

Abstract
The ETS factor ETV2 (aka ER71) is essential for the generation of the blood and vascular system, as ETV2 deficiency leads to a complete block in blood and endothelial cell formation and embryonic lethality in the mouse. However, the ETV2-mediated gene regulatory network and signaling governing hematopoietic and endothelial cell development are poorly understood. Here, we map ETV2 global binding sites and carry out in vitro differentiation of embryonic stem cells, and germ line and conditional knockout mouse studies to uncover mechanisms involved in the hemangiogenic fate commitment from mesoderm. We show that ETV2 binds to enhancers that specify hematopoietic and endothelial cell lineages. We find that the hemangiogenic progenitor population in the developing embryo can be identified as FLK1highPDGFRα−. Notably, these hemangiogenic progenitors are exclusively sensitive to ETV2-dependent FLK1 signaling. Importantly, ETV2 turns on other Ets genes, thereby establishing an ETS hierarchy. Consequently, the hematopoietic and endothelial cell program initiated by ETV2 is maintained partly by other ETS factors through an ETS switching mechanism. These findings highlight the critical role that transient ETV2 expression plays in the regulation of hematopoietic and endothelial cell lineage specification and stability.
Keywords: ChIP-Seq, ER71/ETV2, ETS hierarchy, ETS switching, hemangioblast, VEGFR2/FLK1
Introduction
Functional blood and its conduit vascular system are the first to form during embryogenesis. Blood cells in the developing embryo are generated in close association with endothelial cells. For example, yolk sac blood islands composed of centrally located embryonic blood cells and an outer luminal layer of endothelial cells of the embryo are generated from the extra-embryonic mesoderm presumably through a hemangioblast intermediary 1, 2, 3, 4. While the hemangioblast is believed to be a common hematopoietic and endothelial cell progenitor, a recent study suggests that primitive erythroid cells in the yolk sac are developmentally distinct from the yolk sac endothelium 5. It has also been suggested that hemangioblast is not a progenitor, but represents a competent cell state that can generate either blood or endothelium depending on the surrounding signals 6. Regardless, it is now widely accepted that hemogenic endothelium within the dorsal aorta of the embryo is the cell origin of the definitive hematopoietic system 7, 8, 9, 10. Later in adult life, hematopoietic system is maintained partly through the vascular system. As such, sinusoidal endothelial cells form an important component of the niche in which hematopoietic stem cells reside 11, 12, 13. It is thus noticeable that many transcription factors and signaling pathways are largely shared between blood and endothelial cells. Gene-targeting studies have shown that mutations in any of the shared genes often affect both cell lineages, supporting the notion of common genetic pathways regulating hematopoietic and endothelial cell lineage development and function. Remarkably, Vegfa- or Vegfr2 (Flk1)-deficient animals completely fail to generate blood and blood vessels and die early in embryogenesis, indicating that precise VEGFA signaling via FLK1 is critical for the proper formation of the blood and vascular systems 14, 15, 16. Clearly, molecular mechanisms by which blood and vessel lineages are specified in the developing embryo need to be better elucidated. Such information in turn would be greatly useful for future applications for blood and vascular repair and regeneration as well as for obtaining hematopoietic and endothelial cells from pluripotent stem cells.
ETS transcription factors have emerged as critical regulators of hematopoietic and vascular development 17, 18, 19. The ETS domain, which is composed of a winged helix-turn-helix motif, binds a consensus sequence (GGAA/T) to regulate target gene expression. Many ETS factors are redundantly expressed in blood and endothelial cells. Consistently, mice or zebrafish deficient in Ets factors display differing levels of hematopoietic and vascular defects 20, 21, 22, 23. Distinct from other ETS factors, Etv2 is transiently expressed in the primitive streak, yolk sac blood islands, and large vessels including the dorsal aorta during embryogenesis 24. Remarkably, Etv2-deficient animals display a complete block in blood and blood vessel formation, indicating that ETV2 performs a non-redundant and indispensable function in hematopoietic and vessel development 24, 25, 26, 27. As such, Etv2 inactivation leads to similar hematopoietic and vascular defects to those of Vegfa or Flk1 deficiency. Herein, we characterized germ line and conditional Etv2 knockout mice and performed genomewide ChIP-Seq of ETV2 using in vitro differentiated embryonic stem (ES) cells to better understand how ETV2 can achieve such a non-redundant predominant role in hematopoietic and endothelial cell development. We discover that specification of the hemangiogenic program requires ETV2 activation of the blood and endothelial cell lineage-specifying genes and VEGF signaling. Moreover, ETV2 establishes an ETS hierarchy by directly activating other Ets genes, which then maintain blood and endothelial cell program initiated by ETV2 through an ETS switching mechanism. Collectively, we provide molecular and cellular basis by which ETV2 establishes the hematopoietic and endothelial cell program.
Results
ETV2 ChIP-Seq and target gene identification
To understand ETV2-mediated genetic program regulating hematopoietic and endothelial cell lineage development, we performed ETV2 ChIP-Seq analysis using in vitro differentiated embryonic stem (ES) cells. We previously described A2 ES cells expressing ETV2-V5 in a doxycycline (DOX)-inducible manner 24, 27. DOX addition from day 2 to 3.5, a time frame when Etv2 is normally expressed, in these cells robustly induced hemangioblast formation. To facilitate ETV2 target identification, we additionally generated polyclonal antibodies against ETV2200–219 peptide (ETV2-polyAbs) to pull down ETV2-associated chromatin. Two independent biological replicates from DOX-treated day 3.5 iEtv2 EB cells were subjected to ETV2-polyAbs and V5 ChIP and deep sequencing using IgG as controls. Sequencing reads were mapped to the mouse genome assembly mm9 provided by the UCSC Genome Browser 28. Using MACS2 29 at a P-value cutoff of 1E-6, we identified 8,019 ETV2 binding peaks from the ETV2-polyAbs samples and 11,892 ETV2 binding peaks from the V5 samples (Supplementary Table S1). We focused our analysis on the 3,933 peaks that overlapped between the ETV2-polyAbs samples and the V5 samples (Fig1A and B), as we reasoned that this strategy provided the best balance between sensitivity and specificity given the genomic technology we employed. As such, the ETV2 overexpression system ensures higher sensitivity of detection of ETV2 genomewide target sites than assaying against endogenous protein. Moreover, focusing on reproducible ChIP-seq peaks based on independent antibodies ensures a higher specificity. Indeed, several initial quality assurance analyses suggested that these ETV2 binding peaks had high quality and were connected to ETV2 biology, supporting that they were bona fide biological target sites of ETV2. First, raw read densities within these ETV2 peaks were highly reproducible between experiments (cc = 0.974 for ETV2-polyAbs and 0.969 for V5 between replicates, cc = 0.993 between the two antibodies, Supplementary Fig S1A–C). ChIP-Seq signals were highly enriched in peak centers across samples when compared to surrounding genomic regions or control (Fig1A and B). We also identified the most significant sequence motif associated with the ChIP-Seq peaks (Fig1C). This motif, which represented ∽85% of the peaks, matched perfectly with the known binding specificity of other ETS factors, FLI1, and ERG 30. We additionally identified GATA, SOX, or E-box motifs to be frequently associated with the ETV2 peaks (Supplementary Fig S1D). Genomewide distribution of these binding peaks was far from random expectation, with ∽14% significant enrichment in the promoter regions and ∽70% in introns or intergenic regions, suggesting that ETV2 functions by interacting with both gene promoters and distal enhancers (Fig1D and Supplementary Fig S1F). We next subjected these ETV2 binding peaks to a GREAT analysis 31 to understand the overall ETV2-mediated biological function and found that they were strongly associated with endothelial and hematopoietic cell lineage development and differentiation (Fig1E, for complete result see Supplementary Table S2A). Finally, we examined evolutionary conservation of sequences associated with ETV2 peaks and found that they are much more conserved than their neighboring sequences (Fig1F). Indeed, 2,231 (56.7%) peaks overlapped with conserved elements determined based on 30-way vertebrate alignment 32 from the UCSC Genome Browser (P-value < 2.2e−16, binomial test), suggesting that the identified peaks were strongly enriched for functionally constrained sequences. As the majority of the ETV2 binding peaks were outside gene promoters, we reasoned that some of them could serve as distal enhancers. Connecting enhancers to their target genes is an extremely challenging problem, because enhancers do not necessarily regulate the nearest genes, nor do they necessarily regulate one single target genes. Therefore, we combined GREAT analysis with gene expression analysis to optimize sensitivity and specificity. Using GREAT default parameters, we defined a “basal regulatory domain” for each gene (Methods), and we associated an ETV2 ChIP-seq peak with a target gene if the peak was within the basal regulatory domain of the gene. This approach resulted in 4,580 ETV2 peak-associated genes.
Figure 1. ChIP-Seq analysis of ETV2 binding regions.

- Heatmap of read density from each replicate in a 6-kb region flanking the peak centers.
- Averaged read density profile of each replicate in a 6-kb region flanking the peak center.
- ETV2 motif logo, de novo trained by Homer software using ChIP-Seq peaks.
- Distribution of ETV2 ChIP-Seq peaks in different genomic features. Red line represents genomic background (expectation), and blue bar represents observed peak distribution.
- Functional enrichment analysis of ChIP-Seq peaks.
