

Abstract
Antibodies armed with biologic drugs could greatly expand the therapeutic potential of antibody–drug conjugates for cancer therapy, broadening their application to disease targets currently limited by intracellular delivery barriers. Additional selectivity and new therapeutic approaches could be realized with intracellular protein drugs that more specifically target dysregulated pathways in hematologic cancers and other malignancies. A multifunctional polymeric delivery system for enhanced cytosolic delivery of protein drugs has been developed that incorporates endosomal-releasing activity, antibody targeting, and a biocompatible long-chain ethylene glycol component for optimized safety, pharmacokinetics, and tumor biodistribution. The pH-responsive polymeric micelle carrier, with an internalizing anti-CD22 monoclonal targeting antibody, effectively delivered a proapoptotic Bcl-2 interacting mediator (BIM) peptide drug that suppressed tumor growth for the duration of treatment and prolonged survival in a xenograft mouse model of human B-cell lymphoma. Antitumor drug activity was correlated with a mechanistic induction of the Bcl-2 pathway biomarker cleaved caspase-3 and a marked decrease in the Ki-67 proliferation biomarker. Broadening the intracellular target space by more effective delivery of protein/peptide drugs could expand the repertoire of antibody–drug conjugates to currently undruggable disease-specific targets and permit tailored drug strategies to stratified subpopulations and personalized medicines.
Introduction
The introduction of antibody and antibody–drug conjugate (ADC) therapeutics has had a major impact in the cancer field.1 ADCs, in particular, have shown considerable promise in improving the delivery of potent chemotoxins to tumor lesions while limiting off-target toxicities.2,3 Current ADCs are limited to membrane-permeable small molecule drugs, but expanding their repertoire to biologic drugs could provide therapies directed against currently “undruggable” intracellular targets. A promising example is a peptide that antagonizes prosurvival proteins overexpressed in a wide variety of cancers.4 Overexpression of the B-cell lymphoma-2 (Bcl-2) protein family is a hallmark of numerous malignancies and has been linked to poor prognosis and diminished response to chemotherapeutic and radiation treatment in patients with hematologic and other cancers.5 Patients with tumors near the apoptotic threshold by virtue of mitochondrial priming generally show better clinical responses to chemotherapy.6 Promising clinical trials with small molecule Bcl-2 antagonists including AT-101, GX15-070, ABT-199, and ABT-263 have prompted the development and exploration of protein therapeutics with expanded targets.7,8,9
Bcl-2 antagonists elicit apoptotic activity by binding Bcl-2 family proteins and displacing sequestered proapoptotic factors, including the release of BH3-only activators and the pore-forming BAK and BAX proteins.10 Apoptosis is initiated via BAK and BAX-mediated release of cytochrome c from mitochondria, which activates downstream caspases. BH3-mimetics like ABT-737, which specifically targets Bcl-2, Bcl-XL, and Bcl-w, have shown promising clinical outcomes as single agents and in combination with low-dose chemotherapy.11,12 In particular, the 20 amino acid BIM peptide (derived from the BH3-only proapoptotic BIM protein) has a high binding affinity (KD = 1–5 nM) to all six prosurvival Bcl-2 proteins (including Mcl-1, Bcl-B, and Bfl1), and has been shown to induce cytochrome c release in primed and unprimed mitochondria isolated from a variety of cancer cell lines.13,14 TAT-BIM and BIM-stapled peptide drugs have been shown to induce apoptosis in resistant hematologic cells and tumor models, and to preferentially affect cancer cells compared to normal cells.15,16 Both approaches are under active investigation for in vivo peptide delivery.
These initial studies have highlighted the potential therapeutic activities of BIM-derived peptides, but enhancing preferential tumor uptake, cytosolic delivery, and tumor biodistribution could provide improved therapeutic efficacy. We have earlier developed pH-responsive polymer compositions that increase cytosolic delivery of anti-CD22 and targeting monoclonal antibodies in cell culture, as well as other protein and nucleic acid drugs and vaccines.17,18,19,20 Here, we have generated a new pH-responsive, diblock copolymer with both targeting and intracellular peptide delivery capabilities. The first block of the polymer was designed to conjugate CD22-targeting monoclonal antibodies-linked to streptavidin (αCD22) and the BIM peptide, and to incorporate long-chain ethylene glycol (polyethylene glycol (PEG)) segments to improve pharmacokinetics of the peptide drug and enhance tumor distribution. The PEG segment offers biocompatibility and is well tolerated even at high doses often employed for peptide/protein delivery. The second block contains a pH-responsive composition that drives micelle formation at physiological pH and transitions to membrane disruptive unimers at endosomal pH to promote endosomal escape and intracellular delivery of the BIM peptide drug (Figure 1).21,22 The optimized polymer delivery system enhanced circulation of the BIM peptide and tumor distribution through αCD22 with a good safety profile at high dosages. The αCD22–polymer–BIM system was shown to suppress tumor growth and prolong survival time in combination with low-dose cytotoxic drugs in a human B-cell lymphoma xenograft mouse model.
Figure 1.

Comprehensive drug delivery system utilizing multifunctional polymer. The first block of the polymer contains three functionalized monomers incorporating ethylene glycol segments to optimize safety and tumor biodistribution, biotin to conjugate targeting αCD22-mAb-SA, and pyridyldisulfide moieties for reversible disulfide conjugation of BIM peptide (1 BIM per polymer). The second block contains DEAEMA and BMA, which provide pH-responsive, endosomal-releasing activity. BMA, butyl methacrylate; DEAEMA, diethylaminoethyl methacrylate.
Results
Synthesis of multifunctional diblock copolymers
Two versions of the PEGylated diblock copolymers were designed for in vivo evaluation. The first polymer contained a short 300 Da polyethylene glycol methacrylate (PEGMA) monomer (six ethylene oxide units) that has been investigated widely in the literature.23,24,25 The second incorporated a longer 950 Da PEGMA monomer (19 ethylene oxide units) that we recently described in a different polymer composition for small molecule delivery.26 The longer 950 Da PEGMA monomer had not been characterized in vivo or in the context of a pH-responsive, endosomal-releasing diblock copolymer. The 950 Da PEGMA-containing diblock copolymer (Pol950) was compared to a 300 Da PEGMA-containing diblock copolymer (Pol300) with antibody-targeting and peptide-conjugating elements incorporated into the corona-forming segment (Figure 1). The core-forming segment of each polymer contained a comonomer feed ratio of 60% diethylaminoethyl methacrylate and 40% butyl methacrylate , earlier shown to have strong endosomal-releasing properties.21 In this design, the polymeric micelle contains a hydrophilic corona-forming segment that hydrates and stabilizes the nanoparticle in aqueous solution while the pH-responsive segment makes up the hydrophobic core of the nanoparticle. Under physiological conditions (in D2O), the 1H NMR spectrum of these pH-responsive polymers show a significant peak suppression of the resonances associated with the comonomers found in the hydrophobic block. Resonances associated with comonomers in the hydrophilic block remain visible in D2O. The feed ratios, monomer compositions, molecular weight (Mn), and polydispersity (PDI) for Pol950 and Pol300 are given in Table 1. 1H NMR spectroscopy was employed to confirm copolymer composition by analysis of resonances associated with each of the comonomers (see Supplementary Figure S1). Experimental molecular weights (42,500 Da for Pol950 and 25,500 Da for Pol300) and dispersity values (1.14 for Pol950 and 1.13 for Pol300) were determined via size-exclusion gel-permeation chromatography (SEC-GPC). Excellent agreement existed between the experimentally determined target degree of polymerization and comonomer conversions, and between molar feed ratios and experimentally determined copolymer compositions. For example, the theoretical and experimental Mn for Pol300 was found to be 24,100 Da and 25,500 Da, respectively. SEC-GPC chromatograms demonstrate a clear shift from macro chain transfer agent (macroCTA) or the first polymer block synthesized) to a diblock copolymer for Pol950 and Pol300 (see Supplementary Figure S2a,b).
Table 1. Characterization of Pol950 and Pol300 macroCTAs and diblock copolymers.
