

Abstract
Asc-1 (SLC7A10) is an amino acid transporter whose deletion causes neurological abnormalities and early postnatal death in mice. Using metabolomics and behavioral and electrophysiological methods, we demonstrate that Asc-1 knockout mice display a marked decrease in glycine levels in the brain and spinal cord along with impairment of glycinergic inhibitory transmission, and a hyperekplexia-like phenotype that is rescued by replenishing brain glycine. Asc-1 works as a glycine and L-serine transporter, and its transport activity is required for the subsequent conversion of L-serine into glycine in vivo. Asc-1 is a novel regulator of glycine metabolism and a candidate for hyperekplexia disorders.
Keywords: glycine receptor, GlyT2, hyperekplexia, non-ketotic hyperglycinemia, D-serine
Introduction
Glycine is a classic inhibitory neurotransmitter acting on glycine receptors (GlyRs), which are highly expressed in the spinal cord and pons–medulla 1. Accumulation of glycine in neurons requires the activity of the glycine transporter GlyT2 2. Patients with mutations in GlyR subunit α1 (GLRA1) 3 or β (GLRB) 4, as well as GlyT2 (SLC6A5) 5, 6 display hyperekplexia (OMIM 149400) characterized by pronounced startle responses and stiffness. Although mutations in GLRA1 account for most cases of hyperekplexia, some patients apparently do not display mutations in known target genes 7. Spontaneous mutations in GLRA1 or GLRB in mice cause glycinergic transmission impairment and a hyperekplexia-like phenotype that includes exaggerated startle responses, tremors, and impairment of righting reflex 7, 8, 9. Likewise, impairment of glycine transport by deletion of the pre-synaptic GlyT2 leads to glycinergic dysfunction 2. Recently, pre-synaptic GlyRs were shown to contribute to the hyperekplexia-like phenotype, demonstrating another pre-synaptic mechanism that affects glycinergic transmission 10.
Asc-1 (SLC7A10) is a plasma membrane antiporter present in neurons that has high affinity for small neutral amino acids, such as glycine, L-serine, D-serine, alanine, and cysteine 11, 12. Asc-1 knockout mice exhibit long-lasting tremors and convulsions that start in the second postnatal week and are associated with a shorter life span 13. Since Asc1 has broad substrate specificity, it has been unclear how deletion of this transporter causes the neurological phenotype and how it affects amino acid and neurotransmitter metabolism.
We now investigate the role of Asc-1 in neurotransmitter metabolism by studying Asc-1 knockout (KO) mice. Using metabolomics and electrophysiological and behavioral methods, we show that Asc-1-KO mice exhibit a selective decrease in brain glycine levels, impaired glycinergic inhibitory transmission, and a hyperekplexia-like phenotype that is reversed by replenishing brain glycine. Our data indicate that Asc-1 is a previously undescribed regulator of glycine metabolism and a novel candidate for hyperekplexia.
Results and Discussion
In order to study the role of Asc-1 in brain metabolism, we carried out metabolomics analysis of homozygote Asc-1-KO and WT littermate mice at postnatal day 7 (P7), prior to the appearance of neurological alterations. Changes in metabolites were analyzed using a dual selection criteria (P < 0.050 and ± 1.2-fold change). Strikingly, among 268 metabolites, glycine is the sole metabolite that is significantly lower in the brains of Asc-1-KO mice (50% decrease, P = 0.00002 and false discovery rate-adjusted P = 0.0036) (Fig1A and Supplementary Table S1). Only cysteine-glutathione disulfide (CSSG), a glutathione derivative, appears to increase in Asc-1-KO brains (threefold increase, P = 0.037) (Supplementary Table S1). However, when corrected for multiple comparisons, possible changes in CSSG exhibited an adjusted P-value of 0.75 (Supplementary Table S1). Since other metabolites in the glutathione pathway (GSH and GSSG) are unchanged in the Asc-1-KO mice (Supplementary Table S1), and CSSG appears to be a spurious in vitro oxidation product of GSH and cysteine 14, we did not investigate further possible changes in CSSG.
Figure 1. Asc-1-KO mice exhibit lower levels of glycine in the nervous system.
