

Abstract

We report a versatile dendritic structure based platform for construction of targeted dual-modality imaging probes. The platform contains multiple copies of 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) branching out from a 1,4,7-triazacyclononane-N,N′,N″-triacetic acid (NOTA) core. The specific coordination chemistries of the NOTA and DOTA moieties offer specific loading of 68/67Ga3+ and Gd3+, respectively, into a common molecular scaffold. The platform also contains three amino groups which can potentiate targeted dual-modality imaging of PET/MRI or SPECT/MRI (PET: positron emission tomography; SPECT: single photon emission computed tomography; MRI: magnetic resonance imaging) when further functionalized by targeting vectors of interest. To validate this design concept, a bimetallic complex was synthesized with six peripheral Gd-DOTA units and one Ga-NOTA core at the center, whose ion T1 relaxivity per gadolinium atom was measured to be 15.99 mM–1 s–1 at 20 MHz. Further, the bimetallic agent demonstrated its anticipated in vivo stability, tissue distribution, and pharmacokinetic profile when labeled with 67Ga. When conjugated with a model targeting peptide sequence, the trivalent construct was able to visualize tumors in a mouse xenograft model by both PET and MRI via a single dose injection.
Introduction
Molecular imaging is gaining importance for noninvasive assessment of biological events in animal and human subjects. Through in vivo real-time visualization and imaging quantification of biological targets of interest, a better understanding of the biological processes underlining the initiation and progression of diseases can be obtained for more efficacious diagnosis and treatment.1,2 However, a single imaging technique is often unable to deliver all the necessary information required for a definitive clinical decision. To date, successful attempts have been seen to combine two or more imaging modalities in order to overcome the shortcomings of single modality systems for optimal visualization and better quantitative delineation of targets of interest. Consequently, multimodal imaging techniques have become the norm in both clinical practice and preclinical research as evidenced by the implementation of PET/CT (PET: positron emission tomography; CT: computed tomography) and PET/MRI (MRI: magnetic resonance imaging) procedures to patient care and the increasing use of a variety of combinations of the currently available imaging techniques in translational or basic biomedical research, such as MRI/optical and PET/NIRF (NIRF: near-infrared fluorescence).3−8 The fusion of PET and MRI is especially desirable because technically their physical features are complementary. While radionuclide-based imaging techniques, either PET or SPECT (single photon emission computed tomography), are extremely sensitive and quantitative, which allows the studies of biological events or processes at the molecular and cellular level, their spatial resolution is limited (>1 cm for current clinical scanners).9−11 On the other hand, while MRI is able to provide high spatial resolution (<0.1 cm) with exquisite soft tissue contrast even without exogenous contrast agents, contrast agents are often required to highlight specific biological or physiological processes in an amount a few orders of magnitude higher than PET or SPECT.12−14
Given that more and more PET/MRI systems will likely be used in the practice of diagnostic radiology, it becomes important to develop dual modality imaging probes that can realize the full potential of both modalities.
To take advantage of the strengths of PET and MRI, our approach is to design and synthesize a dual-modality agent scaffold offering a “single pharmacological behavior” for both imaging acquisitions. The merging of PET and MRI probe moieties to a single molecular platform would facilitate colocation and cross-validation of each modality in targeted regions of interest (two measures of one event). While MRI can provide the exact location of the probe, motion artifact correction, and PET partial volume correction, PET can afford better imaging quantification for higher detection sensitivity. Further, given that the proton MRI contrast actually reflects the map of the proton density, a colocalized PET signal distinct from the proton background could make the MRI contrast more identifiable, which further improves the MRI sensitivity.
Indeed several nanoparticle-based PET/MRI agents have been reported with enhanced magnetic relaxivities and considerable promise as dual-modality imaging agents.15−17 However, a nanoparticle platform for PET/MRI probe development presents challenges. First, the nanoparticle-based PET/MRI probes are not single molecular entities and often have questionable in vivo stability or molecular integrity. Second, to serve the purpose of diagnostic imaging, contrast agents are desired to have a reasonable blood circulation half-life for efficient accumulation in targets and minimal nonspecific deposition in the mononuclear phagocyte system (MPS) organs after systemic administration. However, it is well-known that most nanoparticles if not all possess suboptimal in vivo distribution as shown by rapid sequestration into MPS organs and thereafter slow clearance. These properties present potential health hazards18−20 which could impede their translation toward clinical applications. Third, the majority of nanoparticle-based PET/MRI agents provide T2 contrast; only a handful are T1-based agents, which are preferred in the cases of low proton density in the target tissues. To date, a wide range of macromolecules, such as perfluorocarbon emulsions,15 silica nanoparticles,16,21,22 liposomal vesicles,17,23−25 dendrimers,26−32 and polymers,33 have been explored for the development of gadolinium-based T1 contrast agents. Of the macromolecular systems, dendrimers, which possess definitive molecular structures and formula, have shown a promising role. For instance, the dendrimer systems of poly(amido amine) (PAMAM) have been used as blood pool,34 liver,35 renal,36 lymphatic,37 and tumor-specific38 contrast agents. However, the Gd-PAMAM complexes rarely afford an ionic relaxivity greater than 11 mM–1 s–1 per gadolinium ion.39,40 When such dendrimer platforms are used to develop PET/MRI or SPECT/MRI agents, it is challenging to achieve precise control of radioisotope loading into specific chelating moieties. Herein, we present a dual-modality molecular probe design (Figure 1) carrying six 1,4,7,10-tetraazacyclododecane-1,4,7,10-tetraacetic acid (DOTA) moieties branched out from a single 1,4,7-triazacyclononane-N,N′,N″-triacetic acid (NOTA) core structure. In this work, we demonstrate an exclusive loading of Gd3+ into the DOTA units affording a T1 ion relaxivity of 15.99 mM–1 s–1 at 20 MHz, while the NOTA core remains available to form a chelate with 67/68Ga3+ for nuclear imaging. If the targeted accumulation event is rapid, 68Ga (t1/2 = 68 min; β+ = 89%, Eβ+max = 1.92 MeV; EC = 11%) will be used for PET imaging; otherwise, 67Ga (t1/2 = 3.26 d; γ = 184 keV) will be used for SPECT imaging.
Figure 1.
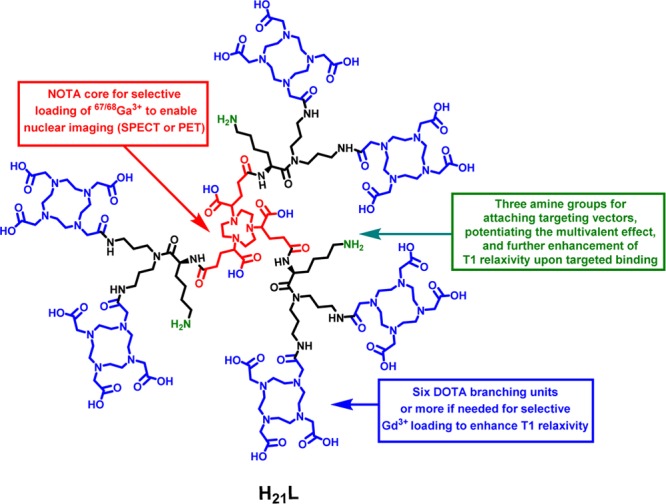
Dual-modality molecular probe design.
Results and Discussion
The dual-modality molecular probe design also possesses three amino groups for potential conjugation with multiple copies of targeting molecules, thereby providing multivalent effect to the construct for better specific binding to the biological target of interest. Of note, the strong interaction between the targeting molecule tethered probe and the corresponding target, triggered by multivalent effect, is expected to further enhance the relaxivity of the probe. For proof of concept, we conjugated each amine with a model integrin αvβ3 targeting peptide, c(RGDyK). The main features of this molecular dual-modality imaging probe design include the apparent in vivo stability (due to the inertness of the Gd-DOTA and Ga-NOTA complexes) and the defined molecular formula with selective loading of two metal ions.
Synthesis
The desired ligand H21L was synthesized in 7 steps with an overall yield of approximately 15%. The structure of each intermediate was verified by 1H NMR and 13C NMR as well as mass spectroscopy. The synthesis route to compound H21L is outlined in Scheme 1 and its functionalization is depicted in Scheme 2. Compound 2 was obtained from bis(3-aminopropyl)amine and carbobenzyloxy (Cbz) protected lysine in three steps (see Supporting Information). The free primary amine of 2 was coupled to the carboxylic acid bearing side arms of NOTA-(OH)3 (Scheme 1) via carbodiimide chemistry to form 3. The N-phthaloyl protected amino groups of 3 were freed by deprotection via hydrazine to afford 4, which contains six amino groups serving as points of attachment for the DOTA units.
Scheme 1. Synthetic Route to Ligand H21L.

Scheme 2. Synthesis of Integrin αvβ3 Targeted Ligand Scaffolds.