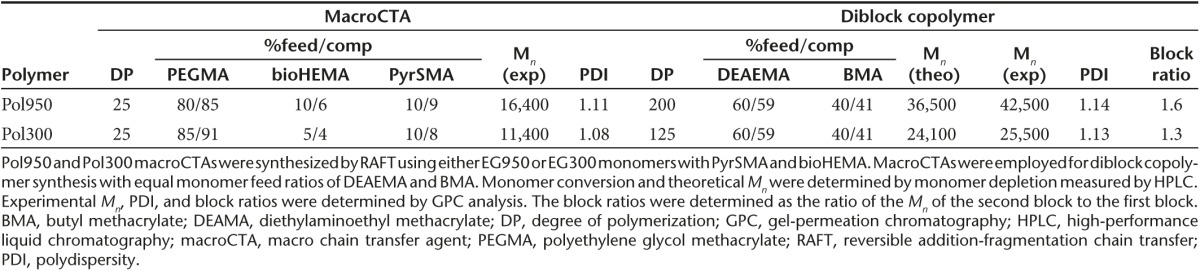
Characterization of micelle conjugates
To formulate micelles, polymers were dissolved in ethanol, diluted into sodium phosphate buffer (PBS), and spin filtered to remove ethanol. The Mn of the Pol950 and Pol300 micelles, measured by static light scattering (SLS), were 873 ± 8 and 643 ± 13 kDa, with 20 and 26 polymer chains per micelle, respectively (see Supplementary Figure S2c,d). The measured extinction coefficients of Pol950 and Pol300 micelles and the BIM peptide were 19,740/M/cm, 22,750/M/cm, and 4,660/M/cm, respectively (see Supplementary Figure S3a,b). The absorbance spectrum of Pol950 and Pol300 was first measured then blanked to quantify pyridyl disulfide (PDS) release (see Supplementary Figure S3c,d). The number of PDS per polymer chain, determined by tris(2-carboxyethyl)phosphine (TCEP) reduction, was 1.4 for Pol950 and 2.5 for Pol300 (see Supplementary Figure S3e,f). Incubation with a 1.1-fold excess of BIM per polymer chain resulted in ~1 BIM for Pol950 and Pol300 (see Supplementary Figure S3g,h). Conjugation of BIM or the inactive control BIM/LE (a single L▵E point mutation to a functionally critical amino acid) to Pol950 and Pol300 micelles, and reduction with TCEP was confirmed by SDS–PAGE (see Supplementary Figure S3i,j). Complexation of streptavidin with the biotin-containing Pol950 and Pol300 micelles was quantified by a 4′-hydroxyazobenzene-2-carboxylic acid (HABA) assay. An equimolar mixture of Pol950 or Pol300 with streptavidin resulted in 1.4 or 0.6 biotin-binding events, respectively (Figure 2a,b). Biotin complexation was unaffected when the polymer was first conjugated to BIM. The fully constructed, antibody-targeted Pol950 and Pol300 delivery systems are termed αCD22–Pol950–BIM and αCD22–Pol300–BIM. Their ability to form stable micelles was determined by dynamic light scattering (DLS) (Figure 2c,d). At pH 7.4, Pol950 and Pol300 micelles were 18 and 17 nm, respectively. No statistical variation in size was observed between Pol950, Pol950–BIM, and αCD22–Pol950–BIM, or the Pol300 equivalents. A spherical particle is assumed for particle size analysis, and the addition of one asymmetric mAb at most per micelle over a distribution of other unconjugated micelles did not have a significant effect on the average size of distribution. As micelles acidified toward endosomal pH a phase transition to unimers was observed around pH 6.6, as was designed by the comonomer feed and block ratios. Previous work by Manganiello et al.21 demonstrated that the pH at which the polymers transition from micelles to unimers could be tuned by varying the ratio of hydrophobic butyl methacrylate to protonatable diethylaminoethyl methacrylate in the polymer. The pH-responsive, membrane destabilizing activity of Pol950 and Pol300 conjugates was evaluated by hemolysis across pH 5.8–7.4 to mimic the endosomal acidification profile (Figures 2e,d).27,28,29 Pol950 conjugates exhibited increased hemolytic activity ≤ pH 6.6, whereas Pol300 conjugates exhibited increased hemolytic activity ≤ pH 7.0. Pol950 and Pol300 micelles retained their particle sizes and pH-responsive hemolytic activity when loaded with BIM and αCD22.
Figure 2.
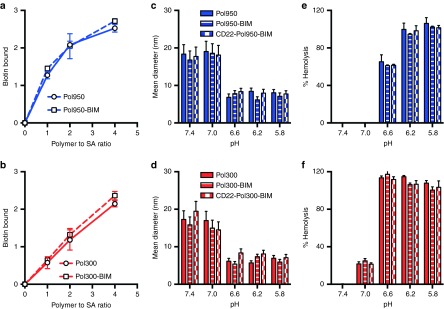
Characterization of αCD22-Pol950-BIM and αCD22-Pol300-BIM conjugates. After formulating polymer–peptide conjugates a HABA assay confirmed streptavidin complexation with (a) Pol950 (squares with blue dashed line) and Pol950-BIM conjugates (circles with solid blue line) and (b) Pol300 (squares with red dashed line) and Pol300-BIM conjugates (circles with solid red line). The number mean diameter of (c) Pol950 (blue solid), Pol950-BIM (blue stripes) and αCD22-Pol950-BIM (blue checkers) and (d) Pol300 (red solid), Pol300-BIM (red stripes), and αCD22-Pol300-BIM (red checkers) micelle particles (n = 3) was measured by DLS at 0.5 mg/ml across a range of five pH values. The hemolytic profile of (e) Pol950 conjugates and (f) Pol300 conjugates (n = 4) at 20 µg/ml across a range of pH values (n = 5) was also evaluated. DLS, dynamic light scattering.
Blood clearance, biodistribution, and toxicity of BIM delivery systems
The pharmacokinetics and biodistribution of dual radiolabeled conjugates were evaluated in Ramos tumor-xenograft mice (Figure 3). The beta half-life for αCD22 (mAb-SA) alone was 12.81 hours (95% confidence interval (CI) 11.63–14.25 hours). The half-life increased to 17.31 hours (95% CI 12.30–29.25 hours) with αCD22–Pol950–BIM and reduced to 6.64 hours (95% CI 6.25–7.03 hours) with αCD22–Pol300–BIM (Figure 3a). Free BIM peptide was cleared from blood within 15 minutes but the half-life increased to 10.85 hours (95% CI 8.66–14.51 hours) with αCD22–Pol950–BIM and 9.47 hours (95% CI 5.50–23.53 hours) with αCD22–Pol300–BIM (Figure 3b). The peptide drug to mAb ratio in the blood decreased from 90:1 to 39:1 for αCD22–Pol950–BIM, and 90:1 to 29:1 for αCD22–Pol300–BIM. Based on area under the clearance curve calculations, the bioavailability of αCD22 increased from 1.089 mg-h/ml to 1.628 mg-h/ml for αCD22–Pol950–BIM and 1.141 mg-h/ml for αCD22–Pol300–BIM. The bioavailability of BIM was 0.018 mg-h/ml, and increased to 1.204 mg-h/ml for αCD22–Pol950–BIM and 0.826 mg-h/ml for αCD22–Pol300–BIM.
Figure 3.
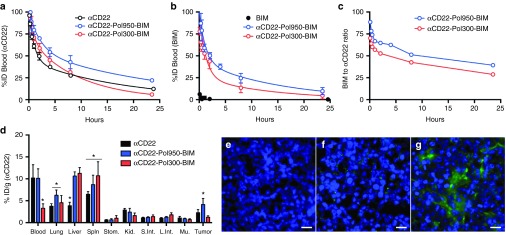
αCD22-Pol950-BIM conjugates enhance blood circulation half-life, peptide stability, and tumor distribution. The blood clearance of dual-radiolabeled αCD22-Pol950-BIM and αCD22-Pol300-BIM conjugates in comparison to free αCD22 or BIM were monitored through (a) 3H-labeled αCD22 and (b) 14C-labeled BIM (n = 3). Solid lines represent a two compartment open model with 1/y2 weighting. (c) Stability of disulfide-linked BIM on Pol950 and Pol300 was measured by following the BIM to αCD22 ratio in the blood (initial ratio = 90:1). Data in plots a and b represent the mean ± SD. (d) Biodistribution of αCD22-Pol950-BIM and αCD22-Pol300-BIM conjugates after 24 hours was measured by following 3H-αCD22 (n = 5). Asterisks represent statistical significance (P < 0.05) by two-way ANOVA. Data represent mean ± SD. (e–f) Representative fluorescence microscopy images of tumor sections 48 hours after administration of Alexa Fluor 647-labeled Pol950 and Pol300 without BIM peptide. Treatments include e PBS, f αCD22-Pol300, and (g) αCD22-Pol950 (n = 3). Nuclei stained with DAPI (blue) and polymer labeled with Alexa Fluor 647 (green). Scale bar = 15 nm.