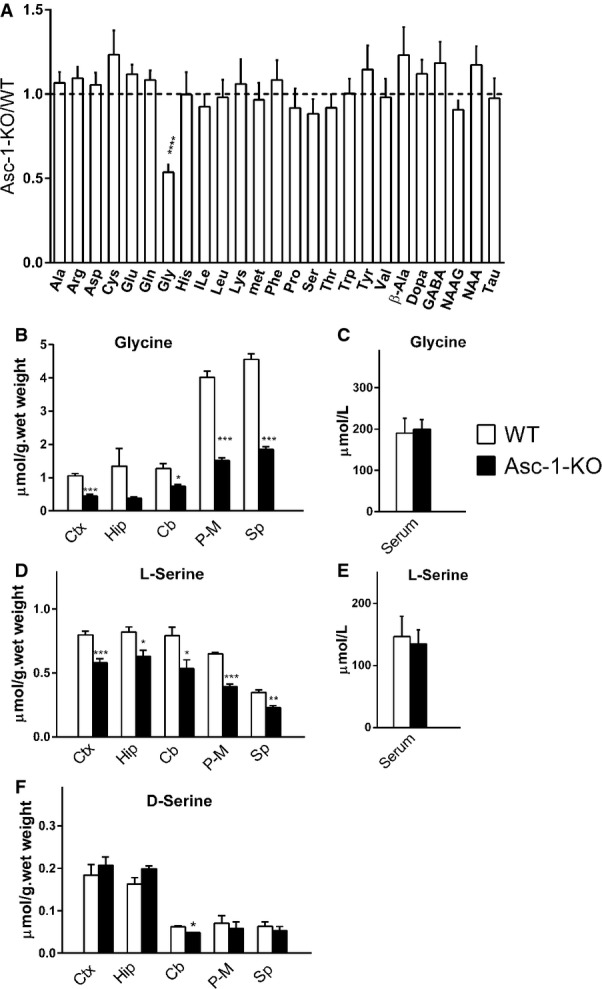
- Metabolomics of selected brain metabolites show a decrease in glycine levels in the brains of Asc-1-KO mice (P7). The values are expressed as Asc-1-KO/WT ratio.
- Glycine levels in different brain areas of WT (open bars) and Asc-1-KO mice (filled bars) at P17–20.
- Glycine levels in the serum of WT and Asc-1-KO mice (P17–20).
- As in (B), but with L-serine determination.
- As in (C), but with L-serine determination.
- As in (B), but with D-serine determination.
Data information: The data represent the average ± SEM obtained from 8 (A) or 4 mice (B–F) in each group. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001, compared to WT (two-tailed unpaired t-test with Welch's correction). Ctx, cerebral cortex; Hip, hippocampus; Cb, cerebellum; P-M, pons–medulla; Sp, spinal cord. Source data are available online for this article.
To ascertain possible regional changes in glycine, we analyzed different brain areas of WT and homozygote Asc-1-KO mice at P17–20, when neurological abnormalities are most evident. We found a 50–60% decrease in glycine levels in all tested brain areas and spinal cord of Asc-1-KO mice, confirming a global dysregulation of brain glycine metabolism (Fig1B).
Glycine is known to be mostly synthesized de novo in the brain, rather than taken up via the blood–brain barrier 15. In agreement, the lower levels of glycine in Asc-1-KO mice brains are not caused by lower glycine availability from the periphery, since serum glycine levels are unchanged in these mice (Fig1C).
Since metabolomics does not distinguish between L- and D-amino acids, and D-serine accounts for up to 30% of brain serine, we investigated whether serine enantiomers are altered in Asc-1-KO mice using HPLC. We detected a 30% decrease in L-serine levels in several areas of the brain and spinal cord of Asc-1-KO mice (Fig1D), whereas serum levels of L-serine are not altered (Fig1E). Brain D-serine levels were little or not affected in Asc-1-KO mice (Fig1F).
Asc-1-KO mice exhibit lower weight, long-lasting tremors, convulsions that start at about P10-12, and the vast majority die within 3 weeks 13. This phenotype, however, was not previously thought to involve changes in glycine metabolism or glycinergic transmission. We found that Asc-1-KO mice display a hyperekplexia-like phenotype due to impairment of inhibitory glycinergic trans-mission, resembling some GlyR mutant mice, especially the oscillator, cincinnati, and Nmf11 types, as well as knockout of GlyT2 (Supplementary Table S2).
While WT mice open their hind legs when suspended by their tails (Fig2A), Asc-1-KO mice display hind-feet clasping (Fig2B), typical of some GlyR mutant mice (Supplementary Table S2). Because Asc-1-KO mice display lower levels of brain glycine, we attempted to reverse the phenotype by replenishing glycine. Administration of glycine (28 mmol/kg, i.p.) to Asc-1-KO mice completely reverses the hind-feet clasping when compared with saline (compare Fig2E and F). This was due to glycine action at strychnine-sensitive receptors, since administration of strychnine at a sub-convulsive dose (1 mg/kg, i.p.) 10 prevented the reversal of the hind-feet clasping by glycine (Supplementary Fig S1). Administration of the glycine precursor L-serine (28 mmol/kg, i.p.) reverses the hind-feet clasping as well (Fig2G), but injection of D-serine, another Asc-1 substrate, and a NMDA receptor co-agonist 11, 16, 17 does not improve the phenotype (Fig2H).
