

Abstract
Endoplasmic reticulum (ER) stress induces the unfolded protein response (UPR), an essential adaptive intracellular pathway that relieves the stress. Although the UPR is an evolutionarily conserved and beneficial pathway, its chronic activation contributes to the pathogenesis of a wide variety of human disorders. The fidelity of UPR activation must thus be tightly regulated to prevent inappropriate signaling. The nonsense-mediated RNA decay (NMD) pathway has long been known to function in RNA quality control, rapidly degrading aberrant mRNAs, and has been suggested to regulate subsets of normal mRNAs. Here, we report that the NMD pathway regulates the UPR. NMD increases the threshold for triggering the UPR in vitro and in vivo, thereby preventing UPR activation in response to normally innocuous levels of ER stress. NMD also promotes the timely termination of the UPR. We demonstrate that NMD directly targets the mRNAs encoding several UPR components, including the highly conserved UPR sensor, IRE1α, whose NMD-dependent degradation partly underpins this process. Our work not only sheds light on UPR regulation, but demonstrates the physiological relevance of NMD's ability to regulate normal mRNAs.
Keywords: cancer, ER stress, IRE1, NMD, UPR
See also: A Carreras-Sureda & C Hetz (May 2015)
Introduction
Cells have evolved elaborate mechanisms to ensure the accuracy with which secreted and membrane proteins are folded and assembled 1. In mammals, three ER transmembrane sensors—inositol requiring enzyme 1 (IRE1), eukaryotic translation initiation factor 2 alpha kinase (PERK, also known as PEK), and activating transcription factor 6 (ATF6)—serve to monitor ER lumen protein folding needs and, if necessary, initiate a set of intracellular signaling pathways, collectively termed the ‘unfolded protein response’ (UPR) 2. These signaling pathways activate transcriptional and translational mechanisms that reduce global protein synthesis, increase ER protein-folding capacity, and promote the degradation of misfolded proteins 3. If ER homeostasis is not achieved by these mechanisms in a timely manner, UPR triggers programmed cell death 3, 4, 5, 6, 7. Because of the important role of UPR in regulating cell life/death decisions, it is critical that mechanisms are in place to prevent inappropriate UPR activation in response to innocuous or low-level stimuli. Little is known about how this is achieved. In this report, we demonstrate that an RNA regulatory mechanism—nonsense-mediated RNA decay (NMD)—serves in this capacity by raising the activation threshold of the UPR and promoting its timely attenuation.
NMD has two broad functions. It was originally identified as an RNA quality control pathway that degrades aberrant transcripts with premature translation termination codons generated as a result of mutations that cause disease 8, 9, 10. Subsequently, it was discovered that NMD also degrades a subset of normal transcripts 11. Approximately 3–10% of mRNAs are directly or indirectly regulated by NMD in eukaryotes spanning the phylogenetic scale 11. While the ‘rules’ dictating whether a normal mRNA is targeted for decay by NMD have not been entirely elucidated, in general, RNA decay is triggered by stop codons that are in a premature context. For example, stop codons in middle exons typically trigger NMD by a mechanism dependent on a set of proteins collectively called the exon-junction complex (EJC) that is recruited just upstream of nearly all exon–exon junctions after RNA splicing 12. Upstream ORFs (uORFs) can also trigger NMD, presumably as a result of translation termination at the 3′ end of the uORF 13. Another NMD-inducing feature in mammalian mRNAs is a long 3′ untranslated region (UTR) 14, 15. The mechanism of how NMD targets mRNAs with long 3′ UTRs to decay is only partially understood; it may involve the capacity to load the NMD factor, UPF1, on the 3′ UTR and/or the physical distance between the stop codon and the poly (A) tail 14, 15, 16, 17, 18.
While it is clear that NMD targets a subset of normal transcripts for decay, the physiological significance of this regulation is not known. This is a challenging problem, as the biological responses influenced by NMD are only just beginning to be understood. Furthermore, hundreds of mRNAs are regulated by NMD in all cell contexts that have been examined, making it difficult to assign functions to specific mRNAs 17, 19, 20, 21, 22, 23, 24. Indeed, it is not known whether a given biological function of NMD depends on the regulation of a single mRNA or whole sets of mRNAs.
In this communication, we demonstrate that NMD has a critical role in shaping the UPR. It prevents inappropriate activation of the UPR and promotes timely termination of the UPR to protect cells from prolonged ER stress. We identify several mRNAs encoding UPR components that are targeted for decay by NMD and demonstrate that the mRNA encoding the most conserved UPR sensor, IRE1α 25, is a direct NMD target and has a role in NMD's ability to shape the UPR.
Results and Discussion
NMD targets transcripts encoding UPR components
Microarray analysis has shown that many mRNAs encoding proteins involved in stress–response pathways are upregulated in cell lines and mice depleted of NMD factors 19, 23, 26. However, it has not been clear whether this is because NMD is important for regulating such mRNAs or because NMD factor depletion merely causes stress, which secondarily induces these transcripts. To assess the latter possibility that NMD perturbation triggers ER stress, we examined whether any of the three branches of the UPR were activated when NMD factors were depleted. We assessed this by measuring the following: (i) XBP1 mRNA splicing, which is catalyzed by the UPR sensor, IRE1α, and thus is specifically induced by the IRE1 branch of the UPR 27, (ii) BIP transcriptional activation, which is primarily induced by the ATF6 branch of the UPR, but also by the IRE1 branch 28, 29, and (iii) CHOP transcriptional activation, which is triggered by the PERK branch of the UPR 30. As a positive control, we used tunicamycin (Tm), a potent ER stress inducer that inhibits N-linked glycosylation of nascent polypeptide chains 6. We found that while spliced XBP1 (XBP1s) was strongly induced by Tm, it was not detectably induced in response to depletion of the NMD factor, UPF3B, which is essential for a branch of the NMD pathway 19 (Supplementary Fig S1A). We also did not detect increased levels of XBP1s mRNA in response to depletion of both UPF3B and the central NMD factor UPF1 (Supplementary Fig S1B). In addition, neither BIP nor CHOP mRNA were statistically significant upregulated in NMD-deficient HeLa cells depleted of NMD factors (Supplementary Fig S1C). Together, these data suggest that depletion of NMD factors does not detectably induce any of the three branches of the UPR and thus perturbation of NMD elicits little or no ER stress, at least in the cells we tested.
