

Abstract
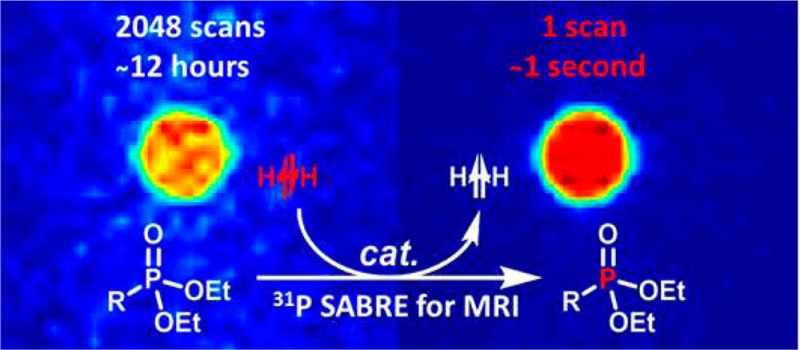
Traditional 31P NMR or MRI measurements suffer from low sensitivity relative to 1H detection and consequently require longer scan times. We show here that hyperpolarization of 31P nuclei through reversible interactions with parahydrogen can deliver substantial signal enhancements in a range of regioisomeric phosphonate esters containing a heteroaromatic motif which were synthesized in order to identify the optimum molecular scaffold for polarization transfer. A 3588-fold 31P signal enhancement (2.34% polarization) was returned for a partially deuterated pyridyl substituted phosphonate ester. This hyperpolarization level is sufficient to allow single scan 31P MR images of a phantom to be recorded at a 9.4 T observation field in seconds that have signal-to-noise ratios of up to 94.4 when the analyte concentration is 10 mM. In contrast, a 12 h 2048 scan measurement under standard conditions yields a signal-to-noise ratio of just 11.4. 31P-hyperpolarized images are also reported from a 7 T preclinical scanner.
Phosphorus containing molecules are essential to the human body.1−5 In addition, a number of widely prescribed pharmaceutical products contain one or more phosphorus atoms.6−8 Due to 31P′s high natural abundance, coupled with a wide chemical shift range that is indicative of molecular environment, 31P signal detection represents an exciting probe for magnetic resonance spectroscopy (MRS) and magnetic resonance imaging (MRI) studies in both inorganic chemistry and biochemistry. For example, recent studies have used 31P MRS for the in vivo monitoring of metabolites in breast cancer,9,10 liver disease,11 schizophrenia,12 and tissue assessment.13,14
The detection of 31P nuclei by magnetic resonance (MR), however, suffers from lower sensitivity than 1H nuclei because of its lower magnetogyric ratio.15 Consequently, high quality 31P NMR spectra or MR images require relatively large amounts of sample in conjunction with signal averaging. Hyperpolarization techniques are seen as a route to overcome this low sensitivity issue which arises from the small population difference (ca. 10–5) that exists between the Zeeman-split energy levels that are probed. Until recently, 31P hyperpolarization has received limited attention, and the development of a simple method to achieve it is desirable.16,17
Parahydrogen induced polarization (PHIP) is a popular method of hyperpolarization that utilizes molecular hydrogen as its source of polarization.18−23 By incorporating p-H2 into an unsaturated molecule its singlet symmetry is broken, and this process unlocks its latent, and normally invisible, polarization. A recent report using PHIP for 13C detection, showed that labeled 1-13C-phospholactate-d2 could be detected at just 5.74 mT.24 PHIP has, however, been widely used to probe reaction mechanisms where it enables the detection of many metal hydride containing complexes.25−27 In these studies magnetization transfer to 31P has proven to be a very important feature of the characterization of many reaction products.28
A new form of PHIP, called Signal Amplification by Reversible Exchange (SABRE), has emerged as a tool for the rapid hyperpolarization of molecules without the need for changing their chemical identity.29 SABRE utilizes a stable metal complex, such as [IrCl(COD)(IMes)] (1) (where IMes = 1,3-bis(2,4,6-trimethylphenyl)imidazolium and COD = cis,cis-1,5-cyclooctadiene), to form what is known as the polarization transfer catalyst (Scheme 1).
Scheme 1. An Active SABRE Catalyst Can Employ Magnetic Inequivalence (e.g. 2a) or Chemical Inequivalence (e.g. 2b) at the Magnetization Transfer Stage.
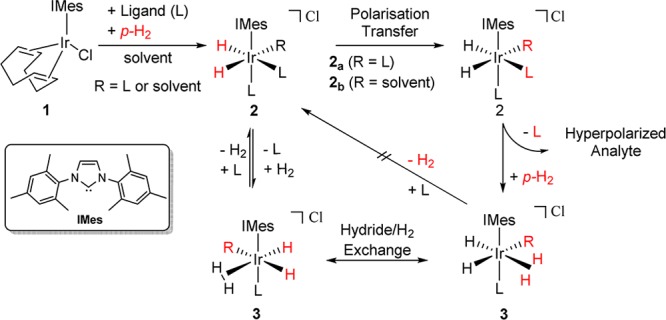
The generation of hyperpolarized ligands (L) that return to bulk solution through ligand exchange enables the production of analytes that are highly visible to NMR and MRI.
The role of catalyst 2 in Scheme 1 is to bring together p-H2, now in the form of a pair of hydride ligands, and the hyperpolarization target (L). Polarization transfer catalysis (PTC) occurs during the short time period for which 2 maintains this ligand arrangement. Polarization is passed through the J-coupling network of the metal complex into the ligand rather than by changing the chemical identity of the ligand through formal p-H2 incorporation.30 H2 loss and rebinding of the analyte are necessary for the delivery of high levels of analyte hyperpolarization in conjunction with high catalyst turnover. In this case, the p-H2 refresh step involves the short-lived iridium dihydrogen dihydride complex 3.31
To date, reported studies using the SABRE approach have focused on the polarization of 1H29,32−34 and 13C29,35 nuclei, although 15N36,3719F29 and 31P28,38 signals have been seen when polarized. Both of the 31P studies focused on PPh3, with Koptyug et al. reporting that the signal for free PPh3 can be enhanced by 2 orders of magnitude, and a single shot image recorded at 9.4 T.38 Importantly, SABRE has been shown to be successful for a growing range of biologically relevant molecules; this has established the potential of the method to produce 1H and 13C hyperpolarized agents for subsequent MRI measurement.29,32,35,39,40
Herein, we report the optimization of SABRE with respect to the hyperpolarization of 31P nuclei within heteroaromatic molecules. We rationalize how the chemical structure affects the observed polarization level and thereby outline a strategy that may subsequently find use in the production of future diagnostic agents for 31P MRI.
Materials and Methods
All reactions utilizing air- and moisture-sensitive reagents were performed in dried glassware under an atmosphere of dry nitrogen. Dry solvents (THF, toluene and DCM) were obtained from a Braun MB-SPS-800. For thin-layer chromatography (TLC) analysis, Merck precoated plates (silica gel 60 F254, Art 5715, 0.25 mm) were used. Column chromatography was performed on Fluka Silica gel (60 Å, 220–440 mesh). 1H NMR, 13C NMR, and 31P NMR spectra were measured on Bruker 400 or 500 MHz spectrometers. Deuterated solvents (methanol-d4 and chlorofrom-d) were obtained from Sigma-Aldrich and used as supplied.
