

Abstract
Reovirus type 3 (Dearing) (RT3D) infection is selective for cells harboring a mutated/activated RAS pathway. Therefore, in a panel of melanoma cell lines (including RAS mutant, BRAF mutant and RAS/BRAF wild-type), we assessed therapeutic combinations that enhance/suppress ERK1/2 signaling through use of BRAF/MEK inhibitors. In RAS mutant cells, the combination of RT3D with the BRAF inhibitor PLX4720 (paradoxically increasing ERK1/2 signaling in this context) did not enhance reoviral cytotoxicity. Instead, and somewhat surprisingly, RT3D and BRAF inhibition led to enhanced cell kill in BRAF mutated cell lines. Likewise, ERK1/2 inhibition, using the MEK inhibitor PD184352, in combination with RT3D resulted in enhanced cell kill in the entire panel. Interestingly, TCID50 assays showed that BRAF and MEK inhibitors did not affect viral replication. Instead, enhanced efficacy was mediated through ER stress-induced apoptosis, induced by the combination of ERK1/2 inhibition and reovirus infection. In vivo, combined treatments of RT3D and PLX4720 showed significantly increased activity in BRAF mutant tumors in both immune-deficient and immune-competent models. These data provide a strong rationale for clinical translation of strategies in which RT3D is combined with BRAF inhibitors (in BRAF mutant melanoma) and/or MEK inhibitors (in BRAF and RAS mutant melanoma).
Introduction
Until relatively recently, there were few effective therapeutic options for patients with metastatic malignant melanoma (MMM). Standard systemic chemotherapy for MMM, mostly using dacarbazine-based regimens, resulted in response rates of only 10–20% and these were likely to be of short duration.1
In the last 5 years, treatment strategies for metastatic malignant melanoma (MMM) have altered radically, such that MMM is now seen as an exemplar of the power of targeted therapy. The discovery that the majority of MMMs harbor an activating mutation in the RAS-RAF-MEK-ERK signaling pathway has led directly to the development of novel agents for therapy. NRASQ61 and BRAFV600 mutations are the most common mutations in MMM (together account for ~70% of cases) but they are mutually exclusive unless put under selective pressure of BRAF-inhibitors (http://cancer.sanger.ac.uk/cancergenome/projects/cosmic/). BRAFV600E/K mutation occurs in ~50% of melanomas2 and can be targeted by potent, selective inhibitors such as vemurafenib (PLX4032) and dabrafenib (GSK2118436).3,4,5,6,7 Randomized studies in patients with BRAF mutant tumors have shown that BRAF inhibition can improve progression-free and overall survival rates, when compared to the previous gold-standard of single-agent dacarbazine.8 In addition, specific inhibitors of downstream components of the RAS-RAF-MEK-ERK pathway (e.g., MEK inhibitor, trametinib, selumetinib) have shown single agent activity in BRAF mutant MMM (clinicaltrials.gov).9 A further refinement of this approach has been to combine both BRAF and MEK inhibition to achieve double blockade on the pathway. Using this approach, the combination of dabrafenib and trametinib was shown to be superior to single-agent dabrafenib in patients with BRAFV600E/D mutant melanoma.10 However, despite the dramatic successes in the context of BRAF mutant melanoma, the vast majority of responding patients will relapse following the development of BRAF inhibitor-resistant disease. Such patients have a very poor prognosis. Moreover, for the 15–20% of patients with NRASQ61 and the 2% of patients with KRASG12 mutant tumors, the therapeutic outlook is not so promising. In such patients, BRAF inhibition leads to paradoxical activation of the RAS-RAF-MEK-ERK pathway11 and it is uncertain if single-agent MEK inhibition can achieve clinically beneficial pathway blockade at maximum tolerated doses.12 MEK inhibitors are currently under evaluation in RAS mutant melanomas and may represent an important therapeutic advance in this genetic context. There is a continued need to develop new treatment approaches that will be able to optimize therapeutic outcomes for patients with RAS-RAF-MEK-ERK pathway-driven MMM.
Oncolytic virotherapy represents a novel therapeutic approach to melanoma. The importance of RAS-RAF-MEK-ERK pathway activation to the biology and natural history of melanoma makes it an excellent model in which to test a RAS-targeted oncolytic virus, such as reovirus Type 3 (Dearing) (RT3D). As a single agent, RT3D is safe and tolerable, but lacks significant antitumor efficacy—either in phase 1 (refs. 13,14) or phase 2 studies.15 In the latter study, we reported no objective responses in 15 patients with metastatic melanoma with median progression-free and overall survivals of only 45 and 165 days, respectively. Importantly, however, RT3D has been extensively tested preclinically and clinically in combination with conventional cytotoxic drugs and these studies have shown it to be safe and tolerable.16,17,18 Critically, this work has shown that by combining RT3D with conventional anticancer drugs or radiation, we can achieve synergistic interactions.19,20,21,22,23
As a direct extension of this work, we explored the possibility that combining RT3D with drugs that target the RAS-RAF-MEK-ERK pathway may be effective in malignant melanoma. We tested interactions between RT3D and agents that enhance or inhibit BRAF and/or MEK in various melanoma genotypes (RAS mutant, BRAF mutant and RAS/BRAF wild-type) with a view to understanding the nature of any combinatorial interactions between the treatments. In planning these studies, we recognized the possibility of a number of different interactions. Specifically, we predicted that the viral and small molecule therapeutics might simply act independently of one another to mediate an additive effect. Alternatively, we considered the possibility that MEK and/or BRAF inhibition might antagonize the activity of RT3D by shutting off RAS pathway-driven viral replication and cytotoxicity. Finally, we hypothesized that a synergistic interaction might occur in RAS mutant cell lines, through BRAF inhibitor-mediated paradoxical activation of MAPK signaling with consequent enhancement of RT3D replication and cytotoxicity. In fact, these studies revealed interactions of increased cell kill between RT3D and inhibition (rather than enhancement) of the RAS-RAF-MEK-ERK signaling pathway. These findings were contrary to our original hypothesis, and provide a clear rationale for translational clinical studies of RT3D with drugs that inhibit RAS-RAF-MEK-ERK signaling in MMM.
Results
RT3D, PLX4720, and PD184352 are selective for melanoma relative to normal skin fibroblasts, and melanoma with a range of genetic backgrounds vary in sensitivity to RT3D
Malme-3 (normal skin fibroblasts) and Malme-3M (BRAF mutant melanoma) are both derived from the same patient, thus providing tumor and normal tissue counterparts for comparative in vitro studies. Following RT3D treatment, maximum levels of cell death were observed in the melanoma cell line at doses as low as multiplicity of infection (MOI) 3. The normal skin fibroblasts were refractory to RT3D, even at MOI 350. Similar tests were carried out for the PLX4720 and PD184352 inhibitors on these cell lines. PLX4720 and PD184352 were toxic at concentrations of 0.4 µmol/l or greater, with no toxicity on normal skin fibroblasts (Figure 1a,b, Supplementary Figure S1). To confirm on-target effect, pERK1/2 levels, downstream of RAS/MEK, were assessed in PMWK, MeWo (RAS/BRAF wild-type), A375, Mel624 (BRAFV600E mutant), WM266.4 (BRAFV600D mutant), DO4 (NRASQ61L mutant) and WM1791c (KRASQ61H) cells treated with inhibitors by western blot (Figure 1c). The BRAF inhibitor PLX4720 switched off ERK1/2 signaling in BRAFV600E mutant cell lines at 0.3 µmol/l, but this was less apparent in the WM266.4 BRAFV600D mutant cell line. In RAS mutant cell lines, PLX4720 at 0.3 µmol/l paradoxically enhanced ERK1/2 signaling, as previously reported.11 The MEK inhibitor PD184352 completely abrogated ERK1/2 signaling in all cell lines at 1 µmol/l.