Given that UPR component mRNAs are not upregulated by NMD perturbation because of ER stress, this raised the possibility that, instead, they are direct NMD target transcripts that are normally degraded by NMD. To test this possibility, we used quantitative polymerase chain reaction (qPCR) analysis to examine the effect of NMD factor depletion on 13 transcripts encoding UPR-related components, most of which have known NMD-inducing features (Supplementary Table S1). We found that eight of the 13 were significantly upregulated by in response to depletion of UPF1 (Fig1A). Six of these 8 mRNAs—ATF3, ATF4, FSD1L, IRE1α, TNRC1, and TRAF2 mRNA—were also upregulated in response to depletion of both UPF3A and UPF3B (Fig1A), which we simultaneously depleted because of the evidence that these two NMD factors can act redundantly 19, 31, 32. Three of these six mRNAs were also upregulated in response to UPF3B depletion alone (Supplementary Fig S1D). Two of these mRNAs—ATF3 and ATF4—were previously suggested to be NMD targets, based on other lines of evidence 33, 34. The two mRNAs significantly upregulated by UPF1 depletion, but not in response to UPF3A/UPF3B depletion—HERP and PERK (Fig1A)—are candidates to be targeted by the UPF3B-independent branch of NMD 19, 35, 36. We also identified transcripts with the converse expression pattern; that is, significant upregulation in response to UPF3A/UPF3B depletion but not UPF1 depletion—ATF6, BAX, and PDRG1 mRNA in Fig1A. This was unexpected given that UPF1 is regarded as a central NMD factor required for all branches of the NMD pathway 36; however, this has not been rigorously tested. These mRNAs may be either NMD target transcripts or regulated by an UPF3A/UPF3B-dependent mechanism not involving NMD. Another unexpected finding was that some mRNAs were downregulated, rather than upregulated, by UPF3A and/or UPF3B depletion (Fig1A and Supplementary Fig S1D). These effects were usually modest. We do not know the underlying basis for the differential responsiveness of UPR transcripts to NMD factor depletion. Heterogeneous responses of NMD substrate mRNAs as a result of depletion of different NMD factors is a common occurrence 19, 22, 35, 36, 37, 38 and is a subject of ongoing investigations in numerous laboratories.
Figure 1. NMD targets several mRNAs encoding UPR components.

- qPCR analysis of HeLa cells stably depleted of UPF3A and UPF3B using shRNAs (as previously described 19) or transiently depleted of UPF1 using a siRNA. A value of 1 (dotted line) indicates expression in control HeLa cells transiently transfected with a luciferase siRNA or stably transfected with a shRNA luciferase construct (shLUC). mRNA levels were normalized against the mRNA encoding ribosomal protein L19, which is not affected by the UPR 6 (n = 3).
- Schematic representation of the Tet-off reporter, pTET2-βGlob-mini, containing the indicated sequences in the 3′ UTR region; β-292 is a NMD-resistant short 3′ UTR 15.
- Pulse-chase mRNA half-life analysis of the indicated Tet-off reporter mRNAs. The reporter constructs were transiently transfected in HeLa Tet-off cells depleted of UPF1 (siUPF1) or luciferase (siLUC; control). Cells were treated with tetracycline to block transcription of the reporter promoter and samples were collected at the indicated time points. Values were normalized as in (A) (n = 6).
- Pulse-chase mRNA half-life analysis of a Tet-off reporter mRNA harboring the full-length IRE1α 3′ UTR (β-IRE1α 3′UTR FL) versus the Tet-off reporter harboring the downstream and upstream deletions of the IRE1α 3′ UTR (β-IRE1α Del 1 and Del 2). Values were normalized as in (A) (n = 6).
- Pulse-chase mRNA half-life analysis performed as in (D) in HeLa cells transiently depleted of UPF1 (siUPF1) or luciferase (siLUC; control) by RNAi. Values were normalized as in (A) (n = 3).
Data information: Statistical analysis by t-test (*P < 0.05). All error bars reflect SEM.
Because the hallmark of NMD substrate RNAs is they are destabilized by NMD, we next performed RNA half-life analysis. If mRNAs encoding UPR components are direct NMD substrates, this predicts that perturbation of NMD will stabilize them. We observed that all UPR transcripts upregulated by UPF1 depletion (8 of 8) were also stabilized by UPF1 depletion (Supplementary Fig S1E). This stabilization effect was specific to UPR mRNAs that were upregulated upon NMD factor depletion; it was not exhibited by UPR mRNAs not upregulated by UPF1 depletion (ATF6 or PDRG1; Fig1A) or housekeeping transcripts we tested (GAPDH and RPL13) (Supplementary Fig S1E). Transcripts upregulated in response to UPF3A and UPF3B depletion (Fig1A) were also stabilized by this double depletion (Supplementary Fig S1F). HERP and UFM1 mRNA, which were not upregulated by this treatment (Fig1A), were not stabilized (Supplementary Fig S1F).
Together, these data constitute strong evidence that 10 transcripts encoding UPR components—ATF3, ATF4, ATF6, FSD1L, HERP, IRE1α, PERK, PRDG1, TNRC1, and TRAF2—are NMD substrates. All but HERP have NMD-inducing features conserved in both humans and mice (Supplementary Tables S1 and S2). The field is still in the process of identifying the complete set of contexts that elicit NMD, and thus, further analysis may elucidate the molecular basis for why HERP mRNA appears to be targeted for decay by NMD.
We elected to study the molecular basis for how the mRNA encoding IRE1α is targeted for decay by NMD. IRE1α is the most highly conserved sensor protein of the three UPR branches 2. We noted that IRE1α mRNA has a conserved long 3′ UTR (958 nt and 922 nt in human and mice, respectively; Supplementary Tables S1 and S2), a feature that, as described above, can trigger the decay of an mRNA by NMD 14, 15, 16, 17, 18. To assess whether this is the case for IRE1α mRNA, we inserted its full-length 3′ UTR into a β-globin mini-gene reporter system controlled in a tetracycline (Tet)-regulated manner (β-IRE1α 3′UTR FL in Fig1B). We transiently transfected this reporter construct into HeLa cells and performed pulse-chase analysis and found that NMD factor knockdown stabilized the transcribed reporter mRNA, but had no effect on a control mini-gene reporter mRNA containing a short (292 nt) β-globin 3′ UTR (Fig1C). To test whether the length of the IRE1α 3′ UTR was the determining factor that dictated its destabilization by NMD, we made deletions in the 3′ UTR (β-IRE1α Del 1 and Del 2 in Fig1B) and observed this stabilized the β-IRE1α Tet-reporter mRNA (Fig1D) and rendered it unresponsive to depletion of UPF1 (Fig1E). Together, these data suggest that IRE1α mRNA is a direct NMD target as a result of its long 3′ UTR. While the underlying mechanism by which long 3′ UTRs trigger NMD is not well understood, evidence suggests that this feature decreases the probability of translation release factors interacting with poly(A)-binding protein, thereby increasing their probability of interacting with UPF1, an event that promotes NMD 14, 15, 16. We note that our results do not rule out that the IRE1α 3′ UTR confers sensitivity to NMD for another reason, such as essential cis elements in both its 5′ and 3′ halves.
NMD shapes the unfolded protein response
We reasoned that NMD may promote the decay of IRE1α and other UPR factor mRNAs to prevent activation of the UPR in response to low, non-pathogenic levels of ER stress. To test this, we treated NMD-deficient and control HeLa cells with different concentrations of Tm. We found that NMD-deficient cells had a dramatically lower UPR activation threshold in response to Tm than did control cells (Fig2A). While NMD-deficient cells reached the same maximal response as control cells, they did this with much lower concentration of Tm. Similar results were obtained with another UPR-inducing drug, thapsigargin, which blocks calcium influx in the ER 6 (Supplementary Fig S2A–C). To evaluate whether this was also the case in vivo, we examined UPR activation in a classic UPR-responsive tissue—liver 39—in NMD-deficient Upf3b-null mice 35. Treatment of these NMD-deficient mice with different doses of Tm by intra-peritoneal (IP) injection revealed that they had a significantly lower UPR activation threshold than did control littermate mice. Thus, Upf3b-null mice exhibited significantly elevated UPR responses to the lower doses of Tm (0.1, 0.25, and 0.5 μg/g) (Fig2B and Supplementary Fig S2D). In contrast, Upf3b-null mice responded normally (or almost normally) to the highest dose of Tm (1 μg/g), consistent with NMD raising the threshold for the UPR, but not affecting the potential to maximally respond to ER stress. Together, these data provided both in vitro and in vivo evidence that NMD increases the activation threshold of the UPR.
