM-encoding plasmid and the RHO construct. We found that the trans-splicing rate was directly correlated with the amount of PTM. These results confirm those obtained by Puttaraju et al. in a different system.19 This has very important implications for in vivo applications, particularly as concerns rhodopsin, which is present in very large amounts in rod photoreceptors.61 For therapeutic benefits in animal models, it will therefore be necessary to express high levels of PTM in the target cells. Recent vector developments have shown that AAVs can transduce rod photoreceptors very efficiently and safely.62 In combination with a strong tissue-specific promoter such as the rhodopsin one, it should be possible to achieve therapeutic levels of PTM expression in vivo.
We then checked that the trans-splicing repair of mutant mRNA led to at least a partial restoration of wild-type protein levels in cells, as assessed on the basis of a phenotypic index. For studies of the subcellular distribution of RHO in a context closer to physiological conditions than transient transfection, we created cell lines with stable protein production, thanks to the integration of the lentiviral genome. Transduced 293T cells proved to be a functional system, with almost all of the cells expressing RHO. The localization phenotype observed in 293T cells was different from that described in rod photoreceptors,63 but this could be accounted for by differences in structure between these two cell types. Indeed, RHO is contained in a very unusual structure in rods: phospholipid bilayers arranged in stacks of discs in the outer segment, which have no equivalent in 293T cells. Nonetheless, the ImageStreamX set-up made it possible to quantify this phenotype and to show that the R135W RHO cell line could be partially rescued by trans-splicing. These results highlight two important points: (i) trans-splicing is efficient and quantifiable in conditions close to physiological conditions, in which PTM must enter the nucleus to be transcribed and to bind and react with the endogenous pre-mRNA before its prospective cis-splicing and (ii) the occurrence of trans-splicing repair at the protein level, leading to the relocalization of the protein to its normal physiological position.
Our results provide the first demonstration that trans-splicing rates can be high enough to confer expected therapeutic benefit in the context of a dominant disease. Indeed, given that most of the patients suffering from dominant diseases are heterozygous, the conversion of 40% of the mutated mRNA to the normal form would decrease the ratio of mutant to normal protein from 1:1 to 1:3. This ratio could be even lower if the mutant protein turns out to be less stable than the normal protein.
Finally, the in vivo study demonstrated that no truncated protein was produced from the PTM alone in PR. The subretinal injection of AAV2/8-bRho-PTM20 at P23 to P30 resulted in a general trans-splicing rate of about 10%. This result is highly encouraging because it is clearly underestimated given that it was measured for the entire retina, rather than just for the transduced area. This hypothesis was confirmed by a trans-splicing rate nearly 2.5 times more important when measured on transduced areas only, which moreover certainly still contained nontransduced photoreceptors. The trans-splicing rate required to prevent disease development will undoubtedly depend on mutation severity. In our case, PTM20 gene transfer was not sufficient to slow PR degeneration. Therapeutic efficacy could be improved by increasing transduction rate and/or PTM efficiency, and/or by using an animal model with less severe degeneration.
In conclusion, the results obtained in this study pave the way for the use of SMaRT technology to prevent retinitis pigmentosa due to RHO mutations, for which there is still no effective treatment. In addition, the use of a 5′ trans-splicing PTM targeting RHO intron 1 would make it possible to correct exon 1 mutations, thereby facilitating the correction of all rhodopsin mutations with only two therapeutic tools. This work also highlights the power of SMaRT as a tool for treating not only recessive diseases, but also dominant diseases, and demonstrates that refinements of PTM design can increase trans-splicing rate.
Materials and Methods
Plasmid construction
For transient transfection and lentivirus production. The plasmid backbone used for RHO expression contained a self-inactivating lentiviral genome and has been described elsewhere.64 Human wild-type (WT) RHO was amplified from the genomic DNA of HEK-293T cells with the following primers: 5′-CACCAGAGTCATCCAGCTGGAGCC-3′ and 5′-GTCCTAGGCAGGTCTTAGGC-3′. The PCR product was then inserted into the pENTR/D-TOPO plasmid according to the manufacturer's instructions (Life Technologies, Saint-Aubin, France), and the sequence of the whole insert was checked. The P23H, R135W and P347L mutations were inserted by site-directed mutagenesis (QuikChange kit, Agilent Technologies, Les Ulis, France). Wild-type or mutant alleles of the human RHO gene were then introduced in the lentiviral shuttle plasmid by Gateway recombination (Life Technologies), under the transcriptional control of the short form of the ubiquitous promoter elongation factor 1 alpha (EF1alpha) promoter. The transgene expression cassette was inserted into the lentiviral genome in the reverse orientation, to prevent rhodopsin intron splicing during vector production.