- Conservation score (phastCon) in a 6-kb region flanking peak centers of all peaks.
To narrow down to a more confident list of direct target genes of ETV2, we integrated our ChIP-Seq data with gene expression pattern of FLK1+ mesoderm isolated from control and iETV2 EB cells that were generated with DOX (from day 2–3.5) as well as FLK1+ mesoderm sorted from Etv2+/+ and Etv2−/− day 3.5 EBs 27. ETV2 peak-associated genes were significantly enriched for genes that exhibited increased expression in the ETV2 overexpression system and/or reduced expression in the ETV2 knockout system (Fig2A, P-value < 2.2E-16, hypergeometric test). On the other hand, there was no significant enrichment of genes with the opposite expression pattern (i.e., upregulated in ETV2 knockout and downregulated in ETV2 overexpression, Supplementary Fig S2A). This expression pattern suggested that ETV2 primarily functions as a transcriptional activator. We identified 425 ETV2 peak-associated genes that exhibiting the expected expression difference. They constitute a group of high confidence, direct target genes of ETV2. Functional enrichment analysis revealed that this group of genes was considerably enriched for hematopoietic and endothelial cell lineage development and differentiation, with the enrichment level markedly improved from genes identified by ChIP-seq peaks alone (Supplementary Table S2B). Many genes that exhibited expression changes upon either ETV2 overexpression or ETV2 knockout did not associate with ETV2 binding peaks. They are potentially downstream but not direct targets of ETV2. Additionally, many ETV2 peak-associated genes did not show expression change, even though as a group they are strongly enriched for endothelial and hematopoietic cell relevant functions. These binding peaks could potentially be false positives, but could also reveal functions of ETV2 other than directly activating target genes. One such potential function could be to modulate chromatin structure to a permissive state that allows genes to express at future developmental stages. We thus examined the epigenetic landscape of ETV2 binding sites across several cell and tissue types. We took advantage of publicly available whole-genome bisulfite sequencing data of mouse embryonic stem cells, neural progenitor cells, as well as adult tissues 33, 34 to examine epigenetic changes co-occurring with ETV2 binding, focusing on distal sites that potentially function as lineage-specific enhancers. Intriguingly, these ETV2 binding sites were largely methylated in ES cells. The relatively high DNA methylation levels were maintained in neuronal progenitor cells and cerebellum, but reduced in heart and bone marrow (Fig2B and Supplementary Fig S2B).
Figure 2. Identification of ETV2 downstream target genes and regulatory network.

- Venn diagram showing the overlap between ChIP-Seq peak-associated genes and ETV2 target genes identified by microarray data analysis. UP (+DOX): upregulated genes by ETV2 overexpression; DN (ETV2 KO): downregulated genes in ETV2 knockout.
- DNA methylation profile of ETV2 distal binding peaks. Averaged DNA methylation level was calculated using publicly available whole-genome bisulfite sequencing data from multiple cell and tissue types and plotted for a 6-kb region flanking peak centers.
- ETV2 gene regulatory network showing Notch/MAPK signaling, VEGF signaling/lineage specification, Rho-GTPase, and ETS transcription factor as downstream targets.
- Genomic snapshots depicting the ETV2 binding regions at the indicated genomic loci.
ETV2 induces hematopoietic and endothelial cell lineage-specifying genes
ETV2 targets can be broadly categorized into hematopoietic and endothelial cell lineage-specifying genes, VEGF, Notch, Rho-GTPase, and MAP kinase pathway and Ets factors (Fig2C). Specifically, Flk1, Fli1, Erg, Gata2, Scl, Meis1, Lmo2, Tie2, VE-cadherin, Dll4, and Notch were among the 425 genes, which play critical roles in hematopoietic and endothelial cell development (Figs2C and D, 3A and 5A). While some of these peaks occur on previously identified regulatory regions, such as VE-cadherin 27, currently identified ChIP-Seq peaks represented unique peaks that have not been reported yet. We additionally compared transcriptional profiling among FLK1+ cells generated by enforced Etv2 expression, Etv2-deficient FLK1+ cells 27, and genes immediately upregulated by Etv2 expression 35. There was a significant enrichment in genes involved in the VEGF and Notch signaling pathways, suggesting the involvement of these pathways in hemangiogenic lineage development (Supplementary Fig S2C).
Figure 3. ETV2 directly regulates VEGF receptors and activate VEGF signaling pathway.

- Genomic snapshots depicting the ETV2 binding peaks associated with the VEGFR genes. Numbers indicate ChIP-Seq peaks with high confidence.
- ChIP-PCR analysis showing ETV2 recruitment to the 15 potential binding sites in (A). ETV2 enrichments were shown by both V5 antibody and ETV2-polyAb pull-down. PCR primers and genomic locations are provided in Supplementary Table S3. Error bars represent SD, n = 4.
- Luciferase reporter assay for 1–15 ETV2 peak regions from (A). The indicated peak regions or selected ETV2 binding motif deletion mutants (2, 7, 10, 11, and 14) were cloned into the pGL4.24[luc2P/minP] vector (left panel). 293T cells were transfected with pGL4.24 control vector, ETV2 wild-type, or mutant luciferase reporter constructs, together with Renilla luciferase vector in the presence (pMSCV-Etv2) or absence of ETV2 expression plasmids. For each ETV2 peak reporter construct, luciferase activity was first normalized to Renilla luciferase values. Luciferase activity value obtained with ETV2 was then compared to that of the –ETV2 value. Error bars represent the SEM obtained from four biological replicates. *P < 0.05, **P < 0.01, ***P < 0.001, two-tailed Student's t-test.
- Mouse VEGF signaling array using RNA from day 3.5 iEtv2 EBs (±DOX from day 2) is shown as scattered plots of fold changes (+DOX versus −DOX). Each gene was normalized to three housekeeping genes. Red dots are upregulated genes by DOX addition.
- RNA obtained from E8.5 Etv2+/+ and Etv2−/− embryos was subjected to VEGF signaling analysis. Key genes with significant changes in expression are shown.
- Mouse Cell Motility PCR array using RNA from day 3.5 iEtv2 EBs (±DOX from day 2) is shown as fold changes (+DOX versus −DOX).
- Gene expression analysis of Rho-GTPase activating proteins 18, 25, and 27 in day 3 iEtv2 (±DOX on day 2) or Etv2+/+ and Etv2−/− EB cells. Genes were normalized to Gapdh. The fold change was obtained from the ratios of +DOX to −DOX or Etv2−/− to Etv2+/+. Error bars represent the SD of four independent biological samples.
Figure 5. Ets genes are direct targets of ETV2.
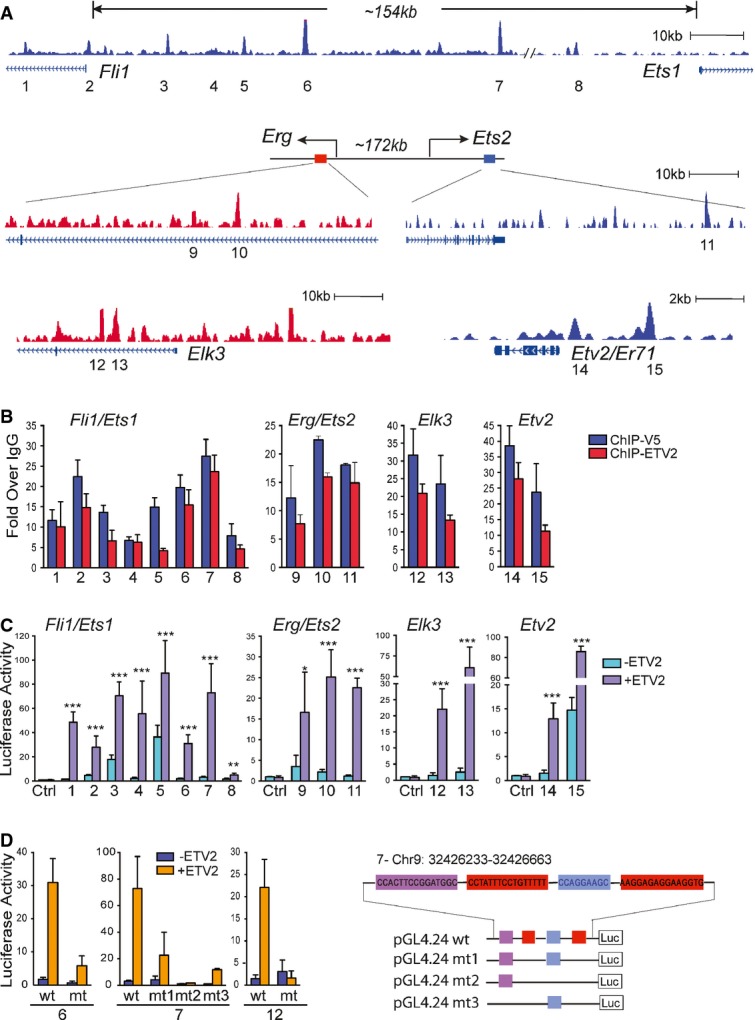
- Genome browser views of the ETV2 binding peaks associated with Ets loci. Numbers (1–15) indicate ETV2 peaks.