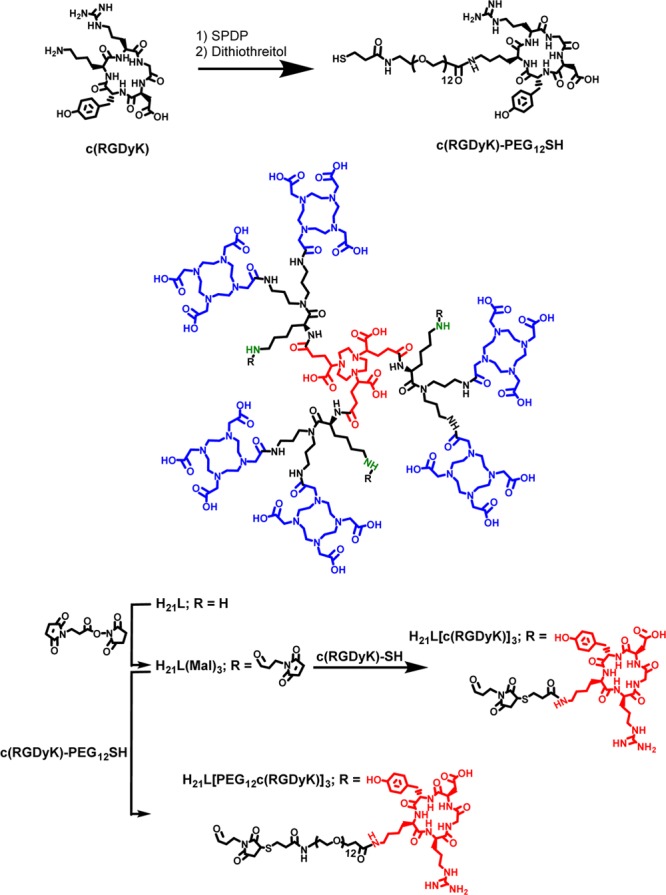
The N-hydroxy succinamide activated DOTA ester, 5, was reacted to the six amino groups forming 6. Attempts to deprotect the Boc groups of compound 6 led to partial deprotection of the Cbz groups. To overcome this problem, the ligand was fully deprotected by 30% HBr in an acetic acid solution. The synthesized ligand, H21L, was fully characterized. The ligand carries six DOTA units intended for chelating Gd3+ and one NOTA unit intended for chelating Ga3+. Since Gd3+ and Ga3+ can be specifically loaded into the DOTA and NOTA moieties, respectively, based on their ion-size preference, the bimetallic molecular complex is expected to carry the two metal ions in a specified ratio so as to facilitate PET imaging based quantification.
Functionalization of ligand H21L was performed by utilizing the previously mentioned amino groups. Maleimido groups were tethered to amines using the commercially available reagent of N-succinimidyl 3-maleimidopropionate (Scheme 2). The model targeting peptide, c(RGDyK), was tethered to the maleimido carrying ligand H21L via thiol–maleimide coupling. Given that targeting molecules attached to the bimetallic complex may not be able to stretch out for specific binding because of the steric hindrance from the construct, additionally we introduced a PEG12 linker between the complex and targeting vector.
The thiol group without a spacer (zero) or with a PEG12 linker was introduced to c(RGDyK) using the commercially available reagent: 2-pyridyldithiol-tetraoxaoctatriacontane-N-hydroxysuccinimide (SPDP). The thiol terminated c(RGDyK) peptides with zero and PEG12 linker were subsequently added to H21L(Mal)3 to afford Gd6H3L[c(RGDyK)]3 and Gd6H3L[PEG12c(RGDyK)]3, respectively.
Complexation with Gd3+ and Ga3+ to form Gd6LGa
The thermodynamic stability constants of Gd-DOTA (log KGd-DOTA = 24.7), Gd-NOTA (log KGd-NOTA = 14.3), and Ga-NOTA (log KGa-NOTA = 31.0)41,42 suggest that when ligand H21L is treated with 6 equiv of Gd3+, its complex can be formed exclusively with the DOTA units. The bimetallic complex of H21L, Gd6LGa, can then be formed by incorporation of Ga3+ into the NOTA core. However, considering that the reaction of L with the stoichiometric amount of Gd3+ may be slow and could lead to partial complexation of the DOTA units, the first complexation was performed with excess of Gd3+ to give complete chelation of both the DOTA and NOTA units forming Gd7L. This was confirmed with mass spectroscopy (see Supporting Information) as well as 1H NMR spectra using Eu3+ instead of Gd3+. Later, Gd3+ was efficiently removed from the NOTA core by treating the complex with excess diethylene triamine pentaacetic acid (DTPA) (the log K of Gd-DTPA42 is approximately 8 orders of magnitude higher than that of Gd-NOTA). The incorporation of either cold Ga3+ or 68Ga3+ into the NOTA core of complex of Gd6H3L was then carried out at pH 3–5 in an HEPES buffer. The cold Gd6LGa complex was purified and characterized by HPLC and mass spectrometry. Various concentration of Gd6LGa were injected to inductively coupled plasma mass spectrometry (ICP-mass) and the ratio of Gd:Ga was calculated to be 6:1 (see Supporting Information). The Gd6L68Ga was purified and characterized by radio-HPLC using the cold complex as reference.
Coordination Chemistry of the NOTA Core of H21L
To prove the efficient removal of Gd3+ from the NOTA core during the complexation procedure, the same complexation procedure was followed using Eu3+ instead of Gd3+. Chemically, Eu3+ and Gd3+ have similar reactivities toward NOTA and DOTA. Since Eu3+ is only weakly paramagnetic, the 1H NMR signals of its complexes are relatively sharp and also paramagnetically shifted. This facilitates relatively straightforward structural characterization by NMR spectroscopy. To accomplish this, the Eu3+ complex of H21L, were synthesized via two procedures, i.e., with and without a challenge by excess DTPA, followed by HPLC purification. Both complexes were characterized by 1H NMR spectroscopy (400 MHz). Figure 2a,b shows the 1H NMR spectra of Eu3+ complexes of H21L with and without the DTPA challenge, respectively. The DTPA treated Eu3+ complex (Figure 2a) showed sharp peaks in the NOTA region (around 3–4 ppm) suggesting a metal free NOTA core as opposed to the untreated complex (Figure 2b), which showed broad peaks indicating the interaction of metal with the NOTA core. This characterization along with the mass spectroscopy results of Gd7L and Gd6H3L indicates the exclusive Gd3+ coordination with the six DOTA moieties of H21L. Further, the formation of Gd6LGa was confirmed by the molar ratio of Gd:Ga determined by ICP-mass (Supporting Information). These results demonstrate that Gd3+ and Ga3+ have been exclusively loaded to the DOTA and NOTA moieties as designed, respectively, for the preparation of the desired dual-modality molecular imaging probe.
Figure 2.
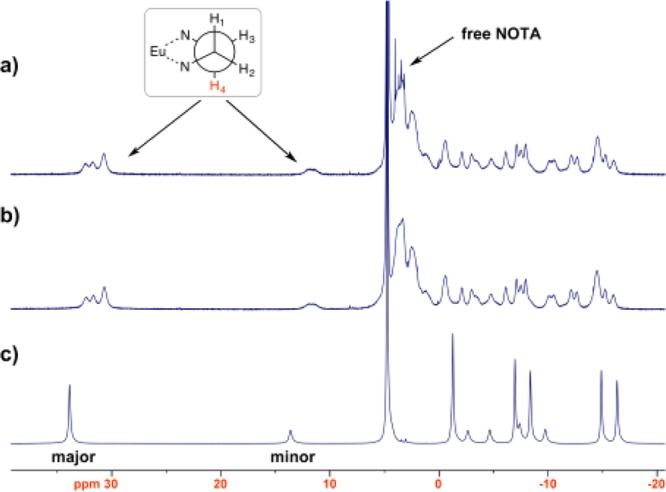
400 MHz 1H NMR spectra of (a) Eu3+ complex of H21L treated with excess DTPA, (b) untreated Eu3+ complex of H21L, and (c) Eu-DOTA complex.
Relaxometric Measurements
The T1 relaxivity of Gd6LGa was measured at 20 MHz, 25 °C, to be 95.98 mM–1 s–1 with R2 value greater than 0.99 (each Gd3+ in the complex accounted for 15.99 mM–1 s–1 (ion relaxivity)). Under the same conditions, the relaxivity of Magnavist was found to be 4.1 mM–1 s–1. Impressively, the T1 relaxivity of Gd6H3L was approximately 4 times higher than that of the Gd-DOTA complex and comparable to the Gd-PAMAM complexes, which have been reported with the ion relaxivity up to 11 mM–1 s–1.39,40 It is noteworthy that a similar relaxivity was also reported for a polylysine-based contrast agent (MW 17 453) with 24 Gd-DOTA units.43,44 At the physiological temperature of 37 °C, the relaxivity of Gd6LGa was unchanged while its relaxivity in rat serum at 25 °C increased to 153.1 mM–1 s–1. These observations suggest two things: first, the lack of temperature dependence between 25 and 37 °C suggests that the water exchange rate in the appended Gd-DO3A-monoamide complexes limits the relaxivity somewhat at 25 °C. Given that water exchange is faster at 37 °C, the expected decline in relaxivity with increasing temperatures is offset by faster water exchange and a more favorable relaxivity at 37 °C. This was an unanticipated yet welcomed added advantage for this probe platform design. The higher relaxivity in serum indicates that Gd6LGa does interact with one or more serum proteins.