The following compounds were synthesized according to literature procedures: 1,41 d10-diethyl phosphite,42 4-pyridyl diphenylphosphine L1,43 4-pyridyl diphenylphosphine oxide L2,44 4-pyridylmethyl diethylphosphonate L8, 3-pyridylmethyl diethylphosphonate L9.45 Detailed synthetic procedures and characterization data can be found in the Supporting Information.
p-H2 was produced by cooling H2 gas over Fe2O3 at 30 K.26 Samples involved the analysis of 5 mM concentrations of 1 and 5 or 6 equiv of substrate (L1-L12) in methanol-d4 (0.6 or 3.0 mL) solution that was located in either a 5 or 8 mm NMR tube fitted with a J. Young’s Tap. The resulting solutions were degassed prior to the introduction of p-H2 at a pressure of 3 bar. Samples were then shaken for 10 s in a specified fringe field of either a 9.4 T Bruker Avance (III) NMR spectrometer or a 7 T Bruker BioSpec preclinical MRI scanner. They were then rapidly transported into the main magnetic field of the instrument for subsequent probing by NMR or MRI methods. The associated polarization transfer experiment data can be found in the Supporting Information.
Results and Discussion
Examining 4-Substituted Pyridine Derivatives
Historically, N-heteroaromatic molecules have been shown to be excellent receptors for polarization transfer to 1H and 13C nuclei under SABRE.29,31,33 We therefore synthesized a phosphine (L1), a phosphine oxide (L2), a phosphine sulfide (L3) and a phosphonate ester (L4) which incorporated a 4-substituted pyridine motif, as detailed in the Supporting Information (Figure 1). We selected the 4-substitution pattern for this study in order to minimize any steric interactions between the hyperpolarization target L and the IMes ligand in the catalyst 2. We expected this restriction to promote activity in the widest range of analytes possible. A series of samples were then prepared in methanol-d4 that contained L1-L4, at a concentration of 5 mM, and the catalyst precursor 1 at a 17% catalyst loading. These samples were probed after SABRE had taken place at 298 K on a 400 MHz NMR spectrometer by the application of a simple 90° rf-pulse. We will discuss the effects of SABRE on the pyridyl 1H polarization, then the 31P polarization, and finally the polarization received by the other substituent groups.
Figure 1.

1H and 31P signal intensity gains for analytes L1-L4 under SABRE conditions using 1 as the catalyst.
When this 90° pulse was applied to the 1H nuclei of these samples, only L3 proved to exhibit weak pyridyl proton polarization. The other three analytes produced good proton signal enhancements. For example, the two 1H NMR signals of L4, that were derived from the pyridine functionality, showed a 1499-fold total signal enhancement when the initial polarization transfer step was undertaken in a 45 G field. This high 1H PTC was reflected in the fact that the corresponding 31P signal exhibited a similarly impressive 545-fold signal enhancement. For 31P, PTC was maximized through transfer at 0.5 G rather than the 45 G field observed for its 1H nuclei. Figure 1 summarizes the corresponding 1H and 31P signal intensity gains determined for L1-L4.
A series of control experiments were then undertaken on triphenylphosphine, triphenylphosphine oxide, and phenyldiethylphosphonate in order to explore the role that the pyridine substituent might play in the hyperpolarization transfer process. These experiments revealed that no polarization transfer took place into the 1H or 31P nuclei of these materials under the same SABRE conditions as used to hyperpolarize L1–4 described above. Based on this information it is clear that the pyridine motif is necessary for successful PTC under these conditions. This is achieved by the formation of an active SABRE catalyst as typified in Scheme 1. We note that this situation differs from that reported when the related [IrCl(H)2(PPh3)3] complex was used to successfully polarize triphenylphosphine at temperatures between 333 and 353 K by Koptyug et al.38 In this case the Ir–P bond proved sufficiently labile that at elevated temperatures SABRE was successful. However, when 1 reacts with an excess of PPh3 a series of new and rapidly relaxing hydride ligand signals are observed at δ −7.33, −7.98, and −8.59 which are indicative of the formation of [Ir(H)2(H2)(IMes)(PPh3)2]Cl. The formation of such species precludes the observation of SABRE in these samples because of their role in the rapid quenching of p-H2 (see the Supporting Information).46
As a consequence of the formation of 2a-L4, when the corresponding 1H NMR spectra are examined under SABRE conditions polarized hydride resonances appear as a singlet in the region around δ −22.15 for the pair of chemically equivalent hydride ligands (see the Supporting Information). Characteristic signals for L4, as a ligand in 2a-L4, are also seen to be polarized in the associated 31P NMR spectra. We illustrate selected regions of these hyperpolarized 1H and 31P NMR spectra after PTC at 0.5 G, in Figures 2 and 3 respectively to detail some of these effects.
Figure 2.

SABRE enhanced 1H NMR spectrum (lower) and corresponding thermal reference spectrum (upper, x64 vertical expansion) of 2a-L4 and L4 (• = equatorial ligand signal, ▲ = axial ligand signal of 2a-L4).
Figure 3.
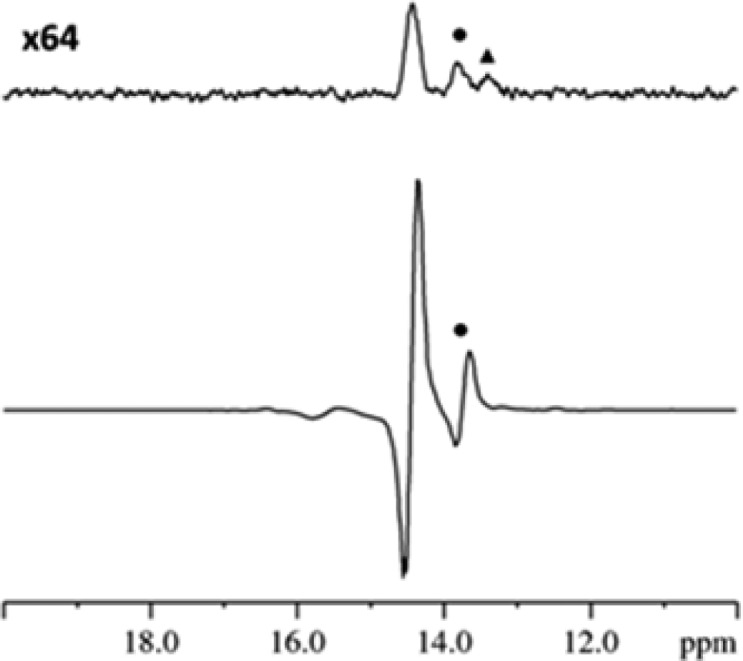
SABRE enhanced 31P NMR spectrum (lower) and thermal reference spectrum (upper, x64 vertical expansion) of 2a-L4 and L4 (• = equatorial ligand, ▲ = axial ligand of 2a-L4).