Biodistribution was quantified 24 hours after administration by following [3H]αCD22 due to insufficient signal from [14C]BIM, with the %-ID in each organ normalized to tissue weight (Figure 3d). In the tumors, the %ID/g of αCD22 increased from 2.3 to 4.1 for αCD22–Pol950–BIM, and decreased from 2.3 to 1.2 for αCD22–Pol300–BIM. These accumulation values are consistent with prior studies utilizing this particular αCD22 mAb.30,31 The αCD22–Pol950–BIM and αCD22–Pol300–BIM delivery systems increased accumulation in the liver and spleen, but at relatively low levels in comparison to those reported for other particulate delivery systems administered intravenously.32,33,34,35 The intratumoral distribution of fluorescent Pol950 and Pol300 was evaluated with αCD22 targeting, 2 days after administration (Figures 3e–g). Deconvolution fluorescence microscopy demonstrated a diffuse accumulation of αCD22–Pol950 along the periphery and through the center of the tumor relative to few punctate spots with αCD22–Pol300.
Finally, an initial organ toxicity screening was conducted with αCD22–Pol950–BIM and αCD22–Pol300–BIM. 72 hours after administration of high polymer doses at equimolar ratios (300 mg/kg for Pol950 and 159 mg/kg for Pol300), a comprehensive metabolic and enzyme panel was performed across the major organs (Figure 4). Overall enzyme levels fell within normal values, with a significant elevation of liver AST and creatine kinase with αCD22–Pol300–BIM.
Figure 4.
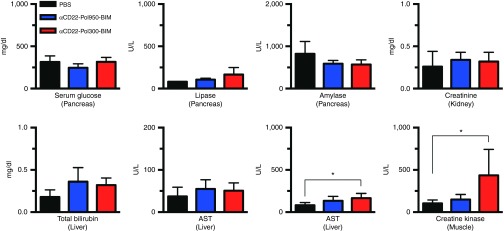
Comprehensive metabolic and enzyme panel demonstrates single dose safety profile of αCD22-Pol950-BIM conjugates. A comprehensive metabolic and enzyme panel on major organs was conducted 72 hours after treatment with PBS (black), αCD22-Pol950-BIM (blue), and αCD22-Pol300-BIM (red). The doses administered were 15 mg/kg for αCD22, 300 mg/kg for Pol950, 159 mg/kg for Pol300, and 18 mg/kg for BIM. Data represents the mean ± SD, n = 5; *indicates statistical significance by two-way ANOVA. PBS, sodium phosphate buffer.
Induction of proapoptotic activity with BIM conjugates in a Bcl tumor model
The superior tumor targeting, pharmacokinetic, and safety properties of αCD22–Pol950–BIM led to the selection of this delivery system for initial tumor activity studies. The mechanistic ability of the BIM peptide drug to antagonize Bcl-2 family targets in a B-cell lymphoma xenograft mouse model was evaluated by measuring levels of a cleaved caspase-3 (CC-3) apoptosis marker and the Ki-67 cell proliferation marker within tumors. Mice were treated with three different drug formulations in combination with low-dose cyclophosphamide and bortezomib. The drug formulations and controls tested included αCD22–Pol950, αCD22–Pol950–BIM/LE, and αCD22–Pol950–BIM. 48 hours after administration, tumors (n = 5) were harvested and stained for CC-3 activity. A representative image of the PBS control (Figure 5a) and αCD22–Pol950–BIM treated tumors (Figure 5b) are shown. The percentage of CC-3+ cells in each treatment group was also quantified (Figure 5c). The αCD22–Pol950 treatments with and without BIM/LE were significantly higher than PBS and chemo with mean values of 2.68% and 2.58%, respectively. The αCD22–Pol950–BIM treatment exhibited nearly double the CC-3 activity at 4.24% and was significantly greater than all other treatments. A second set of tumors (n = 5) were harvested and stained for Ki-67 activity, 72 hours after administration. A representative image of the PBS control (Figure 5d) and αCD22–Pol950–BIM treated tumor (Figure 5e) are shown. The density of Ki-67+ cells/mm2 in tumors was also quantified (Figure 5f). The density in the PBS control was 16,870 Ki-67+ cells/mm2. Chemotherapy alone or in combination with αCD22–Pol950 or αCD22–Pol950–BIM/LE decreased the density to 14,220, 14,290, and 13,916 Ki-67+ cells/mm2, respectively. Tumors treated with αCD22–Pol950–BIM were significantly less dense with 11,526 Ki-67+ cells/mm2 (P < 0.02). Two mice in the αCD22–Pol950–BIM group exhibited lower proliferation densities resulting in a wider downward distribution compared to the other treatments.
Figure 5.

Apoptotic induction in tumors demonstrated by CC-3 and Ki-67 marker analysis. Lymphoma tumor-xenograft mice were treated with a single low-dose chemo (cyclophosphamide and bortezomib) in combination with αCD22-Pol950, αCD22-Pol950-BIM/LE, or αCD22-Pol950-BIM. Tumors (n = 5) were harvested after 48 and 72 hours, processed for immunohistochemistry, stained for CC-3 and Ki-67 markers and imaged on a TissueFAXS microscope (10×). Representative images of (a) PBS control and (b) αCD22-Pol950-BIM-treated tumors stained for CC-3 activity (brown) and nuclei (blue) (scale bar = 100 µm). (c) % CC-3+ cells in tumors quantified using HistoQuest software. Data represents mean ± SD. Representative images of (d) PBS control and (e) αCD22-Pol950-BIM treated tumors stained for Ki-67 activity (brown) and nuclei (blue). (f) Density of Ki-67+ cells/mm2 within entire tumors was measured. Boxes represent the first and third quartiles around the median and whiskers represent the minimum and maximum of each treatment group. For both staining protocols, entire bisected tumor sections were analyzed. Data analyzed with ordinary one-way ANOVA using Tukey's multiple comparison test for significance. CC, cleaved caspase; PBS, sodium phosphate buffer.
Tumor growth suppression and survival
In myc-driven aggressive lymphomas, small molecule Bcl-2 antagonists have been shown to sensitize tumors and synergize with low-dose chemotherapy.12 The therapeutic efficacy of αCD22–Pol950–BIM was therefore evaluated in combination with low-dose chemotherapy (35 mg/kg cyclophosphamide, 0.5 mg/kg bortezomib) in a (c-myc+) Ramos-xenograft mouse model.36,37 Tumor bearing mice (n = 10) were treated with αCD22–Pol950–BIM, αCD22–Pol950–BIM/LE, or αCD22–mCTA–BIM (mCTA denotes the Pol950 macroCTA without the endosomal-releasing segment), on days 6, 9, 12, and 15. Mice were euthanized when tumors reached a volume of 1250 mm3. The therapeutic efficacy was evaluated using a Kaplan–Meier survival curve combining data from three different studies (Figure 6). Median survival times in the first, second, and third quartiles were calculated for each treatment (see Supplementary Table S1). A log-rank (Mantel–Cox) test confirmed that the survival curve for the BIM delivery system was significantly greater than all other treatments (P < 0.0001, χ2 = 60.62). The active BIM delivery system significantly extended the median survival time to 29 days, compared to 16 days with PBS and 20 days with chemotherapy. The inactive BIM/LE point mutant peptide control abrogated the antitumor activity, as did the polymer control where the endosomal-releasing block was removed from the carrier. The Pol950 polymer alone did not show toxicity or change survival time relative to the PBS control. Although no mice in any control group survived past 31 days, 30% of mice in the BIM delivery system survived between 31 and 43 days.