MR Imaging and Relaxivity Measurements of Gd6LGa at 1.0 T
The MR imaging potential of Gd6LGa was evaluated at magnetic field of 1.0 T. Figure 3 shows the T1 weighted imaging of Gd6LGa in comparison with Magnevist at a series of decreasing concentrations (0.4–0.02 mM determined by ICP-mass). The images of Gd6LGa were obviously brighter than those of Magnevist (4.1 mM–1 s–1) at each equivalent concentration. The T1 relaxivity of Gd6LGa was measured at 113.36 mM–1 s–1 (T1 ion relaxivity: 18.89 mM–1 s–1) at 1.0 T (25 °C). Since the agent is intended for conjugation to a targeting moiety for receptor-based imaging, such interactions could further enhance the relaxivity of the agent in vivo.
Figure 3.

T1-weighted MR images of samples recorded at 1.0 T at 25 °C. Samples of Magnevist (a → e) and Gd6H3L (v → z) containing a series of decreasing concentrations from 0.4 mM, 0.2 mM, 0.1 mM, 0.04 mM, to 0.02 mM.
In Vivo Tissue Distribution, Pharmacokinetics, and Stability of Gd6LGa
As compared to other nanoparticle-based dual modality imaging platforms, our molecular probe design is expected to have a reasonable blood circulation half-life and an efficient clearance profile from the organs. To evaluate the in vivo behavior of this probe design, Gd6LH3 was radiolabeled with 67Ga (t1/2 = 3.26 d) so that the tissue biodistribution could be evaluated over an extended period. As shown in Table 1, the bimetallic complex, Gd6L67Ga, indeed displayed a rapid and efficient clearance profile from all major organs except kidneys in normal mice. The high renal uptake can be attributed to the net positive charge on the probe due to the presence of amine groups.45 However, an efficient renal clearance is obvious after the initial high uptake as seen from the decrease of the kidney uptake level from 101.0 ± 4.3%ID/g at 1 h p.i. to 43.0 ± 5.1%ID/g at 24 h p.i. One would anticipate that kidney clearance would be even faster once the positively charged amino groups are removed from the agent through conjugation with a targeting vector. Indeed, when the amine groups of the agent were capped with c(RGDyK), the kidney uptake was impressively reduced to ca. 3%ID/g at 2 h p.i.
Table 1. Tissue Distribution Data and Pharmacokinetic Parameters of Gd6L67Ga in Normal Balb/C Mice (n = 4)a.
| %ID/g |
|||
|---|---|---|---|
| Organ | 1 h | 4 h | 24 h |
| Blood | 3.51 ± 0.22 | 2.63 ± 0.84 | 0.21 ± 0.06 |
| Heart | 1.21 ± 0.13 | 0.94 ± 0.40 | 0.21 ± 0.02 |
| Lung | 2.59 ± 0.5 | 1.78 ± 0.87 | 0.38 ± 0.09 |
| Liver | 1.39 ± 0.11 | 2.13 ± 0.81 | 1.39 ± 0.24 |
| Kidney | 101.32 ± 4.32 | 57.42 ± 17.29 | 43.90 ± 5.12 |
| Spleen | 0.73 ± 0.19 | 0.69 ± 0.07 | 0.42 ± 0.12 |
| Muscle | 0.27 ± 0.09 | 0.38 ± 0.13 | 0.08 ± 0.02 |
| Fat | 0.27 ± 0.10 | 0.27 ± 0.10 | 0.06 ± 0.02 |
| Brain | 0.08 ± 0.01 | 0.07 ± 0.02 | 0.02 ± 0.00 |
| Two-Compartment Clearance Profile (min) | |||
|---|---|---|---|
| t1/2α | 11.33 ± 1.31 | t1/2β | 438.37 ± 15.11 |
Data are presented as mean ± standard deviation.
The pharmacokinetics of Gd6L67Ga showed a two-compartment profile with t1/2α = 11.3 min and t1/2β = 7.3 h, quite different compared to nanoparticle-based imaging agents which can be described by one-compartment models.46,47 The t1/2α reveals that the tissue distribution of Gd6L67Ga was relatively rapid while the reasonably long t1/2β enables the probe to accumulate in its targeted tissues without incurring high background uptake. This indicates that the probe behaves similarly to other clinically available small molecule contrast agents.48 The presence of amine groups on the ligand provides the ease of incorporation of polyethylene glycol (PEG) chains into the probe for optimization of the in vivo kinetics if necessary.
The in vitro and in vivo stabilities of Gd6L67Ga were evaluated through incubation in rat serum and by mouse urine metabolite analysis, respectively. The bimetallic complex remained intact during the entire course of study, which further validates this molecular probe platform design.
Receptor-Binding Assay of Targeted Gd6LGa Complexes
Two integrin αvβ3 targeted Gd6LGa complexes Gd6H3L[c(RGDyK)]3 and Gd6H3L[PEG12c(RGDyK)]3) were prepared to validate that our molecular construct design can be utilized for specific imaging of cancer biomarkers. The αvβ3-binding affinities were determined by a competitive cell-binding assay using 125I-echistatin (PerkinElmer) as the αvβ3-specific radioligand. The best-fit IC50 values (inhibitory concentration where 50% of the 125I-echistatin bound on U87MG cells are displaced) of c(RGDyK), Gd6H3L[c(RGDyK)]3, and Gd6H3L[PEG12c(RGDyK)]3) were measured to be 199 nM, 489 nM, and 37 nM (Supporting Information), respectively. The significantly improved αvβ3 binding affinity of Gd6H3L[PEG12c(RGDyK)]3) clearly indicates the critical role of the PEG12 linker in the desired targeting property of our molecular scaffold design.
PET/MR Imaging Using Gd6L[PEG12c(RGDyK)]368Ga
To overcome the sensitivity difference between PET and MR, the radioactivity of 68Ga was controlled in the labeling of Gd6H3L[PEG12c(RGDyK)]3. The final dose injected into each animal carried about 6.0 mg of the final compound with an activity of 100–150 μCi (Gd: ca. 0.027 mmol/kg; c(RGDyK): 2.0 μmol). Figure 4 shows in vivo dual PET/MR images of integrin ανβ3 positive U87MG tumor enabled by a single dose injection of Gd6L[PEG12c(RGDyK)]368Ga. The average tumor uptake measured by PET was 0.88%ID/g. It should be noted that the tumor uptake value was reduced by roughly 3 times when the injected amount of the agent increased from 0.1 mg to 6.0 mg. This decrease also indicates the integrin ανβ3 imaging specificity. The observed MR contrast enhancement in the tumor was 3.06 (n = 3) when compared to the contrast before injection.
Figure 4.
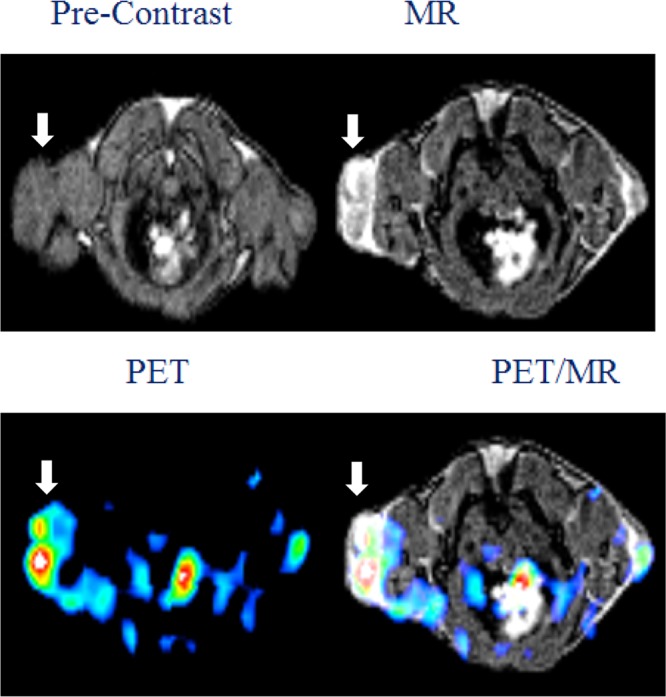
Representative MRI (upper panel), PET (lower panel: left), and fused PET/MR (lower panel: right) images of U87MG tumor xenograft in SCID mice at 1 h post injection of Gd6L[PEG12c(RGDyK)]368Ga. The white arrow indicates the tumor site.