For transient transfection and AAV production. PTM sequences were inserted into a shuttle plasmid for self-complementary AAV vector production, as previously described.65 PTMs were expressed under the control of the ubiquitous cytomegalovirus (CMV) immediate early promoter, with termination by the BGH (bovine growth hormone) polyadenylation signal. Fourteen different binding sequences targeting the rhodopsin intron 1 were synthesized by Eurofins MWG Operon (Les Ulis, France) and inserted into the AAV-PTM expression plasmid via the HindIII/SacII restriction sites. The final PTMs (PTMs 1 to 14) consisted of a binding sequence, followed by an artificial intron sequence and the rhodopsin partial cDNA. The rhodopsin partial cDNAs of PTM1 and PTM8 were modified by the addition of the endogenous rhodopsin intron 3 (PTM19, 22) or another artificial intron sequence, inserted at nucleic acid position 172 (PTM20, 23) or 296 (PTM21, 24), after the start of exon 2. The artificial intron sequence40 consisted of several elements important for splicing: a branch point, a poly-pyrimidine tract and a 3′ acceptor splice site.
Virus production and transduction
Lentivirus. Recombinant lentiviral particles were produced by transient transfection of HEK-293T cells, as previously described.64 Viral supernatants were concentrated by ultracentrifugation at 70,000g for 90 minutes at 4 °C. Finally, viral pellets were resuspended in a minimal volume of PBS to achieve a 1,000-fold concentration of the initial supernatant. Aliquots of 5–10 μl were then stored at −80 °C until further use. The total particle concentration of the viral stocks was estimated by quantifying the p24 capsid protein with the RETRO-TEK HIV-1 p24 Antigen ELISA kit (ZeptoMetrix, Buffalo, NY) according to the manufacturer's instructions. HEK-293T cells were transduced with a quantity of lentivirus corresponding to 2 pg of p24 protein per cell.
AAV. Self-complementary (sc) AAV vectors expressing GFP or PTM under the control of the bovine rhodopsin (bRho) promoter.66 Virions were produced by transfecting HEK293 cells with (i) the adenovirus helper plasmid (pXX6-80), (ii) the AAV packaging plasmid carrying the rep2 and the cap8 genes, and (iii) the AAV2 shuttle plasmid containing the gene encoding GFP or PTM in a sc genome. Recombinant vectors (rAAV) were purified by ultracentrifugation on a discontinuous iodixaniol density gradient followed by dialysis against the formulation buffer of the vector stocks, 0.5 mmol/l MgCl2 and 1.25 mmol/l KCl in phosphate-buffered saline (PBSMK; five buffer changes, 3 hours per round of dialysis). Physical particles were quantified by real-time PCR. Vector titers are expressed as viral genomes per milliliter (vg/ml). Each mouse received in both eyes the same product of injection. Rho−/− mice received 6.1010 vg/eye of AAV (5.1010 vg/eye of AAV2/8-bRho-PTM0 or -PTM20 and 1010 vg/eye of AAV2/8-bRho-GFP), while Rho+/- RHOP347S/- received 8.1010 vg/eye of AAV (7.1010 vg/eye of AAV2/8-bRho-PTM0 or -PTM20 and 1010 vg/eye of AAV2/8-bRho-GFP).
Cell culture and transfection. Human embryonic kidney cells (HEK-293T) were grown in Dulbecco's modified Eagle's medium (Gibco, Life Technologies) supplemented with 10% heat-inactivated fetal bovine serum (Gibco, Life Technologies), 100 U/ml penicillin and 100 µg/ml streptomycin (Gibco, Life Technologies). HEK-293T cells were incubated at 37 °C under an atmosphere containing 5% CO2. For transfection, HEK-293T cells were plated in six-well plates at a density of 500,000 cells/well, transfected with 2.5 µg of plasmid DNA by the CaCl2 precipitation method the following day, and analyzed 48 hours after transfection.
Quantification of the trans-splicing rate at RNA level. A silent point mutation was introduced by site-directed mutagenesis (QuikChange kit, Agilent Technologies) into the partial rhodopsin cDNA of the PTM. This mutation was designed to convert a BglI restriction site into an NheI site. For trans-splicing quantification, HEK-293T cells were lysed, and the mRNAs were collected (Nucleospin RNA II extraction kit, Macherey-Nagel, Düren, Germany) and reverse-transcribed to generate cDNA (Superscript II and OligodT, Life Technologies). The rhodopsin cDNA was then amplified by end-point PCR. The primers used were: 5′-GCACGCCTCTCAACTAC-3′ and 5′-TCTTGCCGCAGCAGATG-3′. The PCR product was separated into two equal batches for restriction digestion with BglI and NheI, respectively. Cis-spliced products are cut by BglI, whereas trans-spliced products are cut by NheI. Partial digestion was then quantified on a Bioanalyzer (2100 Bioanalyzer, Agilent Technologies).