Figure 6.

Intracellular delivery of BIM conjugates extends survival with low dose chemotherapy. Ramos xenograft mice (n = 10) were injected intraperitoneally with cyclophosphamide (35 mg/kg) and bortezomib (0.5 mg/kg) [chemo] 30 minutes before intravenous injection with αCD22 (15 mg/kg), Pol950 (300 mg/kg) or mCTA (115 mg/kg), BIM/LE (18 mg/kg) or BIM (18 mg/kg) on days 6, 9, 12, and 15. A log-rank (Mantel Cox) test was used to determine statistical significance (P < 0.0001) for the Kaplan–Meier survival curve. Mice were euthanized when tumors reached 1250 mm3. The PBS and chemo treatments represent data from three separate studies and chemo+Pol950 from two separate studies. mCTA, macro chain transfer agent.
The corresponding tumor growth curves were plotted on a log scale and fit with a linear curve (see Supplementary Figure S4a–g). While the control treated tumors followed an exponential growth response (a linear fit on the log scale), the BIM delivery system suppressed tumor growth for the first 20 days, causing significant further delay in tumor growth after treatment termination. The doubling time (DT) and tumor growth delay were also calculated for each treatment (see Supplementary Table S1).38,39 The DT for the PBS, Pol950, and chemo groups were 3.15, 3.30, and 4.06 days, respectively. The BIM delivery system increased the DT to 9.07 days with a tumor growth delay of 5.92 days. Replacement of BIM with BIM/LE decreased the DT to 4.63 days, whereas replacing Pol950 with the mCTA decreased the DT to 5.06 days.
Induction of mitochondrial cytochrome c release in cancer cell lines
To explore the potency of the BIM peptide drug, mitochondria were isolated from 10 different malignant hematologic lines and treated with BIM (0.01–100 µmol/l) to induce cytochrome c release (Figure 7). The inactive control BIM/LE did not exhibit any activity in Ramos or Ramos-AW cells. The effective concentration for 50% activity (EC50) was also calculated using a dose–response curve. The BIM peptide had an EC50 value of 673 nmol/l in Ramos cells but was 10–20 times more potent in Granta-519 (51 nmol/l), JVM-13 (40 nmol/l), and K562 (32 nmol/l). These results demonstrate that the BIM peptide is active across a range of cancer cell lines but with variation in potency as expected with cells exhibiting different levels and types of Bcl-2 family targets.
Figure 7.
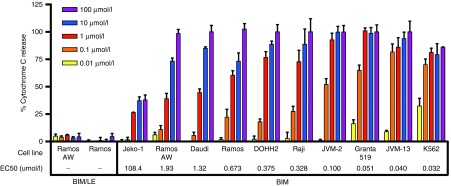
BIM activates cytochrome c release in a range of cancer cells lines with varying potency. Mitochondria isolated from 10 different cancer cell lines were incubated with BIM or BIM/LE for 30 minutes at a range of concentrations (0.01–100 µmol/l). BIM/LE (L62E) has a knockout mutation in the binding interface. The % cytochrome c release in the supernatant was quantified using an ELISA assay. The % cytochrome c release was defined as ((A450, sample)–(A450, DMSO))/((A450, Triton-X100)–(A450, DMSO)) × 100, where DMSO was used as a negative control and 1% Triton-X100 as a positive control. Data represents mean ± SD with n = 4. A nonlinear dose–response curve was fit to each data set using GraphPad Prism to determine the potency (EC50) of BIM in each cell line (italicized in the figure).
Discussion
Non-Hodgkin lymphomas are generally treated with chemotherapy in combination with anti-CD20 mAbs. More recently, radiolabeled mAbs and ADCs have been employed to selectively target hematologic tumors. Current ADCs exploit small molecule drugs that are typically hydrophobic and diffuse broadly inside cells to reach their intracellular targets. In this study, an antibody-targeted, intracellular delivery system has been developed to extend ADCs to protein/peptide drugs that act against disease-specific targets accessed from the cytosolic compartment. By opening stratified subpopulations associated with particular disease targets, intracellular-acting protein drugs could similarly open new personalized medicines. Although exciting in potential, development has been stymied by delivery barriers that make it difficult to achieve sufficient protein quantities to inhibit intracellular targets while maintaining appropriate pharmacokinetic and therapeutic index properties.
We designed a multifunctional carrier with a corona composed primarily of PEG graft segments in a near “bottle brush” density to stabilize and sterically shield the disulfide-linked peptide from degradation in the blood. The biocompatibility of the PEG corona coupled with the potent endosomal releasing technology allowed for a high dose of peptide-based therapy to be employed. Pol950 and Pol300 polymers were synthesized to study the effect of PEGMA monomer length on the polymer stability in vivo. The longer segment PEGMA displayed dramatically better pharmacokinetic and tumor distribution properties. A potential key additional advantage of this design is that it allows the corona segment to achieve a high ethylene glycol weight percentage at relatively low degrees of polymerization (monomer numbers). This is potentially important in regard to polymer clearance and safety properties, because all the monomers are designed with ester junctions at the carbon backbone to degrade down to the minimal backbone segment. Decreasing the number of monomers while maintaining function leads to smaller final poly(methacrylic acid) degradation products after ester hydrolysis that are only on the order of ≤10 kDa in molecular weight. The reversible addition-fragmentation chain transfer (RAFT) polymerization technology also allows for easy scale up of diblock copolymers for clinical grade production.40,41,42
Pharmacokinetic and biodistribution studies revealed that Pol950 not only increased the circulation half-life of αCD22 43%, but also enhanced tumor accumulation by 83%. Although both PEG polymers demonstrated good safety profiles, the Pol950 conjugates were within statistical significance of PBS across the metabolic and enzyme panel. A combination therapy design incorporating concurrent treatment with cyclophosphamide and bortezomib was based on previous literature results where Bcl-2 antisense and small molecule ABT-737 drug studies showed promising synergy responses with these chemotherapeutic agents.43,44 In this study, direct measurement of target-induced activity within tumors after just a single dose revealed a 4.2-fold increase in the percentage of apoptotic cells (CC-3) and a 32% decrease in the density of proliferating cells (Ki-67).
These observed antitumor effects translated into therapeutic responses with the active BIM delivery system in a human B-cell lymphoma xenograft model. The BIM delivery system suppressed tumor growth for the duration of the treatment cycle (6–15 days) and for an additional 5 days post-treatment. BIM treatment significantly extended the median survival by 61% relative to the PBS control in the Kaplan–Meier survival analysis. The polymer delivery controls slightly extended survival in the Kaplan–Meier survival analysis over the chemotherapy alone with a low but significant increase in the DT as well. Low activity of the BIM/LE delivery system suggests that the combination of chemotherapy, αCD22 mAb, and Pol950 elicit some therapeutic effects. A reduction in DT and median survival with the Pol950-mCTA in place of Pol950 provides further evidence to the importance of the endosomal-releasing segment for intracellular delivery of the BIM peptide drug.
These results demonstrate the potential utility of a new antibody-targeted, intracellular delivery system for peptides/proteins that antagonize disease targets located in the cytosolic compartment. Cytochrome c release studies suggest that potentially better results could be achieved in xenograft tumors with a more optimal priming profile (e.g., Granta-519). Characterization of mitochondrial priming could also be a way to identify patients that could benefit from BIM-directed therapies. Current limitations that require future optimization before clinical translation in humans include evolution of the polymer to provide direct conjugation of the targeting antibody to avoid an immunogenic response to streptavidin in patients.45,46 While the polymer delivery system substantially improved the blood half-life and stability of the BIM peptide, further stabilization on the polymer could improve tumor biodistribution. Optimization of the dosing regime could improve activity and synergy and eventually lead to long-term tumor regression and disease-free survival. These preliminary studies help to address a key barrier to intracellular biologic delivery using an antibody-targeted polymer delivery vehicle with promising antitumor activity.