Figure 1.

RT3D, PLX4720, and PD184352 are selective for melanoma relative to normal skin fibroblasts. Driving paradoxical p-ERK signaling using PLX4720 in BRAF wild-type melanoma cell lines does not enhance RT3D cell kill. (a) RT3D, PLX4720, or PD184352 was titrated along Malme 3 (normal skin fibroblast) and Malme-3M (BRAF mutant melanoma) cells, both derived from the same patient. Cell survival was measured 96 hours later by MTT assay. (b) Pictomicrograph of cytopathic effect of RT3D at a low dose (MOI 3) and high dose (MOI 350) on Malme-3 and Malme-3M cells. (c) PMWK and MeWo (wild-type BRAF/RAS), A375 and Mel624 (BRAFV600E mutant), WM266.4 (BRAFV600D mutant), DO4 (NRASQ61L mutant) and WM1791c (KRASQ61H mutant) melanoma cells were treated with PLX4720 (0.3 µmol/l) or PD184352 (0.001–1 µmol/l) for 6 hours before harvesting for western blot and probing for pERK1/2 downstream of RAS/MEK signaling. (d) Melanoma cells were treated with dilutions of RT3D, PLX4720, or PD184352 and cell survival was measured at 96 hours by MTT assay. (e) BRAF wild-type cells (PMWK, MeWo, DO4, WM1791c) were assessed for RT3D cytoxoicity in the presence (red) and absence (black) of PLX4720 (0.3 µmol/l), whereby p-ERK signaling is enhanced in the RAS mutant background (DO4, WM1791c). Cell survival was measured 96 hours later by MTT. Data are derived from three independent experiments ± SEM.
This panel of seven melanoma cell lines with varying genetic backgrounds were analyzed for their sensitivity to RT3D. RT3D sensitivity was not dependent on mutational status. The cell line panel was also assessed for sensitivity to the BRAF inhibitor PLX4720 and the MEK inhibitor PD184352 (Figure 1d, Supplementary Table S1). BRAF mutant cell lines were sensitive to PLX4720 and most sensitive to PD184352 relative to the BRAF wild-type cells tested. In RAS/BRAF wild-type and RAS mutant cell lines, it was not possible to derive an IC50 for PLX4720, as expected.
Further activation of MEK-ERK signaling does not enhance RT3D cytotoxicity in RAS mutant cells
Heidorn et al. demonstrated that treating RAS mutated cell lines (DO4, WM1791c) with the BRAF inhibitor (PLX4720) causes paradoxical enhancement of MEK-ERK signaling, through BRAF:CRAF heterodimerization and CRAF activation (see Figure 1c). We hypothesized that such signaling events would provide a favorable environment for RT3D-induced replication and cytotoxicity and, in effect, represent a situation in which combined RT3D and BRAF inhibition would exert synthetic lethality in RAS mutant melanoma. Thus, RT3D was titrated on DO4 and WM1791c cells (NRASQ61L and KRASQ61H mutant, respectively) in the presence of PLX4720. Interestingly, RT3D-induced cytotoxicity was not enhanced by MEK-ERK up-regulation and, if anything, was reduced in DO4 cells. Similarly, in RAS/BRAF wild-type melanoma cells (PMWK, MeWo), the combination did not mediate increased cytotoxicity (Figure 1e).
Inhibition of MEK-ERK signaling with RT3D enhances cell kill in melanoma
We also hypothesized that abrogation of RAS-RAF-MEK-ERK signaling by PLX4720 in BRAF mutant cell lines could create an adverse environment for RT3D-induced cytotoxicity. Cells with oncogenic BRAF were treated with RT3D and BRAF inhibitor PLX4720, which switches off MEK-ERK signaling, contrary to the RAS mutant setting (see Figure 1c). Surprisingly, the combination of BRAF inhibition and RT3D infection resulted in the greatest levels of cell kill (Figure 2a). When these surviving fractions were entered into calcysyn software to calculate CI (combination index) values, derived by the Chou and Talalay method to assess for synergy, addition or antagonism, a synergistic reaction was recorded in the BRAFV600E mutant (A375 and Mel624) cell lines (Supplementary Figure S2). To confirm this observation, similar experiments combining RT3D with downstream MEK inhibition (with PD184352) were performed in BRAF mutant melanoma (Figure 2b), and in BRAF wild-type melanoma (Figure 2c). These surviving fractions also translated to a synergistic interaction when entered into calcusyn across all cell lines, with the exception of BRAFV600D mutant WM226.4, and PMWK (at doses of PD184352 at 10 µmol/l and above) (Supplementary Figure S2). To confirm that this was a cell-specific phenomenon in PMWK cells, the combination of RT3D and PD184352 was assessed with another RAS/BRAF wild-type melanoma cell line, MeWo. In this cell line, RT3D and PD184352 gave greatest levels of cell kill across all doses (Supplementary Figure S3).
Figure 2.

Combining RT3D with BRAF and/or MEK inhibition enhances cell kill in melanoma cell lines. BRAF mutant melanoma cells were treated with 4×, 2×, 1×, 0.5×, and 0.25× IC50 doses of (a) RT3D, PLX4720, or the combination at equal ratios. (b) RT3D, PD184352, or the combination at equal ratios. (c) BRAF wild-type melanoma cell lines were treated with 4×, 2×, 1×, 0.5×, and 0.25× IC50 doses of RT3D, PD184352, or the combination at equal ratios. (d) Wild-type BRAF/RAS (PMWK), BRAFV600E mutant (A375) and BRAF wild-type/NRASQ61L mutant (DO4) cell lines were treated with RT3D, PLX4720 (PLX), PD184352 (PD), the combination of PLX4720 and PD184352, or the triple combination of RT3D, PLX4720, and PD184352 at 4×, 2×, 1×, 0.5×, and 0.25× IC50 doses. For all experiments, cell survival was measured at 96 hours by MTT assay and data are derived from three independent experiments ± SEM. (e) BRAFV600E mutant (A375 and Mel624) cells were treated with BRAF and/or MEK inhibitors (PLX4720 and PD184352 respectively), followed by infection with RT3D at various MOI. 48 hours later, cells were stained with crystal violet.
It has been previously reported that the combination of both BRAF and MEK inhibition leads to improved progression-free survival in patients with BRAF mutant melanoma.10 We tested the PLX4720/PD184352 combination with RT3D in PMWK (wild-type), A375 (BRAFV600E mutant), and DO4 (NRASQ61L mutant) cell lines. The triple combination again caused most cell death compared to the BRAF plus MEK inhibitor doublet therapy, and the single agent counterparts. These data translated to synergy using the Chou and Talalay method, again with the exception of PMWK cells at high ratios of the IC50 (two- and fourfold) (Figure 2d, Supplementary Figure S2).
Taken together, the BRAFV600E mutant cell lines showed the greatest levels of cell death with BRAF/MEK inhibition and RT3D, as further illustrated by crystal violet assay (Figure 2e). If RT3D is titrated on BRAFV600E mutant melanoma, we estimate almost a log reduction in RT3D particles needed to cause similar cell kill with the presence of PLX4720 (Supplementary Figure S4).