Immunocytochemistry
Single-labeling protocol. Cells were plated on glass coverslips in 24-well plates, incubated for 24 hours and then fixed by incubation for 10 minutes with 4% PFA. Cells were permeabilized by incubation for 15 minutes in PBS-1% NGS-0.2% Triton before incubation for 1 hour with a primary antibody directed against rhodopsin (Rho4D2 antibody MABN15, Merck-Millipore, 1/500 dilution in PBS-1% NGS), then for 45 minutes with Alexa-488–conjugated secondary antibody (Alexa Fluor A11001, Life Technologies, 1/1,000 dilution in PBS-1% NGS) and finally for 5 minutes with PBS-DAPI (1/5,000 dilution).
Double-labeling protocol (before and after permeabilization). Cells were plated on glass coverslips in 24-well plates and incubated for 24 hours. They were then fixed by incubation with 2% PFA for 10 minutes. The coverslips were incubated for 1 hour with a primary antibody against rhodopsin (Rho4D2 antibody MABN15, Merck-Millipore, 1/500 dilution in PBS-1% NGS), and then for 45 minutes with Alexa-594–conjugated secondary antibody (Alexa Fluor A11005, Life Technologies, 1/1,000 dilution in PBS-1% NGS). The cells were then fixed again by incubation in 2% PFA for 10 minutes, and permeabilized by incubation for 15 minutes in PBS-1% NGS-0.2% Triton. The coverslips were once again incubated for one hour with primary antibody against rhodopsin, and then for 45 minutes with an Alexa-488-conjugated secondary antibody (Alexa Fluor A11001, Life Technologies, 1/1,000 dilution in PBS-1% NGS), before incubation for 5 minutes with PBS-DAPI (1/5,000 dilution).
The coverslips were mounted on a glass slide and observed under an Olympus FV1000 laser-scanning confocal microscope.
ImageStreamX imaging. HEK-293T cells were transfected as described above. A reporter plasmid encoding a cerulean fluorescent protein67 was used to label transfected cells. Cells were then pelleted and fixed by incubation for 10 minutes in 4% PFA. The cells were then permeabilized by incubation for 15 minutes in PBS-1% NGS-0.05% Triton. The cells were labeled with a primary antibody against rhodopsin (Rho4D2 antibody MABN15, 1/500 dilution, Merck-Millipore) and an Alexa-647-conjugated secondary antibody (Alexa Fluor A31571, 1/1,000, Life Technologies). They were then analyzed with ImageStreamX technology (AMNIS). The parameters used were: cerulean fluorescence detection: excitation by 488 laser (100 mW), emission in Channel 02; Alexa-647 detection: excitation by 658 laser (120 mW), emission in Channel 11; Magnification: x40; Core diameter: 8 μm; No compensation was necessary.
The minimal number of events considered statistically representative was 500 cells. All experiments were performed with at least 500 cells.
Western blot. Retinal protein extracts were obtained by dissociation in a buffer containing 50 mmol/l HEPES pH 7.4, 150 mmol/l NaCl, 10% glycerol, 1.5 mmol/l MgCl2, 1% Triton X-100. Total protein concentration was determined in a Biorad protein assay, and 10 μg of protein was subjected to electrophoresis in a polyacrylamide gel. Samples were not denatured by heating before electrophoresis. The proteins were transferred to a membrane, which was then probed with antirhodopsin (MAB5356 Millipore 1:2,000), anti-GFP (A11122 Molecular Probes 1:2,000), or antiactin (A5060 Sigma 1:500) primary antibodies and HRP-linked antimouse IgG or antirabbit IgG secondary antibodies. HRP activity was detected with a chemiluminescent HRP substrate (P90719 Millipore).
Animals. C57BL/6JRj mice were purchased from Janvier SA (Le Genest-Saint-Isle, France). Rho−/− knockout mice were provided by Dr Janis Lem (Tufts University) and RHOP347S/P347S mice were provided by Prof Peter Humphries (Trinity College Dublin, Ireland). We used the first generation from a cross between Rho−/− knockout mice and RHOP347S/P347S transgenic mice to obtain Rho+/- RHOP347S/- mice. Mice were maintained at the Institut de la Vision animal facility under pathogen-free conditions. All animals were housed under a 12-hour light/dark cycle, with food and water available ad libitum. All animal manipulations were performed in accordance with the Association for Research in Vision and Ophthalmology (ARVO) Statement for the Use of Animals in Ophthalmic and Vision Research. In addition, all the experimental procedures were approved by the institutional animal care and use committee, the “Comité d'éthique pour l'expérimentation animale Charles Darwin” (ID Ce5/2010/044).