Materials and Methods
Antibody synthesis. HD39 hybridomas were injected into pristine-primed mice to generate ascites and the αCD22 monoclonal antibody was purified as described earlier.31 The αCD22 mAb was conjugated to streptavidin via a succinimidyl-4-(N-maleimidomethyl)-cyclohexane-1-carboxylate heterobifunctional linker to form a covalent chemical conjugate using previously described methods,47 resulting in one streptavidin per αCD22. The αCD22–streptavidin conjugate will be referred to as αCD22 in the following.
Peptide synthesis. Peptides were synthesized by Fmoc solid phase synthesis on a PS3 (Protein Technologies) synthesizer. BIM and BIM-L62E (BIM/LE) were synthesized on Rink Amide-MBHA resin (EMD Millipore, Billerica, MA) with the following sequences: Ac-MRPEIWIAQELRRIGDEFNAC-ON (BIM) and Ac-MRPEIWIAQEERRIGDEFNAC-ON (BIM/LE). Crude peptides were purified by reverse phase-high-performance liquid chromatography (HPLC) on a Jupiter 5-μm C18 300Å column (Phenomenex, Torrance, CA) with an Agilent 1260 HPLC (Agilent, Santa Clara, CA). Final products were confirmed by ion trap mass spectrometry with electrospray.
RAFT synthesis of diblock copolymers. MacroCTAs, defined as the macro chain transfer agents or the first polymer block used for subsequent synthesis of the diblock copolymers, were synthesized via RAFT using azobis(4-cyanopentanoic acid) as the initiator, 4-cyanopentanoic acid dithiobenzoate as the chain transfer agent (CTA), with 20 wt% monomer in DMSO. PDS and bioHEMA monomers were synthesized and characterized by 1H-NMR as described in Supplementary Materials (see Supplementary Figure S5). The initial monomer ([M]0) to CTA ([CTA]0) to initiator ([I]0) ratios for the Pol950 and Pol300 macroCTAs was 25:1:0.1. Feed ratios for each macroCTA can be found in Table 1. Polymerization solutions were vortexed, transferred to septa-sealed round bottom flasks, purged under N2 for 30 minutes and incubated in a 70 °C water bath for 14 hours. Resultant polymers were isolated by precipitation in diethyl ether at room temperature. The precipitated polymers were then dissolved in acetone and reprecipitated into diethyl ether (×6), then dried under vacuum overnight. The macroCTAs were then used for subsequent RAFT copolymerization with azobis(4-cyanopentanoic acid) as the initiator with 50 wt% monomer in 1,4-dioxane. Polymerization solutions were vortexed, transferred to septa-sealed round bottom flasks, purged under N2 for 20 minutes and incubated in a 70 °C water bath for 8 hours. The resultant polymers were isolated by precipitation in petroleum ether. The precipitated polymers were then dissolved in acetone and reprecipitated into petroleum ether (×5). Purity of macroCTAs and diblock copolymers was evaluated by 1H NMR.
Characterization of polymers by SEC-GPC and HPLC. The number average molecular weight (Mn) and PDI for the macroCTAs and the diblock copolymers were measured by GPC using Tosoh SEC TSK GEL α-3000 and α-4000 columns (Tosoh Bioscience, Montgomeryville, PA) connected in series to a 1200 Series liquid chromatography system (Agilent, Santa Clara, CA) and a miniDAWN TREOS three-angle light scattering instrument with an Optilab T-rEX refractive index detector (Wyatt Technology, Santa Barbara, CA). HPLC-grade dimethylformamide (DMF) containing 0.1 wt% LiBr at 60 °C was used as the mobile phase at a flow rate of 1 ml/minute. Monomer conversion and theoretical Mn were quantified by RP-HPLC using aliquots collected at T0 and Tx with an acetonitrile/H2O gradient from 10 to 90%. Monomer peaks were identified by first running pure monomers on the HPLC. Monomer depletion was measured at 220 nm wavelength and calculated by subtracting Tx peaks from T0 peaks in the polymer sample. The %monomer conversion was determined by the equation: (Peak (T0)–Peak (Tx))/(Peak (T0)).
Formulation of polymer micelles. Dried polymers were dissolved in ethanol at 100 mg/ml followed by dilution into PBS (100 mmol/l, pH 7.0) to a final concentration of 10 mg/ml. Serial dilutions were made and measured by UV–Vis to determine the extinction coefficient at 290 nm. Ethanol content was reduced to 0.01% in polymer solutions using Amicon-15 Ultra 30k MWCO spin columns (EMD Millipore). Polymer concentrations were measured by UV–Vis and aliquots were made for peptide conjugation.
Formulation of polymer–peptide conjugates. BIM or BIM/LE was conjugated to the PDS groups on the polymer micelles via disulfide exchange reactions. The extinction coefficient of BIM and BIM/LE was determined by dissolving peptides in anhydrous DMSO, performing serial dilutions and measuring absorbance at 280 nm. A linear regression of the data was used to calculate the extinction coefficient. Concentrated stocks of BIM and BIM/LE in DMSO were added to polymer micelles at a 1.1-molar excess of peptide to polymer chains, vortexed, and allowed to react overnight at room temp. The extent of conjugation was determined via pyridyldisulfide release, nonreducing SDS–PAGE, and reverse-phase HPLC. Conversion of pyridyldisulfide to 2-mercaptopyridine was monitored by UV–Vis at 343 nm to measure PDS concentration with an extinction coefficient, ɛ = 8,080/M/cm. The percent conjugation was determined by the equation C(PDS)/C(BIM) × 100. For SDS–PAGE, conjugates (5 µg of peptide) were run at 137 V, on a 4–20% Tris–glycine PROTEAN TGC precast gel, Bio-Rad) in the presence and absence of a 100-fold excess of soluble TCEP. Peptide conjugation and reduction from the polymer with glutathione was confirmed by reverse-phase HPLC. The disappearance of a peptide peak after conjugation and reappearance in the presence of glutathione were confirmed. Free peptide was run as a positive control.
Formulation of antibody–polymer–peptide conjugates. To quantify the availability of bioHEMA on the polymer micelles, a HABA assay was performed as described earlier.17 In brief, streptavidin and antibody-streptavidin were incubated with an excess of biotin, polymer or polymer-peptide, and a 40-fold molar excess of HABA and allowed to react for 60 minutes. The displacement of HABA by the biotinylated polymer was measured by UV–Vis (500 nm) and the amount of biotin bound was determined with a standard curve. To formulate antibody–polymer–peptide conjugates, antibody was mixed with polymer–peptide conjugates 2 hours prior to characterization or animal studies. For animal studies, an excess of 90 polymer chains per antibody was used to allow for the maximum dose of polymer (300 mg/kg) with a clinically relevant antibody dose (15 mg/kg).
Characterization of micelles by SLS and DLS. The polymer micelles were analyzed by DLS to determine particle mean diameter and by SLS to measure particle molecular weight using a Malvern Zetasizer Nano, equipped with a 5-mW He–Ne laser at 633 nm. DLS measurements were performed in a range of pH buffers at 0.5 mg/ml with the mean diameter defined as the ±half peak width. Measurements were performed in triplicate comparing polymer, polymer–peptide and antibody–polymer–peptide conjugates. For SLS measurements, serial dilutions of the polymer solutions were made (0.125–1 mg/ml) and injected into an Optilab T-rEX refractive index detector to determine dn/dc. The molecular weight (Mw) and second viral coefficient (A2) of the micelles were determined using a Debye plot generated by the SLS measurements using a constant scattering angle (173o) and a Rayleigh equation: KC/Rθ = (1/Mw + 2A2C), where C varies from 1 to 0.125 mg/ml, K is an optical constant, and Rθ is the Rayleigh ratio of the scattered to incident light intensity. The Mw of the micelles was used to determine an aggregation number by dividing the Mw of the micelles by the Mw of the unimers (determined by GPC).
pH-responsive hemolysis assay. The membrane destabilizing activity of the polymer conjugates was measured using a red blood cell hemolysis assay as described earlier.48 Polymer conjugates in quadruplicate were incubated for 1 hour at 37 °C in the presence of human red blood cells at 20 μg/ml in 100 mmol/l PBS (supplemented with 150 mmol/l NaCl) in the pH range of the endosomal trafficking pathway (7.4, 7.0, 6.6, 6.2, and 5.8). The percentage of hemolysis was determined spectrophotometrically by measuring the amount of hemoglobin released (A541,nm) and normalized to a 100% hemolysis control (1% Triton-X100).