To date, few PET/MRI or SPECT/MRI agents with a defined structure have been reported. In vivo pH monitoring PET/MRI agents carrying one Gd-DOTA unit along with 18F or 68Ga as PET contrast have been reported.49 Bimetallic complexes carrying one Gd-DOTA conjugated to 68Ga-NOTA and one Gd-DOTA conjugated to cold Cu or In-DOTA do exist.50,51 However, they were not designed to serve the goal of providing targeted contrast to both imaging techniques. To provide proof of concept, the probe design in this work consists of only six gadolinium chelatable DOTA units. While the anticipated T1 relaxivity enhancement was achieved, we recognize that further amplification of MRI sensitivity is necessary in order to realize the practical application of the molecular probe design as dual-modality imaging agents. Given the versatility of the design concept, we believe that it can be readily modified or adapted to reach the desired MRI sensitivity.
In conclusion, we have successfully demonstrated a molecular platform intended for targeted dual-modality imaging of PET/MRI or SPECT/MRI, which carries quantifiable numbers of 67/68Ga3+ and Gd3+ ions in their specific chelation moieties. This probe design displays a very favorable high T1 relaxivity for MRI contrast enhancement. With the current rapid growth of hybrid PET/MRI systems in diagnostic radiology, our molecular design of dual-modality agents possessing a “single pharmacological behavior” offers a versatile platform for the development of multimodality imaging agents with potential applications for noninvasive molecular profiling of various diseases.
Experimental Procedures
General Materials and Methods
All reactions were carried out under N2 atmosphere in degassed dried solvents. Commercially available starting materials were purchased from commercial vendors and used directly without further purification unless otherwise stated. All aqueous solutions were prepared with Milli-Q water. Bulk solvents were removed using rotary evaporator under reduced pressure at 40 °C. Trace solvents were removed under high vacuum. Matrix-assisted laser desorption/ionization (MALDI) mass spectra were acquired on an Applied Biosystems Voyager-6115 mass spectrometer. Radiolabeled conjugates were purified by Light C-18 Sep-Pak cartridges (Waters, Milford, MA). High performance liquid chromatography (HPLC) was performed on a Waters Xterra Shield RP C-18 semiprep column (250 × 10 mm, 10 μm) and read by a Waters 2996 photodiode array detector and an in-line Shell Jr. 2000 radio-detector. The mobile phase was H2O with 0.1% trifluoroacetic acid (TFA) (solvent A) and acetonitrile with 0.1% TFA (solvent B). The gradient consisted of 0% B to 80% B in 0–40 min at 4.0 mL/min flow rate. The radioactivity of excised tissue samples and radioactive standards were counted by a Wizard2 300 automatic γ-counter (PerkinElmer).
Synthesis Procedures
Compound 3
To a solution of the protected acid 1 (0.10 g, 0.15 mmol) in CH3CN (1.0 mL) were added the deprotected amine 2 (0.40 g, 0.60 mmol), dicyclohexylcarbodiimide (0.15 g, 0.83 mmol), and triethylamine (0.30 g, 0.27 mmol). The resultant solution was stirred for 12 h, filtered, and the solvent evaporated. The crude product was purified by flash chromatography (ethyl acetate) to give 3, a NOTA derivative, as a white solid (0.26 g, 0.10 mmol, 67%). 1H NMR (400 MHz, CDCl3): δ 8.73 (bs, 6H), 7.71 (m, 12H), 7.61 (m, 12H), 7.23 (m, 15H), 5.56 (bs, 2H), 4.99 (m, 5H), 4.74 (m, 3H), 3.79–3.27 (m, 24H), 3.25–2.77 (m, 13H), 2.60–2.17 (m, 4H), 2.15–1.71 (m, 12H), 160 (m, 4H), 1.38 (m, 43H). 13C NMR (100 MHz, CDCl3): δ 172.5, 168.3, 156.8, 136.5, 134.1, 133.9, 131.8, 128.4, 127.9, 123.3, 123.2, 79.0, 69.8, 66.5, 64.8, 49.3, 45.9, 44.3, 40.5, 35.5, 35.3, 325, 32.2, 29.3, 28.3, 27.9, 26.8, 22.6. MS (MALDI) m/z calcd for C141H168N18O30: 2594.2; found: 2595.9 ([M + H]+).
Compound 4
To a solution of 3 (0.26 g, 0.11 mmol) in ethanol (1.0 mL) was added hydrazine monohydrate (0.1 mL, 2.0 mM) and the mixture was stirred for 12 h at room temperature. After the reaction, the precipitate was removed by filtration. The filtrate was evaporated and extracted with CH2Cl2 (3 × 10 mL). The combined organic layers were evaporated to give 4 as light yellow oil (0.15 g, 0.08 mmol, 80%). This compound was used for the next step without further purification. 1H NMR (400 MHz, CD3OD): δ 7.31 (s, 15H), 4.99 (m, 6H), 4.66 (m, 3H), 3.68–3.39 (m, 10H), 3.38–3.31 (m, 6H), 3.20–2.94 (m, 17H), 2.93–2.72 (m, 9H), 2.65–2.30 (m, 7H), 1.95–1.81 (m, 6H), 1.78–1.57 (m, 6H), 1.55–1.20 (m, 44H). 13C NMR (100 MHz, CD3OD): δ 173.7, 173.1, 157.6, 136.9, 128.9, 127.3, 126.8, 126.5, 82.6, 77.3, 65.9, 64.5, 50.2, 48.8, 45.9, 44.5, 42.3, 38.5, 38.1, 36.7, 30.3, 29.1, 27.8, 26.6, 25.2. MS (MALDI) m/z calcd for C93H156N18O18: 1814.1; found: 1816.3 ([M + H]+).
Compound 6
To a solution of 4 (0.15 g, 0.08 mmol) in DMF (1 mL) were added 5 (0.53 g, 0.80 mM), an N-hydroxysuccinimide ester of DOTA, and the mixture was stirred for 24 h at room temperature. The solvent was evaporated and the product purified by reverse phase HPLC using water and acetonitrile solvent mixture to give 6 as a white solid (0.29 g, 0.06 mmol, 70%). 1H NMR (400 MHz, CDCl3): δ 7.40–7.18 (s, 15H), 5.15–4.99 (m, 6H), 4.71 (m, 6H), 4.38–4.83 (m, 26H), 3.82–3.36 (m, 61H), 3.30–2.62 (m, 17H), 2.60–2.30 (m, 12H), 2.23–1.98 (m, 10H), 1.99–1.80 (m, 10H), 1.79–0.96 (m, 174H). 13C NMR (100 MHz, CD3OD): δ 172.8, 172.6, 170.8, 170.6, 157.3, 137.2, 128.2, 127.6, 127.2, 84.5, 81.5, 65.8, 64.7, 53.6, 51.6, 49.2, 48.5, 45.3, 43.6, 42.0, 40.1, 36.5, 31.9, 27.2, 28.5, 27.2, 27.1, 22.7. MS (MALDI) m/z calcd for C261H456N42O60: 5139.4; found: 5138.7 ([M + H]+).
Ligand H21L
To a solution of 6 (0.10 g, 0.02 mmol) was added 30% HBr in AcOH (2 mL) and the solution was allowed to stir for 4 h. The solvent was evaporated, and the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture to give H21L as a white solid (0.06 g, 0.02 mmol, 75%). 1H NMR (400 MHz, CD3OD): δ 4.42–3.62 (m, 89H), 3.59–3.32 (m, 67H), 3.24–2.85 (m, 48H), 2.80–2.30 (m, 22H), 2.34–2.03 (m, 17H), 2.06–1.85 (m, 14H), 1.88–1.65 (m, 20H), 1.65–1.39 (m, 11H), 1.31 (m, 6H). MS (MALDI) m/z calcd for C153H270N42O54: 3561.9; found: 3563.0 ([M + H]+).
Compound H21L(Mal)3
To the free ligand H21L (0.200 g, 0.056 mmol) dissolved in DMF (1 mL) was added triethyl amine (0.022 g, 0.224 mmol) and N-(γ-maleimidobutyryloxy)succinimide and the solution was allowed to stir for 24 h. The solvent was evaporated, the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture to give H21L(Mal)3 as a white solid. (0.102 g, 0.052 mmol, 45.1%). MS (MALDI) m/z calcd for C174H285N45O63: 4015.0; found: 4016.3 ([M + H]+).