Subretinal injection. Animals were anesthetized by an intraperitoneal injection of ketamine (50 mg/kg, Virbac) and xylazine (10 mg/kg, Bayer HealthCare), and their pupils were dilated with tropicamide (Mydriaticum, Théa) and phenylephrine (Néosynephrine, Europhta). Subretinal injections were performed with a 33-gauge blunt needle mounted on a 10 μl syringe (Hamilton). Briefly, a hole was created through the sclera/choroid/retina layers with the sharp tip of a 30-gauge needle, and the blunt needle of the Hamilton syringe was then gently inserted into the vitreous cavity through this hole. The needle was pushed further into the vitreous cavity until it passed through the retina opposite its site of entry into the eye. After injection, the needle was left in place for an additional 10 seconds, to prevent leakage of the injected fluid. The subretinal injection was considered successful if a subretinal bleb could be seen. An anti-inflammatory and antibacterial ointment (Sterdex, Novartis) was then applied locally.
OCT measurements. Pupils were dilated with tropicamide (Mydriaticum, Théa, France) and phenylephrine (Néosynephrine, Europhta, France). Animals were then anesthetized by inhalation of isoflurane (Axience, France) and placed in the front of the SD-OCT imaging device (Bioptigen 840 nm HHP; Bioptigen, NC). Eyes were kept moisturized with 0.9% NaCl throughout the procedure. Image acquisitions and treatment were performed with the parameters reported elsewhere.68
Micron III imaging. Pupils were dilated with tropicamide (Mydriaticum, Théa, France) and phenylephrine (Néosynephrine, Europhta, France). Animals were then anesthetized by inhalation of isoflurane (Axience, France) and fundus photographs were taken with the Micron III retinal imaging microscope from Phoenix Research Laboratories (Phoenix, AZ). Eyes were kept moisturized with Lubrithal gel (Dechra, UK) throughout the procedure.
Microdissection. Retinas were removed and freshly flat-mounted directly on the microdissection slide (Leica, Steel frames, PET-membrane 11505151). For each transduced retina, four transduced areas and four nontransduced areas were defined under the GFP filter of a microscope (Leica LMD 6500), dissected and separately collected in two tubes containing RNA extraction buffer. One area measured around 40,000µm2 and was duplicated to keep the same surface for all dissected areas. Parameters used for the laser dissection were: power: 60, aperture: 5, speed: 5, specimen balance: 6.
Statistics. GraphPad Prism 6 (GraphPad Software, San Diego, CA) was used for data analysis and graphical representation. All values are reported as medians. Details of statistical analyses are given in the legends of the figures. *P < 0.1, **P < 0.01 ***P < 0.001 and ****P < 0.0001.
SUPPLEMENTARY MATERIAL Figure S1. Localization of PTM1 to PTM14 binding domains in the rhodopsin gene sequence. Figure S2. Methodology for trans-splicing quantification at RNA level. Figure S3. Validation of RT-PCR used to quantify trans-splicing rate. Figure S4. Validation of conformity of the rhodopsin mRNA obtained by cis and trans-splicing. Figure S5. Effect of the addition of an intron sequence to the replacement cDNA of the PTM. Figure S6. Visualization of the RHO cell line phenotypes by flow cytometry imaging. Figure S7. Quantification of trans-splicing rate in microdissected transduced retinas.
Acknowledgments
We wish to thank Nicole Boggetto and Luisa Riancho for valuable advice and technical assistance for the use of the ImageStreamX flow cytometer at ImagoSeine Institut Jacques Monod platform (Université Paris 7) and the flow cytometer of the Institut de la Vision (Université Paris 6), respectively. We also sincerely thank Christina Zeitz for her assistance with rhodopsin labeling. We thank Marie-Laure Niepon from the histology platform at the Institut de la Vision for her technical help with the microdissector.
This work was supported by grant from Association Française contre les Myopathies (AFM grant 14814) and was performed in the framework of the LABEX LIFESENSES (reference ANR-10-LABX-65) with the support of French state funds managed by the ANR. Adeline Berger holds a PhD fellowship from Association Française contre les Myopathies.