To support these results, experiments were carried out with an alternative BRAF inhibitor (Dabrafenib) and MEK inhibitor (Trametinib). Similar levels of increased cell kill with combination therapy were observed (Supplementary Figure S5).
To assess whether the ordering of virus and inhibitors had any effect on results, RT3D was added either 6 hours before, at the same time, or after PLX4720. These various schedules had no effect on cell kill (Supplementary Figure S6).
Therapeutic concentrations of BRAF or MEK inhibitors do not affect RT3D replication
To explain the enhanced cell kill with RT3D in combination with PLX4720 and/or PD184352, we explored whether BRAF/MEK inhibition was helping to increase reoviral replication. A RAS/BRAF wild-type (PMWK), a BRAFV600E mutant (A375), and NRASQ61L mutant (DO4) melanoma were analyzed for viral replication in the presence of PLX4720 or PD184352 by one-step growth curves (Figure 3a). The presence of BRAF or MEK inhibitors had no effect on reoviral replication.
Figure 3.
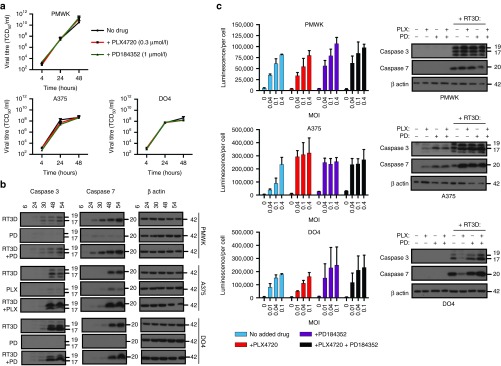
RT3D replication is not affected in the presence of BRAF/MEK inhibition; however, cleaved caspase 3/7 is enhanced in BRAF mutant melanoma. In RAS mutant melanoma, enhancing p-ERK signaling with PLX4720 prevents RT3D-induced caspase 3/7 activation. (a) RT3D replication was measured in the presence of PLX4720 (0.3 µmol/l) or PD1843522 (1 µmol/l) by one step viral growth assay in PMWK, A375 and DO4. (b) Wild-type BRAF (PMWK, DO4) were treated with PD184352 (1 µmol/l) (PD), followed by RT3D (MOI 0.1), and BRAFV600E mutant (A375) were treated with PLX4720 (0.3 µmol/l) (PLX) followed by RT3D (MOI 0.1). At 6, 24, 30, 48, and 54 hours after infection samples were harvested and probed for cleaved caspase 3 and 7 by western blot. (c) Cells were treated with BRAF/MEK inhibition and various doses of RT3D depending on sensitivity. At 72 hours cleaved caspase 3/7 (luminescence) was analyzed by caspase glo assay. Alongside these assays, cells were treated with inhibitors in combination with RT3D at an MOI of 0.04 (PMWK, A375), or 0.01 (DO4), and at 48 hours cells were harvested and probed for cleaved caspase 3 and 7 by western blot.
RT3D-induced caspase 3/7 activation is enhanced in BRAF mutant melanoma in the presence of PLX4720 and/or PD184352
RT3D has been previously shown to induce apoptotic cell death. To explain the observed enhanced cell kill, we wanted to test whether the effects of BRAF or MEK inhibition with RT3D caused any changes in apoptotic events. Samples were collected at 6, 24, 30, 48, and 54 hours, from BRAF wild-type melanoma (PMWK, DO4) treated with RT3D in combination with PD184352, or BRAFV600E mutant melanoma (A375) treated with RT3D in combination with PLX4720. Samples were probed for cleaved caspase 3 and 7 by western blot. By 48 hours, RT3D caused caspase 3 and 7 cleavage in all three cell lines by western blot. However, the kinetics of this activation was unchanged by the addition of a BRAF/MEK inhibitor. Strikingly, cleaved caspase 3 and 7 was dramatically enhanced in BRAFV600E mutant A375 cells treated with the combination of RT3D and BRAF inhibition, compared to single agent treatment (Figure 3b).
To confirm this observation, the same panel of cell lines was treated with a variety of RT3D doses (depending on their RT3D sensitivity), in the presence of either or both BRAF/MEK inhibitors, and cleaved caspase 3/7 was measured by caspase glo assay (Figure 3c). In A375V600E BRAF mutant cells, inhibition of BRAF, MEK or both BRAF and MEK resulted in an increase in caspase 3/7 cleavage, correlating to results observed by western blot. Interestingly, when RT3D is in an environment in which ERK1/2 signaling is paradoxically enhanced instead of switched off (by BRAF inhibitor PLX4720 in RAS mutant DO4 cells), cleaved caspase 3/7 appears to be reduced (Figure 3c).
Enhanced apoptosis is not mediated through p-JNK/TNF-α signaling
We have previously reported that vaccinia virus (GLV-1h68) activates MAPK and JNK/TNF-α (prosurvival) signaling and that inhibition of these pathways during vaccinia viral infection leads to synthetic lethality through enhanced apoptosis, specifically in BRAF mutant melanoma.24 Because RT3D is also capable of activating ERK1/2 signaling (Figure 4a), we tested RT3D's effect on JNK/TNF-α pathway activity and looked for off-target inhibitory effects of BRAF and MEK inhibitors on antiapoptotic p-JNK.
Figure 4.
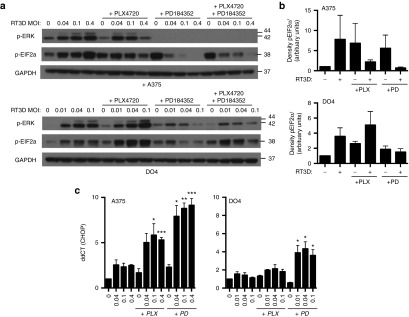
Enhanced apoptosis observed with RT3D and BRAF/MEK inhibition is mediated by ER stress. (a) BRAFV600E mutant (A375) and NRASQ61L mutant (DO4) cell lines were treated with increasing doses of RT3D in the presence of PLX4720 (0.3 µmol/l), PD184352 (1 µmol/l), or the combination of both inhibitors. At 48 hours, lysates were collected and analyzed for p-ERK and p-EIF2α by western blot. (b) Densitometry of p-EIF2α by western analysis is shown for A375 and DO4 cells treated with inhibitors and RT3D (MOI 0.1) at 48 hours. Data are averaged for three independent repeats ± SEM (c) A375 and DO4 cells were treated with PLX4720 and/or PD184352 followed by various doses of RT3D. CHOP gene expression was determined by qRT-PCR at 40 hours. Levels of mRNA were standardized to the expression of beta actin. Mean ± SEM, n = 3.
In contrast to GLV-1h68 vaccinia, RT3D does not induce p-JNK in BRAF mutant melanoma, and TNF-α was also undetectable in PMWK, A375 and DO4 cells treated with RT3D, with and without the addition of BRAF/MEK inhibitors (Supplementary Figure S7). Therefore, we were able to exclude a synthetic lethal interaction between BRAF/MEK inhibition and JNK/TNF-α signaling.
Enhanced apoptosis through RT3D and BRAF/MEK inhibition is due to ER stress-induced apoptosis
BRAFV600E mutant (A375) and NRASQ61L mutant (DO4) melanoma both respond to RT3D by switching on ERK1/2, and antiviral EIF2α (Figure 4a,b).