Maintenance of cell lines. The Ramos, Ramos-AW, JVM-2, JVM-13, Raji, Daudi, and DOHH2 cell lines were maintained in log-phase growth in RPMI 1640 medium containing L-glutamine, 25 mmol/l HEPES supplemented with 1% penicillin–streptomycin (GIBCO) and 10% fetal bovine serum (Invitrogen). Granta-519 and K562 cells were grown in Iscove's DMEM supplemented with 10% fetal bovine serum. All cells were maintained in log-phase growth at 37 °C and 5% CO2.
Formation of Ramos tumor xenografts. For all in vivo studies, BALB/c nu/nu mice (6–8 weeks old) were used from Harlan Sprague–Dawley and housed under protocols approved by the FHCRC Institutional Animal Care and Use Committee. Mice were placed on a biotin-free diet (Purina Feed) for the duration of each study. Ramos cells resuspended in PBS (5 × 107/ml) were injected subcutaneously (200 µl) in the right flank of each mouse. Tumors were allowed to grow for 6 days to a volume of 50 mm3.
Radiolabeling αCD22 mAb and BIM peptide. αCD22 (2 mg/ml) in borate buffer (50 mmol/l sodium borate, 150 mmol/l NaCl, pH 8.5) was reacted with equimolar 3H-N-succinimidyl propionate (5 mCi/ml in DMF) (American Radiolabeled Chemicals) for 2 hours. 3H-αCD22 was purified with a desalting column and concentrated by spin filtration. Nonacetylated BIM peptide in DMSO was reacted with a fivefold excess of 14C-acetic anhydride (in toluene) for 30 minutes, then precipitation into ether. Specific radioactivity of 3H-αCD22 and 14C-BIM was measured by scintillation counting in ULTIMA GOLD (Perkin Elmer) scintillation fluid.
Pharmacokinetics and biodistribution of dual-radiolabeled conjugates. Conjugates were formulated with radiolabeled proteins as described earlier. For whole blood clearance, mice (n = 5) received conjugate formulations at the following quantities: 1.4 nmol of 3H-αCD22, 126 nmol of polymer, and 126 nmol of 14C-BIM via tail vein injection. Serial blood samples were drawn (n = 3) from the retro-orbital venus plexus using a 10-µl capillary tube at the following timepoints: 5 minutes, 15 minutes, 30 minutes, 1 hour, 2 hours, 4 hours, 8 hours, and 24 hours (Exact injection and collection times were recorded for each mouse). Blood was immediately transferred to scintillation vials with 500 µl of Solvable (Perkin Elmer), and 5 ml of ULTIMA GOLD scintillation fluid was added. 3H and 14C activity was measured by scintillation counting using an automated deconvolution program for radioactive signal. Syringe weights were measured before and after injection to calculate total injection volume. Specific radioactivity for each treatment was measured in triplicate from stock solutions. For blood clearance calculations, 80 ml (blood)/kg (mouse) was assumed. Blood concentrations were plotted with GraphPad Prism and fit with a two-compartmental open model (biexponential equation) with 1/y2 weighting. The mice (n = 5) were weighed, sacrificed, and bled by heart stick 24 hours after administration. Tumors, lung, liver, spleen, stomach, kidney, small intestine, large intestine, and muscle were harvested, weighed, and dissolved in Solvable (5–10 ml/g) and processed as recommended by manufacturer (Perkin Elmer). Solutions were diluted into ULTIMA GOLD, vortexed, reacted overnight at room temperature and run on a scintillation counter to determine the percentage injected dose per gram (%ID/g) of tissue.
Intratumoral distribution of fluorescently labeled polymer conjugates. For intratumoral distribution studies, Pol950 was labeled with Alexa Fluor 647. To fluorescently label polymer, a 10-fold molar excess of dithiothreitol (DTT) was added to polymer (10 mg/ml) in PBS to reduce PDS groups. A fourfold molar excess of Alexa Fluor 647 C2 maleimide (10 mmol/l) in anhydrous DMSO was added to the polymer solution for 2 hours to form thioether linkages. Fluorescent polymer was purified using a PD-10 desalting column (GE Life Sciences) to remove unreacted dye. αCD22 was added to fluorescent polymer at a ratio of 1:90 (mAb to polymer chain) to form αCD22–Pol950 fluorescent micelles then injected via tail vein in tumor-bearing mice (n = 3). The mice were euthanized and tumors excised, then fixed in formalin 48 hours after treatment. Tumors were processed by the Histology core at FHCRC, nuclei stained with DAPI, and mounted on microscope slides with coverslips. Tumors slides were imaged using a Nikon Ti-E microscope using a mercury lamp and 60× objective with the following filter sets: 350/400 nm (EX/EM) for DAPI and 650/680 nm (EX/EM) for Alexa Fluor 647 (49000 Series, Chroma Technology, Rockingham, VT). Images were arbitrarily selected based on DAPI staining with four image stacks per tumor slide, 20 slices per Z-stack with a 0.5 μm step size. Z-stacks were deconvolved using object-based measurement software (Volocity, PerkinElmer) using a calculated point-spread function for the blue and far-red channels with 25 iterations or 100% CI.
Comprehensive metabolic and enzyme panel. Mice (n = 5) were injected via tail vein with PBS, αCD22–Pol950–BIM, or αCD22–Pol300–BIM at a dose of 15 mg/kg (αCD22), 300 mg/kg (Pol950) or 159 mg/kg (Pol300), and 18 mg/kg (BIM). After 72 hours, mice were sacrificed and blood was collected. Plasma was separated from red blood cells by centrifugation and samples were submitted to Phoenix Central Laboratory for analysis of metabolic and enzyme levels.
CC-3 and Ki-67 activity in tumor xenografts. Tumored xenograft mice (n = 10) were injected intraperitoneally with 200 μl solution of cyclophosphamide (35 mg/kg) and bortezomib (0.5 mg/kg). After 30 minutes, mice were injected intravenously with conjugates: αCD22 (15 mg/kg), Pol950 (300 mg/kg), BIM/LE or BIM (18 mg/kg). Mice were sacrificed after 48 and 72 hours (n = 5 per time point), tumors were harvested, bisected, fixed in formalin, processed for immunohistochemistry by the FHCRC Histology Departments and stained for CC-3 and Ki-67. Entire tumor sections were imaged with a TissueFAXS microscope at original magnification ×10 then analyzed with HistoQuest software. Nuclei were stained blue and CC-3 or Ki-67 positive cells were stained brown. The % CC-3+ cells for each tumor and the density of Ki-67+ cells per tumor area were calculated with HistoQuest software.
Tumor regression study. For therapy studies, mice (n = 10) were treated with chemotherapy and conjugates on days 6, 9, 12, and 15 after tumor cell innoculation as described earlier. Body weight was monitored and tumor sizes were measured three times a week with calipers in the x, y, and z plane while blinded to treatment group. Mice were euthanized when tumors reached a volume of 1250 mm3.
Cytochrome C release assay. 109 cells were equilibrated in 5 ml homogenization buffer (0.25 mol/l sucrose, 1 mmol/l EGTA, 10 mmol/l HEPES/NaOH, 0.5% BSA, pH 7.4, Roche Complete protease inhibitor) for 5 minutes at 4 °C and remained at 4 °C unless otherwise stated. Cells were homogenized under N2 pressure (400 psi) in a nitrogen bomb (Parr Instrument Company) for 10 minutes. Membrane disruption was confirmed by Trypan Blue staining. Cells were centrifuged (750 g) for 10 minutes in 15 ml round bottom tubes (BD (35)2059) to remove intact cells. Supernatant was centrifuged again (12,000 g) for 12 minutes. The mitochondria pellet was resuspended in 300 μl wash buffer (0.25 mol/l sucrose, 1 mmol/l EDTA, 10 mmol/l Tris–HCl, pH 7.4). BIM and BIMLE peptides were predissolved in DMSO then incubated with 25 µg (based on BCA assay (Sigma)) of mitochondria in 50 μl of experimental buffer (125 mmol/l KCl, 10 mmol/l Tris–MOPS, pH 7.4, 5 mmol/l glutamate, 2.5 mmol/l malate, 1 mmol/l KPO4, 10 μmol/l EGTA–Tris, pH 7.4) for 30 minutes at 37 °C. Reaction solutions were centrifuged (18,000 g) for 10 minutes and cytochrome c release was quantified using a Cytochrome c ELISA kit (Life Technologies). Complete cytochrome c release was quantified by treatment using 1% Triton X-100 and background with DMSO.