- ETV2 enrichment at ETV2 peaks (1–15) by ChIP-qPCR analysis (mean ± SD, n = 4 biological replicates). PCR primers and genomic locations are provided in Supplementary Table S3.
- Luciferase reporter assay for 1–15 ETV2 peak regions from (A). Similar to Fig3C, the graph shows relative luciferase activity over reporter construct alone in the presence or absence of ETV2 expression plasmids (pMSCV-Etv2). Data are represented as mean + SEM. n = 4. *P < 0.05; **P < 0.01; ***P < 0.001, two-tailed Student's t-test.
- Luciferase reporter assay for the deletion mutants of the ETV2 motif in the selected peak regions (6, 7, and 12) is shown on the left (see Supplementary Table S3). Deletions of 2 (mt1, mt2) or 3 (mt3) ETV2 motifs within the peak 7 are shown on the right. Error bars represent the SEM obtained from four biological replicates.
Previous studies have implicated the core gene regulatory network played by the ETS, GATA, and E-box motifs in hematopoietic and endothelial cell development 36. Etv2, Gata2, and Scl can independently modulate hemangioblast development 37, 38, 39. Moreover, coexpression of Etv2, Gata2, and Scl during the time of hemangioblast formation stage can robustly induce hemangioblast cell population 39. Notably, GATA and E-box motifs were frequently associated with ETV2 peaks (Supplementary Fig S1D and E). Thus, we determined whether sequences representing binding sites of these factors occur in ETV2 peaks. We utilized the ChIP-Seq data of GATA2 and SCL from Wilson et al 30, which allowed us to train a positional weight matrix for each of these factors. GATA2 and SCL motifs were significantly enriched within the 3,933 peaks: 2,945, 609, and 484 co-occurrences of ETV2-SCL motifs (8-fold enrichment over random expectation, P = 0 from binomial test), ETV2-GATA2 motifs (14-fold enrichment, P < 1.11E-35), and ETV2-SCL-GATA2 motifs (15-fold enrichment, P = 0) (Supplementary Fig S2D). This observation is highly non-random, suggesting that ETV2 and these factors may interact or cobind to some of these sites. Indeed, ETV2 and GATA2 have been recently reported to form a complex to regulate hematopoietic and endothelial cell gene expression 40. ETV2 presumably specifies the hemangiogenic cell fate by collectively turning on hematopoietic and endothelial cell lineage-specifying genes. The ETS, GATA, and E-box gene regulatory network is integral to this process.
ETV2 enhances VEGF signaling
ChIP-Seq analysis revealed ETV2 binding to VEGF receptors and downstream signaling pathway genes including MAPK. Specifically, ETV2 targets include Flk1, Flt1, Flt4, Nrp1, Nrp2, and Mapk3 genes (Fig3A). Rho-GTPases and adhesion molecules were also identified as potential ETV2 direct targets. We selected 15 peak regions associated with Flk1, Flt1, Nrp1, and Nrp2 genes occupied by ETV2, of which 14 were evolutionarily conserved (Fig3A and Supplementary Table S3), and validated ETV2 binding in day 3.5 iEtv2 EBs using ChIP-qPCR. A significant mean enrichment was observed for ETV2 binding at these genomic locations with chromatin pulled down by V5 antibody or endogenous ETV2 antibody (ETV2-polyAbs) in iEtv2 EB cells (Dox added on day 2) (Fig3B). Importantly, ETV2 binding at these genomic locations was confirmed in R1 wild-type EB cells using ETV2-polyAbs, validating that these are bona fide ETV2 targets (Supplementary Fig S3A). To assess the functional significance of the ETV2 binding, we tested these regions for the response to ETV2 using the luciferase reporter assay. We found that ETV2 could activate the luciferase constructs tested, approximately 5- to 100-folds, compared to the pGL4 vector control (Fig3C). We further selectively deleted the ETV2 binding motif in several reporter vectors and found that the luciferase activity was greatly impaired when ETV2 binding motif was deleted (Fig3C). Collectively, these regions may act as ETV2 cis-regulatory elements for VEGF receptor gene expression. Future in vivo transgenic reporter system would further solidify such notion.
As ChIP-Seq analysis suggested that ETV2 could elevate FLK1 signaling activity, we determined whether ETV2 could directly modulate VEGF signaling. To this end, we performed VEGF signaling and Cell Motility PCR array, which included Rho-GTPases, adhesion and integrin genes, using RNA obtained from iEtv2 EBs (differentiated ES cells) with DOX 27. Strikingly, VEGF signaling pathway genes were upregulated by Etv2 overexpression, which were reciprocally downregulated in Etv2−/− embryos as well as Flk1−/− embryos (Fig3D and E, Supplementary Table S4). Additionally, while Rho-GTPase and its activating protein genes were upregulated by Etv2 overexpression, they were greatly downregulated in E8.5 Flk1−/− embryos 41 (Fig3F and G, Supplementary Table S5). These studies support the notion that hemangiogenic program specification requires ETV2-mediated FLK1 signaling activation.
ETV2 is required for the formation and expansion of FLK1+ hemangiogenic progenitors
Consistent with the data that ETV2 enhances VEGF signaling, previous findings suggested that blood and endothelial cell progenitors express high levels of FLK1 (FLK1high) compared to other FLK1-expressing cardiac or muscle progenitors (FLK1low) 42. To determine whether we can identify differential FLK1 activity associated with hemangiogenic cell population development in the embryo, we subjected embryos to FLK1 and PDGFRα expression analysis. As FLK1+PDGFRα− cells isolated from differentiating ES cells were enriched for the hemangioblast 27 and as FLK1+PDGFRα− cells in the developing embryo have not been characterized yet, we initially analyzed FLK1 and PDGFRα expression kinetics in developing embryos. In wild-type embryos, FLK1+PDGFRα− hemangiogenic progenitors were already detected around embryonic day (E) 7.5 and progressively expanded with time during the course of E7.5–E8.5 (Fig4A). Notably, the mean fluorescence intensity of the FLK1 staining within the FLK1+PDGFRα− cell population became greater as embryos develop, suggesting an elevated FLK1 signaling activity within FLK1+PDGFRα− cells compared to FLK1+PDGFRα+ cells (Fig4A). Remarkably, FLK1highPDGFRα− cell population was exclusively missing in Etv2−/− embryos, indicating that ETV2 was specifically required for the stage of FLK1highPDGFRα− cell generation in the embryo (Fig4A, Supplementary Fig S3B and C).
Figure 4. ETV2 is required for the generation and expansion of hemangioblast.
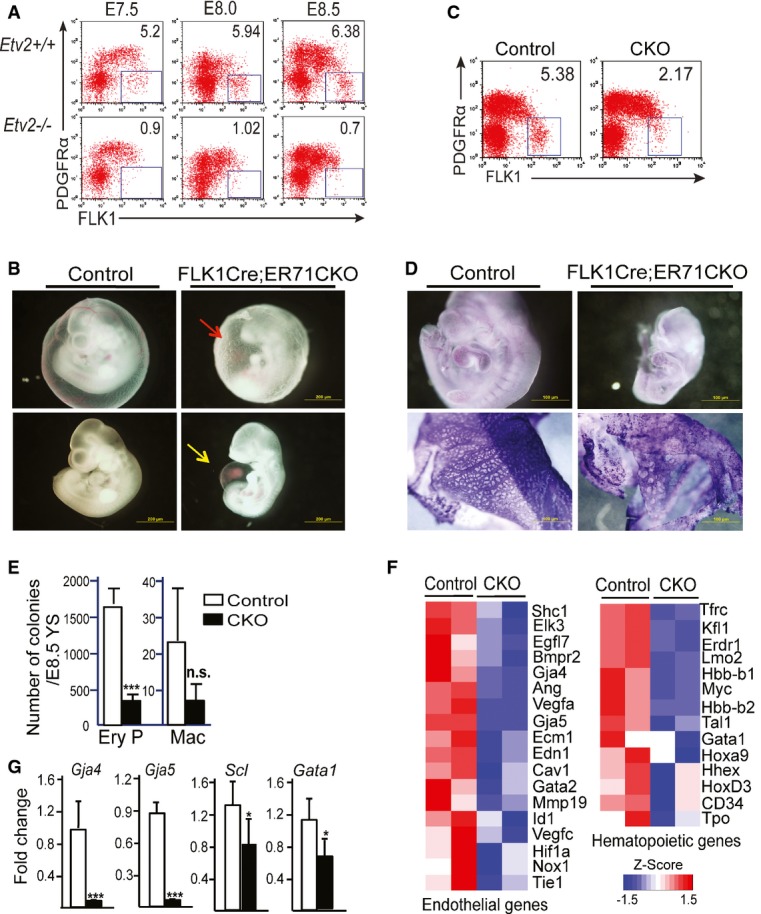
- A Etv2+/+ and Etv2−/− yolk sac and embryo proper were analyzed for FLK1 and PDGFRα expression at E7.5, 8, and 8.5 (n = 6 for each). Representative FACS analysis at each time point is shown.
- B Gross morphology of the E10.5 Flk1Cre;Etv2 CKO embryos is shown. Note the wrinkled yolk sac (red arrow) and the swollen pericardial cavity accumulated with fluid/blood (yellow arrow) in the mutants.
- C FACS analysis of E8.5 Flk1Cre;Etv2 CKO embryos for FLK1 and PDGFRα expression.