EIF2α is also phosphorylated in response to BRAF and/or MEK inhibition in BRAF V600E mutant A375 cells, and with MEK inhibition in NRASQ61L mutant DO4 cells. However, combinations of RT3D with BRAF/MEK inhibition downregulated phosphorylated EIF2α in both A375 and DO4 cells, correlating with a situation that yielded the greatest level of cell death (see Figure 2). In DO4 cells, where ERK1/2 is enhanced paradoxically by PLX4720 treatment, in contrast, an increase in p-EIF2α was observed, correlating with a situation that did not enhance cell kill (see Figure 1e).
EIF2α is phosphorylated in response to ER stress, and dephosphorylated in a proapoptotic fashion when ER stress is sustained and unresolvable, by CHOP-mediated upregulation of GADD34.25 We hypothesized that BRAF or MEK inhibition would sensitize melanoma to ER stress induced by RT3D. RT3D has been previously shown to induce ER stress-mediated apoptosis in pancreatic cancer,26 and MEK inhibition sensitizes melanoma cells to ER stress-induced apoptosis.27
A375 and DO4 cells were treated with various doses of RT3D in combination with BRAF or MEK inhibition, and analyzed for CHOP mRNA by Q-PCR (Figure 4c). Both PLX4720 and PD184352 in combination with RT3D in BRAF mutant A375 cells induced CHOP mRNA levels above those seen with just the single agent counterparts. In DO4 cells, CHOP mRNA levels were elevated significantly with RT3D in combination with the MEK inhibitor.
Enhanced cell kill caused by BRAF/MEK inhibition in combination with RT3D can be reversed by salubrinal in BRAF mutant melanoma
We next assessed if the synergistic interaction between BRAF/MEK inhibition and RT3D could be reversed by salubrinal, an inhibitor of the phosphatases that dephosphorylate EIF2α (and, thus, antagonize ER stress-induced apoptosis). Salubrinal was able to sustain EIF2α phosphorylation throughout combinational therapy in A375 BRAFV600E melanoma, preventing the cells ability to “tip” into ER stress-induced apoptosis, and was able to partially rescue cells from the enhanced cell kill of the combinatorial treatments (Figure 5a,b Supplementary Figure S8).
Figure 5.

Salubrinal can partially rescue cells from enhanced cytotoxicity observed with RT3D and BRAF/MEK inhibition in BRAF mutant melanoma. (a) BRAFV600E mutant (A375) cells were treated with PLX4720 (0.3 µmol/l) or PD184352 (1 µmol/l) prior to infection with RT3D, 24 hours later, cells were treated with 20 µmol/l salubrinal (as to not interfere with replication), and cell survival measured at 72 hours by MTT assay (top panel). In a similar experiment, cells were stained with crystal violet at 48 hours (bottom panel). (b) To confirm on-target effect, cells that were treated with inhibitors, followed by RT3D (MOI 0.1), and salubrinal 24 hours after infection, were also harvested at 40, 44, and 48 hours and probed with p-EIF2α by western blot. (c) A375 cells treated with inhibitors and RT3D (MOI 0.1), and at 40 hours were analyzed by qRT-PCR for PUMA and NOXA, with or without the addition of salubrinal 24 hours after infection. For all qRT-PCR analysis, levels of mRNA were standardized to the expression of beta actin. Mean ± SEM, n = 3. (d) Cells treated with inhibitors, followed by RT3D, and salubrinal 24 hours post infection were analyzed for presence of cleaved caspase 3/7 at 72 hours by caspase glo assay. (e) Cells were treated with the caspase 4 inhibitor Z-YVAD prior to treatment with PLX4720 and RT3D (MOI 3). Cell survival was then measured 72 hours later by MTT assay, or harvested 48 hours later and probed with caspase 3 to confirm on-target effect.
Next, we assessed other pro-apoptotic genes downstream of CHOP, such as PUMA and NOXA, which have previously been implicated in reovirus-mediated ER stress-induced apoptosis,28 and whether the expression of these could be reduced by salubrinal. NOXA was strongly induced by RT3D, and expression was enhanced further by BRAF or MEK inhibition. With the addition of salubrinal, the expression of proapoptotic NOXA, but not PUMA, was reduced (Figure 5c).
To establish a link between EIF2α phosphorylation and caspase activation, caspase 3/7 was measured by luminescence assay with therapeutic RT3D plus BRAF/MEK inhibitor combinations either with or without salubrinal. The addition of salubrinal reduced the amount of cleaved caspase 3/7, which was otherwise increased with combination treatments (Figure 5d). Since caspase 4 activation has been implicated in ER stress-induced apoptosis, we also hypothesized that by using a caspase 4 inhibitor (Z-YVAD), we would be able to rescue cells from RT3D + BRAF/MEK inhibition-induced cytotoxicity. Cell survival of BRAFV600E mutant A375 melanoma treated with the combination of RT3D and the BRAF inhibitor PLX4720 was partially restored by the addition of the caspase 4 inhibitor (Figure 5e).
Taken together, these data show that the enhanced cytotoxicity of RT3D with BRAF/MEK inhibition is due to enhanced ER stress-induced apoptosis, mediated through EIF2α, in BRAF mutant melanoma.
In vivo combination of RT3D and BRAF inhibition is therapeutic in BRAF mutant melanoma
The therapeutic efficacy of BRAF inhibition in combination with RT3D was assessed in A375 (BRAFV600E mutant) xenografts in CD1 nude mice. Animals were treated with PLX4720 daily by oral gavage, either alone or in combination with a single intratumoral injection of RT3D. Combined treatment reduced tumor burden and survival was 100% by day 60 (Figure 6a, Supplementary Figure S9). At the termination of the experiment (day 60), tumors were harvested and analyzed for RT3D by TCID50 assay. Despite increased therapeutic efficacy in tumors treated with RT3D and PLX4720, viral titres per mg of tumor were similar to those treated with RT3D alone at the end of the experiment (Figure 6b). This finding supports the in vitro data suggesting that the increased therapy seen with combinations of RT3D and BRAF inhibition is not due to enhanced viral replication. Next, we assessed therapeutic efficacy of BRAF inhibition in combination with RT3D in immune-competent C57BL/6 mice bearing 4434 murine BRAF mutant melanoma cells. Combination therapy resulted in the lowest tumor burden compared to the single agent treatments (Figure 6c). The effects of pharmaceutical inhibitors and RT3D on this cell line were also analyzed in vitro, with combination treatment resulting in enhanced cell death over and above that observed in single agent counterparts (Figure 6d, Supplementary Figure S2).
Figure 6.
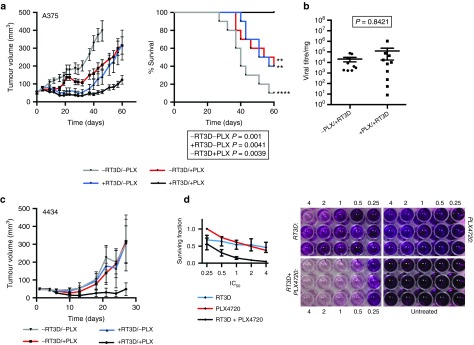
PLX4720 enhances RT3D-mediated antitumor activity in BRAF mutant tumors. (a) CD1 nude mice bearing A375 (BRAFV600E mutant) tumors received PLX4720 (20 mg/kg) administered daily or vehicle, followed by a single intratumoral injection of 5 × 107 pfu RT3D or PBS sham 3 days later (day 1). Data show tumor volumes per treatment group (n = 10 for each group) and survival rates. A log-Rank (Mantel-Cox) test was used to compare groups to the doublet therapy (RT3D plus PLX4720) (b) At day 60, tumors were analyzed for RT3D viral titre by TCID50 assay. Data shown are for tumors treated with RT3D or combination treatments of RT3D with PLX4720 and are relative to tumor weight (viral titre per mg). Titres were compared by ranked Mann-Whitney test. (c) Immune-competent C57BL/6 mice bearing 4434 (BRAF mutant murine) tumors were treated daily with PLX4720 (PLX 40 mg/kg) followed by a single intra-tumoral injection of 5x106 pfu RT3D or PBS sham 3 days later. Data show tumor volumes per treatment group (n = 10 for each group). (d) In vitro, 4434 cells were treated with 4×, 2×, 1×, 0.5× and 0.25× IC50 doses of RT3D, PLX4720 or the combination at equal ratios. Data show surviving fractions (left panel) and photography of representative cell survival (right panel) measured at 96 hours by MTT assay where presence of purple crystals that have been solubilized denotes working mitochondria. Surviving fractions are derived from three independent experiments ± SEM.