The hyperpolarized 1H NMR trace shown in Figure 2 confirms that both of the two pairs of equivalent ring protons of bound L4 receive magnetization, while those of the axial group remain unaffected. This is manifested in the detection of emission signals for the free analyte at δ 8.76 and for the equatorial ligand at δ 8.50. The magnetic state that produces this emission effect is single spin order in nature and associated with an enhanced but inverted Zeeman population.47 In contrast, the corresponding pair of 3,5-ring protons in L4 produce antiphase peaks that are separated by 13.5 Hz. This splitting corresponds to the value of 3JPH in L4 and requires that a heteronuclear two-spin order proton-phosphorus term is created through SABRE. Application of a 90° rf-pulse to the 1H nuclei of this term creates the visible antiphase signal that is seen; the creation of homonuclear proton terms of this type is a well-established characteristic of PHIP.47 For L4, the total 1H signal enhancement was 1499-fold.
The hyperpolarized 31P NMR spectrum of the L4 sample, collected as a single transient, also showed two antiphase peaks, at δ 14.44 and 13.73 for free and equatorially bound L4 respectively (Figure 3). The signal for free L4 showed a 545-fold increase in size after PTC at 0.5 G relative to the corresponding thermally polarized trace. The separation between the point of maximum displacement and minimum displacement for the signal due to L4 in this NMR trace is 27.0 Hz (smaller 4JPH couplings are hidden within the peak envelope) and twice that of the separation shown in the 1H NMR spectrum. This difference confirms the creation of a proton-phosphorus two spin order term, which leads to a triplet when excited wherein the central feature has zero intensity (−1:0: +1). Related behavior in PHIP derived trihydride complexes have been described previously.25,48−50 Application of the multinuclear OPSY protocol confirmed this deduction.35,51
Interestingly, when 2a-L4 is probed by EXSY methods both the axial and equatorial L4 ligands are observed to undergo exchange with the free ligand pool present in bulk solution. The exchange profile seen for axial-L4 suggests that there is some in-cage recombination of this ligand, a process which is not observed for equatorial-L4 (see the Supporting Information). The axial ligand loss rate is 0.645 ± 0.010 s–1 at 283 K and slightly more rapid than equatorial ligand loss rate which is 0.522 ± 0.007 s–1. The observed hyperpolarization seen in the equatorial-L41H and 31P signals is therefore a consequence of SABRE prior to ligand loss.
We note that pyridylphosphonates find widespread use within a biological setting as lysophosphatidic acid receptor antagonists,52,53 nucleotide analogues,54,55 and enzyme inhibitors.56,57 Importantly, related compounds have been shown to exhibit low toxicity in rat studies (LD50 of 3.2 g/kg (oral)).58 As a consequence of these facts and the observation that L4 delivers high levels of visible 31P hyperpolarization we further optimized the ability of pyridylphosphonates to respond to PTC. This was achieved by the synthesis of the phosphonate derivatives L5-L12 which are shown in Table 1, alongside the levels of 1H and 31P hyperpolarization that results from their analysis under SABRE.
Table 1. 1H and 31P NMR Total Signal Intensity Gains for L5-L12 in a Methanol-d4 Solution under 3 bar of p-H2 Achieved through PTC by 1 under SABRE in the Specified Magnetic Fielda.
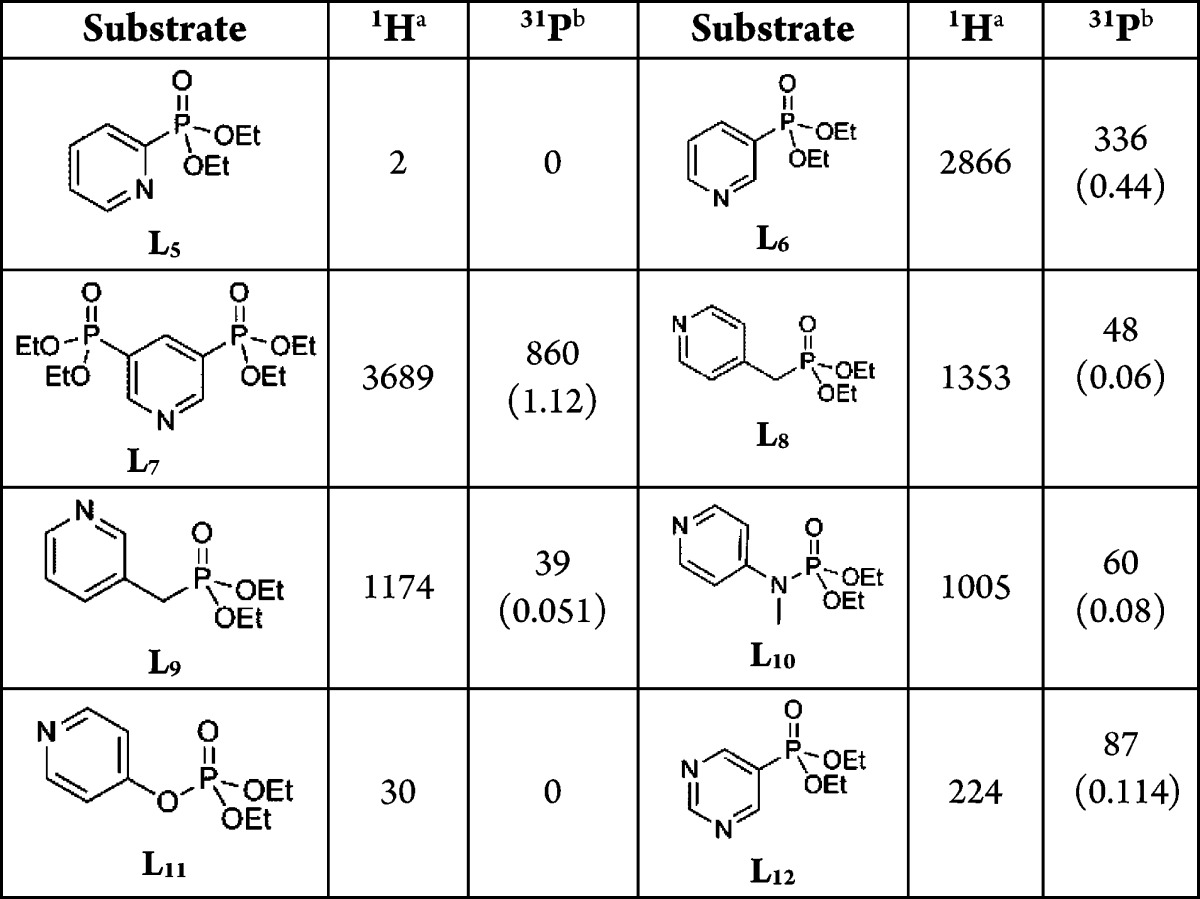
For 31P, % polarization indicated in brackets. 5 mM concentrations of 1 with 5 equiv of L7 and 6 of equivalents of L5-L12 were employed at (a) 45 G and (b) 0.5 G.