- D CD31/PECAM1 whole-mount staining of the E9.5 embryos (up: embryos; down: yolk sacs).
- E Hematopoietic colony assay of the E8.5 wild-type and mutant yolk sacs. Mean ± SD, n = 4 biological replicates. ***P < 0.001, two-tailed Student's t-test.
- F, G Microarray analysis (F) and qRT–PCR analysis (G) of the Flk1Cre;Etv2 CKO yolk sacs. Mean ± SD, n = 3 biological replicates. *P < 0.05, ***P < 0.001, two-tailed Student's t-test.
Etv2 is transiently expressed in developing embryos and ES/EBs 24. To determine whether Etv2 was still required once FLK1+ hemangioblast was formed, we conditionally deleted Etv2 within Flk1-expressing cells, Flk1Cre;Etv2 CKO, by crossing Flk1+/Cre;Etv2+/− (or Flk1+/Cre;Etv2f/+) and Etv2f/f mice to generate Flk1+/Cre;Etv2f/− (or Flk1+/Cre;Etv2f/f) mice. No live Flk1Cre;Etv2 CKO mice were obtained at weaning. Timed matings between Flk1+/Cre;Etv2+/− (or Flk1+/Cre;Etv2f/+) and Etv2flf mice were performed, and embryos were analyzed at different time points. Flk1Cre;Etv2 CKO embryos died around E10.5, later than Etv2−/− mice that die around E9.5, and exhibited wrinkled yolk sacs with dispersed blood (Fig4B). At E8.5, FLK1+PDGFRα− cells were detected but at much reduced levels within E8.5 Flk1Cre;Etv2 CKO embryos (Fig4C, Supplementary Fig S3D). Consequently, the embryo proper of the mutants was relatively pale and small and showed fluid and blood accumulation in the pericardial cavities. Whole-mount PECAM1/CD31 staining revealed disorganized vasculatures in the mutants (Fig4D). E8.5 mutant yolk sacs contained fewer hematopoietic progenitor cells (Fig4E). CD31-expressing cells also appeared to be reduced in E9.5 yolk sacs (Supplementary Fig S3E). Consistent with the phenotypic defects, expression of hematopoietic and endothelial genes in Flk1Cre;Etv2 CKO yolk sacs was also greatly reduced (Fig4F and G). Collectively, we conclude that ETV2 is required until sufficient FLK1highPDGFRα− cells are generated to guarantee proper blood and endothelial cell development.
ETS genes are ETV2 direct targets
Many ETS factors are potential ETV2 direct targets (Fig5A and Supplementary Table S3). Importantly, Etv2 was also identified as a target, suggesting that there is a positive auto-feedback regulation involving Etv2 expression. We selected 15 peak regions associated with Fli1, Ets1, Erg and Ets2 genes, of which 12 were conserved (Supplementary Table S3), and validated ETV2 binding on these genes (Fig5B). We next constructed luciferase reporters using the validated peak regions of the Ets genes and found that all the tested fragments enhanced the luciferase activity by ETV2 (Fig5C). Importantly, deletion of the respective ETV2 binding motifs resulted in substantially reduced luciferase activities (Fig5D). In addition, we observed cooperative action among the potential ETV2 motifs. For example, while the deletion of the two binding sites (mt1) in the Fli1 regulatory region (peak 7 in Fig5A) still conferred the luciferase activity in response to ETV2, deletion of all three ETV2 binding motifs (mt2 or mt3) in this region almost completely abrogated ETV2 responsiveness in the luciferase activity (Fig5D). Collectively, we conclude that ETV2 positively regulates its own expression and that of other Ets genes.
Having confirmed that Ets genes are ETV2 targets, we validated the kinetics of the Ets gene expression in developing EBs and embryos. Indeed, Etv2 expression precedes that of other Ets genes in differentiating ES cells. Specifically, Fli1 expression was detected shortly after Etv2, followed by Erg, Elk3, Ets1, and Ets2 (Fig6A). Moreover, in the DOX-inducible Etv2 expression system (iEtv2 ES), where Etv2 expression becomes visible within 6 h of DOX addition, expression of Fli1, Erg, and Elk3 was prominently induced by 12–24 h after DOX addition (Fig6B). Ets1 and Ets2 were also induced, although at much modest levels (Fig6B). Importantly, expression of Fli1, Erg, and Elk3 was greatly reduced in Etv2-null embryos (Fig6C). Expression of Ets1 and Ets2 was also reduced in Etv2-null embryos (Fig6C). If Fli1 is truly a direct target of Etv2 in hematopoietic and endothelial cell development, it was expected that Etv2 expression would be unaffected by Fli1 manipulation. Indeed, expression of Etv2, Erg, and Elk3 was not impaired in Fli1-null ES cells differentiated in culture (Fig6D) or in embryos (Fig6E). Moreover, enforced Fli1 expression in differentiating ES cells (iFli1 ES cells) did not alter Etv2 expression, although other Ets genes were upregulated (Fig6F).
Figure 6. ETV2 functions at the top of the ETS hierarchy in development.

- Kinetic analysis of Brachyury and Ets gene expression in wild-type EBs, day 0 to day 5, is shown. Gene expression was normalized to Gapdh. Error bars represent the SD from four independent experiments.
- Inducible Etv2 ES cells were differentiated in serum-free conditions with DOX added on day 2. Ets gene expression was analyzed at indicated times by qRT–PCR. Mean ± SD, n = 4 biological experiments. Genes were normalized to Gapdh, and the ratio of the +DOX gene quantity to −DOX gene quantity was determined to yield fold changes shown on the y-axis.
- qRT–PCR analysis of Ets gene expression in E8.5 embryos of Etv2+/+ (n = 10), Etv2+/− (n = 18) and Etv2−/− (n = 9) (embryos were obtained from 4 to 5 pregnant mice). Gene expression was normalized to Gapdh. Mean values are shown ± SD. Student's t-test *P < 0.05; **P < 0.01; ***P < 0.001; ns, not significant.
- Fli1+/+ and Fli1−/− ES cells were differentiated on OP9 feeder cells and the expression of Etv2, Erg, and Elk3 genes was analyzed from day 0 to day 6. Mean ± SD, n = 4 experiments.
- Analysis of Etv2 expression in E8.5 embryos of Fli1+/+ (n = 10), Fli1+/− (n = 14) and Fli1−/− (n = 8) (embryos were obtained from 4 to 5 pregnant mice). Gene expression was normalized to β-actin. Mean values are shown ± SD.
- Expression of Ets genes was analyzed on day 3 iFli1 EB cells with DOX added on day 2 (mean ± SD, n = 4). Student's t-test *P < 0.05; **P < 0.01; ***P < 0.001.
Hierarchical ETS function in hemangioblast, hematopoietic, and endothelial cell lineage development
One possibility for the non-redundant role of ETV2 in the FLK1highPDGFRα− hemangioblast commitment could be the timing of the ETS factor availability. As such, ETV2 happens to be expressed before the other ETS factors and thus carries out such a robust function. To determine whether ETS factors are inherently redundant or unique in their function in the hematopoietic and endothelial cell specification, we analyzed the hemangiogenic output by Fli1, the immediate ETV2 target. Specifically, we determined whether Fli1−/− embryos showed defects in the FLK1+PDGFRα− hemangiogenic progenitor generation. As shown, FLK1highPDGFRα− progenitor cell population still developed in the Fli1-null embryos and ES cells, similar to wild-type control levels, suggesting that hemangioblast formation does not require FLI1 function (Fig7A and B, Supplementary Fig S4A and B). However, enforced Fli1 expression was able to skew mesoderm, although at a lesser degree than ETV2, into the hemangiogenic fate in iFli1 ES cells (Fig7C and Supplementary Fig S4C). Ets1 or Ets2 cannot skew mesoderm into the hemangioblast 39. This supports the notion that Fli1 is not necessary, but sufficient for hemangioblast generation. Ets1 or Ets2 cannot induce hemangioblast, even if overexpressed at the same time frame as Etv2.
Figure 7. ETS switching mechanism in maintaining hematopoietic and endothelial cell program.

- A–C A representative FACS profile of FLK1 and PDGFRα expression of E8.5 Fli1+/+ (n = 9), Fli1+/− (n = 11), and Fli1−/− (n = 6) embryos (A) (embryos were obtained from 4 to 5 pregnant mice); Fli1+/+ and Fli1−/− EBs (B, four independent biological samples); and iFli1 EBs (± DOX) (C, four independent biological samples) at indicated times.
- D iEtv2-GS (=iEtv2-2A-Gata2-2A-Scl), iFli1-GS, iErg-GS, and iEts1-GS EBs were treated with DOX from day 2 and analyzed for FLK1 and PDGFRα at indicated times. Numbers indicate the percentage within each quadrant. Representative results from four independent biological samples are shown.
- E The enrichment of ETV2 and FLI1 binding at the indicated gene loci by ChIP-qPCR analysis in wild-type EBs is shown. qPCR primers and genomic locations are provided in Supplementary Table S3. Error bars represent SD, n = 4. *P < 0.05, **P < 0.01, ***P < 0.001 by Student's two-tailed t-test.
- F A model showing ETV2 regulation of the hematopoietic and endothelial cell program.