Discussion
This study addresses the important issue of how oncolytic virotherapy can be used in combination with targeted therapy (kinase inhibitors) to increase efficacy of single agent treatments in BRAF and RAS mutant melanoma.
First, we evaluated the potential tumor specificity of RT3D, PLX4720 and PD184352 treatments on benign melanocytes versus malignant melanoma cells derived from the same patient. All treatments were essentially inactive against the non-malignant Malme-3 cells, such that IC50 values could not be derived (precluding subsequent combination studies) (Figure 1a,b). In contrast, the melanoma cells (Malme-3M) were sensitive to all three agents. Moreover, assessment of single agent activities indicated that malignant melanoma cell lines were sensitive to RT3D-induced cytotoxicity, but exhibited an almost 4-log range of IC50 values that was not clearly associated with any specific genotype (Figure 1d). Interestingly, in the panel of seven cell lines, the least and most sensitive cell lines were KRAS and NRAS mutated cell lines, respectively. The BRAF mutant cell lines were more similar in their sensitivities, although even they showed a 40-fold difference in IC50 values for RT3D. For PLX4720 and PD184352, the BRAF mutant cell lines were most sensitive to each of the drugs and, as expected, it was not possible to define IC50 values for PLX4720 in the RAS mutant or RAS/BRAF wild-type cell lines. Our data corroborate previous reports on BRAF inhibitor-induced reduced ERK1/2 signaling in BRAFV600E mutant cell lines, but increased signaling in RAS mutant cells (Figure 1c).11
As BRAF inhibitors are known to paradoxically increase RAS-RAF-MEK-ERK signaling downstream of oncogenic RAS in RAS mutant melanoma cell lines, and RT3D has been shown to replicate selectively in cells with mutated or activated RAS signaling, we expected to see enhanced RT3D cytotoxicity in this context. However, to the contrary, RT3D in combination with PLX4720 in a RAS mutant cell line produced neither the anticipated “synthetic lethal” interaction, nor increased viral replication in response to paradoxical activation of ERK1/2 signaling (Figures 1e and 3a). In contrast, there appeared to be a reduction in RT3D-induced apoptosis (Figure 3c). In fact, the greatest degree of cell kill correlated to suppression of ERK1/2 signaling rather than its paradoxical induction. The combination of RT3D and BRAF inhibition led to greater cell kill in the BRAFV600E mutant cell lines (Figure 2a,e), with the combination of MEK inhibition and RT3D resulting in the greatest levels of cell death in both BRAF and RAS mutants (Figure 2b,c).
Given the success of combined BRAF and MEK inhibition in clinical studies in BRAF mutant melanoma, we next studied double pathway blockade in combination with RT3D. Similarly, greatest levels of cell kill were seen with the triple therapy in the RAS (DO4) and BRAF (A375) mutant cells across all ratios of IC50—and in the PMWK wild-type cells at IC50 ratios of 1 or lower (Figure 2d). Again, correlation was observed between enhanced cell kill and suppression of ERK1/2 signaling, with MEK inhibition able to suppress paradoxical activation of ERK by BRAF inhibition in RAS mutant cell lines (Figure 4a).
Whilst the presence of inhibitors had no effect on viral replication, apoptosis was enhanced with the combination of RT3D with BRAF/MEK inhibition, most strikingly in BRAF mutant melanoma (Figure 3b,c). We observed that levels of p-EIF2α closely followed those of p-ERK1/2, with cell kill due to RT3D plus BRAF/MEK inhibition corresponding to a decrease in both. EIF2α phosphorylation by RT3D is normally associated with an anti-viral response mediated by PKR that acts to halt viral protein production. However, constitutive activation of RAS/RAF is linked to PKR inactivation.29 However, without any observed changes in viral replication, we investigated a potential role of EIF2α phosphorylation in ER stress and the unfolded protein response (UPR).
The unfolded protein response (UPR) is initially a prosurvival response, acting to reduce the burden of unfolded proteins and restore normal ER function. In the initial phase of ER stress, PERK activation (through GRP78 dissociation) leads to phosphorylation of EIF2α, which inhibits protein translation and aids survival by reducing the protein load in the ER. However, if the ER stress persists and cannot be resolved, signaling becomes pro-apoptotic.30 In this instance, EIF2α is dephosphorylated by CHOP-mediated upregulation of GADD34.
Recently RT3D has been shown to induce ER stress and induce the UPR in multiple myeloma in vitro and in vivo. RT3D induced canonical upregulation of the ER stress linked proteins GRP78, CHOP, and GADD34, splicing of the UPR transcriptional regulator XBP1, swelling of the ER and apoptotic signaling through upregulation of NOXA and PUMA, effects that have been shown in multiple myeloma cells but not PBMCs.28 Our observed RT3D-associated phosphorylation of EIF2α and upregulation of CHOP in both NRAS and BRAF mutant melanoma (Figure 4) is in keeping with this previously observed ER stress response.
The toxicity of RT3D in multiple myeloma and KRAS mutant pancreatic cancer was further enhanced by blocking the degradation of misfolded proteins using the proteosomal inhibitor bortezomib both in vitro and in vivo.26,28 The mechanism of action of bortezomib in combination with RT3D is likely due to a backlog of unfolded viral proteins, which eventually tip the UPR into a proapoptotic signaling state.
Existing evidence also supports a role for MEK-ERK signaling in responding to ER stress. The MEK inhibitor U0126, as well as MEK siRNA, is able to sensitize cell lines, including melanoma, to the ER stress agents tunicamycin and thapsigargin.27,31 Tolerance to ER stress due to upregulation of the BCL-2 family member MCL-1 has also been shown in melanoma.32 Suppression of ERK1/2 signaling corresponds to a reduction in active XBP-1, GRP78 abundance and MCL-1 expression in melanoma cell lines.32,33 This is in keeping with the idea that signaling through ERK1/2 mediates an anti-apoptotic adaptive response to ER stress. Conversely, reduced ERK1/2 signaling sensitizes melanoma cells to apoptosis induced by ER stress.