Figure 2. Asc-1-KO mice exhibit phenotypic abnormalities that are rescued by glycine and L-serine.

- WT mice spread their hind legs when suspended by the tail.
- Typical hind-feet clasping in Asc-1-KO mice.
- WT mice quickly right themselves after they are placed on their backs.
- Asc-1-KO mice display impaired righting response.
- Hind-feet clasping in Asc-1-KO mice 3 h after i.p. injection of saline.
- Reversal of hind-feet clasping in Asc-1-KO mice 3 h after i.p. injection of glycine.
- Reversal of hind-feet clasping in Asc-1-KO mice 2 h after i.p. injection of L-serine.
- Lack of effect of D-serine injection in Asc-1-KO mice hind-feet posture.
- WT mice display righting latency times less than a second. Most saline-treated Asc-1-KO mice exhibit righting times longer than 120 s, which are restored to near normal values after administration of glycine or L-serine.
- Asc-1-KO mice display tremors that are restored to WT levels at 3 and 6 h after i.p. injection of glycine.
- Open-field activity is reduced in Asc-1-KO mice and is restored by i.p. injection of glycine.
- Acoustic startle response in WT and Asc-1-KO mice at baseline, 3 and 6 h after i.p. injection of glycine.
Data information: Mice ages were P14 (A–D), P17–20 (E–H), P15 (I), and P17–19 (J–L). The results are average ± SEM of 9 WT and 7 Asc-1-KO mice (J–L). *P < 0.05, **P < 0.01, ***P < 0.01, compared to control (two-way ANOVA with post hoc t-test). Source data are available online for this article.
We found that Asc-1-KO mice exhibit impaired righting response when placed on their backs (compare Fig2C and D; Supplementary Movie S1). Righting latency times in Asc-1-KO mice exceed 2 min in most cases as compared to less than a second in WT mice (Fig2I). Glycine or L-serine administration (28 mmol/kg, i.p.) promotes nearly complete normalization of the righting times measured 2–3 h after the injection (Fig2I). Additionally, tremors are significantly higher in Asc-1-KO mice when compared to WT littermates (Fig2J) and were triggered or exacerbated by handling (Supplementary Movie S1). This was associated with an extremely low level of locomotor activity in the open field displayed by Asc-1-KO mice when compared to WT (Fig2K). Glycine replenishment rescues the tremors (Fig2J) and improves the locomotor activity of Asc-1-KO mice to levels indistinguishable from WT littermates for several hours (Fig2K). Asc-1-KO mice also display intense myoclonic episodes that are prevented by glycine, but not by D-serine administration (Supplementary Movie S2).
Different from most GlyR mice mutants (Supplementary Table S2), we found that Asc-1-KO mice exhibit normal baseline acoustic startle responses at P17–19 (Fig2L). Interestingly, glycine replenishment promotes a significant decrease in the startle responses of the Asc-1-KO mice for several hours, but has no effect on WT littermates (Fig2L). The sensitivity of Asc-1-KO mice to glycine administration is in agreement with their hypoglycinergic phenotype, but their normal baseline acoustic startle reflex points to compensatory effects not observed in GlyR mutant mice (Supplementary Table S2). Furthermore, since glycine also stimulates NMDA receptors, its deficiency may not cause the same abnormalities observed in GlyR mutant mice. For instance, NMDA receptors are involved in complex modulation of startle responses 18. In addition to affect the pre-pulse inhibition of the startle response, administration of the NMDAR antagonist MK-801 has a biphasic effect on the acoustic startle reactivity, in which low doses increase and high doses decrease it 18.
As control, we found that glycine or L-serine at doses that rescued the Asc-1-KO phenotype effectively increases glycine levels both in the cortex and in brainstem of Asc-1-KO mice (Supplementary Fig S2). Brain L-serine also increases following glycine or L-serine injection because of metabolic interconversion (Supplementary Fig S2). Levels of GABA and glutamate are unchanged by glycine or L-serine treatments, except for a small increase in glutamate at the pons–medulla upon L-serine injection (Supplementary Fig S2). In agreement with its inability to improve Asc-1-KO phenotype, D-serine administration to Asc-1-KO mice (9 mmol/kg, i.p.) fails to increase brain glycine levels, while increasing several fold the levels of D-serine itself (Supplementary Fig S2).
The rescue of the phenotype by glycine administration was not due to non-specific sedative effects, which are typically observed at a higher dose 19 and are transient 20. We found that glycine restoration of Asc-1-KO hind-feet posture is preventable by sub-convulsive doses of strychnine, indicating a specific effect on GlyRs (Supplementary Fig S1). Glycine also completely normalizes the locomotor activity and tremors of Asc-1-KO mice to WT levels for at least 6 h (Fig2J and K). Furthermore, the lack of effect of glycine on the acoustic startle reflex in WT mice indicates that glycine effects are not due to sedation, since startle responses are known to be attenuated by sedatives 21.