To further elicit functional hierarchy among ETS factors, we additionally determined the hemangioblast skewing effect by different ETS factors in the context of Gata2 and Scl coexpression. We previously reported that Etv2, when coexpressed with Gata2 and Scl, robustly skewed mesoderm toward the hemangioblast while suppressing the cardiac output from ES cells 39. Thus, we compared the coexpression effect of Ets genes with Gata2 and Scl on the FLK1+PDGFRα− hemangioblast formation. We specifically generated doxycycline-inducible ES cells coexpressing Fli1-Gata2-Scl, Erg-Gata2-Scl, or Ets1-Gata2-Scl. As previously reported 39, Etv2-Gata2-Scl coexpression during mesoderm formation stage robustly induced FLK1+PDGFRα− hemangioblast from ES cells. While FLK1+PDGFRα− hemangioblast population was increased by Fli1-Gata2-Scl or Erg-Gata2-Scl, the level of the FLK1+PDGFRα− cells generated by Fli1-Gata2-Scl or Erg-Gata2-Scl was not as robust as that by the Etv2-Gata2-Scl (Fig7D, Supplementary Fig S4D). Importantly, there was incomplete skewing of mesoderm toward FLK1+PDGFRα− hemangioblast by Fli1-Gata2-Scl or Erg-Gata2-Scl, as judged by FLK1+PDGFRα+ cardiac mesoderm production (Fig7D, Supplementary Fig S4D). Ets1-Gata2-Scl was least effective in inducing FLK1+PDGFRα− hemangioblast from ES cells (Fig7D, Supplementary Fig S4D). Collectively, ETV2 carries out a uniquely distinct role in the hemangioblast generation. No other ETS factors can replace ETV2 in the hemangioblast formation.
Hematopoietic and endothelial cell program is maintained by an ETS switching mechanism
ETV2 ChIP-Seq identified key hematopoietic and endothelial cell genes as ETV2 direct targets. However, the mechanism that allows these genes to be continuously expressed in mature hematopoietic and endothelial cells even when Etv2 is no longer expressed remains to be elucidated. We postulated that ETV2 target gene expression could be maintained within mature hematopoietic and endothelial cells by other ETS factors, which were induced by ETV2. To this end, we analyzed key target genes, Lmo2, Gata2, Cdh5, Tie2/Tek, and Scl/Tal1 for the ETS factor occupancy using day 3 and day 4–5 EB cells. Specifically, we hypothesized that while the ETS sites of the target genes would be occupied by ETV2 in days 2–3 EBs, when ETV2 expression is robust, the same target sites would be occupied by other ETS factors when ETV2 is no longer expressed, that is, days 4–5. To test this idea, we collected wild-type EB cells at day 3, 4, and 5 and performed ChIP-qPCR using ETV2-polyAbs and FLI1 Ab. As shown, while the evolutionary conserved potential ETS binding sites of these genes were occupied by ETV2 in day 3 EBs, the same regions were occupied by FLI1 in later developmental stages, that is, days 4 and 5. FLI1 occupancy on these genes was minimal in day 3 EBs (Fig7E). Collectively, ETV2-initiated blood and endothelial cell program is maintained by other ETS factors through an ETS switching mechanism.
Discussion
Studies so far have established a non-redundant and essential function of ETV2 in blood and vascular development. However, mechanistic understanding of how ETV2 performs such a profound function in blood and vessel formation has been lacking. We demonstrate that FLK1highPDGFRα− cell population enriched for hemangiogenic progenitors is uniquely sensitive to ETV2 function, as this population was specifically missing in Etv2-deficient embryos. The formation of the FLK1highPDGFRα− cell population would require simultaneous activation of the hematopoietic and endothelial cell lineage specifying genes and FLK1 signaling, which are coordinated by ETV2. As Flk1Cre;Etv2CKO mice generate suboptimal levels of FLK1highPDGFRα− progenitor, we propose that ETV2 is required up to the timeframe when FLK1highPDGFRα− hemangiogenic progenitor population is sufficiently generated to ensure proper hematopoietic and endothelial cell generation. Based on studies that VEGF-FLK1 signaling can induce Etv2 expression 26, 43, we propose that ETV2-FLK1 feed forward mechanism is responsible for FLK1highPDGFRα− hemangiogenic progenitor expansion (Fig7F). Collectively, we provide novel insights into how the FLK1highPDGFRα− hemangiogenic progenitor specification is achieved by ETV2.
Consistent with our data that Etv2 was still required downstream of Flk1, Etv2 deletion using tamoxifen injection from E8.5, but not E9.5, in ROSACreER;Etv2f/f mice results in similar vascular development defects as ours 44. Intriguingly, Flk1Cre;Etv2 CKO mice were reported to be born without any phenotypic defects 35. Given that Etv2 expression is transient and that Etv2 deletion using the Tie2-Cre or tamoxifen injection in ROSACreER;Etv2f/f mice from E9.5 leads to normal live birth 44, the discrepancy in the embryonic lethality between the two studies might be due to different deletion efficiency or the accessibility of the floxed Etv2 alleles. For example, while exons 5–6 were floxed in our study, the entire Etv2 gene was floxed in the reported study, which might have contributed to different CRE accessibility of the floxed alleles in the chromatin context. We rule out the possibility that our strategy might have generated a dominant negative protein and contributed to the embryonic lethal phenotype, since Etv2 message corresponding to exons 2–3 in purified endothelial cells (CD45−CD31+) from the Tie2Cre;Etv2f/− (or Tie2Cre;Etv2f/f) mice was virtually undetectable (not shown). Additionally, genetic backgrounds often affect the severity of the knockout phenotypes. In our studies, Flk1Cre, Etv2+/−, and Etv2fl/f mice have all been backcrossed to and maintained on a pure C57BL/6 background.
As ETV2 function is transiently required, there must be a mechanism maintaining the hematopoietic and endothelial cell program that was initiated by ETV2. We propose that ETV2 drives lineage-specific epigenetic landscape in blood and vascular systems. As ETV2 target loci remained unmethylated in the heart and bone marrow (Fig2B and Supplementary Fig S1G), ETV2 binding could directly result in demethylation of its binding sites, thereby shaping the lineage-specific epigenome 33. Hypomethylation of these binding sites is maintained in blood and vascular systems as an epigenetic memory 45, potentially serving as the future tissue-specific enhancer elements of ETV2 downstream factors. We provide ETS switching mechanism as one such mode. Similar to the GATA switching mechanism controlling the erythroid cell lineage differentiation 46, 47, ETV2 induces expression of other Ets genes, thereby creating an ETS hierarchy. Specifically, we show that hematopoietic and endothelial cell program is initiated by ETV2, but is maintained by other ETS factors (Fig7F). However, timing of different Ets factor expression alone cannot explain the non-redundant ETV2 function, as we demonstrated that FLI1, ERG, or ETS1 could not replace ETV2 function even if they were expressed at the same time frame as ETV2, in the hemangioblast induction from ES cells. To this end, structure and function of ETV2 in relationship to other ETS factors may elucidate the unique role of ETV2 in the hematopoietic and endothelial cell development. Additionally, ETV2 may require a unique cofactor(s). Recent finding of OVOL2 as ETV2, not ETS1 or ETS2, cofactor further suggests such non-redundant function played by ETV2 48.
While ETV2 is a potent inducer of the hemangiogenic program, it is transiently expressed, suggesting that there might be a strong pressure to keep this gene off once hematopoietic and vascular systems are established. Indeed, sustained Etv2 expression during development results in hematopoietic and vessel defects as manifested by dilated yolk sac vessels 49. Future studies on Etv2 gene regulation are warranted. Presumably, Etv2 reactivation in mature endothelial cells might be a helpful strategy for hematopoietic and endothelial cell regeneration or diseases requiring angiogenesis, such as peripheral arterial disease and wound healing. To this end, it is worth noting that Etv2, alone or in combination with other endothelial cell factors, can reprogram somatic cells to functional endothelial cells 50, 51. It will also be important to determine whether Etv2 is aberrantly expressed in pathologic conditions, such as tumor angiogenesis. If so, mechanisms involved in Etv2 downregulation may have a direct efficacy in the pathologic angiogenesis.
Materials and Methods
Antibodies
Antibodies against V5 (ab15828), control rabbit IgG (ab31475), and FLI1 (ab15289) were purchased from Abcam (Cambridge, MA). Polyclonal antibodies against endogenous ETV2 (ETV2-polyAbs) were produced by YenZym Antibodies (South San Francisco, CA). Briefly, rabbits were immunized three times with a synthetic peptide corresponding to the mouse ETV2200–219 (EGHQSPAFTTPSKSNKQSDR). Polyclonal antibody was affinity purified using ETV2 peptide-conjugated affinity matrix. Antibody specificity was further confirmed by antigen-specific ELISA and immunoprecipitation followed by Western blot.