Compound c(RGDyK)-SH
To the DMF solution (1 mL) of the commercially available integrin αvβ3 targeting peptide c(RGDyK) (0.030 g, 0.048 mmol) (Peptides International Inc., Kentucky) was added N-succinimidyl 3-(2-pyridyldithio)-propionate (0.020 g, 0.064 mmol) (Thermo Scientific, IL) and the solution was allowed to stir for 6 h. The solvent was evaporated, the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture and lyophilized. The resultant white solid was dissolved in DMF (1.0 mL) and dithiothreitol (0.010 g, 0.065 mmol) was then added. The solution was allowed to stir for 3 h. The solvent was evaporated and the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture to give c(RGDyK)-SH as a white solid (0.103 g, 0.014 mmol, 29.5%). MS (MALDI) m/z calcd for C30H45N9O9S: 707.3; found: 708.3 ([M + H]+).
Compound H21L[c(RGDyK)]3
To the malemide carrying ligand, H21L(Mal)3, (0.020 g, 0.005 mmol) dissolved in PBS (1×) was added the thiol carrying c(RGDyK)-SH (0.014 g, 0.029 mmol), and the solution was allowed to stir for 18 h. The solution was purified by reverse phase HPLC using water and acetonitrile solvent mixture to give H21L[c(RGDyK)]3 as a white solid. (0.010 g, 0.002 mmol, 33.3%). MS (MALDI) m/z calcd for C264H420N72O90S3: 6136.9; found: 6139.1 ([M + H]+).
Compound c(RGDyK)PEG12SH
To the DMF solution (1 mL) of the integrin αvβ3 targeting peptide c(RGDyK) (0.040 g, 0.064 mmol) (Peptides International Inc., Kentucky) dissolved in DMF (1.0 mL) was added N-2-pyridyldithiol-tetraoxaoctatriacontane-N-hydroxysuccinimide (0.060 g, 0.065 mmol) (Thermo Scientific, IL) and the solution was allowed to stir for 6 h. The solvent was evaporated and the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture and lyophilized. The resultant white solid was dissolved in DMF (1 mL) and dithiothreitol (0.010 g, 0.065 mmol) was then added and the solution was allowed to stir for 3 h. The solvent was evaporated and the product neutralized and purified by reverse phase HPLC using water and acetonitrile solvent mixture to give c(RGDyK)PEG12SH as a colorless viscous liquid. (0.025 g, 0.019 mmol, 30.2%). MS (MALDI) m/z calcd for C57H98N10O22S: 1306.6; found: 1307.5 ([M + H]+).
Compound H21L[PEG12c(RGDyK)]3
To the malemide carrying ligand, H21L(Mal)3 (0.030 g, 0.007 mmol), dissolved in PBS (1×) was added the thiol carrying peptide c(RGDyK)-SH (0.038 g, 0.028 mmol) and the solution was allowed to stir for 18 h. The solution was purified by reverse phase HPLC using water and acetonitrile solvent mixture to give H21L[PEG12c(RGDyK)]3 as a white solid (0.019 g, 0.002 mmol, 35.3%). MS (MALDI) m/z calcd for C345H579N75O129S3: 7936.0; found: 7936.7 ([M + H]+).
Compound Gd7L
The free ligand H21L (0.060 g, 0.013 mmol) was dissolved in water (1 mL) and the pH was adjusted to 7.0 with NaOH (0.1 M). To this solution was added an excess amount of GdCl3·6H2O and the pH was readjusted to 6.5 and allowed to stir at room temperature overnight. The pH of the resultant solution was raised above 8 using 1 M aqueous NaOH, causing the excess Gd3+ to precipitate as Gd(OH)3. The solution was filtered and the pH was readjusted to 7.0 using 1 N HCl. The solution was purified using HPLC and the fractions pooled together and lyophilized to give a white solid (0.051 g, 0.011 mmol, 84.6%). MS (MALDI) m/z calcd for C153H261Gd7N42O60: 4644.2; found: 4645.1 ([M + H]+).
Compound Gd6H3L
To the solution of Gd7L (0.05 g, 0.011 mmol), DTPA (0.1 mM, 1 mL) was added. The solution was stirred at room temperature for 2 h and then purified using HPLC. The desired fractions were pooled together and lyophilized to give Gd6H3L as a white solid (0.04 g, 0.01 mmol, 81%). MS (MALDI) m/z calcd for C153H264Gd6N42O60: 4489.3; found: 4634.9 ([M + K+ 6H2O]+).
Compound Eu6H3L
The free ligand H21L (0.030 g, 0.008 mmol) was dissolved in water (1 mL) and the pH was adjusted to 7.0 with NaOH (0.1 M). To this solution was added an excess amount of EuCl3·6H2O and the pH was readjusted to 6.5 and allowed to stir at room temperature overnight. The pH was raised above 8.0 using 1.0 N aqueous NaOH, which caused the excess Eu3+ to precipitate as Eu(OH)3. The solution was filtered and the pH was readjusted to 7.0 using 1.0 N HCl. To the resulting solution, DTPA (0.1 mM, 1 mL) was added and the solution was purified using HPLC to give the desired complex. The desired fractions were pooled together and lyophilized to give Eu6LH3 as a white solid. (0.042 g, 0.009 mmol, 54%). MS (MALDI) m/z calcd for C153H252Eu6N42O54: 4453.4; found: 4454.5 ([M + H]+).
Compound Cold Gd6LGa
The gadolinium complex, Gd6H3L (0.001 g, 0.225 μmol), was dissolved in a solution of 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES, pH = 6.5, 1.0 M, 1.0 mL). To the resulting solution was added a solution of GaCl3 (0.0001 g, 0.567 μmol) in 0.6 N HCl (0.3 mL) and the resulting solution was stirred for 1 h. To the mixture was added 500 μL of 5.0 mM ethylendiaminetetraacetic acid (EDTA) and the mixture was stirred for another 5 min at room temperature (EDTA was used to remove nonspecifically bound or free GaCl3 from the complex). The purification of the bimetallic complex was carried out by passing the mixture through a preconditioned Sep-Pak C-18 heavy cartridge. After thorough rinsing (3 × 5 mL, water) of the cartridge, the bimetallic complex, Gd6LGa, was eluted by an ethanol–water mixture (70:30). The product was purified with HPLC and characterized by mass spec. MS (MALDI) m/z calcd for C153H264GaGd6N42O60: 4555.28; found: 4556.9 ([M + H]+).
Compound Gd6L68Ga
To a 1.5 mL eppendorf tube containing 30 μg Gd6H3L complex in 1.0 mL of HEPES (pH = 6.5) solution was added a solution of 8.0 mCi of 68GaCl3 in 0.6 N HCl. The reaction mixture was incubated at 75 °C for 0.5 h on a shaker. To this solution was added DTPA (5.0 mM, 5.0 μL) and the reaction mixture was stirred for 5 min at room temperature (DTPA was used to remove nonspecifically bound or free 68Ga from Gd6L68Ga). The Gd6L68Ga complex was purified by passing the mixture through a preconditioned Sep-Pak C-18 light cartridge. After thorough rinsing (3 × 3 mL, water) of the cartridge, Gd6L68Ga was eluted by an ethanol–water mixture (70:30) to give 3.6 mCi of labeled compound. The product was first analyzed by a Rita Star Radioisotope TLC Analyzer (Straubenhardt, Germany) on instant thin-layer chromatography (ITLC-SG) plates (Pall Life Sciences, East Hills, NY) and then by radio-HPLC to determine the radiochemical purity of the product. The compound was determined to have more than 95% purity.
Compound Gd6L67Ga
To a 1.5 mL eppendorf tube containing 30 μg Gd6H3L complex in 1.0 mL of HEPES (pH = 6.5) solution was added a solution of 4.0 mCi of 67GaCl3 in 0.6 N HCl. The reaction mixture was shaken and incubated at 75 °C for 0.5 h. To this solution was added DTPA (5.0 mM, 5.0 μL) and the reaction mixture was incubated for 5 min at room temperature. The Gd6L67Ga complex was purified by passing the mixture through a preconditioned Sep-Pak C-18 light cartridge. After thorough rinsing (3 × 3 mL, water) of the cartridge, Gd6L67Ga was eluted by an ethanol–water mixture (70:30) to give 2.1 mCi of the labeled compound. The product was first analyzed by a TLC Analyzer on ITLC-SG plates and then by radio-HPLC to determine the radiochemical purity of the product. The compound was determined to have more than 95% purity.
Compound Gd6H3L[c(RGDyK)]3
The c(RGDyK) modified ligand H21L[c(RGDyK)]3 (0.010 g, 0.002 mmol) was dissolved in water (1 mL) and the pH was adjusted to 7 with NaOH (0.1 M). To this solution was added an excess of GdCl3·6H2O and the pH was again adjusted to 6.5 and allowed to stir at room temperature overnight. The pH was raised above 8.0 using 1.0 N aqueous NaOH, which caused the excess Gd3+ to precipitate as Gd(OH)3. The solution was filtered and the pH was readjusted to 7.0 using 1.0 N HCl. To the resulting solution, DTPA (0.1 mM, 1 mL) was added and the solution was purified using HPLC to give the desired complex. The desired fractions were pooled together and lyophilized to give Gd6H3L[c(RGDyK)]3 as a white solid. (0.005 g, 0.001 mmol, 48%). MS (MALDI) m/z calcd for C264H402Gd6N72O90S3: 7064.4; found: 7085.0 ([M + Na]+).