Extension to 2 and 3 Substitution
In order to examine the effect that the regiochemistry of substitution played on the polarization level we replaced the 4- substitution pattern exhibited by L1-L4 with 2- and 3- substitutions in the form of substrates L5 and L6. Polarization transfer into 2-pyridylphosphonate L5 proved to be unsuccessful, and this is due to the formation of a complex that yields a very broad hydride resonance at δ −31.3 which does not show PHIP. Upon cooling the solution to 233 K this hydride ligand signal separates into two resonances at δ −30.2 and δ −32.1 which confirms that this complex is a dihydride. The corresponding hydride ligand signals yield an average T1 value of 0.56 s at 253 K and are therefore classically bonded and terminal in character.46 Nonetheless, the presence of this complex in solution provides a pathway to rapidly destroy the p-H2 present, presumably via the formation of a dihydrogen-dihydride complex, with the result that there is very little observable polarization transfer into L5.
In contrast, 3-substitution proved to result in good PTC. The measured 1H signal enhancement at 45 G for L6 was 2866-fold, and the corresponding 31P signal enhancement was 336-fold. Clearly, the change to a 3-substitution pattern has improved the level of 1H PTC relative to 4-substitution but surprisingly decreased the efficiency for 31P transfer. The synthesis of the bis-phosphonate L7 was therefore undertaken, as this material contains two chemically equivalent 31P nuclei and one fewer pyridyl proton than the monosubstituted derivatives. We speculated that better 31P-PTC might result as a consequence of this change on the basis that each p-H2 molecule contains a specific amount of latent polarization, and consequently when shared with fewer acceptor sites a net gain in the level of observable transfer might result. The corresponding 1H and 31P signal enhancements for L7 were 2271- and 741-fold respectively when 6 equiv of the analyte was employed, and therefore this hypothesis is confirmed. However, one further and somewhat unexpected observation was made, the enhanced steric bulk of L7 results in a change in the active complex such that 2b-L7 results wherein one coordination site is occupied by chloride (see the Supporting Information). The two hydride signals for this complex appear at δ −23.74 and −24.06.59 Because of this change in catalyst form we found that reducing the number of equivalents of L7 to 5 led to an improvement in PTC, giving 1H and 31P signal enhancements of 3689- and 860-fold after transfer at 45 and 0.5 G respectively.
In order to further test the efficiency of polarization transfer to 31P in these systems we added a series of spacers between the pyridyl carbon and the phosphorus center. In L8 and L9 this corresponds to the introduction of a −CH2– group, while for L10 it is −NMe– and for L11 it is −O–. These analytes were SABRE active, but while the pyridyl ring protons remained strongly enhanced the spacer acted to substantially reduce the levels of 31P polarization. We also prepared a phosphonate using a pyrimidine motif (L12). This change again results in a reduction in the number of proton environments able to accept magnetization. While successful, the levels of both 1H and 31P signal enhancement were again lower than those achieved for the pyridylphosphonates.
We now address the potential of these substrates to act as MRI agents for which in-phase 31P signals are desirable. As we have indicated, all of the described 31P signals that were created through PTC in a 0.5 G appear in antiphase. While this effect can be refocused to produce an in-phase signal, we proposed that changing the polarization transfer field (PTF) might achieve a similar result. This would be achieved by changing the relative proportions of single spin (in-phase) and two spin (antiphase) components and result in a predominantly in-phase signal being obtained. A series of experiments were therefore performed in which the PTF was increased using a variable field polarizer35 and the resulting 31P signal monitored. We completed this process for both L6 and L7 (Figure 4). Significant in-phase contributions were observed after transfer at 45 G without any significant reduction in the overall 31P signal enhancement (216-fold vs 336-fold for L6 and 703-fold vs 860-fold for L7 at 45 and 0.5 G respectively). While PTFs above 45 G did result in an increase in the relative single spin contribution, a sharp decline in the overall signal enhancement was noted. For L6 the relative proportions of the single spin and two spin terms are 0.08:1 and 0.23:1 when the PTF is 0.5 and 45 G respectively. In contrast, when L7 is examined, the relative proportions are 0.06:1 at 0.5 G and 0.88:1 at 45 G respectively. We conclude from this that L7 is potentially the better MRI probe for these model studies. This is because the signal for L4 proved to remain predominately antiphase at both 0.5 G (0.15:1) and 45 G (0.17:1), and it is therefore less suitable for such measurements.
Figure 4.

SABRE hyperpolarized 31P NMR traces for L4 [a], L6 [b], and L7 [c] that result after polarization transfer catalysis in a 45 or 0.5 G field.
Optimizing the Level of 31P Hyperpolarization
Further examination of the hyperpolarized 1H NMR spectra revealed that the signal enhancements described so far also extend into the ethyl groups of the phosphonate. Typically these 1H signal enhancements were 50-fold per ethyl group and reflect a relayed 1H-31P-1H transfer process. It was therefore reasoned that deuteration of the phosphonate ethyl esters would increase the level of retained 31P polarization as transfer to 2H is less efficient due to the large frequency difference that exists between them. Therefore, we synthesized d10-L4, d10-L6, and d20-L7 and analyzed them under analogous conditions to their protio-counterparts (Figure 5). The levels of hyperpolarization seen in the aromatic proton signals of d10-L4, d10-L6, and d20-L7 proved to remain comparable to those of their protio analogues, but the 31P signals were found to dramatically increase in intensity.
Figure 5.
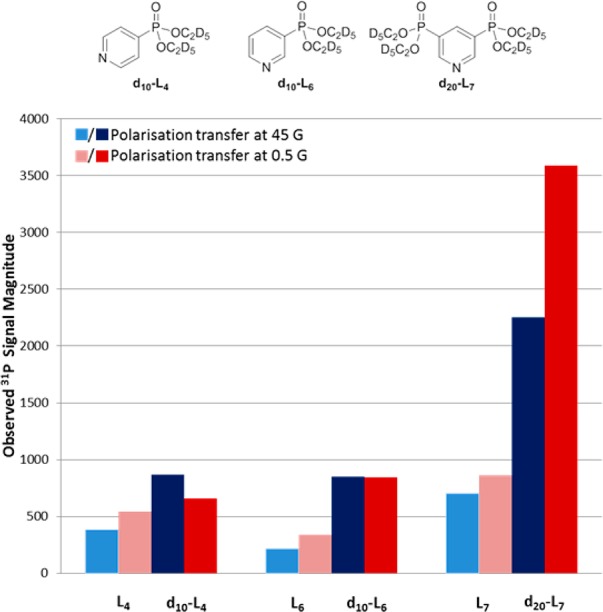
Structure of substrates d10-L4, d10-L6, and d20-L7 and their signal intensity gains, alongside those of their protio counterparts L4, L6, and L7, under SABRE after PTC at the specified PTF.