Our data further confirm that cytotoxicity through RT3D plus BRAF/MEK inhibition was mediated by ER stress was revealed by the use of salubrinal, an inhibitor of EIF2α dephosphorylation. Knockdown or pharmacological inhibition of EIF2α dephosphorylation that maintains EIF2α in a phosphorylated state, preventing entry of newly synthesized proteins into an already stressed ER, has been shown to protect cells from ER stress-mediated apoptosis.33,34,35 Our observation of salubrinal-mediated reversal of enhanced cell death due to combined BRAF/MEK inhibition and RT3D was consistent with the concept of this combination therapy as an ER stressing agent (Figure 5a). Furthermore, salubrinal appeared to reduce gene expression of downstream ER stress linked pro-apoptotic NOXA, and caspase 3/7 activation, which was otherwise enhanced with RT3D plus BRAF/MEK inhibition (Figure 5c,d). We also report that by inhibiting Caspase 4, which initiates the ER stress triggered caspase cascade, we could partially rescue cells from the enhanced cell death resulting from RT3D plus BRAF/MEK inhibition (Figure 5e)
The potential role of increased ERK1/2 signaling as a protective response to ER stress27,31,33 was also supported by the observation that paradoxical activation of mutant RAS signaling by BRAF inhibition in BRAF wild-type cells decreased RT3D induced apoptosis (Figure 3c), and increased p-EIF2α (Figure 4a). This is the first indication that paradoxical activation of mutant RAS may also enhance tolerance of ER stress through increased downstream ERK1/2 signaling.
In a proof-of-principle therapeutic experiment in A375 BRAF mutant xenografts, the combination of RT3D plus PLX4720 was significantly more potent than either of the single agents—consistent with the in vitro demonstration of enhanced cell kill (Figure 6a). Importantly, to study the effect of this combination treatment in conditions more closely resembling the clinical situation, we performed therapy-allograft experiments using immune-competent mice and obtained similar results.
Taken together, these data provide a very strong rationale for biomarker-led clinical translational studies of RT3D with BRAF and/or MEK inhibition in malignant melanoma. From first principles, in BRAF mutant melanomas, there are good reasons to assess RT3D either with BRAF inhibition alone or double pathway blockade with BRAF and MEK inhibition. Instead, in RAS mutant melanoma, combining RT3D with MEK inhibition appears attractive. However, even though there was no evidence of synthetic lethality when BRAF inhibition was used with RT3D in RAS mutant cells, the triple combination showed very significant cell death in NRAS mutant DO4 cells. Therefore, it would be interesting to further evaluate this treatment combination in preclinical and clinical studies.
Materials and Methods
Cell lines. PMWK, MeWo, (both RAS and BRAF wild-type), A375, Mel624 (both BRAFV600E mutant), WM266.4 (BRAFV600D mutant), DO4 (NRASQ61L mutant) and WM1791c (KRASQ61H mutant) melanoma cell lines, and 4434 (murine BRAF mutant melanoma)36 were obtained from Prof. Richard Marais (Cancer Research UK Manchester Institute). L929 (mouse fibroblast; Oncolytics Biotech, Calgary, Alberta, Canada) were used as reovirus-sensitive target cells. Malme-3 (normal skin fibroblasts, ATCC, Teddington, UK) and Malme-3M (melanoma) are cell lines both derived from the same patient. PMWK, MeWo, A375, Mel624, WM266.4, and L929 cells were cultured in DMEM. Media was supplemented with 5% (v/v) FCS, 1% (v/v) glutamine, and 0.5% (v/v) penicillin/streptomycin. DO4, WM1791c and Malme-3M cells were cultured in RPMI, supplemented with 10% (v/v) FCS, 1% (v/v) glutamine, and 0.5% (v/v) penicillin/streptomycin. Malme-3 were cultured in McCoy's 5a Medium, with 15% (v/v) FCS, 1% (v/v) glutamine, and 0.5% (v/v) penicillin/streptomycin.
Reagents. For western blotting the following antibodies were used: Phospho-p44/42 MAPK (ERK1/2), Caspase 3, Caspase 7, p-EIF2α, GAPDH (Cell signaling Technology, Danvers), Mouse anti-human GAPDH (AbD Serotec, Kidlington, UK), and Anti-β-actin (Abcam). PLX4720 (SelleckBio, Houston), PD184352 (LC Laboratories, Woburn), Dabrafenib, Trametinib (both from Selleckchem, Suffolk, UK), Salubrinal (Enzo Life Sciences), and Z-YVAD (R&D Systems, Abingdon, UK) were prepared in DMSO.
RT3D stocks. Reovirus (type 3 Dearing) stocks at 4.6 × 109 pfu/ml were obtained from Oncolytics Biotech and stored at −80 °C in PBS. 1:10 dilutions were made in DMEM with 2% (v/v) FCS, 1% (v/v) glutamine, and 0.5% (v/v) penicillin/streptomycin, and titred by TCID50 assay on L929 cells.
Cell survival assays. Cells were plated at 5 × 103 per well in 96-well plates and incubated at 37 °C for 24 hours before treatment with inhibitors or RT3D, in 200 µl. Cell survival was measured by 3-(4,5-dimethythiazol-2-yl)-2,5-diphenyl tetrazolium bromide (MTT) assay according to the manufacturer's instructions. For crystal violet assays, cells were plated at 3 × 105 per well in six-well plates, and then treated the next day with inhibitors in 2 ml for 1–2 hours before infection. Cells were stained 48 after infection. For salubrinal experiments, salubrinal was added 24 hours after infection as to not interfere with replication. For caspase 4 inhibitor experiments, Z-YVAD was added 1 hour before therapeutic treatment commenced.
Western blotting. Cells were plated at 1 × 106 per 10 cm dish, 5 × 105 per 6 cm dish, or 3 × 105 per six-well plate and incubated at 37 °C for 24 hours before treatment with PLX4720, PD184352 followed by RT3D infection 1–2 hours later. Samples were harvested for western blotting 6 hours or 48 hours after treatment. For caspase 4 inhibitor experiments, Z-YVAD was added 1 hour before therapeutic treatment commenced. Density readings of bands were measured using ImageJ software.
Synergy testing. Cells were plated at 5 × 103 per well in 96-well plates and incubated at 37 °C for 24 hours before treatment with PLX4720, PD184352 or RT3D as single agents or in combinations at various IC50 doses at equal ratios in 200 µl. Cell survival was measured at 96 hours by MTT assay. Interactions were assessed by the method of Chou and Talalay, and CI values were generated using CalcuSyn software (Biosoft, Cambridge, UK).
One-step viral growth assays. Cells were plated at 1 × 105 in 24-well plates and treated the following day with PLX4720 (0.3 µmol/l) or PD184352 (1 µmol/l) and incubated at 37 °C for 1–2 hours. RT3D was then added at MOI 5 (to ensure infection of all cells). After another 2 hours, cells were washed twice with media and the inhibitors replaced. At 4, 24, and 48 hours after infection, cells were harvested into the media and the lysate was subjected to 3× freeze/thawing between −80 and 37 °C, centrifuged at 13,000 rpm for 5 minutes and stored at −80 °C. The supernatant was used to titre for RT3D by TCID50 assay on L929 cells.
qRT-PCR experiments. RNA was extracted from samples using Qiagen RNeasy kit, and cDNA synthesized using SensiFAST cDNA synthesis kit (Bioline). Samples were then amplified against transcripts by qRT-PCR with SYBR green. Primers used (CHOP, PUMA, NOXA) were commercially available QuantiTect primer assays (Qiagen). Primers for beta actin were as follows: beta actin Forward 5'-GGCACCCAGCACAATGAA-3', beta actin Reverse 5'-GCCGATCCACACGGAGTACT-3'. Relative gene expression was calculated with the 2-ddCT method using beta actin as a house keeping gene. All kits were used as per manufacturers' instructions.
Caspase-Glo assay. Cells were plated at 5 × 103 cells per well in 96-well plates and treated the next day with inhibitors 1–2 hours before infection. For salubrinal experiments, salubrinal was added 24 hours after infection as to not interfere with replication. At 72 hours post treatment, caspase 3/7 activity was measured by the luminescence based reporter assay Caspase-Glo 3/7 (Promega, Southampton, UK) following the protocol stated by the manufacturer.