Compound Gd6L[c(RGDyK)]368Ga
To a 1.5 mL eppendorf tube containing 80 μg Gd6H3L[c(RGDyK)]3 complex in 1 mL of HEPES (pH = 6.5) solution was added a solution of 15.0 mCi of 68GaCl3 in 0.6 N HCl. The reaction mixture was incubated at 75 °C for 0.5 h on a shaker. To this solution was added DTPA (5.0 mM, 5.0 μL) and the reaction mixture was incubated for 5 min at room temperature. The 68Ga-labeled conjugate, Gd6L[c(RGDyK)]368Ga, was purified by passing the mixture through a preconditioned Sep-Pak C-18 light cartridge. After thorough rinsing (3 × 5 mL, water) of the cartridge, Gd6L[c(RGDyK)]368Ga was eluted by an ethanol–water mixture (70:30) to give 9.0 mCi of the labeled compound. The product was analyzed by radio-HPLC to determine the radiochemical purity of the product. The radiochemical purity of the compound was determined to be higher than 95%.
Compound Gd6H3L[PEG12c(RGDyK)]3
The c(RGDyK) modified ligand H21L[PEG12c(RGDyK)]3 (0.019 g, 0.002 mmol) was dissolved in water (1.0 mL) and the pH was adjusted to 7.0 with NaOH (0.1 M). To this solution was added an excess of GdCl3·6H2O and the pH was again adjusted to 6.5 and allowed to stir at room temperature overnight. The pH was raised above 8.0 using 1.0 N aqueous NaOH, which caused the excess Gd3+ to precipitate as Gd(OH)3. The solution was filtered and the pH was readjusted to 7.0 using 1.0 N HCl. To the resulting solution DTPA (0.1 mM, 1.0 mL) was added and the solution was purified using HPLC to give the desired complex. The desired fractions were pooled together and lyophilized to give Gd6H3L[PEG12c(RGDyK)]3 as a white solid. (0.006 g, 0.001 mmol, 51%). MS (MALDI) m/z calcd for C345H561Gd6N75O129S3: 8864.3; found: 8864.7 ([M + H]+).
Compound Gd6L[PEG12c(RGDyK)]3Ga
The gadolinium complex, Gd6H3L[PEG12c(RGDyK)]3 (0.001 g, 0.112 μmol), was dissolved in an HEPES solution (pH = 6.5, 1.0 M, 1.0 mL). To the resulting solution was added a solution of GaCl3 (0.001 g, 0.567 μmol) in 0.6 N HCl (0.3 mL) and the resulting solution was stirred for 1 h. To the mixture was added 500 μL of 5.0 mM of EDTA, and the mixture was allowed to incubate for another 5 min at room temperature (EDTA was used to remove nonspecifically bound or free GaCl3). The purification of Gd6L[PEG12c(RGDyK)]3Ga was carried out by passing the mixture through a preconditioned Sep-Pak C-18 heavy cartridge. After thorough rinsing (3 × 5 mL, water) of the cartridge, Gd6L[PEG12c(RGDyK)]3Ga was eluted by an ethanol–water mixture (70:30). The product was characterized by mass spec. MS (MALDI) m/z calcd for C345H558GaGd6N75O129S3: 8930.9; found: 8954.8 ([M + Na]+).
Compound Gd6L[PEG12c(RGDyK)]368Ga
For PET/MR imaging, a slightly different labeling procedure was followed. To a 1.5 mL eppendorf tube containing 6.0 mg of Gd6H3L[PEG12c(RGDyK)]3 complex in 1.0 mL of HEPES (pH = 6.5) solution was added a solution of 300 μCi of 68GaCl3 in 0.6 N HCl. The reaction mixture was incubated at 75 °C for 10 min on a shaker. To this solution was added DTPA (5.0 mM, 5.0 μL) and the reaction mixture was incubated for 1 min at room temperature. The 68Ga-labeled conjugate was purified by passing the mixture through a preconditioned Sep-Pak C-18 plus cartridge. After thorough rinsing (3 × 5 mL, water) of the cartridge, Gd6L[PEG12c(RGDyK)]368Ga was eluted by an ethanol–water mixture (70:30) to give 180–200 μCi of the labeled compound. The product was analyzed by radio-HPLC to determine the radiochemical purity of the product. The radiochemical purity of the compound was determined to be higher than 95%.
Biodistribution and Pharmacokinetics Studies of Gd6L67Ga
Male BALB/C mice were injected with 300 μCi of Gd6L67Ga complex to evaluate the tissue distribution of the tracer in mice. Mice were sacrificed 1, 4, and 24 h post injection (p.i.). The organs of interest (blood, heart, lung, liver, spleen, kidney, stomach, muscle, fat, small intestine, large intestine, and brain) were harvested and weighed, and radioactivity was quantified using a γ-counter. Standards were prepared and counted along with the tissue samples to calculate the percentage-injected dose per gram (%ID/g). To determine the pharmacokinetic parameters, mice injected with the tracer were blood sampled from the retro-orbital sinus at 2, 5, 10, and 30 min, and 1, 2, 24, and 48 h p.i. and quantified using a γ-counter. The pharmacokinetic parameters were calculated based on a two-compartment open model.
In Vitro and Ex Vivo Stability of Gd6L67Ga
The in vitro stability test was performed in rat serum. Briefly, Gd6L67Ga complex (100 μCi, 5 μL) was added into 100 μL of rat serum (n = 3). The solution was incubated for 1, 4, 24, and 48 h at 37 °C, respectively. The solution was vortexed and centrifuged for 5 min at the speed of 21 000 g. The supernatant was then analyzed by HPLC. For the in vivo stability evaluation, each male mouse was injected with 600 μCi of Gd6L67Ga complex in 100 μL of saline via the tail vein. The urine samples were collected at 1, 4, 24, and 48 h p.i. and then analyzed by HPLC.
Integrin αvβ3 Receptor-Binding Assay
The integrin αvβ3 binding affinities of c(RGDyK), Gd6H3L[c(RGDyK)]3, and Gd6H3L[PEG12c(RGDyK)]3 were determined by a competitive cell-binding assay using 125I-echistatin (PerkinElmer) as the αvβ3-specific radioligand. The experiments were performed on U87MG human glioblastoma cells following a previously reported method.52 Briefly, U87MG cells were grown in RPMI 1640 medium supplemented with penicillin, streptomycin, and 10% (v/v) fetal bovine serum (FBS) at 37 °C under 5% CO2. Suspended U87MG cells in the binding buffer (20 mM Tris, pH 7.4, 150 mM NaCl, 2.0 mM CaCl2, 1.0 mM MgCl2, 1.0 mM MnCl2, 0.1% bovine serum albumin) were seeded on multiwell DV plates (Millipore) with 5 × 104 cells per well and then incubated with 125I-echistatin (10 000 cpm/well) in the presence of increasing concentrations (0–5000 nM) of each c(RGDyK) peptide conjugate for 2 h. The final volume in each well was maintained at 200 μL. At the end of incubation, unbound 125I-echistatin was removed by filtration followed by three rinses with cold binding buffer. The retentate was collected and the radioactivity was measured using a γ-counter. The best-fit IC50 values (inhibitory concentration where 50% of the 125I-echistatin bound on U87MG cells are displaced) of c(RGDyK), Gd6H3L[c(RGDyK)]3, and Gd6H3L[PEG12c(RGDyK)]3 were calculated by fitting the data with nonlinear regression using GraphPad Prism (GraphPadSoftware, Inc.). Experiments were repeated with quintuplicate samples.
Relaxivity Measurements at 0.5 T (T)
The T1 values were recorded at 23 MHz (0.5 T) at 25 °C by using a Maran Ultra relaxometer (Oxford Instruments, UK). Longitudinal relaxation times were measured by using the inversion–recovery pulse sequence (180°-t-90°). The T1 relaxivities were determined by the linear regression analysis of the water proton relaxation rates in solutions ranging in concentration from 0.005 to 12.0 mM, in Millipore water in triplicate.
MRI Imaging and Relaxivity Measurements at 1.0 T
The T1-weighted MR images of samples in 0.5 mL microfuge tubes were collected using Aspect Imaging M2TM 1.0 T (43.5–45 MHz, 60 mm diameter volume coil) Gradient Echo Spoiled External Averaging (GRE-EXT) sequence. For imaging, the following parameters were used: TR = 14.2 ms; effective echo time (TE) = 2.8 ms; FOV 64 × 100 mm2, data matrix = 256 × 256, averaging = 4, slice = 1 mm. The T1-maps of the samples at 1.0 T were determined from a series of multi-T1 (0.01 ms to 5 s) inversion recovery spin echo sequence (IR-SE with minimum TE = 7.0 ms); TR = 10 s; FOV = 64 × 100 mm2; matrix = 128 × 128; averaging = 1, steady state scans = 10), fitted and calculated using Matlab code. All the fits for T1 values used to calculate the longitudinal relaxivity, r1, had fitting coefficients, R2 ≥ 0.99. Three trials were performed. The Gd and Ga metal concentrations of samples were determined by inductively coupled plasma mass spectrometry (ICP-MS).