Substrate d20-L7 yielded a 31P signal gain of 3588 (2.3% polarization) after PTC at 0.5 G. A 2251-fold signal enhancement (1.4% polarization) is, however, achieved after PTC at 45 G with significant in-phase character being visible in the detected signal. We note this is an order of magnitude larger than previously reported for 31P enhancements using this technique and can be achieved without the need for elevated temperatures during the polarization transfer step.38 In a further development, we completed magnetization transfer by inserting the sample into a μ-magnetic shield, which attenuates the Earth’s field by a factor of 3000-fold, prior to moving it into the 9.4 T magnet for examination. While this created in-phase 31P magnetization,30 the observed signal enhancement decreased to 381-fold (0.497%) under these conditions. It would therefore appear that simply changing the polarization transfer field to a readily accessible value between 0.5 and 130 G reflects the optimal route to achieve in-phase signal. We also note that complexes of type 1 are known to catalyze hydrogen–deuterium exchange,60 and, as such, there is the potential to detect 1-proton-PHIP in such analytes.61,62 Hence, we prepared a sample of d20-L7 and 1 in methanol-d4 under 3-bar of H2 and followed it by 1H NMR for a period of 10 days. At this point, 90% of the 2,6-protons had been replaced by deuterium. Subsequent SABRE catalysis led to a large reduction in the 31P polarization transfer level. If the 31P enhancements that were seen here were due to a 1-proton-PHIP, then the enhancement levels should remain comparable to those observed at day 1. This suggests that when such samples are examined they should be prepared shortly before their use.
Effect of Relaxation on Hyperpolarization Level
We have thus far exemplified the ability to hyperpolarize 31P, and we next evaluated the spin–lattice relaxation times (T1) of the analytes L4, d10-L4, L6, d10-L6, L7, and d20-L7. The T1 relaxation values of the Zeeman based 31P polarization are 8.3, 7.6, 9.2, 9.3, 5.3, and 6.0 s respectively in methanol-d4. For d20-L7 they increase to 6.5 s in ethanol-d6 and decrease to 3.6 s in D2O. These T1 values, although short, do offer the opportunity to obtain images using the SABRE technique.
Imaging at High Field
With a number of these substrates displaying favorable results we were hopeful that it would be possible to collect MRI with improved spatial resolution and short acquisition times. A series of hyperpolarized samples were therefore interrogated in a 9.4 T vertical bore scanner using the rapid acquisition with relaxation enhancement (RARE) pulse sequence. We employed an echo train length of 32 and a matrix size 32 × 32 with zero filling to 128 × 128. This resulted in a total single scan acquisition time of 500 ms. For comparison purposes, thermal images of L6, L7, and d20-L7 were recorded with 2048 averages, as shown in Figure 6. Under hyperpolarization conditions, the largest gains in signal-to-noise ratio (SNR) were observed for d20-L7 (94.4) and reflected a >8-fold improvement on that of the thermal control (11.4). Improvements in SNR of >4-fold were also seen for L7.
Figure 6.

[a]-[c] Thermally polarized 31P images of L6, L7, and d20-L7, respectively collected over 2048 averages. The flip angles used were 86° for L6 and 90° for L7 and d20-L7. The repetition time was 20 s. [d]-[f] Hyperpolarized images of L6, L7, and d20-L7, respectively collected in a single shot after PTC at 45 G. (TE/FOV/slice thickness were 3.2 ms/4 × 4 cm/5 mm respectively). [g] 2048-Average thermal image and [h] 1 scan hyperpolarized 31P image of d20-L7 collected on a 7 T BioSpec 70/30 preclinical scanner. (TE/FOV/slice thickness were 12 ms/3 × 3 cm/10 mm respectively). Samples employed 5 mM concentrations of 1 in methanol-d4 (3 mL) with L6 (6 equiv), L7 or d20-L7 (5 equiv) and 3 bar of p-H2. SNR values were calculated by dividing the mean signal intensity value in the region with signal by the standard deviation of the residual signal seen in an analogously sized noise region.
Similar images were also recorded on a 7 T BioSpec 70/30 preclinical scanner using d20-L7 as the imaging agent (Figures 6g and 6h). A >6-fold improvement in SNR was achieved in the single scan hyperpolarized image (SNR = 27.7) when compared to the corresponding 2048-average thermal average (SNR = 4.5). We note that insertion of the sample tube into the horizontal scanner caused more residual motion of the sample than was apparent in the analogous measurements on the vertical bore scanner. In addition, the time taken to transfer the sample to the magnet is ca. 5 s longer for the horizontal scanner, and the magnetic fields experienced by the sample during transfer differ. A combination of these effects could account for the reduction in observed SNR that is observed in the hyperpolarized images that were recorded on the BioSpec. Nonetheless, these results confirm the value of these signal enhancements.
Conclusions
We have demonstrated that the 1H and 31P nuclei of a series of pyridyl substituted phosphonate esters, a phosphine, and a phosphine oxide can be efficiently hyperpolarized using SABRE. Substitution at the 3-position delivered the greatest efficiency in polarization transfer catalysis to 1H and 31P, compared to substitution at the 2- and 4- positions. In the ligands that showed high performance, the phosphorus center was connected directly to the pyridyl ring such that a strong 3JPH coupling of 13.5 Hz exists between protons in the pyridyl ring and the 31P nucleus.
The phosphonate esters used in this study contained ethyl substituents. These were shown to receive polarization via a relay mechanism which first polarized the pyridyl protons, then the 31P nucleus, and finally the ethyl protons. The net gain for the ethyl protons was of the order of 50-fold per ethyl group in comparison to the 2–3 thousand level shown by the aromatic protons. The effect of leakage of the 31P magnetization to the 1H nuclei of these ethyl groups was shown to reduce the level of retained 31P polarization.
When the 1H and 31P polarization levels are compared between the protio forms of L4 and L6 and their deuterated ethyl counterparts d10-L4 and d10-L6, it could be seen that the best levels of 1H polarization result for three-substituted d10-L6. Furthermore, the 31P polarization levels are broadly similar for these two substrates at 1%. However, this raw observation masks the fact that for symmetrical d10-L4 antiphase signal dominates at all monitored polarization transfer fields. In addition, the T1 values for d10-L4 are much less than those for four-substituted d10-L6 in the presence of 2. The result of the small T1 of d10-L4 is that it must actually be better 31P polarized under SABRE than d10-L6; we observe the magnetization ca. 3 s after polarization transfer has ceased and relaxation has reduced the hyperpolarized signal amplitude. These observations confirm that 3-substituted pyridines are more suitable candidates for the future rational design of MRI contrast agents that exploit SABRE. Deuterium incorporation into the ethyl groups has also been shown to increase the level of 31P signal enhancement.
The symmetric bis-3-5-substituted derivative L7 was also synthesized, which proved to deliver strong 31P polarization. We have shown that this is a consequence of more efficient transfer into the three remaining pyridyl protons and that this synthetic strategy would enable the most atom efficient route to delivering such a pyridyl substituted agent. As a consequence, for d20-L7, the corresponding 31P signal gain exceeds 3500-fold (2.3% polarization) which is an order of magnitude larger than that obtained using other approaches.28,38
We also prepared systems where a C, N, or O based spacer was introduced to separate the 1H and 31P functionalities. While the 1H nuclei of the pyridyl groups in these analytes still retained high levels of hyperpolarization, the level of transfer into the 31P center was reduced by an order of magnitude. We concluded that the addition of a spacer was therefore undesirable.