SUPPLEMENTARY MATERIAL Figure S1. 1H-NMR spectrum of (a) Pol950 and (b) Pol300 confirms the chemical composition of the diblock copolymers. Figure S2. GPC-SEC and SLS characterization of polymers. Figure S3. Characterization of polymer-peptide loading. Figure S4. Intracellular delivery of BIM conjugates suppresses tumor growth and reduces tumor growth rates. Figure S5. 1H-NMR spectrum of functional (a) pyridyldisulfide methacrylate (PyrSMA) monomer and (b) biotin-hydroxyethyl methacrylate (bioHEMA) monomer. Table S1. Median survival, doubling time and tumor growth delay.
Acknowledgments
The authors would like to thank F. Guoyong at WuXi AppTec and J. Wilson for their insightful scientific discussion, and M. Manganiello for instrumentation support. This work was supported by the NIH Grant R21EB014572, Grant K08 CA163603 (M.C.P-W.), Grant P50 CA083636 (M.C.P-W.), Grant P50 CA138293 (D.H.M. and D.H.), Washington State Life Sciences Discovery Fund (Grant 2496490 to the Center for Intracellular Delivery of Biologics), and the Wayne D. Kuni & Joan E. Kuni Foundation and the Kuni family through the 3725 Fund of the Oregon Community Foundation (M.C.P-W.). G.Y.B. was a graduate research fellow of the National Science Foundation. P.S.S. and O.W.P. are cofounders of PhaseRX, Inc, that has licensed some of the delivery technology from the University of Washington described in this article. This work was conducted completely independent of the company under the support of the aforementioned grants.
Supplementary Material
References
- Senter PD. Potent antibody drug conjugates for cancer therapy. Curr Opin Chem Biol. 2009;13:235–244. doi: 10.1016/j.cbpa.2009.03.023. [DOI] [PubMed] [Google Scholar]
- Ricart AD. Immunoconjugates against solid tumors: mind the gap. Clin Pharmacol Ther. 2011;89:513–523. doi: 10.1038/clpt.2011.8. [DOI] [PubMed] [Google Scholar]
- Johannes L, Decaudin D. Protein toxins: intracellular trafficking for targeted therapy. Gene Ther. 2005;12:1360–1368. doi: 10.1038/sj.gt.3302557. [DOI] [PubMed] [Google Scholar]
- Susnow N, Zeng L, Margineantu D, Hockenbery DM. Bcl-2 family proteins as regulators of oxidative stress. Semin Cancer Biol. 2009;19:42–49. doi: 10.1016/j.semcancer.2008.12.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lessene G, Czabotar PE, Colman PM. BCL-2 family antagonists for cancer therapy. Nat Rev Drug Discov. 2008;7:989–1000. doi: 10.1038/nrd2658. [DOI] [PubMed] [Google Scholar]
- Ni Chonghaile T, Sarosiek KA, Vo TT, Ryan JA, Tammareddi A, Moore Vdel G, et al. Pretreatment mitochondrial priming correlates with clinical response to cytotoxic chemotherapy. Science. 2011;334:1129–1133. doi: 10.1126/science.1206727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu G, Kelly WK, Wilding G, Leopold L, Brill K, Somer B. An open-label, multicenter, phase I/II study of single-agent AT-101 in men with castrate-resistant prostate cancer. Clin Cancer Res. 2009;15:3172–3176. doi: 10.1158/1078-0432.CCR-08-2985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- O'Brien SM, Claxton DF, Crump M, Faderl S, Kipps T, Keating MJ, et al. Phase I study of obatoclax mesylate (GX15-070), a small molecule pan-Bcl-2 family antagonist, in patients with advanced chronic lymphocytic leukemia. Blood. 2009;113:299–305. doi: 10.1182/blood-2008-02-137943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Hann CL, Garon EB, Ribeiro de Oliveira M, Bonomi PD, Camidge DR, et al. Phase II study of single-agent navitoclax (ABT-263) and biomarker correlates in patients with relapsed small cell lung cancer. Clin Cancer Res. 2012;18:3163–3169. doi: 10.1158/1078-0432.CCR-11-3090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas S, Quinn BA, Das SK, Dash R, Emdad L, Dasgupta S, et al. Targeting the Bcl-2 family for cancer therapy. Expert Opin Ther Targets. 2013;17:61–75. doi: 10.1517/14728222.2013.733001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yecies D, Carlson NE, Deng J, Letai A. Acquired resistance to ABT-737 in lymphoma cells that up-regulate MCL-1 and BFL-1. Blood. 2010;115:3304–3313. doi: 10.1182/blood-2009-07-233304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mason KD, Vandenberg CJ, Scott CL, Wei AH, Cory S, Huang DC, et al. In vivo efficacy of the Bcl-2 antagonist ABT-737 against aggressive Myc-driven lymphomas. Proc Natl Acad Sci USA. 2008;105:17961–17966. doi: 10.1073/pnas.0809957105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letai A, Bassik MC, Walensky LD, Sorcinelli MD, Weiler S, Korsmeyer SJ. Distinct BH3 domains either sensitize or activate mitochondrial apoptosis, serving as prototype cancer therapeutics. Cancer Cell. 2002;2:183–192. doi: 10.1016/s1535-6108(02)00127-7. [DOI] [PubMed] [Google Scholar]
- Certo M, Del Gaizo Moore V, Nishino M, Wei G, Korsmeyer S, Armstrong SA, et al. Mitochondria primed by death signals determine cellular addiction to antiapoptotic BCL-2 family members. Cancer Cell. 2006;9:351–365. doi: 10.1016/j.ccr.2006.03.027. [DOI] [PubMed] [Google Scholar]
- LaBelle JL, Katz SG, Bird GH, Gavathiotis E, Stewart ML, Lawrence C, et al. A stapled BIM peptide overcomes apoptotic resistance in hematologic cancers. J Clin Invest. 2012;122:2018–2031. doi: 10.1172/JCI46231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashiwagi H, McDunn JE, Goedegebuure PS, Gaffney MC, Chang K, Trinkaus K, et al. TAT-Bim induces extensive apoptosis in cancer cells. Ann Surg Oncol. 2007;14:1763–1771. doi: 10.1245/s10434-006-9298-z. [DOI] [PubMed] [Google Scholar]
- Berguig GY, Convertine AJ, Shi J, Palanca-Wessels MC, Duvall CL, Pun SH, et al. Intracellular delivery and trafficking dynamics of a lymphoma-targeting antibody-polymer conjugate. Mol Pharm. 2012;9:3506–3514. doi: 10.1021/mp300338s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palanca-Wessels MC, Convertine AJ, Cutler-Strom R, Booth GC, Lee F, Berguig GY, et al. Anti-CD22 antibody targeting of pH-responsive micelles enhances small interfering RNA delivery and gene silencing in lymphoma cells. 2009;19:1–9. doi: 10.1038/mt.2011.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duvall CL, Convertine AJ, Benoit DS, Hoffman AS, Stayton PS. Intracellular delivery of a proapoptotic peptide via conjugation to a RAFT synthesized endosomolytic polymer. Mol Pharm. 2010;7:468–476. doi: 10.1021/mp9002267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson JT, Keller S, Manganiello MJ, Cheng C, Lee CC, Opara C, et al. pH-Responsive nanoparticle vaccines for dual-delivery of antigens and immunostimulatory oligonucleotides. ACS Nano. 2013;7:3912–3925. doi: 10.1021/nn305466z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manganiello MJ, Cheng C, Convertine AJ, Bryers JD, Stayton PS. Diblock copolymers with tunable pH transitions for gene delivery. Biomaterials. 2012;33:2301–2309. doi: 10.1016/j.biomaterials.2011.11.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng C, Convertine AJ, Stayton PS, Bryers JD. Multifunctional triblock copolymers for intracellular messenger RNA delivery. Biomaterials. 2012;33:6868–6876. doi: 10.1016/j.biomaterials.2012.06.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang C, Neoh KG, Xu L, Kang ET, Chiong E. Polymeric nanoparticles with encapsulated superparamagnetic iron oxide and conjugated cisplatin for potential bladder cancer therapy. Biomacromolecules. 2012;13:2513–2520. doi: 10.1021/bm300739w. [DOI] [PubMed] [Google Scholar]