PET/MR Imaging Using Gd6L[PEG12c(RGDyK)]368Ga
The MR and PET imaging studies were performed on a 1.0 T MR scanner (Aspect Imaging, Shoham, Israel) using a 35 mm mouse body coil, and a Siemens Inveon Multimodality PET/CT system (Siemens Medical Solutions Inc., Knoxville, TN, USA), respectively. Ten minutes prior to imaging, the animals were anesthetized using 3% isofluorane at room temperature until stable vitals were established. Once the animals were sedated, they were placed onto a custom-made interchangeable bed between the MR and PET scanners under 2% isofluorane anesthesia for the duration of the imaging. A T1-weighted gradient echo, spoiled sequence (echo time/repetition time [TE/TR] 3/15 ms; field of view [FOV] 80 × 100 mm; slice thickness 0.8 mm; matrix 256 × 256; number of excitations [NEX] = 3; flip angle = 25°) was performed for each mouse. Each mouse was injected with 100–150 μCi (Gd: ca. 0.027 mmol/kg) via the tail-vein. MR scans were obtained precontrast and 1 h after contrast medium injection.
PET imaging was acquired with the mouse mounted on the interchangeable bed directly following the MR acquisition. A 15 min PET static scan was performed and reconstructed using Fourier rebinning and ordered subsets expectation maximization 3D (OSEM3D) algorithm. Reconstructed MR and PET images were fused and analyzed using the VivoQuant software (Invicro LLC, Boston, USA). For quantification, regions of interest were placed on the tumor and muscle. The latter was used for normalization. The resulting quantitative data were expressed in intensity and %ID/g for MR and PET images.
Supporting Information Available
Supporting schemes, synthesis of compounds, ICP quantification. This material is available free of charge via the Internet at http://pubs.acs.org.
Author Contributions
The manuscript was written through contributions of all authors. All authors have given approval to the final version of the manuscript.
This work was partially supported by the Prostate Cancer Research Program of the United States Army Medical Research and Materiel Command (W81XWH-12–1–0336), the Dr. Jack Krohmer Professorship Funds, and the National Institutes of Health (CA115531 and EB0158908).
The authors declare no competing financial interest.
Supplementary Material
References
- Weissleder R. (2006) Molecular imaging in cancer. Science 312, 1168–1171. [DOI] [PubMed] [Google Scholar]
- Cheon J.; Lee J.-H. (2008) Synergistically integrated nanoparticles as multimodal probes for nanobiotechnology. Acc. Chem. Res. 41, 1630–1640. [DOI] [PubMed] [Google Scholar]
- Jennings L. E.; Long N. J. (2009) ’Two is better than one’-probes for dual-modality molecular imaging. Chem. Commun. 28, 3511–3524. [DOI] [PubMed] [Google Scholar]
- Ntziachristos V.; Yodh A. G.; Schnall M.; Chance B. (2000) Concurrent MRI and diffuse optical tomography of breast after indocyanine green enhancement. Proc. Natl. Acad. Sci. U.S.A. 97, 2767–2772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer T.; Townsend D. W.; Brun T.; Kinahan P. E.; Charron M.; Roddy R.; Jerin J.; Young J.; Byars L.; Nutt R. (2000) A combined PET/CT scanner for clinical oncology. J. Nucl. Med. 41, 1369–1379. [PubMed] [Google Scholar]
- Murray C. B.; Norris D. J.; Bawendi M. G. (1993) Synthesis and characterization of nearly monodisperse CdE (E = sulfur, selenium, tellurium) semiconductor nanocrystallites. J. Am. Chem. Soc. 115, 8706–8715. [Google Scholar]
- Link S.; El-Sayed M. A. (1999) Spectral properties and relaxation dynamics of surface plasmon electronic oscillations in gold and silver nanodots and nanorods. J. Phys. Chem. B 103, 8410–8426. [Google Scholar]
- Alivisatos A. P. (1996) Semiconductor clusters, nanocrystals, and quantum dots. Science 271, 933–937. [Google Scholar]
- Catana C.; Wu Y.; Judenhofer M. S.; Qi J.; Pichler B. J.; Cherry S. R. (2006) Simultaneous acquisition of multislice PET and MR images: initial results with a MR-compatible PET scanner. J. Nucl. Med. 47, 1968–1976. [PubMed] [Google Scholar]
- Cherry S. R. (2006) The 2006 Henry N. Wagner lecture: of mice and men (and positrons)--advances in PET imaging technology. J. Nucl. Med. 47, 1735–1745. [PubMed] [Google Scholar]
- Cherry S. (2001) Fundamentals of positron emission tomography and applications in preclinical drug development. J. Clin. Pharmacol. 41, 482–491. [DOI] [PubMed] [Google Scholar]
- Caravan P.; Ellison J. J.; McMurry T. J.; Lauffer R. B. (1999) Gadolinium(III) chelates as MRI contrast agents: structure, dynamics, and applications. Chem. Rev. 99, 2293–2352. [DOI] [PubMed] [Google Scholar]
- Raymond K. N.; Pierre V. r. C. (2004) Next generation, high relaxivity gadolinium MRI agents. Bioconjugate Chem. 16, 3–8. [DOI] [PubMed] [Google Scholar]
- Seo W. S.; Lee J. H.; Sun X.; Suzuki Y.; Mann D.; Liu Z.; Terashima M.; Yang P. C.; McConnell M. V.; Nishimura D. G.; et al. (2006) FeCo/graphitic-shell nanocrystals as advanced magnetic-resonance-imaging and near-infrared agents. Nat. Mater. 5, 971–976. [DOI] [PubMed] [Google Scholar]
- Morawski A. M.; Winter P. M.; Crowder K. C.; Caruthers S. D.; Fuhrhop R. W.; Scott M. J.; Robertson J. D.; Abendschein D. R.; Lanza G. M.; Wickline S. A. (2004) Targeted nanoparticles for quantitative imaging of sparse molecular epitopes with MRI. Magn. Reson. Med. 51, 480–486. [DOI] [PubMed] [Google Scholar]
- Santra S.; Bagwe R. P.; Dutta D.; Stanley J. T.; Walter G. A.; Tan W.; Moudgil B. M.; Mericle R. A. (2005) Synthesis and characterization of fluorescent, radio-opaque, and paramagnetic silica nanoparticles for multimodal bioimaging applications. Adv. Mater. 17, 2165–2169. [Google Scholar]
- Cheng Z.; Tsourkas A. (2008) Paramagnetic porous polymersomes. Langmuir 24, 8169–8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng L.; Yang K.; Li Y.; Chen J.; Wang C.; Shao M.; Lee S.-T.; Liu Z. (2011) Facile preparation of multifunctional upconversion nanoprobes for multimodal imaging and dual-targeted photothermal therapy. Angew. Chem., Int. Ed. 123, 7523–7528. [DOI] [PubMed] [Google Scholar]
- Chen J.; Wiley B.; Li Z. Y.; Campbell D.; Saeki F.; Cang H.; Au L.; Lee J.; Li X.; Xia Y. (2005) Gold nanocages: engineering their structure for biomedical applications. Adv. Mater. 17, 2255–2261. [Google Scholar]
- Gao X.; Cui Y.; Levenson R. M.; Chung L. W. K.; Nie S. (2004) In vivo cancer targeting and imaging with semiconductor quantum dots. Nat. Biotechnol. 22, 969–976. [DOI] [PubMed] [Google Scholar]
- Lin Y.-S.; Hung Y.; Su J.-K.; Lee R.; Chang C.; Lin M.-L.; Mou C.-Y. (2004) Gadolinium(III)-incorporated nanosized mesoporous silica as potential magnetic resonance imaging contrast agents. J. Phys. Chem. B 108, 15608–15611. [Google Scholar]
- Rieter W. J.; Kim J. S.; Taylor K. M. L.; An H.; Lin W.; Tarrant T.; Lin W. (2007) Hybrid silica nanoparticles for multimodal imaging. Angew. Chem., Int. Ed. 119, 3754–3756. [DOI] [PubMed] [Google Scholar]
- Hak S.; Sanders H. M. H. F.; Agrawal P.; Langereis S.; Grüll H.; Keizer H. M.; Arena F.; Terreno E.; Strijkers G. J.; Nicolay K. (2009) A high relaxivity Gd(III)DOTA-DSPE-based liposomal contrast agent for magnetic resonance imaging. Eur. J. Pharm. Biopharm. 72, 397–404. [DOI] [PubMed] [Google Scholar]