In these studies, the process of SABRE was found to result in the creation of two types of 31P hyperpolarization. These are single-spin Zeeman magnetization and longitudinal two-spin heteronuclear H–P magnetization. When the corresponding terms were probed by a radio frequency pulse to 31P they yield in-phase and antiphase signals, respectively. As an in-phase signal is desirable when investigating these hyperpolarized substrates using traditional MRI sequences we demonstrated that if the magnetic field experienced by the sample at the point of polarization transfer catalysis is 45 G, optimal in-phase 31P signal enhancement results under SABRE in these systems. Interrogation of the resulting samples on a 9.4 T vertical bore scanner proved to allow images to be collected in a single scan at a 5 mM concentration. The SNR of a typical image was 94.4 and far exceeds that of a similar 12 h measurement under normal conditions which employed 2048 averages. Although the T1 values for these 31P signals proved to be around 6 s, we have demonstrated that we can obtain good images on phantoms. We are working toward creating further phosphorus containing molecules that have longer T1 values which might ultimately be used for in vivo applications. We expect that the methods of Levitt and Warren will be particularly relevant here.63−67
Acknowledgments
This work was supported by The Wellcome Trust (Grant 092506 and 098335), Bruker Biospin UK, and the University of York. We also thank Professor V. H. Perry, Dr. Alexandra M. Olaru, and Dr. Richard A. Green for useful discussion.
Supporting Information Available
Full synthetic procedures and spectroscopic data, NMR and MRI polarization transfer experiments, T1 data and characterization data. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
‡ These authors contributed equally.
The authors declare no competing financial interest.
Supplementary Material
References
- Takeda E.; Taketani Y.; Sawada N.; Sato T.; Yamamoto H. The Regulation and Function of Phosphate in the Human Body. BioFactors 2004, 21, 345–355. [DOI] [PubMed] [Google Scholar]
- Wang H.; Oster G. Energy Transduction in the F1Motor of Atp Synthase. Nature 1998, 396, 279–282. [DOI] [PubMed] [Google Scholar]
- Boyer P. D. The Atp Synthase - a Splendid Molecular Machine. Annu. Rev. Biochem. 1997, 66, 717–749. [DOI] [PubMed] [Google Scholar]
- Hawthorne J. N.; Pickard M. R. Phospholipids in Synaptic Function. J. Neurochem. 1979, 32, 5–14. [DOI] [PubMed] [Google Scholar]
- Dowhan W.The Role of Phospholipids in Cell Function. In Advances in Lipobiology; Gross R. W., Ed.; Elsevier Science: 1997; Vol. 2, pp 79–107. [Google Scholar]
- Abdool Karim Q.; et al. Effectiveness and Safety of Tenofovir Gel, an Antiretroviral Microbicide, for the Prevention of Hiv Infection in Women. Science 2010, 329, 1168–1174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Clercq E. Acyclic Nucleoside Phosphonates: Past, Present and Future: Bridging Chemistry to Hiv, Hbv, Hcv, Hpv, Adeno-, Herpes-, and Poxvirus Infections: The Phosphonate Bridge. Biochem. Pharmacol. 2007, 73, 911–922. [DOI] [PubMed] [Google Scholar]
- Reid I. R.; et al. Comparison of a Single Infusion of Zoledronic Acid with Risedronate for Paget’s Disease. N. Engl. J. Med. 2005, 353, 898–908. [DOI] [PubMed] [Google Scholar]
- Stehouwer B. L.; Kemp W. J. M.; Luijten P. R.; Bosch M. A. A. J.; Veldhuis W. B.; Wijnen J. P.; Klomp D. W. J. 31P Magnetic Resonance Spectroscopy of the Breast and the Influence of the Menstrual Cycle. Breast Cancer Res. Treat. 2014, 144, 583–589. [DOI] [PubMed] [Google Scholar]
- Esmaeili M.; Moestue S. A.; Hamans B. C.; Veltien A.; Kristian A.; Engebråten O.; Mælandsmo G. M.; Gribbestad I. S.; Bathen T. F.; Heerschap A. In Vivo 31P Magnetic Resonance Spectroscopic Imaging (MRSI) for Metabolic Profiling of Human Breast Cancer Xenografts. J. Magn. Reson. Imaging 2015, 41, 601–609. [DOI] [PubMed] [Google Scholar]
- Chmelik M.; Považan M.; Krššák M.; Gruber S.; Tkačov M.; Trattnig S.; Bogner W. In Vivo 31P Magnetic Resonance Spectroscopy of the Human Liver at 7 T: An Initial Experience. NMR Biomed. 2014, 27, 478–485. [DOI] [PubMed] [Google Scholar]
- Nenadic I.; Dietzek M.; Langbein K.; Rzanny R.; Gussew A.; Reichenbach J. R.; Sauer H.; Smesny S. Effects of Olanzapine on 31P MRS Metabolic Markers in Schizophrenia. Hum. Psychopharmacol. 2013, 28, 91–93. [DOI] [PubMed] [Google Scholar]
- Frey M. A.; Michaud M.; VanHouten J. N.; Insogna K. L.; Madri J. A.; Barrett S. E. Phosphorus-31 MMR of Hard and Soft Solids Using Quadratic Echo Line-Narrowing. Proc. Natl. Acad. Sci. U. S. A. 2012, 109, 5190–5195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y.; Reese T. G.; Cao H.; Hrovat M. I.; Toddes S. P.; Lemdiasov R. A.; Ackerman J. L. Bone Mineral Imaged in Vivo by 31P Solid State Mri of Human Wrists. J. Magn. Reson. Imaging 2011, 34, 623–633. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cady E. B.In Vivo Cerebral 31P Magnetic Resonance Spectroscopy. Neural Metabolism in Vivo; Choi I.-Y., Gruetter R., Eds.; Springer US: New York, 2012; Vol. 4, pp 149–179. [Google Scholar]
- McCamey D. R.; van Tol J.; Morley G. W.; Boehme C. Fast Nuclear Spin Hyperpolarization of Phosphorus in Silicon. Phys. Rev. Lett. 2009, 102, 027601. [DOI] [PubMed] [Google Scholar]
- Yang A.; Steger M.; Sekiguchi T.; Thewalt M. L. W.; Ladd T. D.; Itoh K. M.; Riemann H.; Abrosimov N. V.; Becker P.; Pohl H. J. Simultaneous Subsecond Hyperpolarization of the Nuclear and Electron Spins of Phosphorus in Silicon by Optical Pumping of Exciton Transitions. Phys. Rev. Lett. 2009, 102, 257401. [DOI] [PubMed] [Google Scholar]