Figure 2. NMD raises the activation threshold of the UPR and promotes its attenuation.

- qPCR analysis of HeLa cells stably depleted of the NMD factor UPF3B (shUPF3B) and treated with increasing concentrations of tunicamycin (Tm) for 4 h. HeLa cells stably transfected with a construct expressing an shRNA against luciferase (shLUC) serves as a negative control (n = 6).
- qPCR analysis of liver from Upf3b-null (n = 3) and matched control littermate (Wt) mice (n = 3) injected IP with the indicated concentrations of Tm for 12 h.
- qPCR analysis of the stably transfected HeLa cells described in (A) treated with a low dose of Tm (0.25 μg/ml) for the times indicated (n = 6).
- qPCR analysis of the stably transfected HeLa cells described in (A) and incubated with a high dose of Tm (2 μg/ml) for the time points indicated (n = 3).
- qPCR analysis of liver from Upf3b-null (n = 3) and control littermate (Wt) mice (n = 3) injected IP with Tm (1 μg/g) for the time points indicated.
Data information: Values were normalized as in Fig1A and statistically analyzed by t-test (*P < 0.05). All error bars reflect SEM.
To further test whether NMD is responsible for dampening the UPR, we examined the temporal kinetics of the UPR in NMD-deficient cells in response to a low dose of Tm that only minimally triggers ER stress in normal cells. We observed that NMD-deficient cells exhibited more rapid and increased expression of XBP1s mRNA and stronger induction of the ATF6 downstream transcriptional target gene, BIP, relative to control cells (Fig2C). CHOP mRNA was also rapidly and strongly induced in NMD-deficient cells, whereas it was only modestly induced at early time points in control cells (Fig2C). These results suggested that by dampening the UPR, NMD serves to prevent an over-exuberant response to innocuous ER stress.
Prolonged ER stress leads to attenuation of IRE1 signaling as a result of reduced IRE1α-mediated splicing of Xbp1 mRNA 6. The molecular mechanism responsible for this attenuation response is poorly understood. Because NMD destabilizes and downregulates IRE1α mRNA (Fig1 and Supplementary Fig S1D–F), we hypothesized that NMD has a role in this attenuation response. As a first test of this hypothesis, we examined the effect of NMD deficiency on HeLa cells treated with a dose of Tm (2 μg/ml) sufficient to elicit a severe UPR at 12 h that wanes by 18 h in control cells (Fig2D). Like control cells, NMD-deficient cells had elevated levels of XBP1s, BIP, and CHOP mRNA during the plateau phase (12 h), but cells depleted of either UPF3B alone or both UPF3A and UPF3B failed to normally downregulate these UPR mRNAs during the termination phase (Fig2D and Supplementary Fig S2E–G). To determine whether NMD also promotes UPR termination in vivo, we examined NMD-deficient Upf3b-null mice. We found that Upf3b-null mice responded to a high dose of Tm virtually the same as control littermate mice during the peak phase of the UPR (12 h post treatment), but they had an impaired downregulatory response during the termination phase (24 h post treatment). Upf3b-null mice exhibited 1.8-, 1.8-, and 4.6-fold depressed downregulation of Xbp1s, Bip, and Chop mRNA, respectively, relative to littermate control mice (Fig2E; note that Bip and Chop mRNA levels are depicted on a log scale because of their large induction). Together, these in vivo and in vitro data support the notion that NMD promotes the timely termination of the UPR, with the caveat that it is supported by in vivo data from only a single time point.
To further test the role of NMD in the UPR, we assayed activation of the UPR sensors, IRE1α and ATF6. A low dose of Tm was used to simulate innocuous stress. We examined phosphorylated IRE1α protein as a measure of IRE1 branch activation 40. Control cells did not have detectable phosphorylated IRE1α at early time points and only trace levels of phosphorylated IRE1α at later time points (Fig3A, shLUC lanes). In contrast, NMD-deficient cells exhibited rapid and strong induction of IRE1α phosphorylation after 2 h of treatment, which increased in level at later time points (Fig3A, shUPF3B lanes). As another indication of UPR's IRE1 branch activation, NMD-deficient cells exhibited rapid and strong induction of XBP1 splicing (Fig3B), a reaction triggered by activated IRE1α 27. Control cells exhibited only modest (and delayed) XBP1 splicing (Fig3B). The ATF6 branch of the UPR was also strongly induced in NMD-deficient cells, as shown by (i) the increased level of cleaved ATF6 (Fig3C and E), a well-established marker of ATF6 activation 41 and (ii) stronger expression of the ATF6 downstream transcriptional target gene, BIP, than in control cells (Fig3D). CHOP protein was also rapidly and strongly induced in NMD-deficient cells, not control cells (Fig3D). Together, these results indicate that by dampening the UPR, NMD serves to prevent an over-exuberant activation of the response to innocuous ER stress.
Figure 3. NMD suppresses UPR activation in response to endoplasmic reticulum stress.
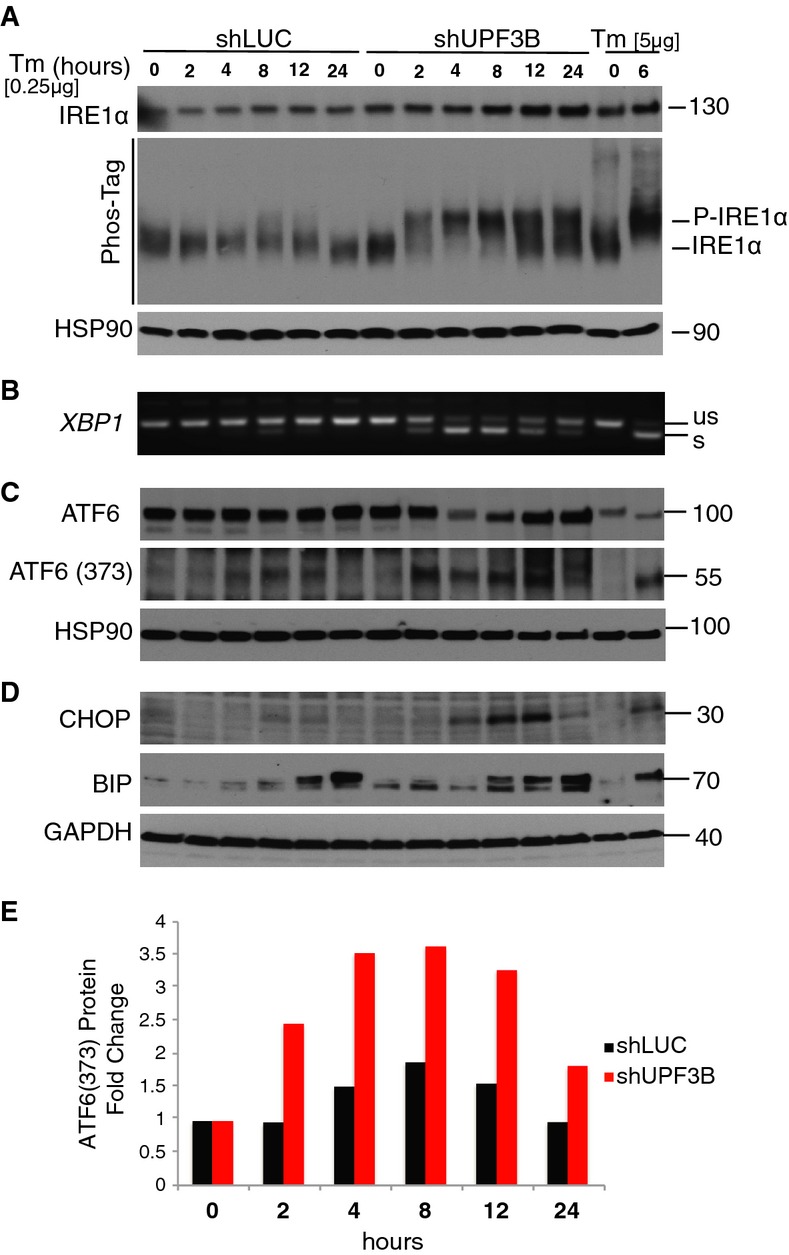
- Western blot analysis of total IRE1α (top) and phospho-IRE1α (bottom) in HeLa cells stably depleted of the NMD factor UPF3B (shUPF3B) treated with a low dose of Tm (0.25 μg/ml) for the times indicated. HeLa cells stably transfected with a luciferase shRNA construct (shLUC) serve as a negative control. Phospho-IRE1α was detected by a mobility shift assay, as described 60. HSP-90 is the loading control, and the positive control is HEK293T cells treated with Tm (5 μg/ml) for 6 h.