- Terreno E.; Delli Castelli D.; Cabella C.; Dastrù W.; Sanino A.; Stancanello J.; Tei L.; Aime S. (2008) Paramagnetic liposomes as innovative contrast agents for magnetic resonance (MR) molecular imaging applications. Chem. Biodivers. 5, 1901–1912. [DOI] [PubMed] [Google Scholar]
- Unger E. C.; MacDougall P.; Cullis P.; Tilcock C. (1989) Liposomal Gd-DTPA: Effect of encapsulation on enhancement of hepatoma model by MRI. Magn. Reson. Imaging 7, 417–423. [DOI] [PubMed] [Google Scholar]
- Boswell C. A.; Eck P. K.; Regino C. A. S.; Bernardo M.; Wong K. J.; Milenic D. E.; Choyke P. L.; Brechbiel M. W. (2008) Synthesis, characterization, and biological evaluation of integrin αvβ3-targeted PAMAM dendrimers. Mol. Pharmaceutics 5, 527–539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi H.; Brechbiel M. W. (2005) Nano-sized MRI contrast agents with dendrimer cores. Adv. Drug Delivery Rev. 57, 2271–2286. [DOI] [PubMed] [Google Scholar]
- Langereis S.; de Lussanet Q. G.; van Genderen M. H. P.; Meijer E. W.; Beets-Tan R. G. H.; Griffioen A. W.; van Engelshoven J. M. A.; Backes W. H. (2006) Evaluation of Gd(III)DTPA-terminated poly(propylene imine) dendrimers as contrast agents for MR imaging. NMR Biomed. 19, 133–141. [DOI] [PubMed] [Google Scholar]
- Langereis S.; Dirksen A.; Hackeng T. M.; van Genderen M. H. P.; Meijer E. W. (2007) Dendrimers and magnetic resonance imaging. New J. Chem. 31, 1152–1160. [Google Scholar]
- Rudovský J.; Botta M.; Hermann P.; Hardcastle K. I.; Lukeš I.; Aime S. (2006) PAMAM dendrimeric conjugates with a Gd–DOTA phosphinate derivative and their adducts with polyaminoacids: the interplay of global motion, internal rotation, and fast water exchange. Bioconjugate Chem. 17, 975–987. [DOI] [PubMed] [Google Scholar]
- Bolskar R. D. (2008) Gadofullerene MRI contrast agents. Nanomedicine 3, 201–213. [DOI] [PubMed] [Google Scholar]
- Zhu W.; Okollie B.; Bhujwalla Z. M.; Artemov D. (2008) PAMAM dendrimer-based contrast agents for MR imaging of Her-2/neu receptors by a three-step pretargeting approach. Magn. Reson. Med. 59, 679–685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duarte M. G.; Gil M. H.; Peters J. A.; Colet J. M.; Elst L. V.; Muller R. N.; Geraldes C. F. G. C. (2001) Synthesis, characterization, and relaxivity of two linear Gd(DTPA)–polymer conjugates. Bioconjugate Chem. 12, 170–177. [DOI] [PubMed] [Google Scholar]
- Brasch R. C. (1991) Rationale and applications for macromolecular Gd-based contrast agents. Magn. Reson. Med. 22, 282–287. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Kawamoto S.; Saga T.; Sato N.; Hiraga A.; Ishimori T.; Akita Y.; Mamede M. H.; Konishi J.; Togashi K.; et al. (2001) Novel liver macromolecular MR contrast agent with a polypropylenimine diaminobutyl dendrimer core: Comparison to the vascular MR contrast agent with the polyamidoamine dendrimer core. Magn. Reson. Med. 46, 795–802. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Kawamoto S.; Jo S. K.; Sato N.; Saga T.; Hiraga A.; Konishi J.; Hu S.; Togashi K.; Brechbiel M. W.; et al. (2002) Renal tubular damage detected by dynamic micro-MRI with a dendrimer-based magnetic resonance contrast agent. Kidney Int. 61, 1980–1985. [DOI] [PubMed] [Google Scholar]
- Sugaya M.; Watanabe T.; Yang A.; Starost M. F.; Kobayashi H.; Atkins A. M.; Borris D. L.; Hanan E. A.; Schimel D.; Bryant M. A.; et al. (2005) Lymphatic dysfunction in transgenic mice expressing KSHV k-cyclin under the control of the VEGFR-3 promoter. Blood 105, 2356–2363. [DOI] [PubMed] [Google Scholar]
- Wiener E. C.; Konda S.; Shadron A.; Brechbiel M.; Gansow O. (1997) Targeting dendrimer-chelates to tumors and tumor cells expressing the high-affinity folate receptor. Invest. Radiol. 32, 748–754. [DOI] [PubMed] [Google Scholar]
- Kobayashi H.; Brechbiel M. W. (2003) Dendrimer-based macromolecular MRI contrast agents: characteristics and application. Mol. Imaging 2, 1–10. [DOI] [PubMed] [Google Scholar]
- Venditto V. J.; Regino C. A. S.; Brechbiel M. W. (2005) PAMAM dendrimer based macromolecules as improved contrast agents. Mol. Pharmaceutics 2, 302–311. [DOI] [PubMed] [Google Scholar]
- Kumar K.; Chang C. A.; Francesconi L. C.; Dischino D. D.; Malley M. F.; Gougoutas J. Z.; Tweedle M. F. (1994) Synthesis, stability, and structure of gadolinium(III) and yttrium(III) macrocyclic poly(amino carboxylates). Inorg. Chem. 33, 3567–3575. [Google Scholar]
- Reichert D. E.; Hancock R. D.; Welch M. J. (1996) Molecular mechanics investigation of gadolinium(III) complexes. Inorg. Chem. 35, 7013–7020. [DOI] [PubMed] [Google Scholar]
- Dong Q.; Hurst D. R.; Weinmann H. J.; Chenevert T. L.; Londy F. J.; Prince M. R. (1998) Magnetic resonance angiography with gadomer-17: an animal study. Invest. Radiol. 33, 699–708. [DOI] [PubMed] [Google Scholar]
- Nicolle G. M.; Tóth É.; Schmitt-Willich H.; Radüchel B.; Merbach A. E. (2002) The impact of rigidity and water exchange on the relaxivity of a dendritic MRI contrast agent. Chem.—Eur. J. 8, 1040–1048. [DOI] [PubMed] [Google Scholar]
- Behr T. M.; Goldenberg D. M.; Becker W. (1998) Reducing the renal uptake of radiolabeled antibody fragments and peptides for diagnosis and therapy: present status, future prospects and limitations. Eur. J. Nucl. Med. 25, 201–212. [DOI] [PubMed] [Google Scholar]
- Ma H. L.; Xu Y. F.; Qi X. R.; Maitani Y.; Nagai T. (2008) Superparamagnetic iron oxide nanoparticles stabilized by alginate: Pharmacokinetics, tissue distribution, and applications in detecting liver cancers. Int. J. Pharm. 354, 217–226. [DOI] [PubMed] [Google Scholar]
- Lux F.; Mignot A.; Mowat P.; Louis C.; Dufort S.; Bernhard C.; Denat F.; Boschetti F.; Brunet C.; Antoine R.; et al. (2011) Ultrasmall rigid particles as multimodal probes for medical applications. Angew. Chem., Int. Ed. 123, 12507–12511. [DOI] [PubMed] [Google Scholar]
- Iznaga-Escobar N.; Arocha L. A. T.; Morales A. M.; Suzarte M. R.; Mesa N. R.; Rodríguez R. P. (1998) Technetium-99m-antiepidermal growth factor-receptor antibody in patients with tumors of epithelial origin: part II. pharmacokinetics and clearances. J. Nucl. Med. 39, 1918–1927. [PubMed] [Google Scholar]
- Vologdin N.; Rolla G. A.; Botta M.; Tei L. (2013) Orthogonal synthesis of a heterodimeric ligand for the development of the Gd-III-Ga-III ditopic complex as a potential pH-sensitive MRI/PET probe. Org. Biomol. Chem. 11, 1683–1690. [DOI] [PubMed] [Google Scholar]
- Notni J.; Hermann P.; Dregely I.; Wester H.-J. (2013) Simultaneous in vivo PET/MRI imaging with a bi-metal bimodal tracer: Towards Ga-68-labeled responsive gadolinium contrast agents. J. Labelled Comp. Radiopharm. 56, S62–S62. [Google Scholar]
- Suchy M.; Bartha R.; Hudson R. H. E. (2013) “Click” chemistry toward bis(DOTA-derived) heterometallic complexes: potential bimodal MRI/PET(SPECT) molecular imaging probes. RSC Adv. 3, 3249–3259. [Google Scholar]
- Liu W.; Hao G. Y.; Long M. A.; Anthony T.; Hsieh J. T.; Sun X. K. (2009) Imparting multivalency to a bifunctional chelator: a scaffold design for targeted PET imaging probes. Angew. Chem., Int. Ed. 48, 7346–7349. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.