Knockout and conditional knockout mouse analysis
Heterozygous Etv2+/− or Fli1+/− mice were subjected to brother–sister matings to generate Etv2−/− or Fli1−/− embryos. Flk1Cre;Etv2 CKO mice were generated by crossing Flk1+/Cre;Etv2+/− (or Flk1+/Cre;Etv2f/+) and Etv2f/f mice. The generation of Flk1Cre, Etv2+/−, Etv2f/f, and Fli1+/− mice has been previously described 20, 24, 52, 53. Information on genotyping primers was previously described 24, 27, 38, 39. Embryos from E7.5 to E9.5 were dissected and disassociated with 0.1% trypsin for 5 min or 0.25% collagenase for 30 min at 37°C into single cells and then subjected to FACS analysis or RNA extraction using RNeasy micro Kit (Qiagen) for gene expression. All mouse experiments were done in accordance with protocols approved by the Institutional Animal Care and Use Committee of Washington University in St. Louis Medical School.
ES cell generation and differentiation
Fli1−/− ES cells were generated from blastocysts obtained from the Fli1+/− brother–sister matings. Inducible ES cell lines (iEtv2 and iEGS) were previously described 24, 27, 37, 38. Inducible Fli1 (iFli1) ES cells were generated by targeting Fli1 into the tet-responsive locus of A2Lox cells 38. To generate inducible Fli1-2A-Gata2-2A-Scl (iFli1-GS), Erg-2A-Gata2-2A-Scl (iErg-GS), or Ets1-2A-Gata2-2A-Scl (iEts1-GS) ES cells, coding sequence of Ets (Fli1, Erg and Ets1), Gata2, and Scl, linked by 2A peptides 54, were targeted into the tet-responsive locus of A2 Lox cells. The correct targeting event was confirmed by a tet-responsive locus/cDNA vector-specific PCR. Doxycycline (DOX) was typically added to the differentiating ES cells from day 2 to induce the genes.
Chromatin immunoprecipitation (ChIP)-sequencing
Wild-type or iEtv2 EBs from day 3–5 were subjected to ChIP assay. Briefly, EBs were disassociated into single cells by trypsin, fixed with 1% formaldehyde at room temperature for 10 min, and quenched by adding glycine to 125 mM final concentration. The cross-linked cell pellets (1–2 × 107) were resuspended in lysis buffer (1% SDS, 5 mM EDTA, 50 mM Tris–HCl [pH 8.1], plus protease inhibitor) and gently rocked at 4°C for 10 min, then sonicated to 200- to 600-bp fragments using an AFA Focused-ultrasonicator or a Diagenode Bioruptor. Sonicated lysates were cleared by pelleting insoluble material at 20,000 × g for 20 min at 4°C. The soluble lysate was diluted by adding 9 volumes of IP buffer (1% Triton X-100, 2 mM EDTA, 150 mM NaCl, 20 mM Tris–HCl [pH 8.1], 1× protease inhibitor cocktail). Subsequently, the lysate was precleared with 30 μl of protein A- or G- Sepharose beads for 2 h at 4°C under rocking, followed by incubation with specific antibody overnight at 4°C. Subsequently, 45 μl protein A- or G- Sepharose beads were added and incubated for 2–3 h at 4°C. The beads were then washed three times serially with cold wash buffer TSE I (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 150 mM NaCl), TSE II (0.1% SDS, 1% Triton X-100, 2 mM EDTA, 20 mM Tris–HCl, pH 8.1, 500 mM NaCl), buffer III (0.25 M LiCl, 1% NP-40, 1% deoxycholate, 1 mM EDTA, 10 mM Tris–HCl, pH 8.1) and then TE buffer. Precipitated chromatin complexes were eluted in 100 μl elution buffer (1% SDS, 0.1 M NaHCO3) at room temperature for 10 min and uncross-linked overnight at 65°C, followed by RNase A treatment for 2 h and then proteinase K for 2 h. DNA was extracted with QiaQuick PCR purification kit (Qiagen).
Immunoprecipitated DNA yield was determined via Quant IT fluorescence assay (Invitrogen), and transcription factor enrichment was evaluated by qPCR. Illumina sequencing libraries were generated as following. ChIP DNA was blunt-ended, “A” base was added to 3' end, and sequencing adapters were ligated to the ends. The fragments were size-selected to 200–600 base pairs and underwent amplification for 15 cycles. The resulting libraries were sequenced using the Illumina HiSeq-2000 as single reads extending 50 bases. The raw data were demultiplexed and aligned to the reference genome using Bowtie. MACS was used to call peaks.
Sequence alignment and peak calling
Sequences reads were submitted to NCBI Gene Expression Omnibus (GEO; http://www.ncbi.nlm.nih.gov/geo/) under Accession Number GSE59402. Sequence reads were aligned to mouse genome assembly mm9 28 using BWA aligner 55 for alignment and methylQA 56 for initial process. MACS2 29 was used for peak calling against IgG input using P-value threshold 1E-6.
GREAT analysis
Peak regions were submitted to GREAT (http://bejerano.stanford.edu/great/public/html/, version 2.0.2) using default “Basal plus extension” settings. Enrichment terms less than FDR threshold 0.05 were regarded as statistical significant.
Motif analysis
The ETV2 motif was de novo trained from peaks we identified using Homer software 57. We also used Homer for motif scanning on peak regions. Statistic tests were performed using R environment (http://www.r-project.org/). ETV2 and GATA2 motif came from the de novo training results; SCL (E-box) motif was generated using seq2profile.pl utility from Homer software using consensus sequence CANNTG. Homer was also used for motif scanning in peaks. Peaks with all 3 motifs occurring were used for calculating the nearest distance of ETV2-SCL and SCL-GATA2 using custom script. The ETV2-SCL-GATA2 logo was generated by WebLogo 3 (http://weblogo.threeplusone.com/create.cgi).
Microarray analysis
We analyzed microarray data following instructions as previously described 58. Genes shown 1.5-fold downregulated in Etv2 knock out and DOX+ upregulated, or 1.5-fold upregulated in Etv2 knock out and DOX+ downregulated were used to overlap with genes identified from ChIP-Seq experiments.
RNA obtained from controls and Flk1Cre;Etv2CKO E9.5 yolk sacs was used to gene expression profiling analyses with Illumina MouseWG-6 v2.0 Expression BeadChip Kits at the Northwestern University Genomic Core, Chicago.
Evolutionary analysis
The 30-way vertebrate alignment conservation data were downloaded from UCSC Genome Browser; for each peak, we extend 3 kb from the peak center and split this 6-kb region to 50-bp bins, we assigned each bin with an average conservation score using custom script.
Analysis of whole-genome bisulfite sequencing data
We downloaded published whole-genome bisulfite sequencing (WGBS) data from GEO 33, 34 and assigned the CpG sites around 6-kb region centered each peak center with methylation values for these tissues including ESC (GSE30202), heart (GSM1051154), bone marrow (GSM1051150), neuronal progenitors (GSE30202), and cerebellum (GSM1051151). These 6-kb regions were split to 50-bp bins, and methylation values for each bin were averaged from all ChIP-Seq peaks. Profile of peaks, which were distal (> 1 kb distance) to nearest TSS, was plotted, and a generalized linear model was applied to smooth the methylation changes.
Luciferase reporter assay
ETV2 bound sequences (Supplementary Table S3) obtained through MACS peak-calling algorithm were PCR-amplified and cloned into the pGL4.24[luc2P/minP] vector containing the minimal promoter and the luciferase reporter gene luc2P. Selected peak regions containing ETV2 binding motif were deleted as shown in Supplementary Table S3. For luciferase assays, 293T cells cultured in DMEM supplemented with 10% fetal bovine serum were plated onto 24-well plates (3 × 104 cells) and transfected the next day with the various pGL4.24[luc2P/minP] vector constructs with or without the ETV2 expression plasmid (pMSCV-Etv2). Renilla luciferase vector (Promega) was cotransfected to normalize transfection efficiency. Transfection was performed using Lipofectamine 2000 (Invitrogen, Carlsbad, CA). Cells were harvested 48 h after transfection, and luciferase activity was measured using the Dual-Luciferase Reporter Assay kit and GloMax™ Systems (Promega). Reporter gene activities were first normalized to the Renilla luciferase value and further compared to that of the control pGL4.24 reporter. All experiments were repeated four biological replicates.
Flow cytometry
Embryoid bodies (EBs) or mouse embryos were harvested and dissociated into single cells using 0.1% trypsin (EBs) or 0.25% collagenase (embryos) for staining. Primary Abs included biotin-α-FLK1 (1:200, Avas12, BioLegend), APC-α-PDGFRα (1 μg/ml, APA5, eBioscience), and PE-α-CD31 (1 μg/ml, MEC13.3, Biolegend). Streptavidin (SA)/PE (0.5 μg/ml, eBioscience) was used as secondary antibody. Data were acquired on a FACSCalibur flow cytometer (Becton Dickinson) and analyzed using FlowJo (Treestar) software.
Signaling pathway RT2 profiler PCR array
The RT2 Profiler PCR Array that profiles the expression of 84 key genes in the mouse VEGF signaling (PAMM-091ZA) and the Cell Motility (PAMM-128ZA) pathway were purchased from SA-Bioscience (Qiagen). Total RNA was isolated from day 3.5 iEtv2 EBs (±DOX on day 2–3.5) and a pool of E8.5 Etv2+/+ or Etv2−/− embryos, converted to cDNAs, and used for screening by real-time PCR following the manufacturer's instructions. qRT–PCR assays performed in triplicate, normalized to 4 housekeeping genes (Gusb, Hsp90ab1, Gapdh, and Actb), and analyzed according to the ΔΔCT method with the SA-Biosciences proprietary software.