- Bonhoeffer K. F.; Harteck P. Characteristics of parahydrogen. Z. Elektrochem. Angew. Phys. Chem. 1929, 35, 621–623. [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Transformation of Symmetrization Order to Nuclear-Spin Magnetization by Chemical Reaction and Nuclear Magnetic Resonance. Phys. Rev. Lett. 1986, 57, 2645–2648. [DOI] [PubMed] [Google Scholar]
- Bowers C. R.; Weitekamp D. P. Parahydrogen and Synthesis Allow Dramatically Enhanced Nuclear Alignment. J. Am. Chem. Soc. 1987, 109, 5541–5542. [Google Scholar]
- Morran P. D.; Colebrooke S. A.; Duckett S. B.; Lohman J. A. B.; Eisenberg R. Reaction of Iodocarbonylbis(Trimethylphosphine)Rhodium(I) with Parahydrogen Leads to the Observation of Five Characterisable H2 Addition Products. J. Chem. Soc., Dalton Trans. 1998, 3363–3366. [Google Scholar]
- Eisenschmid T. C.; Kirss R. U.; Deutsch P. P.; Hommeltoft S. I.; Eisenberg R.; Bargon J.; Lawler R. G.; Balch A. L. Para Hydrogen Induced Polarization in Hydrogenation Reactions. J. Am. Chem. Soc. 1987, 109, 8089–8091. [Google Scholar]
- Green R. A.; Adams R. W.; Duckett S. B.; Mewis R. E.; Williamson D. C.; Green G. G. R. The Theory and Practice of Hyperpolarization in Magnetic Resonance Using Parahydrogen. Prog. Nucl. Magn. Reson. Spectrosc. 2012, 67, 1–48. [DOI] [PubMed] [Google Scholar]
- Shchepin R. V.; Coffey A. M.; Waddell K. W.; Chekmenev E. Y. Parahydrogen Induced Polarization of 1-13c-Phospholactate-D2 for Biomedical Imaging with >30,000,000-Fold NMR Signal Enhancement in Water. Anal. Chem. 2014, 86, 5601–5605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox D. J.; Duckett S. B.; Flaschenriem C.; Brennessel W. W.; Schneider J.; Gunay A.; Eisenberg R. A Model Iridium Hydroformylation System with the Large Bite Angle Ligand Xantphos: Reactivity with Parahydrogen and Implications for Hydroformylation Catalysis. Inorg. Chem. 2006, 45, 7197–7209. [DOI] [PubMed] [Google Scholar]
- Godard C.; Lopez-Serrano J.; Galvez-Lopez M. D.; Rosello-Merino M.; Duckett S. B.; Khazal I.; Lledos A.; Whitwood A. C. Detection of Platinum Dihydride Bisphosphine Complexes and Studies of Their Reactivity through Para-Hydrogen-Enhanced Nmr Methods. Magn. Reson. Chem. 2008, 46, S107–S114. [DOI] [PubMed] [Google Scholar]
- Giernoth R.; Heinrich H.; Adams N. J.; Deeth R. J.; Bargon J.; Brown J. M. Phip Detection of a Transient Rhodium Dihydride Intermediate in the Homogeneous Hydrogenation of Dehydroamino Acids. J. Am. Chem. Soc. 2000, 122, 12381–12382. [Google Scholar]
- Fekete M.; Bayfield O.; Duckett S. B.; Hart S.; Mewis R. E.; Pridmore N.; Rayner P. J.; Whitwood A. Iridium(III) Hydrido N-Heterocyclic Carbene–Phosphine Complexes as Catalysts in Magnetization Transfer Reactions. Inorg. Chem. 2013, 52, 13453–13461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams R. W.; Aguilar J. A.; Atkinson K. D.; Cowley M. J.; Elliott P. I. P.; Duckett S. B.; Green G. G. R.; Khazal I. G.; López-Serrano J.; Williamson D. C. Reversible Interactions with Para-Hydrogen Enhance Nmr Sensitivity by Polarization Transfer. Science 2009, 323, 1708–1711. [DOI] [PubMed] [Google Scholar]
- Adams R. W.; Duckett S. B.; Green R. A.; Williamson D. C.; Green G. G. R. A Theoretical Basis for Spontaneous Polarization Transfer in Non-Hydrogenative Parahydrogen-Induced Polarization. J. Chem. Phys. 2009, 131, 194505. [DOI] [PubMed] [Google Scholar]
- Cowley M. J.; Adams R. W.; Atkinson K. D.; Cockett M. C. R.; Duckett S. B.; Green G. G. R.; Lohman J. A. B.; Kerssebaum R.; Kilgour D.; Mewis R. E. Iridium N-Heterocyclic Carbene Complexes as Efficient Catalysts for Magnetization Transfer from Para-Hydrogen. J. Am. Chem. Soc. 2011, 133, 6134–6137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zeng H.; et al. Optimization of Sabre for Polarization of the Tuberculosis Drugs Pyrazinamide and Isoniazid. J. Magn. Reson. 2013, 237, 73–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Weerdenburg B. J. A.; et al. Ligand Effects of Nhc-Iridium Catalysts for Signal Amplification by Reversible Exchange (Sabre). Chem. Commun. (Cambridge, U. K.) 2013, 49, 7388–7390. [DOI] [PubMed] [Google Scholar]
- Eshuis N.; Hermkens N.; van Weerdenburg B. J. A.; Feiters M. C.; Rutjes F. P. J. T.; Wijmenga S. S.; Tessari M. Toward Nanomolar Detection by Nmr through Sabre Hyperpolarization. J. Am. Chem. Soc. 2014, 136, 2695–2698. [DOI] [PubMed] [Google Scholar]
- Mewis R. E.; et al. Probing Signal Amplification by Reversible Exchange Using an Nmr Flow System. Magn. Reson. Chem. 2014, 52, 358–369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkinson K. D.; Cowley M. J.; Elliott P. I. P.; Duckett S. B.; Green G. G. R.; López-Serrano J.; Whitwood A. C. Spontaneous Transfer of Parahydrogen Derived Spin Order to Pyridine at Low Magnetic Field. J. Am. Chem. Soc. 2009, 131, 13362–13368. [DOI] [PubMed] [Google Scholar]
- Theis T.; Truong M.; Coffey A. M.; Chekmenev E. Y.; Warren W. S. Light-Sabre Enables Efficient in-Magnet Catalytic Hyperpolarization. J. Magn. Reson. 2014, 248, 23–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhivonitko V. V.; Skovpin I. V.; Koptyug I. V. Strong 31P Nuclear Spin Hyperpolarization Produced Via Reversible Chemical Interaction with Parahydrogen. Chem. Commun. (Cambridge, U. K.) 2015, 51, 2506–2509. [DOI] [PubMed] [Google Scholar]
- Hövener J.-B.; et al. Toward Biocompatible Nuclear Hyperpolarization Using Signal Amplification by Reversible Exchange: Quantitative in Situ Spectroscopy and High-Field Imaging. Anal. Chem. 2014, 86, 1767–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dücker E. B.; Kuhn L. T.; Münnemann K.; Griesinger C. Similarity of Sabre Field Dependence in Chemically Different Substrates. J. Magn. Reson. 2012, 214, 159–165. [DOI] [PubMed] [Google Scholar]
- Vazquez-Serrano L. D.; Owens B. T.; Buriak J. M. The Search for New Hydrogenation Catalyst Motifs Based on N-Heterocyclic Carbene Ligands. Inorg. Chim. Acta 2006, 359, 2786–2797. [Google Scholar]