- RT–PCR analysis of XBP1 splicing under the same conditions as in (A) (representative of three independent experiments).
- Western blot analysis of total ATF6 (top) and its cleavage product (ATF6-373) in cells under the same conditions as in (A).
- Western blot analysis of CHOP and BIP in cells under the same conditions as in (A).
- Quantification of cleaved ATF6 protein [ATF6 (373)] relative to the loading control, HSP90, from the Western blot data in (C).
Data information: Data in (A, C and D) are representative of two independent experiments.
Our finding that NMD promotes UPR attenuation raised the possibility that NMD protects cells from ER stress-induced apoptosis, a consequence of sustained UPR 6. In support of this, we observed that UPF3B depletion strongly sensitized cells to undergo apoptosis 48 h after Tm treatment, a response that was exacerbated by additional depletion of UPF3A (Fig4A). We also observed a modest increase in apoptosis in cells stably depleted of UPF3B or both UPF3A and UPF3B in the absence of ER stress.
Figure 4. NMD acts through IRE1α to shape the UPR.

- FACS analysis indicating the percentage of apoptotic (annexin-V positive/PI negative) HeLa cells in response to Tm (2 μg/ml) treatment for 48 h. HeLa cells were stably depleted of UPF3B (shUPF3B) or both UPF3A and UPF3B (shUPF3AB). HeLa cells stably transfected with a luciferase shRNA construct (shLUC) serve as a negative control. The values shown are the average (mean ± SEM) from three experiments relative to control (shLUC). The percentage of apoptotic cells in non-Tm-treated shLUC, shUPF3B, and shUPF3AB cells was 1.0, 1.8, and 2.1%, respectively (n = 3).
- Quantification of apoptotic (TUNEL-positive) cells in liver tissue sections. Shown is the average number of TUNEL-positive cells in a field surrounding a hepatic portal area in Upf3b-null (n = 3) and control littermate (Wt) mice (n = 3) injected IP with Tm (1 μg/g) for the times indicated. Scoring was performed without knowledge of genotype.
- qPCR analysis of HeLa cells (described in A) treated with a single low dose of tunicamycin (0.25 μg/ml) for the time points indicated (n = 3).
- qPCR analysis of liver from Upf3b-null (n = 6) and control littermate (Wt) mice (n = 6).
- qPCR analysis of the liver of Upf3b-null (n = 3) and control littermate mice (n = 3) injected IP with Tm (1 μg/g) for the times indicated.
- qPCR analysis of the stably transfected HeLa cells described in (A) and transiently transfected with an IRE1α siRNA (siIRE1) or a control siRNA (siControl) and incubated with Tm (0.25 μg/ml) for 10 h (n = 3).
- qPCR analysis of the stably transfected HeLa cells described in (A) that were incubated with IRE inhibitor STF-083010 (60 μM) and Tm (0.25 μg/ml) for 10 h (n = 3).
- Model depicting the activity of the UPR and NMD pathways during the initiation (Init.), plateau (Plat.), and termination (Term.) phases of the UPR pathway.
- Model depicting the NMD-UPR circuit defined by our data that amplifies the signal-to-noise ratio of endoplasmic reticulum stress responses.
Data information: Statistical analysis by t-test (*P < 0.05). In (C–G), values were normalized as in Fig1A. All error bars reflect SEM.
We next tested whether NMD protects cells from ER stress-induced apoptosis in vivo. We analyzed the apoptosis of liver cells by TUNEL staining 5 and found greatly increased numbers of apoptotic cells in NMD-deficient mice, both 48 and 96 h after Tm treatment as compared to control mice (Fig4B). In contrast, NMD-deficient mice liver cells did not exhibit significantly higher apoptosis than control mice in the absence of Tm treatment, providing evidence that NMD specifically protects cells from death in response to stress.
While our study demonstrated that the UPR is shaped by NMD, Sakaki et al provided evidence for another regulatory relationship between the UPR and NMD 42. In particular, they found that debilitating mutations in the SMG1, 4, and 6 NMD factor genes in C. elegans triggered ER stress and the UPR, leading these investigators to suggest that loss of NMD causes the accumulation of misfolded proteins, which, in turn, activates the UPR. In contrast to these results in worms, we did not observe activation of the UPR when we depleted several different core NMD components in mammalian cells or knocked out the NMD factor UPF3B in vivo (Fig2 and Supplementary Figs S1A–C and S2). While Sakaki et al showed that depletion of the NMD factor, SMG6, elicits UPR activation in HeLa cells, this may be because of loss of SMG6's non-NMD functions, such as telomere maintenance 43. Indeed, Sakaki et al found that knockdown of another NMD factor, SMG1, did not significantly induce the UPR 42. We suggest that the most parsimonious explanation for these seemingly divergent results is that the UPR and NMD pathways impact each other in varied ways, depending on context and species. In mammals, NMD is crucial for shaping the UPR so that it is not unnecessarily triggered by innocuous stimuli or undergoes prolonged activation, thereby avoiding toxicity. In C. elegans, NMD (or another process dependent on SMG6) is essential to avoid intrinsically activating the UPR response, perhaps because of the necessity of NMD's ability to sense genomic noise in worms.
NMD acts, in part, through IRE1α to repress the UPR
We next investigated the underlying mechanism by which NMD suppresses the UPR. This was challenging given that NMD regulates hundreds of mRNAs, any combination of which could contribute to its ability to suppress the UPR. Despite this likely complexity, several findings suggested that one molecule, IRE1α, was a particularly good candidate to be one player that acts downstream of NMD to suppress the UPR. First, our results indicated that IRE1α mRNA is among the UPR component transcripts most strongly upregulated by NMD (Fig1A). Second, NMD-deficient cells not only had increased levels of IRE1α mRNA, but also increased IRE1α protein and IRE1α phosphorylation (Fig3A). Third, we showed that IRE1α mRNA is a direct NMD target (Fig1 and Supplementary Fig S1D–F), making it a more likely candidate to act in an NMD-based circuit. Finally, IRE1α mRNA levels were elevated in NMD-deficient cells in both human cells and mice, regardless of which NMD factor was depleted and whether ER stress was present or not (Figs1A and 4C–E and Supplementary Fig S3A–C), suggesting that the ability of NMD to repress IRE1α expression is conserved and omnipresent in mammals.