Quantitative real-time reverse transcription PCR (qRT–PCR)
Total RNA from embryos and EB cells were prepared with RNeasy Micro/Mini Kit, and reverse-transcribed into cDNAs according to manufacturer's protocol. Expression of genes was measured by quantitative real-time RT–PCR with primers indicated in Supplementary Table S6. Gene expression levels were normalized to Gapdh or β-actin.
Statistical analysis
Student's t-test (Prism5, GraphPad Software, La Jolla, CA) was used for statistical analysis. A P-value of < 0.05 was considered as significant.
Acknowledgments
This work was supported by grants from the National Institutes of Health, NHLBI, HL63736, and HL55337 (K.C.); NHGRI, HG007354, HG007175, ES024992 (T.W.); American Cancer Society, RSG-14-049-01-DMC (T.W.); HL119291 (C.P.); American Heart Association, 11SDG7390074 (C.P.); the March of Dimes Foundation, #5-FY12-44 (C.P.).
Author contributions
FL and KC conceived the experiments, analyzed the data, and wrote the manuscript. FL, YY, IK, and MC performed experiments. JK and CP performed Etv2 conditional knockout studies. DW provided Fli1 knockout mice. DL and TW performed bioinformatics analysis and wrote the manuscript.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Figure and Table Legends
Review Process File
References
- Choi K, Kennedy M, Kazarov A, Papadimitriou JC, Keller G. A common precursor for hematopoietic and endothelial cells. Development. 1998;125:725–732. doi: 10.1242/dev.125.4.725. [DOI] [PubMed] [Google Scholar]
- Murray PDF. The development in vitro of the blood of the early chick embryo. Proc Roy Soc London. 1932;11:497–521. [Google Scholar]
- Nishikawa SI, Nishikawa S, Hirashima M, Matsuyoshi N, Kodama H. Progressive lineage analysis by cell sorting and culture identifies FLK1+VE-cadherin+ cells at a diverging point of endothelial and hemopoietic lineages. Development. 1998;125:1747–1757. doi: 10.1242/dev.125.9.1747. [DOI] [PubMed] [Google Scholar]
- Sabin FR. Studies on the Origin of Blood-vessels and of Red Blood-corpuscles as Seen in the Living Blastoderm of Chicks During the Second Day of Incubation. Contrib Embryol. 1920;9:213–262. [Google Scholar]
- Padron-Barthe L, Temino S, Villa del Campo C, Carramolino L, Isern J, Torres M. Clonal analysis identifies hemogenic endothelium as the source of the blood-endothelial common lineage in the mouse embryo. Blood. 2014;124:2523–2532. doi: 10.1182/blood-2013-12-545939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Myers CT, Krieg PA. BMP-mediated specification of the erythroid lineage suppresses endothelial development in blood island precursors. Blood. 2013;122:3929–3939. doi: 10.1182/blood-2013-03-490045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertrand JY, Chi NC, Santoso B, Teng S, Stainier DY, Traver D. Haematopoietic stem cells derive directly from aortic endothelium during development. Nature. 2010;464:108–111. doi: 10.1038/nature08738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisset JC, van Cappellen W, Andrieu-Soler C, Galjart N, Dzierzak E, Robin C. In vivo imaging of haematopoietic cells emerging from the mouse aortic endothelium. Nature. 2010;464:116–120. doi: 10.1038/nature08764. [DOI] [PubMed] [Google Scholar]
- Kissa K, Herbomel P. Blood stem cells emerge from aortic endothelium by a novel type of cell transition. Nature. 2010;464:112–115. doi: 10.1038/nature08761. [DOI] [PubMed] [Google Scholar]
- Lam EY, Hall CJ, Crosier PS, Crosier KE, Flores MV. Live imaging of Runx1 expression in the dorsal aorta tracks the emergence of blood progenitors from endothelial cells. Blood. 2010;116:909–914. doi: 10.1182/blood-2010-01-264382. [DOI] [PubMed] [Google Scholar]
- Butler JM, Nolan DJ, Vertes EL, Varnum-Finney B, Kobayashi H, Hooper AT, Seandel M, Shido K, White IA, Kobayashi M, et al. Endothelial cells are essential for the self-renewal and repopulation of Notch-dependent hematopoietic stem cells. Cell Stem Cell. 2010;6:251–264. doi: 10.1016/j.stem.2010.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kiel MJ, Yilmaz OH, Iwashita T, Terhorst C, Morrison SJ. SLAM family receptors distinguish hematopoietic stem and progenitor cells and reveal endothelial niches for stem cells. Cell. 2005;121:1109–1121. doi: 10.1016/j.cell.2005.05.026. [DOI] [PubMed] [Google Scholar]
- Morrison SJ, Scadden DT. The bone marrow niche for haematopoietic stem cells. Nature. 2014;505:327–334. doi: 10.1038/nature12984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalaby F, Rossant J, Yamaguchi TP, Gertsenstein M, Wu XF, Breitman ML, Schuh AC. Failure of blood-island formation and vasculogenesis in Flk-1-deficient mice. Nature. 1995;376:62–66. doi: 10.1038/376062a0. [DOI] [PubMed] [Google Scholar]
- Carmeliet P, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Abnormal blood vessel development and lethality in embryos lacking a single VEGF allele. Nature. 1996;380:435–439. doi: 10.1038/380435a0. [DOI] [PubMed] [Google Scholar]
- Ferrara N, Carver-Moore K, Chen H, Dowd M, Lu L, O'Shea KS, Powell-Braxton L, Hillan KJ, Moore MW. Heterozygous embryonic lethality induced by targeted inactivation of the VEGF gene. Nature. 1996;380:439–442. doi: 10.1038/380439a0. [DOI] [PubMed] [Google Scholar]
- De Val S, Chi NC, Meadows SM, Minovitsky S, Anderson JP, Harris IS, Ehlers ML, Agarwal P, Visel A, Xu SM, et al. Combinatorial regulation of endothelial gene expression by ets and forkhead transcription factors. Cell. 2008;135:1053–1064. doi: 10.1016/j.cell.2008.10.049. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wythe JD, Dang LT, Devine WP, Boudreau E, Artap ST, He D, Schachterle W, Stainier DY, Oettgen P, Black BL, et al. ETS factors regulate Vegf-dependent arterial specification. Dev Cell. 2013;26:45–58. doi: 10.1016/j.devcel.2013.06.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oikawa T, Yamada T. Molecular biology of the Ets family of transcription factors. Gene. 2003;303:11–34. doi: 10.1016/s0378-1119(02)01156-3. [DOI] [PubMed] [Google Scholar]
- Spyropoulos DD, Pharr PN, Lavenburg KR, Jackers P, Papas TS, Ogawa M, Watson DK. Hemorrhage, impaired hematopoiesis, and lethality in mouse embryos carrying a targeted disruption of the Fli1 transcription factor. Mol Cell Biol. 2000;20:5643–5652. doi: 10.1128/mcb.20.15.5643-5652.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pham VN, Lawson ND, Mugford JW, Dye L, Castranova D, Lo B, Weinstein BM. Combinatorial function of ETS transcription factors in the developing vasculature. Dev Biol. 2007;303:772–783. doi: 10.1016/j.ydbio.2006.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abedin MJ, Nguyen A, Jiang N, Perry CE, Shelton JM, Watson DK, Ferdous A. Fli1 acts downstream of Etv2 to govern cell survival and vascular homeostasis via positive autoregulation. Circ Res. 2014;114:1690–1699. doi: 10.1161/CIRCRESAHA.1134303145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dejana E, Taddei A, Randi AM. Foxs and Ets in the transcriptional regulation of endothelial cell differentiation and angiogenesis. Biochim Biophys Acta. 2007;1775:298–312. doi: 10.1016/j.bbcan.2007.05.003. [DOI] [PubMed] [Google Scholar]
- Lee D, Park C, Lee H, Lugus JJ, Kim SH, Arentson E, Chung YS, Gomez G, Kyba M, Lin S, et al. ER71 acts downstream of BMP, Notch, and Wnt signaling in blood and vessel progenitor specification. Cell Stem Cell. 2008;2:497–507. doi: 10.1016/j.stem.2008.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferdous A, Caprioli A, Iacovino M, Martin CM, Morris J, Richardson JA, Latif S, Hammer RE, Harvey RP, Olson EN, et al. Nk2–5 transactivates the Ets-related protein 71 gene and specifies an endothelial/endocardial fate in the developing embryo. Proc Natl Acad Sci USA. 2009;106:814–819. doi: 10.1073/pnas.0807583106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, Hayashi M, Nakagawa R, Tanaka Y, Izumi N, Nishikawa S, Jakt ML, Tarui H. Etv2/ER71 induces vascular mesoderm from Flk1+PDGFRalpha+ primitive mesoderm. Blood. 2011;118:6975–6986. doi: 10.1182/blood-2011-05-352658. [DOI] [PubMed] [Google Scholar]
