

Abstract
The unfolded protein response (UPR) is a major signaling cascade that determines cell fate under conditions of endoplasmic reticulum (ER) stress. The kinetics and amplitude of UPR responses are tightly controlled by several feedback loops and the expression of positive and negative regulators. In this issue of EMBO Reports, the Wilkinson lab uncovers a novel function of nonsense-mediated RNA decay (NMD) in fine-tuning the UPR 1. NMD is an mRNA quality control mechanism known to destabilize aberrant mRNAs that contain premature termination codons. In this work, NMD was shown to determine the threshold of stress necessary to activate the UPR, in addition to adjusting the amplitude of downstream responses and the termination phase. These effects were mapped to the control of the mRNA stability of IRE1, a major ER stress transducer. This study highlights the dynamic crosstalk between mRNA metabolism and the proteostasis network demonstrating the physiological relevance of normal mRNA regulation by the NMD pathway.
See also: R Karam et al (May 2015)
Approximately 30% of monogenic diseases are originated from nonsense mutations resulting from single nucleotide changes that introduce a premature termination codon (PTC). These mutations result in the translation of proteins that lack the carboxy-terminal region, leading to the expression of non-functional or even deleterious proteins. Nonsense-mediated RNA decay (NMD) operates as an efficient quality control mechanism to degrade nonsense-codon-containing mutant RNAs prior to the production of aberrant products. NMD detects mRNA substrates at the pioneer-round of translation assisted by distinct NMD components, including SMG and UPF proteins 2. NMD also functions as a post-transcriptional regulatory mechanism that controls the levels of a large variety of normal gene transcripts, between ∽3 and 20% of total mRNAs—including a cluster of genes involved in the unfolded protein response (UPR) and stress-related genes 3. The UPR is a major pathway that orchestrates cellular adaptation under conditions of endoplasmic reticulum (ER) stress 4. Karam et al 1 now report a novel bidirectional crosstalk between NMD and the UPR, whereby NMD determines the threshold of UPR activation, shaping its timely termination to control cell fate under ER stress.
The UPR is initiated by three main stress sensors, IRE1α, PERK, and ATF6, which transduce information about the protein-folding status in the ER to the cytosol and nucleus, to induce an adaptation program 5. Cell injury secondary to chronic ER stress is increasingly implicated in a wide range of human diseases, including diabetes, neurodegeneration, inflammatory and metabolic disorders, and cancer 6. The activation of PERK leads to a global reduction of translation through the direct phosphorylation of the initiation factor eIF2α. This triggers the selective expression of the transcription factor ATF4, which controls a variety of genes involved in redox control, folding, and apoptosis 5. IRE1α is a kinase and endoribonuclease that initiates the most conserved UPR signaling branch. Upon activation, IRE1α catalyses the splicing of the mRNA encoding for the transcription factor XBP1, leading to the expression of a stable protein that upregulates a variety of UPR target genes 5. IRE1α activity is also involved in RNA degradation (a process known as regulated IRE1-dependent decay or RIDD) of ER-localized mRNAs, ribosomal RNA, and micro-RNAs 4, 5. IRE1α is a central controller of cell fate under ER stress, since its downstream outputs engage both stress adaptation and cell death programs. The amplitude and kinetics of IRE1α signaling are tightly controlled through its binding to several negative and positive regulators 4.
NMD is emerging as a relevant pathway modulated by cellular stress. Reactive oxygen species, hypoxia, nutrient deprivation, ER stress, and viral infections, among other perturbations, can repress NMD 3. A common mechanism that contributes to this inhibition is the phosphorylation of eIF2α in response to the activation of PERK and other related kinases 7. Since NMD fully requires protein translation to recognize PTCs, the repression of translation under stress may explain the inhibitory activity on the pathway. The link between mRNA stability and UPR operates on a bidirectional mode. Genetic inhibition of NMD has been shown to stabilize a variety of mRNAs encoding for components of the PERK/eIF2α pathway, increasing the amplitude of downstream responses 7. A pioneering study uncovered a functional link between the UPR and NMD in vivo. An RNAi unbiased screen in C. elegans, identified genes that are required for the development of ire1 mutant worms, and uncovered an unexpected connection with the NMD pathway 8. This study showed that targeting NMD results in spontaneous ER stress, in addition to sensitizing animals to the lethal effects of experimental ER stress. In this context, the occurrence of ER stress in NMD-deficient cells may involve the accumulation of misfolded truncated proteins, suggesting that NMD operates as a translation-dependent surveillance mechanism to reduce basal levels of unfolded proteins at the ER.