- Jiang X, Vogel EB, Smith MR, Baker GL. ‘Clickable' polyglycolides: tunable synthons for thermoresponsive, degradable polymers. Macromolecules. 2008;41:1937–1944. [Google Scholar]
- Lutz J-F. Polymerization of oligo(ethylene glycol) (meth)acrylates: toward new generations of smart biocompatible materials. J Polym Sci A Polym Chem. 2008;46:3459–3470. [Google Scholar]
- Roy D, Berguig GY, Ghosn B, Lane D, Braswell S, Stayton PS, et al. Synthesis and characterization of transferrin-targeted chemotherapeutic delivery systems prepared via RAFT copolymerization of high molecular weight PEG macromonomers. Polym Chem. 2014;5:1791–1799. doi: 10.1039/C3PY01404E. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Sayed ME, Hoffman AS, Stayton PS. Rational design of composition and activity correlations for pH-sensitive and glutathione-reactive polymer therapeutics. J Control Release. 2005;101:47–58. doi: 10.1016/j.jconrel.2004.08.032. [DOI] [PubMed] [Google Scholar]
- Convertine AJ, Benoit DS, Duvall CL, Hoffman AS, Stayton PS. Development of a novel endosomolytic diblock copolymer for siRNA delivery. J Control Release. 2009;133:221–229. doi: 10.1016/j.jconrel.2008.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Convertine AJ, Diab C, Prieve M, Paschal A, Hoffman AS, Johnson PH, et al. pH-responsive polymeric micelle carriers for siRNA drugs. Biomacromolecules. 2010;11:2904–2911. doi: 10.1021/bm100652w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pantelias A, Pagel JM, Hedin N, Saganic L, Wilbur S, Hamlin DK, et al. Comparative biodistributions of pretargeted radioimmunoconjugates targeting CD20, CD22, and DR molecules on human B-cell lymphomas. Blood. 2007;109:4980–4987. doi: 10.1182/blood-2006-11-056895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pagel JM, Pantelias A, Hedin N, Wilbur S, Saganic L, Lin Y, et al. Evaluation of CD20, CD22, and HLA-DR targeting for radioimmunotherapy of B-cell lymphomas. Cancer Res. 2007;67:5921–5928. doi: 10.1158/0008-5472.CAN-07-0080. [DOI] [PubMed] [Google Scholar]
- Nishiyama N, Kato Y, Sugiyama Y, Kataoka K. Cisplatin-loaded polymer-metal complex micelle with time-modulated decaying property as a novel drug delivery system. Pharm Res. 2001;18:1035–1041. doi: 10.1023/a:1010908916184. [DOI] [PubMed] [Google Scholar]
- Bae Y, Nishiyama N, Fukushima S, Koyama H, Yasuhiro M, Kataoka K. Preparation and biological characterization of polymeric micelle drug carriers with intracellular pH-triggered drug release property: tumor permeability, controlled subcellular drug distribution, and enhanced in vivo antitumor efficacy. Bioconjug Chem. 2005;16:122–130. doi: 10.1021/bc0498166. [DOI] [PubMed] [Google Scholar]
- Gu F, Zhang L, Teply BA, Mann N, Wang A, Radovic-Moreno AF, et al. Precise engineering of targeted nanoparticles by using self-assembled biointegrated block copolymers. Proc Natl Acad Sci USA. 2008;105:2586–2591. doi: 10.1073/pnas.0711714105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng J, Teply BA, Sherifi I, Sung J, Luther G, Gu FX, et al. Formulation of functionalized PLGA-PEG nanoparticles for in vivo targeted drug delivery. Biomaterials. 2007;28:869–876. doi: 10.1016/j.biomaterials.2006.09.047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habel ME, Jung D. c-Myc over-expression in Ramos Burkitt's lymphoma cell line predisposes to iron homeostasis disruption in vitro. Biochem Biophys Res Commun. 2006;341:1309–1316. doi: 10.1016/j.bbrc.2006.01.097. [DOI] [PubMed] [Google Scholar]
- Campone M, Noël B, Couriaud C, Grau M, Guillemin Y, Gautier F, et al. c-Myc dependent expression of pro-apoptotic Bim renders HER2-overexpressing breast cancer cells dependent on anti-apoptotic Mcl-1. Mol Cancer. 2011;10:110. doi: 10.1186/1476-4598-10-110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demidenko E. Three endpoints of in vivo tumour radiobiology and their statistical estimation. Int J Radiat Biol. 2010;86:164–173. doi: 10.3109/09553000903419304. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heitjan DF, Manni A, Santen RJ. Statistical analysis of in vivo tumor growth experiments. Cancer Res. 1993;53:6042–6050. [PubMed] [Google Scholar]
- Chiefari J, Chong YK, Ercole F, Krstina J. Living free-radical polymerization by reversible addition-fragmentation chain transfer: the RAFT process. Macromolecules. 1998;31:5559–5562. [Google Scholar]
- Moad G, Chiefari J, Krstina J. Living free radical polymerization with reversible addition–fragmentation chain transfer (the life of RAFT) Polym Int. 2000;49:993–1001. [Google Scholar]
- Convertine AJ, Ayres N, Scales CW, Lowe AB, McCormick CL. Facile, controlled, room-temperature RAFT polymerization of N-isopropylacrylamide. Biomacromolecules. 2004;5:1177–1180. doi: 10.1021/bm049825h. [DOI] [PubMed] [Google Scholar]
- Klasa RJ, Bally MB, Ng R, Goldie JH, Gascoyne RD, Wong FM. Eradication of human non-Hodgkin's lymphoma in SCID mice by BCL-2 antisense oligonucleotides combined with low-dose cyclophosphamide. Clin Cancer Res. 2000;6:2492–2500. [PubMed] [Google Scholar]
- Vandenberg CJ, Cory S. ABT-199, a new Bcl-2-specific BH3 mimetic, has in vivo efficacy against aggressive Myc-driven mouse lymphomas without provoking thrombocytopenia. Blood. 2013;121:2285–2288. doi: 10.1182/blood-2013-01-475855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster GJ, Santos EB, Smith-Jones PM, Zanzonico P, Larson SM. Pretargeted radioimmunotherapy with a single-chain antibody/streptavidin construct and radiolabeled DOTA-biotin: strategies for reduction of the renal dose. J Nucl Med. 2006;47:140–149. [PubMed] [Google Scholar]
- Wilbur DS, Park SI, Chyan MK, Wan F, Hamlin DK, Shenoi J, et al. Design and synthesis of bis-biotin-containing reagents for applications utilizing monoclonal antibody-based pretargeting systems with streptavidin mutants. Bioconjug Chem. 2010;21:1225–1238. doi: 10.1021/bc100030q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hylarides MD, Mallett RW, Meyer DL. A robust method for the preparation and purification of antibody/streptavidin conjugates. Bioconjug Chem. 2001;12:421–427. doi: 10.1021/bc0001286. [DOI] [PubMed] [Google Scholar]
- Murthy N, Campbell J, Fausto N, Hoffman AS, Stayton PS. Design and synthesis of pH-responsive polymeric carriers that target uptake and enhance the intracellular delivery of oligonucleotides. J Control Release. 2003;89:365–374. doi: 10.1016/s0168-3659(03)00099-3. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.