To directly test whether NMD acts through IRE1α to repress the UPR, we first performed a gain-of-function experiment. We examined whether forced expression of IRE1α mRNA to a level similar to that in NMD-deficient cells (Supplementary Fig S3A–C) was sufficient to decrease UPR activation threshold. Indeed, we found that this modest overexpression of IRE1α increased the level of the spliced form of its direct substrate (XBP1s mRNA) and decreased the UPR activation threshold, as measured by both BIP and CHOP mRNA induction (Supplementary Fig 3D).
As a complementary approach to examine whether the ability of NMD to degrade IRE1α mRNA has a role in NMD's ability to suppress the UPR, we performed a rescue experiment. We first did titration studies to define a dose of siRNA against IRE1α that prevented the upregulation of IRE1α that normally occurs in response to NMD perturbation. This defined a dose of IRE1α siRNA that was effective in blocking NMD-deficient cells from expressing elevated levels of both IRE1α and XBP1s, a key direct product of IRE1α action (Fig4F). To elucidate whether blocking IRE1α upregulation impacted the UPR, we examined the UPR effectors BIP and CHOP. We found that preventing IRE1α upregulation in NMD-deficient cells significantly suppressed BIP and CHOP induction in response to Tm (Fig4F). This provided direct evidence that one mechanism by which NMD suppresses the UPR is by degrading IRE1α mRNA.
As another approach to address whether NMD acts through IRE1α to repress the UPR, we used a covalent inhibitor of IRE1α, STF-083010, which forms a selective Schiff's base with a catalytic lysine in the RNase active site of IRE1α, thereby blocking its function 44. We identified a dose of STF-083010 that largely prevented the upregulation of XBP1s mRNA that normally occurs in response to ER stress specifically in NMD-deficient cells. This significantly reduced ER stress-induced UPR activation, as judged by both BIP and CHOP expression (Fig4G). Indeed, BIP mRNA levels were similar in STF-083010-treated NMD-deficient cells as in control cells, suggesting that the ability of NMD to degrade IRE1α mRNA is a major mechanism by which it suppresses BIP gene activation. CHOP mRNA levels, while suppressed by STF-083010, did not reach control levels, consistent with the fact that CHOP is induced by PERK while BIP is induced by both the IRE1α and ATF6 branches of the UPR 28, 29, 30. While these results indicate that NMD acts through its ability to degrade IRE1α mRNA to suppress the UPR, we found that NMD does not use this mechanism to suppress apoptosis. Thus, suppressing the upregulation of IRE1α mRNA that normally occurs in response to NMD perturbation did not significantly prevent the increased apoptosis caused by this perturbation (Supplementary Fig S3E).
The finding that IRE1α mRNA destabilization by NMD plays a role in shaping the UPR is important given that the physiological significance of NMD's ability to degrade normal mRNAs has not been clear since genome-wide approaches first revealed that NMD engendered this function 20, 21, 23. Several studies have shown that loss or depletion of NMD factors causes developmental defects in organisms spanning the phylogenetic scale, which has led to the suggestion that NMD regulates normal transcripts to drive or modulate specific development processes and homeostatic mechanisms 13, 45, 46, 47. However, little is known regarding the specific transcripts NMD acts on to control such processes. Here, we showed using both gain- and loss-of-function approaches that a specific normal mRNA—IRE1α mRNA—is degraded by NMD to shape the UPR. This discovery strongly supports a recently advanced model that posits that IRE1α has a critical threshold concentration, which when reached, leads to UPR activation 40.
The UPR suppresses NMD to allow a strong ER stress response
While the ability of NMD to suppress the UPR provides the benefit of preventing UPR activation in response to innocuous ER stress, this attribute would be predicted to prevent an optimal UPR activation in response to bona fide ER stress. To avoid this, an optimal system would be one that suppresses NMD in response to strong ER stress so that IRE1α mRNA and other NMD-targeted UPR component mRNAs can be stabilized, allowing for strong UPR activation (Fig4H and I). One potential means by which ER stress could achieve this is by downregulating NMD factors. Our analysis of 12 NMD factor mRNAs showed that they were either not affected or only modestly affected by Tm treatment of HeLa cells (Supplementary Fig S4A). The NMD factor mRNA exhibiting the strongest decrease in expression (∽60% decrease) was that encoding the core EJC factor eIF4AIII. The mRNAs encoding UPF2, UPF3B, and SMG1 were also modestly downregulated by Tm. While these downregulatory responses could have a role in downregulating NMD, it should be noted that this would only be the case if the degree of downregulation of a given NMD factor is sufficient to reach a rate-limiting level of that factor. Our previous overexpression analysis of eight NMD factors showed that the endogenous level of only SMG1 was rate limiting for NMD in HeLa cells 35.
A mechanism that has been reported to suppress NMD in response to ER stress is eukaryotic initiation factor 2-α (eIF2α) phosphorylation. A wide variety of cellular stresses, not only ER stress, but also amino acid deprivation, oxygen deprivation, infection, reactive oxygen species, and double-stranded RNA, all are known to trigger the phosphorylation of eIF2α 33, 48, 49, 50. We confirmed that ER stress inhibits NMD activity in human and mice cells (Supplementary Fig S4B). We also found that NMD repression is abrogated by forced expression of the phosphatase GADD34 (Supplementary Fig S4B), which is known to dephosphorylate eIF2α 30, 51, or by mutation of the eIF2α phosphorylation site (Supplementary Fig S4C). While the mechanism by which eIF2α phosphorylation inhibits NMD is not known, an obvious possibility is that it acts by inhibiting translation 52. This follows from the fact that eIF2α phosphorylation is a potent inhibitor of translation 53 and NMD absolutely depends on translation 54. However, some evidence suggests that eIF2α phosphorylation inhibits NMD independently of translation inhibition 48, 50, and thus, the underlying mechanism remains to be determined.
Conclusions and future directions
Here, we report that the highly conserved RNA degradation pathway, NMD, is crucial for avoiding inappropriate and prolonged inactivation of the UPR. We identify several mRNAs encoding UPR components that are targeted for decay by NMD and demonstrate that the mRNA encoding the most conserved sensor of the UPR, IRE1α, plays a crucial role in the ability of NMD to regulate the UPR. We suggest that the NMD-UPR circuit we have uncovered is likely to impact many biological processes. For example, the finding that NMD is downregulated during brain and neural development 22, 37 raises the possibility that this allows the UPR to be activated under low (physiologic) levels of ER stress that occur during neuron differentiation to accommodate fluctuations in the demand for protein synthesis and secretion 2. The ability of NMD to suppress the UPR also has potential biomedical impact, as chronic ER stress occurs in many diseases, including cancer, diabetes, pro-inflammatory disorders, and diseases associated with neural degeneration 55.