- Allen K. J. H.; Nicholls-Allison E. C.; Johnson K. R. D.; Nirwan R. S.; Berg D. J.; Wester D.; Twamley B. Lanthanide Complexes of the Kläui Metalloligand, CpCo(P=O(OR)2)3: An Examination of Ligand Exchange Kinetics between Isotopomers by Electrospray Mass Spectrometry. Inorg. Chem. 2012, 51, 12436–12443. [DOI] [PubMed] [Google Scholar]
- Weiner M. A.; Schwartz P. Cobalt(II) and Nickel(II) Complexes of Methyldiphenyl-4-Pyridylphosphonium Bromide. Effects of a Cationic Pyridyl Ligand. Inorg. Chem. 1975, 14, 1714–1716. [Google Scholar]
- Zhao Y.-L.; Wu G.-J.; Han F.-S. Ni-Catalyzed Construction of C-P Bonds from Electron-Deficient Phenols Via the in Situ Aryl C-O Activation by Pybrop. Chem. Commun. (Cambridge, U. K.) 2012, 48, 5868–5870. [DOI] [PubMed] [Google Scholar]
- Xue J.; Diao J.; Cai G.; Deng L.; Zheng B.; Yao Y.; Song Y. Antimalarial and Structural Studies of Pyridine-Containing Inhibitors of 1-Deoxyxylulose-5-Phosphate Reductoisomerase. Med. Chem. Lett. 2012, 4, 278–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kubas G. J. Activation of Dihydrogen and Coordination of Molecular H-2 on Transition Metals. J. Organomet. Chem. 2014, 751, 33–49. [Google Scholar]
- Natterer J.; Bargon J. Parahydrogen Induced Polarization. Prog. Nucl. Magn. Reson. Spectrosc. 1997, 31, 293–315. [Google Scholar]
- Hasnip S.; Duckett S. B.; Taylor D. R.; Taylor M. J. Parahydrogen Enhanced Nmr Studies on Thermally and Photochemically Generated Products from [IrH3(CO)(PPh3)2]. Chem. Commun. (Cambridge, U. K.) 1998, 923–924. [Google Scholar]
- Millar S. P.; Zubris D. L.; Bercaw J. E.; Eisenberg R. On the Mechanism of Dihydrogen Addition to Tantalocene Complexes. J. Am. Chem. Soc. 1998, 120, 5329–5330. [Google Scholar]
- Bregel D. C.; Oldham S. M.; Eisenberg R. Mechanistic Studies on the Addition of Dihydrogen to Tantalocene Complexes. J. Am. Chem. Soc. 2002, 124, 13827–13832. [DOI] [PubMed] [Google Scholar]
- Aguilar J. A.; Elliott P. I. P.; Lopez-Serrano J.; Adams R. W.; Duckett S. B. Only Para-Hydrogen Spectroscopy (Opsy), a Technique for the Selective Observation of Para-Hydrogen Enhanced Nmr Signals. Chem. Commun. (Cambridge, U. K.) 2007, 1183–1185. [DOI] [PubMed] [Google Scholar]
- Heasley B. H.; Jarosz R.; Carter K. M.; Jenny Van S.; Lynch K. R.; Macdonald T. L. A Novel Series of 2-Pyridyl-Containing Compounds as Lysophosphatidic Acid Receptor Antagonists: Development of a Nonhydrolyzable Lpa3 Receptor-Selective Antagonist. Bioorg. Med. Chem. Lett. 2004, 14, 4069–4074. [DOI] [PubMed] [Google Scholar]
- Heasley B. H.; Jarosz R.; Lynch K. R.; Macdonald T. L. Initial Structure–Activity Relationships of Lysophosphatidic Acid Receptor Antagonists: Discovery of a High-Affinity Lpa1/Lpa3 Receptor Antagonist. Bioorg. Med. Chem. Lett. 2004, 14, 2735–2740. [DOI] [PubMed] [Google Scholar]
- Kers A.; Stawiński J. A New Type of Nucleotide Analogue with 4-Pyridylphosphonate Internucleotide Linkage. Tetrahedron Lett. 1999, 40, 4263–4266. [Google Scholar]
- Zmudzka K.; Johansson T.; Wojcik M.; Janicka M.; Nowak M.; Stawinski J.; Nawrot B. Novel DNA Analogues with 2-, 3- and 4-Pyridylphosphonate Internucleotide Bonds: Synthesis and Hybridization Properties. New J. Chem. 2003, 27, 1698–1705. [Google Scholar]
- Sawa M.; et al. New Type of Metalloproteinase Inhibitor: Design and Synthesis of New Phosphonamide-Based Hydroxamic Acids. J. Med. Chem. 2002, 45, 919–929. [DOI] [PubMed] [Google Scholar]
- Close J., et al. Phosphorus Derivatives as Histone Deacetylase Inhibitors. WO/2008/010985, 16 Jul. 2007, 2008.
- Boenigk W.; Hägele G. Über Die Michaelis-Arbuzov-Reaktion Perhalogenierter Pyridine. Chem. Ber. 1983, 116, 2418–2425. [Google Scholar]
- Lloyd L. S.; et al. Hyperpolarisation through Reversible Interactions with Parahydrogen. Catal. Sci. Technol. 2014, 4, 3544–3554. [Google Scholar]
- Cochrane A. R.; Irvine S.; Kerr W. J.; Reid M.; Andersson S.; Nilsson G. N. Application of Neutral Iridium(I) N-Heterocyclic Carbene Complexes in Ortho-Directed Hydrogen Isotope Exchange. J. Labelled Compd. Radiopharm. 2013, 56, 451–454. [DOI] [PubMed] [Google Scholar]
- Permin A. B.; Eisenberg R. One-Hydrogen Polarization in Hydroformylation Promoted by Platinum–Tin and Iridium Carbonyl Complexes: A New Type of Parahydrogen-Induced Effect. J. Am. Chem. Soc. 2002, 124, 12406–12407. [DOI] [PubMed] [Google Scholar]
- Barskiy D. A.; et al. The Feasibility of Formation and Kinetics of Nmr Signal Amplification by Reversible Exchange (Sabre) at High Magnetic Field (9.4 T). J. Am. Chem. Soc. 2014, 136, 3322–3325. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pileio G.; Bowen S.; Laustsen C.; Tayler M. C. D.; Hill-Cousins J. T.; Brown L. J.; Brown R. C. D.; Ardenkjaer-Larsen J. H.; Levitt M. H. Recycling and Imaging of Nuclear Singlet Hyperpolarization. J. Am. Chem. Soc. 2013, 135, 5084–5088. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Theis T.; Liang X.; Wang Q.; Zhou P.; Warren W. S. Storage of Hydrogen Spin Polarization in Long-Lived C-13(2) Singlet Order and Implications for Hyperpolarized Magnetic Resonance Imaging. J. Am. Chem. Soc. 2013, 135, 9632–9635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tayler M. C. D.; Levitt M. H. Accessing Long-Lived Nuclear Spin Order by Isotope-Induced Symmetry Breaking. J. Am. Chem. Soc. 2013, 135, 2120–2123. [DOI] [PubMed] [Google Scholar]
- Feng Y.; Theis T.; Wu T. L.; Claytor K.; Warren W. S. Long-Lived Polarization Protected by Symmetry. J. Chem. Phys. 2014, 141, 134307. [DOI] [PubMed] [Google Scholar]
- Stevanato G.; Hill-Cousins J. T.; Hakansson P.; Roy S. S.; Brown L. J.; Brown R. C. D.; Pileio G.; Levitt M. H. A Nuclear Singlet Lifetime of More Than One Hour in Room-Temperature Solution. Angew. Chem., Int. Ed. 2015, 54, 3740–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.