TNF-α ELISA assays. Cells were plated at 3 × 105 per well in six-well plates, and then treated the next day with inhibitors in 2 ml for 1–2 hours before infection. 48 hours later cells were collected and ELISA assay was performed according to manufacturers protocol (DTA00C, R&D systems).
In vivo assays. CD1 nude mice (Charles Rivers, Kent, UK) were subcutaneously injected with 3 × 106 A375 cells suspended in PBS in the right flank. C57BL/6 mice (Charles Rivers) were subcutaneously injected with 4 × 106 4434 cells suspended in PBS in the right flank. Once tumors were established to ~6 mm in diameter, mice were allocated treatment groups stratified by tumor size. Mice bearing A375 tumors were treated daily with 20 mg/kg PLX4720 and C57BL/6 mice bearing 4434 tumors were treated daily with 40 mg/kg PLX4720 (or vehicle: 2.5% DMSO, 97.5% water) by oral gavage. 5 × 107 pfu RT3D (A375) or 5 × 106 pfu RT3D (4434) dissolved in PBS (or a PBS sham) was administered as an intra-tumoral injection 3 days after drug administration commenced.
Established A375/4434 tumor volumes were measured at least twice-weekly using Vernier calipers and the tumor volume was estimated from the formula: V = 0.5 × (length × width2). Tumors were harvested by dissection and snap frozen and stored at −80 °C. Samples were homogenized in 400 µl PBS containing protease cocktail inhibitor (Roche, South San Francisco, 1 tablet dissolved in 50 ml PBS). Samples were then centrifuged at 1,000 rcf for 5 minutes and the supernatant was used to titre for RT3D by TCID50 assay on L929 cells. All experiments were carried out in compliance with the NCRI guidelines, with animals judged to have failed treatment if tumor diameter exceeded 10 mm.
Statistical analysis. T tests or one-way ANOVA tests were used to make comparisons between groups. Survival curves were compared using the Kaplan–Meier method and significance was assessed using the χ2 test. P values were derived where P > 0.05 ns, *P ≤ 0.05, **P ≤ 0.01, ***P ≤ 0.001, and ****P ≤ 0.0001.
SUPPLEMENTARY MATERIAL Figure S1. RT3D, PLX4720 and PD184352 are selectively toxic to melanoma cells. Figure S2. Combining RT3D with BRAF or MEK inhibition is synergistic in melanoma cell lines. Figure S3. MEK inhibition with RT3D enhances cell kill in wild-type MeWo cells. Figure S4. RT3D causes almost 10-fold greater cell kill in the presence of PLX4720 in BRAFV600E mutant melanoma. Figure S5. RT3D in combination with Dabrafenib or Trametinib leads to increased cell kill compared to their single agent counterparts. Figure S6. Various scheduling of RT3D and BRAF inhibitor does not alter cell kill. Figure S7. RT3D does not mediate enhanced cell death with BRAF/MEK inhibition through abrogation of p-JNK/TNF-α signaling. Figure S8. Salubrinal trends towards rescuing cells from enhanced cytotoxicity observed with RT3D and MEK inhibition in RAS mutant melanoma. Figure S9. PLX4720 enhances RT3D-mediated anti-tumor activity in BRAF mutant tumors in vivo. Table S1. RT3D, PLX4720 and PD184352 IC50 doses for a panel of melanoma cell lines of various mutation status.
Acknowledgments
We thank Oncolytics Biotech for providing reovirus. Matt Coffey is a shareholder and employee of Oncolytics Biotech Inc. Richard Vile, Alan Melcher and Kevin Harrington received funding from Oncolytics Biotech Inc in support of laboratory research. Victoria Roulstone is supported by the Mark Donegan Foundation and Rosetrees Trust. Kevin Harrington is supported by the RM/ICR NIHR Biomedical Research Centre.
Supplementary Material
RT3D, PLX4720 and PD184352 IC50 doses for a panel of melanoma cell lines of various mutation status.
References
- Mandarà M, Nortilli R, Sava T, Cetto GL. Chemotherapy for metastatic melanoma. Expert Rev Anticancer Ther. 2006;6:121–130. doi: 10.1586/14737140.6.1.121. [DOI] [PubMed] [Google Scholar]
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature. 2002;417:949–954. doi: 10.1038/nature00766. [DOI] [PubMed] [Google Scholar]
- Chapman PB, Hauschild A, Robert C, Haanen JB, Ascierto P, Larkin J, et al. BRIM-3 Study Group Improved survival with vemurafenib in melanoma with BRAF V600E mutation. N Engl J Med. 2011;364:2507–2516. doi: 10.1056/NEJMoa1103782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bollag G, Tsai J, Zhang J, Zhang C, Ibrahim P, Nolop K, et al. Vemurafenib: the first drug approved for BRAF-mutant cancer. Nat Rev Drug Discov. 2012;11:873–886. doi: 10.1038/nrd3847. [DOI] [PubMed] [Google Scholar]
- Chandra S, Pavlick AC. Targeted therapies for metastatic melanoma. Dermatol Clin. 2012;30:517–524. doi: 10.1016/j.det.2012.05.001. [DOI] [PubMed] [Google Scholar]
- Young K, Minchom A, Larkin J. BRIM-1, -2 and -3 trials: improved survival with vemurafenib in metastatic melanoma patients with a BRAF(V600E) mutation. Future Oncol. 2012;8:499–507. doi: 10.2217/fon.12.43. [DOI] [PubMed] [Google Scholar]
- Sosman JA, Kim KB, Schuchter L, Gonzalez R, Pavlick AC, Weber JS, et al. Survival in BRAF V600-mutant advanced melanoma treated with vemurafenib. N Engl J Med. 2012;366:707–714. doi: 10.1056/NEJMoa1112302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- da Rocha Dias S, Salmonson T, van Zwieten-Boot B, Jonsson B, Marchetti S, Schellens JH, et al. The European Medicines Agency review of vemurafenib (Zelboraf®) for the treatment of adult patients with BRAF V600 mutation-positive unresectable or metastatic melanoma: summary of the scientific assessment of the Committee for Medicinal Products for Human Use. Eur J Cancer. 2013;49:1654–1661. doi: 10.1016/j.ejca.2013.01.015. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Robert C, Hersey P, Nathan P, Garbe C, Milhem M, et al. METRIC Study Group Improved survival with MEK inhibition in BRAF-mutated melanoma. N Engl J Med. 2012;367:107–114. doi: 10.1056/NEJMoa1203421. [DOI] [PubMed] [Google Scholar]
- Flaherty KT, Infante JR, Daud A, Gonzalez R, Kefford RF, Sosman J, et al. Combined BRAF and MEK inhibition in melanoma with BRAF V600 mutations. N Engl J Med. 2012;367:1694–1703. doi: 10.1056/NEJMoa1210093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidorn SJ, Milagre C, Whittaker S, Nourry A, Niculescu-Duvas I, Dhomen N, et al. Kinase-dead BRAF and oncogenic RAS cooperate to drive tumor progression through CRAF. Cell. 2010;140:209–221. doi: 10.1016/j.cell.2009.12.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McArthur GA, Ribas A. Targeting oncogenic drivers and the immune system in melanoma. J Clin Oncol. 2013;31:499–506. doi: 10.1200/JCO.2012.45.5568. [DOI] [PubMed] [Google Scholar]