Given that we showed that NMD promotes cell survival under UPR-inducing conditions, interventions that modulate the magnitude of NMD could reduce the severity of such diseases by increasing cell survival. Further therapeutic options stem from the fact that NMD is a branched pathway 19, 31, 38. Thus, drugs that target a specific NMD branch have the potential to greatly increase specificity and decrease side effects. In this regard, we demonstrated herein that the UPF3B-dependent branch of NMD is specifically involved in regulating the UPR since we found that loss or depletion of UPF3B was sufficient to disrupt the normal activation threshold, magnitude, and attenuation of the UPR both in vitro and in vivo. This discovery, coupled with the fact that mutations in the UPF3B gene cause intellectual disability in humans and are associated with autism and schizophrenia 56, 57, raises the possibility that the cognitive and psychiatric pathologies that occur in these UPF3B patients result from aberrant UPR activation. Because we showed that NMD greatly raises the threshold for triggering the UPR in response to ER stress-inducing agents (Fig2 and Supplementary Fig S2), these NMD-deficient individuals may suffer from far worse symptoms if they encounter ER stress. Indeed, we showed that NMD-deficient (Upf3b-null) mice had elevated liver apoptosis in response to external ER stress (Fig4A and B). We note that loss of UPF3B only ablates a branch of the NMD pathway 19 and thus complete loss of NMD is likely to cause more severe UPR-related defects. In summary, our discovery that NMD serves as a post-transcriptional pathway that shapes the UPR provides a foundation for developing strategies for treating the many diseases characterized by chronic ER stress.
Materials and Methods
Animals and cell culture
Cell culture and transfections were performed as previously described 31. Unless otherwise noted, cells were harvested 2 days after transfection. The siRNAs targeting UPF1 and Luciferase were obtained from Ambion (Carlsbad, CA, USA). The siRNAs targeting IRE1 and a scramble sequence were obtained from Thermo Scientific Dharmacon (Lafayette, CO, USA). Luminometry was performed as previously described 58. Apoptosis was determined by FACS analysis of cells stained with annexin-V FITC/propidium iodide as previously described 5. Tunicamycin and thapsigargin were obtained from Sigma (Saint Louis, MO, USA). All experiments with mice were performed in accordance with National Institutes of Health guidelines for care and use of animals. The Upf3b-null mice 35 and control littermate mice were given a single intraperitoneal injection of tunicamycin in 150 mM dextrose, as previously described 5, 39. At various times thereafter, the mice were killed by CO2 narcosis, followed by cervical dislocation. Livers were removed and RNA was isolated for analysis, as described below. To detect liver apoptosis, the sections were stained with the DeadEnd fluorometric TUNEL system (Promega, Madison, WI, USA), following the recommendations of the manufacturer.
RNA analysis
Total cellular RNA was extracted from cells using TRIzol Reagent (Sigma), and cDNA was generated using the iScript cDNA synthesis kit (Bio-Rad, Hercules, CA, USA). [γ32P]-end-labeled RT–PCR analysis was done as previously described 59. qPCR analysis was performed using the relative quantification method (ΔΔCT) in a ABI Step-One thermal cycler system (ABI). Endogenous mRNA half-life analysis and pulse-chase mRNA half-life analysis of the Tet-off reporter was done as previously described 35.
Protein analysis
Western blot analyses, including cell and tissue preparation, were done as previously described 6. The following antibodies and dilutions were used for immunoblotting: anti-ATF6 at 1:1,000 (#ab122897; Abcam, Cambridge, MA, USA); anti-GRP78/BiP at 1:1,000 (#GTX113340; GeneTex, Irvine, CA, USA); anti-CHOP at 1:1,000 [#GTX112827 (N1C3); GeneTex]; anti-GAPDH at 1:5,000 [#sc-25778 (FL-335); Santa Cruz Biotechnologies, Dallas, TX, USA]; and anti-HSP90 at 1:5,000 (#GTX101448; GeneTex). In addition to conventional gels, Phos-tag gels were run that differed in that: (i) the 5% SDS–PAGE gel contained 25 μM Phos-tag and 50 μM MnCl2 (Sigma); (ii) the gel was run at 100 V for 3 h, soaked in 1 mM EDTA for 10 min, and then transferred onto a PVDF membrane. Phos-tag™ Acrylamide AAL-107 was purchased from Wako (NARD Institute).
Oligonucleotides sequences
The primers used in this experiment are provided in Supplementary Table S3.
Acknowledgments
We are grateful to Dr. Jens Lykke-Andersen (UCSD) for kindly providing the tetracycline-regulated β-globin mini-gene plasmids, as well as polyclonal antibodies against human UPF1 and UPF3B. We thank Maho Niwa (UCSD) for providing the IRE1 inhibitor STF-083010 and Andreas Kulozik (University of Heidelberg) for the NMD Luciferase reporter.
Author contributions
RK and MFW conceived, designed, and interpreted the main body of experiments. RK performed the bulk of the experiments. C-HL and HK performed additional experiments and greatly contributed to figure generation and data interpretation. LH generated the Upf3b-null mice and performed experiments in early project development. The manuscript was primarily written by RK and MFW, with contributions from others, including C-HL and JHL.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figures and Tables
Review Process File
References
- Buchberger A, Bukau B, Sommer T. Protein quality control in the cytosol and the endoplasmic reticulum: brothers in arms. Mol Cell. 2010;40:238–252. doi: 10.1016/j.molcel.2010.10.001. [DOI] [PubMed] [Google Scholar]
- Moore KA, Hollien J. The unfolded protein response in secretory cell function. Annu Rev Genet. 2012;46:165–183. doi: 10.1146/annurev-genet-110711-155644. [DOI] [PubMed] [Google Scholar]
- Walter P, Ron D. The unfolded protein response: from stress pathway to homeostatic regulation. Science. 2011;334:1081–1086. doi: 10.1126/science.1209038. [DOI] [PubMed] [Google Scholar]
- Breckenridge DG, Germain M, Mathai JP, Nguyen M, Shore GC. Regulation of apoptosis by endoplasmic reticulum pathways. Oncogene. 2003;22:8608–8618. doi: 10.1038/sj.onc.1207108. [DOI] [PubMed] [Google Scholar]
- Hetz C, Bernasconi P, Fisher J, Lee AH, Bassik MC, Antonsson B, Brandt GS, Iwakoshi NN, Schinzel A, Glimcher LH, et al. Proapoptotic BAX and BAK modulate the unfolded protein response by a direct interaction with IRE1alpha. Science. 2006;312:572–576. doi: 10.1126/science.1123480. [DOI] [PubMed] [Google Scholar]
- Lin JH, Li H, Yasumura D, Cohen HR, Zhang C, Panning B, Shokat KM, Lavail MM, Walter P. IRE1 signaling affects cell fate during the unfolded protein response. Science. 2007;318:944–949. doi: 10.1126/science.1146361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lisbona F, Rojas-Rivera D, Thielen P, Zamorano S, Todd D, Martinon F, Glavic A, Kress C, Lin JH, Walter P, et al. BAX inhibitor-1 is a negative regulator of the ER stress sensor IRE1alpha. Mol Cell. 2009;33:679–691. doi: 10.1016/j.molcel.2009.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bhuvanagiri M, Schlitter AM, Hentze MW, Kulozik AE. NMD: RNA biology meets human genetic medicine. Biochem J. 2010;430:365–377. doi: 10.1042/BJ20100699. [DOI] [PubMed] [Google Scholar]