Supplementary Material
References
- Birch DG, Anderson JL, Fish GE. Yearly rates of rod and cone functional loss in retinitis pigmentosa and cone-rod dystrophy. Ophthalmology. 1999;106:258–268. doi: 10.1016/S0161-6420(99)90064-7. [DOI] [PubMed] [Google Scholar]
- Hartong DT, Berson EL, Dryja TP. Retinitis pigmentosa. Lancet. 2006;368:1795–1809. doi: 10.1016/S0140-6736(06)69740-7. [DOI] [PubMed] [Google Scholar]
- Wilson JH, Wensel TG. The nature of dominant mutations of rhodopsin and implications for gene therapy. Mol Neurobiol. 2003;28:149–158. doi: 10.1385/MN:28:2:149. [DOI] [PubMed] [Google Scholar]
- Yang Y, Mohand-Said S, Danan A, Simonutti M, Fontaine V, Clerin E, et al. Functional cone rescue by RdCVF protein in a dominant model of retinitis pigmentosa. Mol Ther. 2009;17:787–795. doi: 10.1038/mt.2009.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaVail MM, Yasumura D, Matthes MT, Lau-Villacorta C, Unoki K, Sung CH, et al. Protection of mouse photoreceptors by survival factors in retinal degenerations. Invest Ophthalmol Vis Sci. 1998;39:592–602. [PubMed] [Google Scholar]
- Sieving PA, Caruso RC, Tao W, Coleman HR, Thompson DJ, Fullmer KR, et al. Ciliary neurotrophic factor (CNTF) for human retinal degeneration: phase I trial of CNTF delivered by encapsulated cell intraocular implants. Proc Natl Acad Sci USA. 2006;103:3896–3901. doi: 10.1073/pnas.0600236103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leonard KC, Petrin D, Coupland SG, Baker AN, Leonard BC, LaCasse EC, et al. XIAP protection of photoreceptors in animal models of retinitis pigmentosa. PLoS ONE. 2007;2:e314. doi: 10.1371/journal.pone.0000314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorbatyuk M, Justilien V, Liu J, Hauswirth WW, Lewin AS. Suppression of mouse rhodopsin expression in vivo by AAV mediated siRNA delivery. Vision Res. 2007;47:1202–1208. doi: 10.1016/j.visres.2006.11.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Reilly M, Palfi A, Chadderton N, Millington-Ward S, Ader M, Cronin T, et al. RNA interference-mediated suppression and replacement of human rhodopsin in vivo. Am J Hum Genet. 2007;81:127–135. doi: 10.1086/519025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao H, Gorbatyuk MS, Rossmiller B, Hauswirth WW, Lewin AS. Long-term rescue of retinal structure and function by rhodopsin RNA replacement with a single adeno-associated viral vector in P23H RHO transgenic mice. Hum Gene Ther. 2012;23:356–366. doi: 10.1089/hum.2011.213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin JN, Wolken N, Brown T, Dauer WT, Ehrlich ME, Gonzalez-Alegre P. Lethal toxicity caused by expression of shRNA in the mouse striatum: implications for therapeutic design. Gene Ther. 2011;18:666–673. doi: 10.1038/gt.2011.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y, Du H, Xie B, Wu N, Wang J, Wu G, et al. Cerebellum abnormalities in idiopathic generalized epilepsy with generalized tonic-clonic seizures revealed by diffusion tensor imaging. PLoS ONE. 2010;5:e15219. doi: 10.1371/journal.pone.0015219. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang FQ, Aleman TS, Dejneka NS, Dudus L, Fisher KJ, Maguire AM, et al. Long-term protection of retinal structure but not function using RAAV.CNTF in animal models of retinitis pigmentosa. Mol Ther. 2001;4:461–472. doi: 10.1006/mthe.2001.0473. [DOI] [PubMed] [Google Scholar]
- McGee Sanftner LH, Abel H, Hauswirth WW, Flannery JG. Glial cell line derived neurotrophic factor delays photoreceptor degeneration in a transgenic rat model of retinitis pigmentosa. Mol Ther. 2001;4:622–629. doi: 10.1006/mthe.2001.0498. [DOI] [PubMed] [Google Scholar]
- Rodger J, Drummond ES, Hellström M, Robertson D, Harvey AR. Long-term gene therapy causes transgene-specific changes in the morphology of regenerating retinal ganglion cells. PLoS ONE. 2012;7:e31061. doi: 10.1371/journal.pone.0031061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touchard E, Heiduschka P, Berdugo M, Kowalczuk L, Bigey P, Chahory S, et al. Non-viral gene therapy for GDNF production in RCS rat: the crucial role of the plasmid dose. Gene Ther. 2012;19:886–898. doi: 10.1038/gt.2011.154. [DOI] [PubMed] [Google Scholar]