- Liu F, Kang I, Park C, Chang LW, Wang W, Lee D, Lim DS, Vittet D, Nerbonne JM, Choi K. ER71 specifies Flk-1+ hemangiogenic mesoderm by inhibiting cardiac mesoderm and Wnt signaling. Blood. 2012;119:3295–3305. doi: 10.1182/blood-2012-01-403766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waterston RH, Lindblad-Toh K, Birney E, Rogers J, Abril JF, Agarwal P, Agarwala R, Ainscough R, Alexandersson M, An P, et al. Initial sequencing and comparative analysis of the mouse genome. Nature. 2002;420:520–562. doi: 10.1038/nature01262. [DOI] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. Model-based analysis of ChIP-Seq (MACS) Genome Biol. 2008;9:R137. doi: 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wilson NK, Foster SD, Wang X, Knezevic K, Schutte J, Kaimakis P, Chilarska PM, Kinston S, Ouwehand WH, Dzierzak E, et al. Combinatorial transcriptional control in blood stem/progenitor cells: genome-wide analysis of ten major transcriptional regulators. Cell Stem Cell. 2010;7:532–544. doi: 10.1016/j.stem.2010.07.016. [DOI] [PubMed] [Google Scholar]
- McLean CY, Bristor D, Hiller M, Clarke SL, Schaar BT, Lowe CB, Wenger AM, Bejerano G. GREAT improves functional interpretation of cis-regulatory regions. Nat Biotechnol. 2010;28:495–501. doi: 10.1038/nbt.1630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchette M, Kent WJ, Riemer C, Elnitski L, Smit AF, Roskin KM, Baertsch R, Rosenbloom K, Clawson H, Green ED, et al. Aligning multiple genomic sequences with the threaded blockset aligner. Genome Res. 2004;14:708–715. doi: 10.1101/gr.1933104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stadler MB, Murr R, Burger L, Ivanek R, Lienert F, Scholer A, van Nimwegen E, Wirbelauer C, Oakeley EJ, Gaidatzis D, et al. DNA-binding factors shape the mouse methylome at distal regulatory regions. Nature. 2011;480:490–495. doi: 10.1038/nature10716. [DOI] [PubMed] [Google Scholar]
- Hon GC, Rajagopal N, Shen Y, McCleary DF, Yue F, Dang MD, Ren B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat Genet. 2013;45:1198–1206. doi: 10.1038/ng.2746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wareing S, Mazan A, Pearson S, Gottgens B, Lacaud G, Kouskoff V. The Flk1-Cre-mediated deletion of ETV2 defines its narrow temporal requirement during embryonic hematopoietic development. Stem Cells. 2012;30:1521–1531. doi: 10.1002/stem.1115. [DOI] [PubMed] [Google Scholar]
- Pimanda JE, Ottersbach K, Knezevic K, Kinston S, Chan WY, Wilson NK, Landry JR, Wood AD, Kolb-Kokocinski A, Green AR, et al. Gata2, Fli1, and Scl form a recursively wired gene-regulatory circuit during early hematopoietic development. Proc Natl Acad Sci USA. 2007;104:17692–17697. doi: 10.1073/pnas.0707045104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lugus JJ, Chung YS, Mills JC, Kim SI, Grass J, Kyba M, Doherty JM, Bresnick EH, Choi K. GATA2 functions at multiple steps in hemangioblast development and differentiation. Development. 2007;134:393–405. doi: 10.1242/dev.02731. [DOI] [PubMed] [Google Scholar]
- Ismailoglu I, Yeamans G, Daley GQ, Perlingeiro RC, Kyba M. Mesodermal patterning activity of SCL. Exp Hematol. 2008;36:1593–1603. doi: 10.1016/j.exphem.2008.07.005. [DOI] [PubMed] [Google Scholar]
- Liu F, Bhang SH, Arentson E, Sawada A, Kim CK, Kang I, Yu J, Sakurai N, Kim SH, Yoo JJ, et al. Enhanced hemangioblast generation and improved vascular repair and regeneration from embryonic stem cells by defined transcription factors. Stem Cell Rep. 2013;1:166–182. doi: 10.1016/j.stemcr.2013.06.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shi X, Richard J, Zirbes KM, Gong W, Lin G, Kyba M, Thomson JA, Koyano-Nakagawa N, Garry DJ. Cooperative interaction of Etv2 and Gata2 regulates the development of endothelial and hematopoietic lineages. Dev Biol. 2014;389:208–218. doi: 10.1016/j.ydbio.2014.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takase H, Matsumoto K, Yamadera R, Kubota Y, Otsu A, Suzuki R, Ishitobi H, Mochizuki H, Kojima T, Takano S, et al. Genome-wide identification of endothelial cell-enriched genes in the mouse embryo. Blood. 2012;120:914–923. doi: 10.1182/blood-2011-12-398156. [DOI] [PubMed] [Google Scholar]
- Ema M, Takahashi S, Rossant J. Deletion of the selection cassette, but not cis-acting elements, in targeted Flk1-lacZ allele reveals Flk1 expression in multipotent mesodermal progenitors. Blood. 2006;107:111–117. doi: 10.1182/blood-2005-05-1970. [DOI] [PubMed] [Google Scholar]
- Rasmussen TL, Shi X, Wallis A, Kweon J, Zirbes KM, Koyano-Nakagawa N, Garry DJ. VEGF/Flk1 signaling cascade transactivates Etv2 gene expression. PLoS ONE. 2012;7:e50103. doi: 10.1371/journal.pone.0050103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kataoka H, Hayashi M, Kobayashi K, Ding G, Tanaka Y, Nishikawa S. Region-specific Etv2 ablation revealed the critical origin of hemogenic capacity from Hox6-positive caudal-lateral primitive mesoderm. Exp Hematol. 2013;41:567–581. doi: 10.1016/j.exphem.2013.02.009. e569. [DOI] [PubMed] [Google Scholar]
- Zhang B, Zhou Y, Lin N, Lowdon RF, Hong C, Nagarajan RP, Cheng JB, Li D, Stevens M, Lee HJ, et al. Functional DNA methylation differences between tissues, cell types, and across individuals discovered using the M&M algorithm. Genome Res. 2013;23:1522–1540. doi: 10.1101/gr.156539.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grass JA, Jing H, Kim SI, Martowicz ML, Pal S, Blobel GA, Bresnick EH. Distinct functions of dispersed GATA factor complexes at an endogenous gene locus. Mol Cell Biol. 2006;26:7056–7067. doi: 10.1128/MCB.01033-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresnick EH, Lee HY, Fujiwara T, Johnson KD, Keles S. GATA switches as developmental drivers. J Biol Chem. 2010;285:31087–31093. doi: 10.1074/jbc.R110.159079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim JY, Lee RH, Kim TM, Kim DW, Jeon YJ, Huh SH, Oh SY, Kyba M, Kataoka H, Choi K, et al. OVOL2 is a critical regulator of ER71/ETV2 in generating FLK1+, hematopoietic, and endothelial cells from embryonic stem cells. Blood. 2014;124:2948–2952. doi: 10.1182/blood-2014-03-556332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayashi M, Pluchinotta M, Momiyama A, Tanaka Y, Nishikawa S, Kataoka H. Endothelialization and altered hematopoiesis by persistent Etv2 expression in mice. Exp Hematol. 2012;40:738–750. doi: 10.1016/j.exphem.2012.05.012. e711. [DOI] [PubMed] [Google Scholar]
- Morita R, Suzuki M, Kasahara H, Shimizu N, Shichita T, Sekiya T, Kimura A, Sasaki K, Yasukawa H, Yoshimura A, et al. ETS transcription factor ETV2 directly converts human fibroblasts into functional endothelial cells. Proc Natl Acad Sci USA. 2015;112:160–165. doi: 10.1073/pnas.1413234112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ginsberg M, James D, Ding BS, Nolan D, Geng F, Butler JM, Schachterle W, Pulijaal VR, Mathew S, Chasen ST, et al. Efficient direct reprogramming of mature amniotic cells into endothelial cells by ETS factors and TGFbeta suppression. Cell. 2012;151:559–575. doi: 10.1016/j.cell.2012.09.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Motoike T, Markham DW, Rossant J, Sato TN. Evidence for novel fate of Flk1+ progenitor: contribution to muscle lineage. Genesis. 2003;35:153–159. doi: 10.1002/gene.10175. [DOI] [PubMed] [Google Scholar]
- Lee D, Kim T, Lim DS. The Er71 is an important regulator of hematopoietic stem cells in adult mice. Stem Cells. 2011;29:539–548. doi: 10.1002/stem.597. [DOI] [PubMed] [Google Scholar]
- de Felipe P. Polycistronic viral vectors. Curr Gene Ther. 2002;2:355–378. doi: 10.2174/1566523023347742. [DOI] [PubMed] [Google Scholar]
- Li H, Durbin R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics. 2009;25:1754–1760. doi: 10.1093/bioinformatics/btp324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li D, Zhang B, Xing X, Wang T. Combining MeDIP-seq and MRE-seq to investigate genome-wide CpG methylation. Methods. 2015;72:29–40. doi: 10.1016/j.ymeth.2014.10.032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, Glass CK. Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol Cell. 2010;38:576–589. doi: 10.1016/j.molcel.2010.05.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lockstone HE. Exon array data analysis using Affymetrix power tools and R statistical software. Brief Bioinform. 2011;12:634–644. doi: 10.1093/bib/bbq086. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Table S3
Supplementary Table S4
Supplementary Table S5
Supplementary Table S6
Supplementary Figure and Table Legends
Review Process File