- Chang YF, Imam JS, Wilkinson MF. The nonsense-mediated decay RNA surveillance pathway. Annu Rev Biochem. 2007;76:51–74. doi: 10.1146/annurev.biochem.76.050106.093909. [DOI] [PubMed] [Google Scholar]
- Holbrook JA, Neu-Yilik G, Hentze MW, Kulozik AE. Nonsense-mediated decay approaches the clinic. Nat Genet. 2004;36:801–808. doi: 10.1038/ng1403. [DOI] [PubMed] [Google Scholar]
- Rehwinkel J, Raes J, Izaurralde E. Nonsense-mediated mRNA decay: target genes and functional diversification of effectors. Trends Biochem Sci. 2006;31:639–646. doi: 10.1016/j.tibs.2006.09.005. [DOI] [PubMed] [Google Scholar]
- Le Hir H, Izaurralde E, Maquat LE, Moore MJ. The spliceosome deposits multiple proteins 20-24 nucleotides upstream of mRNA exon-exon junctions. EMBO J. 2000;19:6860–6869. doi: 10.1093/emboj/19.24.6860. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Wilkinson MF. Regulation of nonsense-mediated mRNA decay. Wiley Interdiscip Rev RNA. 2012;3:807–828. doi: 10.1002/wrna.1137. [DOI] [PubMed] [Google Scholar]
- Eberle AB, Stalder L, Mathys H, Orozco RZ, Muhlemann O. Posttranscriptional gene regulation by spatial rearrangement of the 3′ untranslated region. PLoS Biol. 2008;6:e92. doi: 10.1371/journal.pbio.0060092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh G, Rebbapragada I, Lykke-Andersen J. A competition between stimulators and antagonists of Upf complex recruitment governs human nonsense-mediated mRNA decay. PLoS Biol. 2008;6:e111. doi: 10.1371/journal.pbio.0060111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hogg JR, Goff SP. Upf1 senses 3′UTR length to potentiate mRNA decay. Cell. 2010;143:379–389. doi: 10.1016/j.cell.2010.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hurt JA, Robertson AD, Burge CB. Global analyses of UPF1 binding and function reveal expanded scope of nonsense-mediated mRNA decay. Genome Res. 2013;23:1636–1650. doi: 10.1101/gr.157354.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zünd D, Gruber AR, Zavolan M, Muhlemann O. Translation-dependent displacement of UPF1 from coding sequences causes its enrichment in 3′ UTRs. Nat Struct Mol Biol. 2013;20:936–943. doi: 10.1038/nsmb.2635. [DOI] [PubMed] [Google Scholar]
- Chan WK, Huang L, Gudikote JP, Chang YF, Imam JS, MacLean JA, 2nd, Wilkinson MF. An alternative branch of the nonsense-mediated decay pathway. EMBO J. 2007;26:1820–1830. doi: 10.1038/sj.emboj.7601628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He F, Li X, Spatrick P, Casillo R, Dong S, Jacobson A. Genome-wide analysis of mRNAs regulated by the nonsense-mediated and 5′ to 3′ mRNA decay pathways in yeast. Mol Cell. 2003;12:1439–1452. doi: 10.1016/s1097-2765(03)00446-5. [DOI] [PubMed] [Google Scholar]
- Lelivelt MJ, Culbertson MR. Yeast Upf proteins required for RNA surveillance affect global expression of the yeast transcriptome. Mol Cell Biol. 1999;19:6710–6719. doi: 10.1128/mcb.19.10.6710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lou CH, Shao A, Shum EY, Espinoza JL, Huang L, Karam R, Wilkinson MF. Posttranscriptional control of the stem cell and neurogenic programs by the nonsense-mediated RNA decay pathway. Cell Rep. 2014;6:748–764. doi: 10.1016/j.celrep.2014.01.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mendell JT, Sharifi NA, Meyers JL, Martinez-Murillo F, Dietz HC. Nonsense surveillance regulates expression of diverse classes of mammalian transcripts and mutes genomic noise. Nat Genet. 2004;36:1073–1078. doi: 10.1038/ng1429. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, Waage J, Tian G, Zhao J, Damgaard I, Jakobsen JS, Kristiansen K, Krogh A, Wang J, Porse BT. Mammalian tissues defective in nonsense-mediated mRNA decay display highly aberrant splicing patterns. Genome Biol. 2012;13:R35. doi: 10.1186/gb-2012-13-5-r35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron D, Walter P. Signal integration in the endoplasmic reticulum unfolded protein response. Nat Rev Mol Cell Biol. 2007;8:519–529. doi: 10.1038/nrm2199. [DOI] [PubMed] [Google Scholar]
- Weischenfeldt J, Damgaard I, Bryder D, Theilgaard-Monch K, Thoren LA, Nielsen FC, Jacobsen SE, Nerlov C, Porse BT. NMD is essential for hematopoietic stem and progenitor cells and for eliminating by-products of programmed DNA rearrangements. Genes Dev. 2008;22:1381–1396. doi: 10.1101/gad.468808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yoshida H, Matsui T, Yamamoto A, Okada T, Mori K. XBP1 mRNA is induced by ATF6 and spliced by IRE1 in response to ER stress to produce a highly active transcription factor. Cell. 2001;107:881–891. doi: 10.1016/s0092-8674(01)00611-0. [DOI] [PubMed] [Google Scholar]
- Lee AH, Iwakoshi NN, Glimcher LH. XBP-1 regulates a subset of endoplasmic reticulum resident chaperone genes in the unfolded protein response. Mol Cell Biol. 2003;23:7448–7459. doi: 10.1128/MCB.23.21.7448-7459.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shoulders MD, Ryno LM, Genereux JC, Moresco JJ, Tu PG, Wu C, Yates JR, 3rd, Su AI, Kelly JW, Wiseman RL. Stress-independent activation of XBP1s and/or ATF6 reveals three functionally diverse ER proteostasis environments. Cell Rep. 2013;3:1279–1292. doi: 10.1016/j.celrep.2013.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreno JA, Radford H, Peretti D, Steinert JR, Verity N, Martin MG, Halliday M, Morgan J, Dinsdale D, Ortori CA, et al. Sustained translational repression by eIF2alpha-P mediates prion neurodegeneration. Nature. 2012;485:507–511. doi: 10.1038/nature11058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan WK, Bhalla AD, Le Hir H, Nguyen LS, Huang L, Gecz J, Wilkinson MF. A UPF3-mediated regulatory switch that maintains RNA surveillance. Nat Struct Mol Biol. 2009;16:747–753. doi: 10.1038/nsmb.1612. [DOI] [PubMed] [Google Scholar]