- Lem J, Krasnoperova NV, Calvert PD, Kosaras B, Cameron DA, Nicolò M, et al. Morphological, physiological, and biochemical changes in rhodopsin knockout mice. Proc Natl Acad Sci USA. 1999;96:736–741. doi: 10.1073/pnas.96.2.736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan E, Wang Q, Quiambao AB, Xu X, Qtaishat NM, Peachey NS, et al. The relationship between opsin overexpression and photoreceptor degeneration. Invest Ophthalmol Vis Sci. 2001;42:589–600. [PubMed] [Google Scholar]
- Puttaraju M, Jamison SF, Mansfield SG, Garcia-Blanco MA, Mitchell LG. Spliceosome-mediated RNA trans-splicing as a tool for gene therapy. Nat Biotechnol. 1999;17:246–252. doi: 10.1038/6986. [DOI] [PubMed] [Google Scholar]
- Murphy WJ, Watkins KP, Agabian N. Identification of a novel Y branch structure as an intermediate in trypanosome mRNA processing: evidence for trans splicing. Cell. 1986;47:517–525. doi: 10.1016/0092-8674(86)90616-1. [DOI] [PubMed] [Google Scholar]
- Sutton RE, Boothroyd JC. Evidence for trans splicing in trypanosomes. Cell. 1986;47:527–535. doi: 10.1016/0092-8674(86)90617-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caudevilla C, Serra D, Miliar A, Codony C, Asins G, Bach M, et al. Natural trans-splicing in carnitine octanoyltransferase pre-mRNAs in rat liver. Proc Natl Acad Sci USA. 1998;95:12185–12190. doi: 10.1073/pnas.95.21.12185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Finta C, Zaphiropoulos PG. Intergenic mRNA molecules resulting from trans-splicing. J Biol Chem. 2002;277:5882–5890. doi: 10.1074/jbc.M109175200. [DOI] [PubMed] [Google Scholar]
- Flouriot G, Brand H, Seraphin B, Gannon F. Natural trans-spliced mRNAs are generated from the human estrogen receptor-alpha (hER alpha) gene. J Biol Chem. 2002;277:26244–26251. doi: 10.1074/jbc.M203513200. [DOI] [PubMed] [Google Scholar]
- Romani A, Guerra E, Trerotola M, Alberti S. Detection and analysis of spliced chimeric mRNAs in sequence databanks. Nucleic Acids Res. 2003;31:e17. doi: 10.1093/nar/gng017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu CS, Yu CY, Chuang CY, Hsiao M, Kao CF, Kuo HC, et al. Integrative transcriptome sequencing identifies trans-splicing events with important roles in human embryonic stem cell pluripotency. Genome Res. 2014;24:25–36. doi: 10.1101/gr.159483.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mansfield SG, Kole J, Puttaraju M, Yang CC, Garcia-Blanco MA, Cohn JA, et al. Repair of CFTR mRNA by spliceosome-mediated RNA trans-splicing. Gene Ther. 2000;7:1885–1895. doi: 10.1038/sj.gt.3301307. [DOI] [PubMed] [Google Scholar]
- Liu X, Jiang Q, Mansfield SG, Puttaraju M, Zhang Y, Zhou W, et al. Partial correction of endogenous DeltaF508 CFTR in human cystic fibrosis airway epithelia by spliceosome-mediated RNA trans-splicing. Nat Biotechnol. 2002;20:47–52. doi: 10.1038/nbt0102-47. [DOI] [PubMed] [Google Scholar]
- Liu X, Luo M, Zhang LN, Yan Z, Zak R, Ding W, et al. Spliceosome-mediated RNA trans-splicing with recombinant adeno-associated virus partially restores cystic fibrosis transmembrane conductance regulator function to polarized human cystic fibrosis airway epithelial cells. Hum Gene Ther. 2005;16:1116–1123. doi: 10.1089/hum.2005.16.1116. [DOI] [PubMed] [Google Scholar]
- Coady TH, Baughan TD, Shababi M, Passini MA, Lorson CL. Development of a single vector system that enhances trans-splicing of SMN2 transcripts. PLoS ONE. 2008;3:e3468. doi: 10.1371/journal.pone.0003468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coady TH, Shababi M, Tullis GE, Lorson CL. Restoration of SMN function: delivery of a trans-splicing RNA re-directs SMN2 pre-mRNA splicing. Mol Ther. 2007;15:1471–1478. doi: 10.1038/sj.mt.6300222. [DOI] [PubMed] [Google Scholar]
- Coady TH, Lorson CL. Trans-splicing-mediated improvement in a severe mouse model of spinal muscular atrophy. J Neurosci. 2010;30:126–130. doi: 10.1523/JNEUROSCI.4489-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chao H, Mansfield SG, Bartel RC, Hiriyanna S, Mitchell LG, Garcia-Blanco MA, et al. Phenotype correction of hemophilia A mice by spliceosome-mediated RNA trans-splicing. Nat Med. 2003;9:1015–1019. doi: 10.1038/nm900. [DOI] [PubMed] [Google Scholar]