Karam et al 1 propose a novel form of control of the UPR by NMD. The authors examined the mRNA levels of a variety of UPR components when the NMD pathway was altered. Remarkably, Ire1α was one of the most induced genes upon NMD disruption at the mRNA and protein levels. The authors were also the first to demonstrate that ER stress and NMD are interconnected in vivo in mammals, using mice lacking Upf3b. Gain- and loss-of-function assays indicated that NMD represses the UPR in an IRE1α-dependent manner. It is well known that under prolonged ER stress, IRE1 signaling is attenuated, leading to a reduction in XBP1 expression, which sensitizes cells to apoptosis 4, 5. Interestingly, NMD activity not only affected the threshold of UPR activation under low doses of ER stressors, but it also shaped its amplitude and kinetics, having a significant role in attenuating XBP1 mRNA splicing (Fig1). As a consequence, NMD inhibition has an impact on the susceptibility of cells to undergo ER stress-dependent apoptosis, both in cell culture and in mouse models. Overall, the Wilkinson lab describes a complex bidirectional circuit where UPR mRNAs are targeted for decay, whereas NMD activity is repressed by ER stress. The authors speculate that NMD may prevent UPR activation by innocuous ER stress, whereas ER stress signaling represses NMD to ensure the establishment of a robust adaptive program (Fig1). In contrast to previous studies in C. elegans 8, no basal ER stress was observed in cells and animals depleted of NMD components, suggesting that NMD and the UPR are connected as a regulatory checkpoint involving signaling events, rather than altered ER physiology.
Figure 1. Bidirectional control between NMD and the UPR.
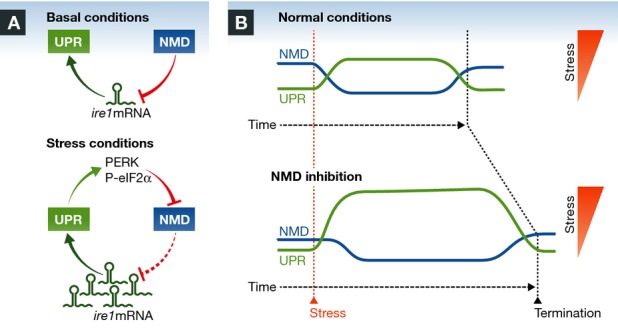
(A) In basal conditions, NMD constantly represses the UPR via the degradation of ire1 mRNA and other components of the pathway. Under ER stress, the UPR inhibits NMD via eIF2α phosphorylation, which in turn promotes the accumulation of ire1 mRNA, resulting in a robust UPR activation. (B) NMD determines the threshold for the establishment of a UPR reaction and thus impacts cell fate under ER stress. NMD modulates the amplitude and dynamics of UPR signaling, affecting the termination phase under chronic ER stress.
Perturbations to the NMD pathway, and more specifically of UFP3B, are involved in cognitive disorders, including autism and schizophrenia 9, 10. NMD promotes the degradation of mutant mRNA related to several disorders, including thalassemia, cystic fibrosis, and muscular dystrophy 2. Recent drug discovery efforts led to the development of therapeutic approaches—including the use of aminoglycoside antibiotics and ataluren—to allow translational read-through of the PTC and generate full-length functional proteins 2. Since ER stress is a salient feature of most neurodegenerative diseases involving abnormal protein aggregation 6, the alterations in the NMD pathway observed in cognitive disorders could be caused by abnormal ER stress levels. In this line, pharmacological modulation of NMD may also impact the adaptive capacity of a cell, increasing UPR responses. Based on these new studies, side effects related to ER stress signaling deregulation due to NMD inhibition are also predicted in the clinic.
Overall, a novel concept is emerging in which mRNA metabolism and protein quality control are merged into an interconnected pathway that globally monitors the proteostasis status of the cell, even before any protein is produced. Since IRE1α regulates mRNA and miRNA stability through RIDD, and NMD controls IRE1 levels, a interesting scenario arises where different layers of interactions occur between mRNA metabolism and protein homeostasis.
References
- Karam R, Lou C, Kroeger H, et al. EMBO Rep. 2015;16:599–609. doi: 10.15252/embr.201439696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, Wengrod J, Gardner LB, et al. Biochim Biophys Acta - Gene Regul Mech. 2013;1829:624–633. doi: 10.1016/j.bbagrm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp MW-L, Maquat LE. Annu Rev Genet. 2013;47:139–165. doi: 10.1146/annurev-genet-111212-133424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hetz C. Nat Rev Mol Cell Biol. 2012;13:89–102. doi: 10.1038/nrm3270. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Hetz C, Chevet E, Harding HP. Nat Rev Drug Discov. 2013;12:703–719. doi: 10.1038/nrd3976. [DOI] [PubMed] [Google Scholar]
- Wang D, Zavadil J, Martin L, et al. Mol Cell Biol. 2011;31:3670–3680. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sakaki K, Yoshina S, Shen X, et al. Proc Natl Acad Sci. 2012;109:8079–8084. doi: 10.1073/pnas.1110589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, et al. Nat Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Laumonnier F, Shoubridge C, Antar C, et al. Mol Psychiatry. 2010;15:767–776. doi: 10.1038/mp.2009.14. [DOI] [PubMed] [Google Scholar]