- Nguyen LS, Jolly L, Shoubridge C, Chan WK, Huang L, Laumonnier F, Raynaud M, Hackett A, Field M, Rodriguez J, et al. Transcriptome profiling of UPF3B/NMD-deficient lymphoblastoid cells from patients with various forms of intellectual disability. Mol Psychiatry. 2012;17:1103–1115. doi: 10.1038/mp.2011.163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner LB. Hypoxic inhibition of nonsense-mediated RNA decay regulates gene expression and the integrated stress response. Mol Cell Biol. 2008;28:3729–3741. doi: 10.1128/MCB.02284-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oren YS, McClure ML, Rowe SM, Sorscher EJ, Bester AC, Manor M, Kerem E, Rivlin J, Zahdeh F, Mann M, et al. The unfolded protein response affects readthrough of premature termination codons. EMBO Mol Med. 2014;6:685–701. doi: 10.1002/emmm.201303347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang L, Lou CH, Chan W, Shum EY, Shao A, Stone E, Karam R, Song HW, Wilkinson MF. RNA homeostasis governed by cell type-specific and branched feedback loops acting on NMD. Mol Cell. 2011;43:950–961. doi: 10.1016/j.molcel.2011.06.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Metze S, Herzog VA, Ruepp MD, Muhlemann O. Comparison of EJC-enhanced and EJC-independent NMD in human cells reveals two partially redundant degradation pathways. RNA. 2013;19:1432–1448. doi: 10.1261/rna.038893.113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bruno IG, Karam R, Huang L, Bhardwaj A, Lou CH, Shum EY, Song HW, Corbett MA, Gifford WD, Gecz J, et al. Identification of a microRNA that activates gene expression by repressing nonsense-mediated RNA decay. Mol Cell. 2011;42:500–510. doi: 10.1016/j.molcel.2011.04.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gehring NH, Kunz JB, Neu-Yilik G, Breit S, Viegas MH, Hentze MW, Kulozik AE. Exon-junction complex components specify distinct routes of nonsense-mediated mRNA decay with differential cofactor requirements. Mol Cell. 2005;20:65–75. doi: 10.1016/j.molcel.2005.08.012. [DOI] [PubMed] [Google Scholar]
- Zhang K, Kaufman RJ. Identification and characterization of endoplasmic reticulum stress-induced apoptosis in vivo. Methods Enzymol. 2008;442:395–419. doi: 10.1016/S0076-6879(08)01420-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner BM, Walter P. Unfolded proteins are Ire1-activating ligands that directly induce the unfolded protein response. Science. 2011;333:1891–1894. doi: 10.1126/science.1209126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye J, et al. ER stress induces cleavage of membrane-bound ATF6 by the same proteases that process SREBPs. Mol Cell. 2000;6:1355–1364. doi: 10.1016/s1097-2765(00)00133-7. [DOI] [PubMed] [Google Scholar]
- Sakaki K, Yoshina S, Shen X, Han J, DeSantis MR, Xiong M, Mitani S, Kaufman RJ. RNA surveillance is required for endoplasmic reticulum homeostasis. Proc Natl Acad Sci USA. 2012;109:8079–8084. doi: 10.1073/pnas.1110589109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lai X, Beilharz T, Au WC, Hammet A, Preiss T, Basrai MA, Heierhorst J. Yeast hEST1A/B (SMG5/6)-like proteins contribute to environment-sensing adaptive gene expression responses. G3 (Bethesda) 2013;3:1649–1659. doi: 10.1534/g3.113.006924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Papandreou I, Denko NC, Olson M, Van Melckebeke H, Lust S, Tam A, Solow-Cordero DE, Bouley DM, Offner F, Niwa M, et al. Identification of an Ire1alpha endonuclease specific inhibitor with cytotoxic activity against human multiple myeloma. Blood. 2011;117:1311–1314. doi: 10.1182/blood-2010-08-303099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hwang J, Maquat LE. Nonsense-mediated mRNA decay (NMD) in animal embryogenesis: to die or not to die, that is the question. Curr Opin Genet Dev. 2011;21:422–430. doi: 10.1016/j.gde.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, Wilkinson M. A conserved microRNA/NMD regulatory circuit controls gene expression. RNA Biol. 2012;9:22–26. doi: 10.4161/rna.9.1.18010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vicente-Crespo M, Palacios IM. Nonsense-mediated mRNA decay and development: shoot the messenger to survive? Biochem Soc Trans. 2010;38:1500–1505. doi: 10.1042/BST0381500. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karam R, Wengrod J, Gardner LB, Wilkinson MF. Regulation of nonsense-mediated mRNA decay: implications for physiology and disease. Biochim Biophys Acta. 2013;1829:624–633. doi: 10.1016/j.bbagrm.2013.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Wengrod J, Gardner LB. Over-expression of the c-myc oncogene inhibits nonsense mediated RNA decay in B-lymphocytes. J Biol Chem. 2011;286:40038–40043. doi: 10.1074/jbc.M111.266361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang D, Zavadil J, Martin L, Parisi F, Friedman E, Levy D, Harding H, Ron D, Gardner LB. Inhibition of nonsense-mediated RNA decay by the tumor microenvironment promotes tumorigenesis. Mol Cell Biol. 2011;31:3670–3680. doi: 10.1128/MCB.05704-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Novoa I, Zeng H, Harding HP, Ron D. Feedback inhibition of the unfolded protein response by GADD34-mediated dephosphorylation of eIF2alpha. J Cell Biol. 2001;153:1011–1022. doi: 10.1083/jcb.153.5.1011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Q, Ye J, Cao J. Translational regulator eIF2alpha in tumor. Tumour Biol. 2014;35:6255–6264. doi: 10.1007/s13277-014-1789-0. [DOI] [PubMed] [Google Scholar]
- Joshi M, Kulkarni A, Pal JK. Small molecule modulators of eukaryotic initiation factor 2alpha kinases, the key regulators of protein synthesis. Biochimie. 2013;95:1980–1990. doi: 10.1016/j.biochi.2013.07.030. [DOI] [PubMed] [Google Scholar]
- Carter MS, Doskow J, Morris P, Li S, Nhim RP, Sandstedt S, Wilkinson MF. A regulatory mechanism that detects premature nonsense codons in T-cell receptor transcripts in vivo is reversed by protein synthesis inhibitors in vitro. J Biol Chem. 1995;270:28995–29003. doi: 10.1074/jbc.270.48.28995. [DOI] [PubMed] [Google Scholar]
- Wang S, Kaufman RJ. The impact of the unfolded protein response on human disease. J Cell Biol. 2012;197:857–867. doi: 10.1083/jcb.201110131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addington AM, Gauthier J, Piton A, Hamdan FF, Raymond A, Gogtay N, Miller R, Tossell J, Bakalar J, Inoff-Germain G, et al. A novel frameshift mutation in UPF3B identified in brothers affected with childhood onset schizophrenia and autism spectrum disorders. Mol Psychiatry. 2011;16:238–239. doi: 10.1038/mp.2010.59. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tarpey PS, Raymond FL, Nguyen LS, Rodriguez J, Hackett A, Vandeleur L, Smith R, Shoubridge C, Edkins S, Stevens C, et al. Mutations in UPF3B, a member of the nonsense-mediated mRNA decay complex, cause syndromic and nonsyndromic mental retardation. Nat Genet. 2007;39:1127–1133. doi: 10.1038/ng2100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boelz S, Neu-Yilik G, Gehring NH, Hentze MW, Kulozik AE. A chemiluminescence-based reporter system to monitor nonsense-mediated mRNA decay. Biochem Biophys Res Commun. 2006;349:186–191. doi: 10.1016/j.bbrc.2006.08.017. [DOI] [PubMed] [Google Scholar]
- Karam R, Carvalho J, Bruno I, Graziadio C, Senz J, Huntsman D, Carneiro F, Seruca R, Wilkinson MF, Oliveira C. The NMD mRNA surveillance pathway downregulates aberrant E-cadherin transcripts in gastric cancer cells and in CDH1 mutation carriers. Oncogene. 2008;27:4255–4260. doi: 10.1038/onc.2008.62. [DOI] [PubMed] [Google Scholar]
- Sha H, He Y, Chen H, Wang C, Zenno A, Shi H, Yang X, Zhang X, Qi L. The IRE1alpha-XBP1 pathway of the unfolded protein response is required for adipogenesis. Cell Metab. 2009;9:556–564. doi: 10.1016/j.cmet.2009.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figures and Tables
Review Process File