- Vidal L, Pandha HS, Yap TA, White CL, Twigger K, Vile RG, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- Gollamudi R, Ghalib MH, Desai KK, Chaudhary I, Wong B, Einstein M, et al. Intravenous administration of Reolysin, a live replication competent RNA virus is safe in patients with advanced solid tumors. Invest New Drugs. 2010;28:641–649. doi: 10.1007/s10637-009-9279-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galanis E, Markovic SN, Suman VJ, Nuovo GJ, Vile RG, Kottke TJ, et al. Phase II trial of intravenous administration of Reolysin(®) (Reovirus Serotype-3-dearing Strain) in patients with metastatic melanoma. Mol Ther. 2012;20:1998–2003. doi: 10.1038/mt.2012.146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16:5564–5572. doi: 10.1158/1078-0432.CCR-10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lolkema MP, Arkenau HT, Harrington K, Roxburgh P, Morrison R, Roulstone V, et al. A phase I study of the combination of intravenous reovirus type 3 Dearing and gemcitabine in patients with advanced cancer. Clin Cancer Res. 2011;17:581–588. doi: 10.1158/1078-0432.CCR-10-2159. [DOI] [PubMed] [Google Scholar]
- Karapanagiotou EM, Roulstone V, Twigger K, Ball M, Tanay M, Nutting C, et al. Phase I/II trial of carboplatin and paclitaxel chemotherapy in combination with intravenous oncolytic reovirus in patients with advanced malignancies. Clin Cancer Res. 2012;18:2080–2089. doi: 10.1158/1078-0432.CCR-11-2181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roulstone V, Twigger K, Zaidi S, Pencavel T, Kyula JN, White C, et al. Synergistic cytotoxicity of oncolytic reovirus in combination with cisplatin-paclitaxel doublet chemotherapy. Gene Ther. 2013;20:521–528. doi: 10.1038/gt.2012.68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Twigger K, Vidal L, White CL, De Bono JS, Bhide S, Coffey M, et al. Enhanced in vitro and in vivo cytotoxicity of combined reovirus and radiotherapy. Clin Cancer Res. 2008;14:912–923. doi: 10.1158/1078-0432.CCR-07-1400. [DOI] [PubMed] [Google Scholar]
- Pandha HS, Heinemann L, Simpson GR, Melcher A, Prestwich R, Errington F, et al. Synergistic effects of oncolytic reovirus and cisplatin chemotherapy in murine malignant melanoma. Clin Cancer Res. 2009;15:6158–6166. doi: 10.1158/1078-0432.CCR-09-0796. [DOI] [PubMed] [Google Scholar]
- Sei S, Mussio JK, Yang QE, Nagashima K, Parchment RE, Coffey MC, et al. Synergistic antitumor activity of oncolytic reovirus and chemotherapeutic agents in non-small cell lung cancer cells. Mol Cancer. 2009;8:47. doi: 10.1186/1476-4598-8-47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinemann L, Simpson GR, Boxall A, Kottke T, Relph KL, Vile R, et al. Synergistic effects of oncolytic reovirus and docetaxel chemotherapy in prostate cancer. BMC Cancer. 2011;11:221. doi: 10.1186/1471-2407-11-221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kyula JN, Khan AA, Mansfield D, Karapanagiotou EM, McLaughlin M, Roulstone V, et al. Synergistic cytotoxicity of radiation and oncolytic Lister strain vaccinia in (V600D/E)BRAF mutant melanoma depends on JNK and TNF-a signaling. Oncogene. 2014;33:1700–1712. doi: 10.1038/onc.2013.112. [DOI] [PubMed] [Google Scholar]
- Marciniak SJ, Yun CY, Oyadomari S, Novoa I, Zhang Y, Jungreis R, et al. CHOP induces death by promoting protein synthesis and oxidation in the stressed endoplasmic reticulum. Genes Dev. 2004;18:3066–3077. doi: 10.1101/gad.1250704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carew JS, Espitia CM, Zhao W, Kelly KR, Coffey M, Freeman JW, et al. Reolysin is a novel reovirus-based agent that induces endoplasmic reticular stress-mediated apoptosis in pancreatic cancer. Cell Death Dis. 2013;4:e728. doi: 10.1038/cddis.2013.259. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang CC, Chen LH, Gillespie S, Wang YF, Kiejda KA, Zhang XD, et al. Inhibition of MEK sensitizes human melanoma cells to endoplasmic reticulum stress-induced apoptosis. Cancer Res. 2007;67:9750–9761. doi: 10.1158/0008-5472.CAN-07-2047. [DOI] [PubMed] [Google Scholar]
- Kelly KR, Espitia CM, Mahalingam D, Oyajobi BO, Coffey M, Giles FJ, et al. Reovirus therapy stimulates endoplasmic reticular stress, NOXA induction, and augments bortezomib-mediated apoptosis in multiple myeloma. Oncogene. 2012;31:3023–3038. doi: 10.1038/onc.2011.478. [DOI] [PubMed] [Google Scholar]
- Strong JE, Coffey MC, Tang D, Sabinin P, Lee PW. The molecular basis of viral oncolysis: usurpation of the Ras signaling pathway by reovirus. EMBO J. 1998;17:3351–3362. doi: 10.1093/emboj/17.12.3351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tabas I, Ron D. Integrating the mechanisms of apoptosis induced by endoplasmic reticulum stress. Nat Cell Biol. 2011;13:184–190. doi: 10.1038/ncb0311-184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu P, Han Z, Couvillon AD, Exton JH. Critical role of endogenous Akt/IAPs and MEK1/ERK pathways in counteracting endoplasmic reticulum stress-induced cell death. J Biol Chem. 2004;279:49420–49429. doi: 10.1074/jbc.M407700200. [DOI] [PubMed] [Google Scholar]
- Jiang CC, Lucas K, Avery-Kiejda KA, Wade M, deBock CE, Thorne RF, et al. Up-regulation of Mcl-1 is critical for survival of human melanoma cells upon endoplasmic reticulum stress. Cancer Res. 2008;68:6708–6717. doi: 10.1158/0008-5472.CAN-08-0349. [DOI] [PubMed] [Google Scholar]
- Tay KH, Luan Q, Croft A, Jiang CC, Jin L, Zhang XD, et al. Sustained IRE1 and ATF6 signaling is important for survival of melanoma cells undergoing ER stress. Cell Signal. 2014;26:287–294. doi: 10.1016/j.cellsig.2013.11.008. [DOI] [PubMed] [Google Scholar]
- Boyce M, Bryant KF, Jousse C, Long K, Harding HP, Scheuner D, et al. A selective inhibitor of eIF2alpha dephosphorylation protects cells from ER stress. Science. 2005;307:935–939. doi: 10.1126/science.1101902. [DOI] [PubMed] [Google Scholar]
- Tsaytler P, Harding HP, Ron D, Bertolotti A. Selective inhibition of a regulatory subunit of protein phosphatase 1 restores proteostasis. Science. 2011;332:91–94. doi: 10.1126/science.1201396. [DOI] [PubMed] [Google Scholar]
- Dhomen N, Reis-Filho JS, da Rocha Dias S, Hayward R, Savage K, Delmas V, et al. Oncogenic Braf induces melanocyte senescence and melanoma in mice. Cancer Cell. 2009;15:294–303. doi: 10.1016/j.ccr.2009.02.022. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
RT3D, PLX4720 and PD184352 IC50 doses for a panel of melanoma cell lines of various mutation status.