- Rodriguez-Martin T, Anthony K, Garcia-Blanco MA, Mansfield SG, Anderton BH, Gallo JM. Correction of tau mis-splicing caused by FTDP-17 MAPT mutations by spliceosome-mediated RNA trans-splicing. Hum Mol Genet. 2009;18:3266–3273. doi: 10.1093/hmg/ddp264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez-Martin T, Garcia-Blanco MA, Mansfield SG, Grover AC, Hutton M, Yu Q, et al. Reprogramming of tau alternative splicing by spliceosome-mediated RNA trans-splicing: implications for tauopathies. Proc Natl Acad Sci USA. 2005;102:15659–15664. doi: 10.1073/pnas.0503150102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wally V, Brunner M, Lettner T, Wagner M, Koller U, Trost A, et al. K14 mRNA reprogramming for dominant epidermolysis bullosa simplex. Hum Mol Genet. 2010;19:4715–4725. doi: 10.1093/hmg/ddq405. [DOI] [PubMed] [Google Scholar]
- Tahara M, Pergolizzi RG, Kobayashi H, Krause A, Luettich K, Lesser ML, et al. Trans-splicing repair of CD40 ligand deficiency results in naturally regulated correction of a mouse model of hyper-IgM X-linked immunodeficiency. Nat Med. 2004;10:835–841. doi: 10.1038/nm1086. [DOI] [PubMed] [Google Scholar]
- Zayed H, Xia L, Yerich A, Yant SR, Kay MA, Puttaraju M, et al. Correction of DNA protein kinase deficiency by spliceosome-mediated RNA trans-splicing and sleeping beauty transposon delivery. Mol Ther. 2007;15:1273–1279. doi: 10.1038/sj.mt.6300178. [DOI] [PubMed] [Google Scholar]
- Chen HY, Kathirvel P, Yee WC, Lai PS. Correction of dystrophia myotonica type 1 pre-mRNA transcripts by artificial trans-splicing. Gene Ther. 2009;16:211–217. doi: 10.1038/gt.2008.150. [DOI] [PubMed] [Google Scholar]
- Lorain S, Peccate C, Le Hir M, Griffith G, Philippi S, Précigout G, et al. Dystrophin rescue by trans-splicing: a strategy for DMD genotypes not eligible for exon skipping approaches. Nucleic Acids Res. 2013;41:8391–8402. doi: 10.1093/nar/gkt621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monjaret F, Bourg N, Suel L, Roudaut C, Le Roy F, Richard I, et al. Cis-splicing and translation of the pre-trans-splicing molecule combine with efficiency in spliceosome-mediated RNA trans-splicing. Mol Ther. 2014;22:1176–1187. doi: 10.1038/mt.2014.35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gruber C, Koller U, Murauer EM, Hainzl S, Hüttner C, Kocher T, et al. The design and optimization of RNA trans-splicing molecules for skin cancer therapy. Mol Oncol. 2013;7:1056–1068. doi: 10.1016/j.molonc.2013.08.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakayama K, Pergolizzi RG, Crystal RG. Gene transfer-mediated pre-mRNA segmental trans-splicing as a strategy to deliver intracellular toxins for cancer therapy. Cancer Res. 2005;65:254–263. [PubMed] [Google Scholar]
- Wang J, Mansfield SG, Cote CA, Jiang PD, Weng K, Amar MJ, et al. Trans-splicing into highly abundant albumin transcripts for production of therapeutic proteins in vivo. Mol Ther. 2009;17:343–351. doi: 10.1038/mt.2008.260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wally V, Murauer EM, Bauer JW. Spliceosome-mediated trans-splicing: the therapeutic cut and paste. J Invest Dermatol. 2012;132:1959–1966. doi: 10.1038/jid.2012.101. [DOI] [PubMed] [Google Scholar]
- Puttaraju M, DiPasquale J, Baker CC, Mitchell LG, Garcia-Blanco MA. Messenger RNA repair and restoration of protein function by spliceosome-mediated RNA trans-splicing. Mol Ther. 2001;4:105–114. doi: 10.1006/mthe.2001.0426. [DOI] [PubMed] [Google Scholar]
- Mansfield SG, Clark RH, Puttaraju M, Kole J, Cohn JA, Mitchell LG, et al. 5' exon replacement and repair by spliceosome-mediated RNA trans-splicing. RNA. 2003;9:1290–1297. doi: 10.1261/rna.5101903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saliba RS, Munro PM, Luthert PJ, Cheetham ME. The cellular fate of mutant rhodopsin: quality control, degradation and aggresome formation. J Cell Sci. 2002;115 Pt 14:2907–2918. doi: 10.1242/jcs.115.14.2907. [DOI] [PubMed] [Google Scholar]
- Sung CH, Davenport CM, Nathans J. Rhodopsin mutations responsible for autosomal dominant retinitis pigmentosa. Clustering of functional classes along the polypeptide chain. J Biol Chem. 1993;268:26645–26649. [PubMed] [Google Scholar]
- Sung CH, Schneider BG, Agarwal N, Papermaster DS, Nathans J. Functional heterogeneity of mutant rhodopsins responsible for autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:8840–8844. doi: 10.1073/pnas.88.19.8840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berson EL, Rosner B, Weigel-DiFranco C, Dryja TP, Sandberg MA. Disease progression in patients with dominant retinitis pigmentosa and rhodopsin mutations. Invest Ophthalmol Vis Sci. 2002;43:3027–3036. [PubMed] [Google Scholar]
- Dryja TP, Hahn LB, Cowley GS, McGee TL, Berson EL. Mutation spectrum of the rhodopsin gene among patients with autosomal dominant retinitis pigmentosa. Proc Natl Acad Sci USA. 1991;88:9370–9374. doi: 10.1073/pnas.88.20.9370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dryja TP, McGee TL, Reichel E, Hahn LB, Cowley GS, Yandell DW, et al. A point mutation of the rhodopsin gene in one form of retinitis pigmentosa. Nature. 1990;343:364–366. doi: 10.1038/343364a0. [DOI] [PubMed] [Google Scholar]
- Audo I, Manes G, Mohand-Saïd S, Friedrich A, Lancelot ME, Antonio A, et al. Spectrum of rhodopsin mutations in French autosomal dominant rod-cone dystrophy patients. Invest Ophthalmol Vis Sci. 2010;51:3687–3700. doi: 10.1167/iovs.09-4766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kornblihtt AR, de la Mata M, Fededa JP, Munoz MJ, Nogues G. Multiple links between transcription and splicing. RNA. 2004;10:1489–1498. doi: 10.1261/rna.7100104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lorain S, Peccate C, Le Hir M, Garcia L. Exon exchange approach to repair Duchenne dystrophin transcripts. PLoS ONE. 2010;5:e10894. doi: 10.1371/journal.pone.0010894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murauer EM, Koller U, Hainzl S, Wally V, Bauer JW. A reporter-based screen to identify potent 3' trans-splicing molecules for endogenous RNA repair. Hum Gene Ther Methods. 2013;24:19–27. doi: 10.1089/hgtb.2012.180. [DOI] [PubMed] [Google Scholar]
- Bentley DL. Coupling mRNA processing with transcription in time and space. Nat Rev Genet. 2014;15:163–175. doi: 10.1038/nrg3662. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeitlin S, Efstratiadis A. In vivo splicing products of the rabbit beta-globin pre-mRNA. Cell. 1984;39 3 Pt 2:589–602. doi: 10.1016/0092-8674(84)90466-5. [DOI] [PubMed] [Google Scholar]
- Koller U, Wally V, Mitchell LG, Klausegger A, Murauer EM, Mayr E, et al. A novel screening system improves genetic correction by internal exon replacement. Nucleic Acids Res. 2011;39:e108. doi: 10.1093/nar/gkr465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung CH, Chuang JZ. The cell biology of vision. J Cell Biol. 2010;190:953–963. doi: 10.1083/jcb.201006020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Allocca M, Mussolino C, Garcia-Hoyos M, Sanges D, Iodice C, Petrillo M, et al. Novel adeno-associated virus serotypes efficiently transduce murine photoreceptors. J Virol. 2007;81:11372–11380. doi: 10.1128/JVI.01327-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendes HF, van der Spuy J, Chapple JP, Cheetham ME. Mechanisms of cell death in rhodopsin retinitis pigmentosa: implications for therapy. Trends Mol Med. 2005;11:177–185. doi: 10.1016/j.molmed.2005.02.007. [DOI] [PubMed] [Google Scholar]
- Bemelmans AP, Bonnel S, Houhou L, Dufour N, Nandrot E, Helmlinger D, et al. Retinal cell type expression specificity of HIV-1-derived gene transfer vectors upon subretinal injection in the adult rat: influence of pseudotyping and promoter. J Gene Med. 2005;7:1367–1374. doi: 10.1002/jgm.788. [DOI] [PubMed] [Google Scholar]
- McCarty DM, Monahan PE, Samulski RJ. Self-complementary recombinant adeno-associated virus (scAAV) vectors promote efficient transduction independently of DNA synthesis. Gene Ther. 2001;8:1248–1254. doi: 10.1038/sj.gt.3301514. [DOI] [PubMed] [Google Scholar]
- Kumar R, Chen S, Scheurer D, Wang QL, Duh E, Sung CH, et al. The bZIP transcription factor Nrl stimulates rhodopsin promoter activity in primary retinal cell cultures. J Biol Chem. 1996;271:29612–29618. doi: 10.1074/jbc.271.47.29612. [DOI] [PubMed] [Google Scholar]
- Goedhart J, von Stetten D, Noirclerc-Savoye M, Lelimousin M, Joosen L, Hink MA, et al. Structure-guided evolution of cyan fluorescent proteins towards a quantum yield of 93% Nat Commun. 2012;3:751. doi: 10.1038/ncomms1738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berger A, Cavallero S, Dominguez E, Barbe P, Simonutti M, Sahel JA, et al. Spectral-domain optical coherence tomography of the rodent eye: highlighting layers of the outer retina using signal averaging and comparison with histology. PLoS ONE. 2014;9:e96494. doi: 10.1371/journal.pone.0096494. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.