In order to directly evaluate the glycinergic inhibitory neurotransmission in Asc-1-KO mice, we monitored glycinergic spontaneous inhibitory post-synaptic currents (sIPSCs) in hypoglossal motoneurons from brainstem slices 2, 22. We found that Asc-1-KO mice display a strong 90% decrease in the frequency of sIPSCs compared to age-matched WT mice (WT: 3.04 ± 0.86 vs Asc-1-KO: 0.32 ± 0.18 Hz, P = 0.0043) (Fig3A and B). Although the average amplitude of the sIPSCs in Asc-1-KO mice was not different from WT mice (WT: 34.5 ± 1.4 vs Asc-1-KO: 30.1 ± 5.7 pA, n.s.) (Fig3C), analysis of the cumulative distribution of the sIPSCs amplitudes and areas demonstrate a slight but significant increase in the occurrence of smaller IPSCs in Asc-1-KO when compared to WT mice (Fig3D and E, P = 0.006 and 0.005, Kolmogorov–Smirnov test). Altogether, the data demonstrate impairment of glycinergic inhibitory neurotransmission in Asc-1-KO mice as indicated by a striking lower sIPSC frequency, which is in agreement with their hypoglycinergic phenotype.
Figure 3. Impairment of glycinergic inhibitory neurotransmission in Asc-1-KO mice.
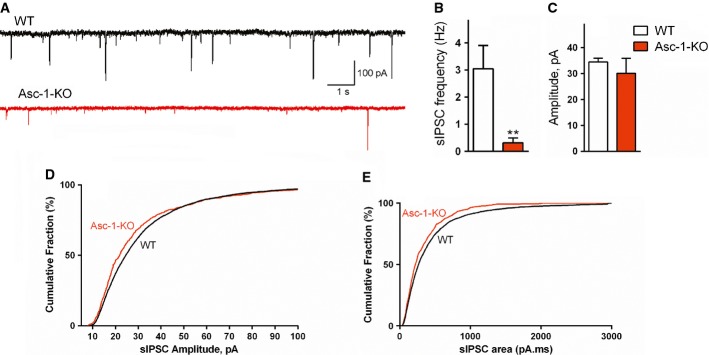
- A Example glycinergic sIPSCs recorded in whole-cell voltage clamp mode from hypoglossal neurons in brainstem slices of WT (black) and Asc-1-KO (red) mice at postnatal day 11.
- B The frequency of glycinergic sIPSCs is decreased in hypoglossal motoneurons of Asc-1-KO mice.
- C Average amplitudes of siPSCs in WT and Asc-1-KO mice.
- D, E Cumulative distribution of sIPSC amplitudes (D) and areas (E) in WT and Asc-1-KO mice.
Data information: The results are average ± SEM of 6 (WT) and 5 (Asc-1-KO) slices obtained from 3 mice each (ages P8–14). The cumulative distribution of sIPSC amplitude and area was calculated from 4,281 (WT) and 474 sIPSC (Asc-1-KO) mice. **P = 0.0043, compared to WT mice (Mann–Whitney test). Source data are available online for this article.
The phenotype of Asc-1-KO is not caused by changes in the expression of other components of the glycinergic inhibitory transmission. We found no changes in the expression levels of GLRA1 and GLRB, plasma membrane, and vesicular glycine transporters or the GlyR scaffolding protein gephyrin (Supplementary Fig S3). Likewise, no changes were detectable in the expression of key metabolic enzymes for glycine or L-serine, such as the glycine cleavage system H and aminomethyltransferase proteins, serine hydroxymethyl transferase isoforms 1 and 2 (SHMT 1 and 2), and D-3 phosphoglycerate dehydrogenase (Supplementary Fig S3).
It is conceivable that changes in glycine and L-serine homeostasis underlie the glycine deficiency in Asc-1-KO mice. Accordingly, we detected a significant 50% decrease in the uptake of [3H]Gly in cortical synaptosomes from Asc-1-KO mice when compared to WT littermates (Fig4A). We also investigated the effect of D-isoleucine (D-Ile), which is a selective artificial substrate of Asc-1 that competes with endogenous substrates 16. We found that D-Ile inhibits by 50% the uptake of [3H]Gly in cortical synaptosomes of WT mice, but has no effect on synaptosomes from Asc-1-KO mice (Fig4A). The data indicate that Asc-1 is a major transporter for glycine in the cerebral cortex and also confirms the selectivity of D-Ile for this transporter.
Figure 4. Asc-1 transports both glycine and L-serine and is required for the subsequent conversion of L-serine into glycine in vivo.
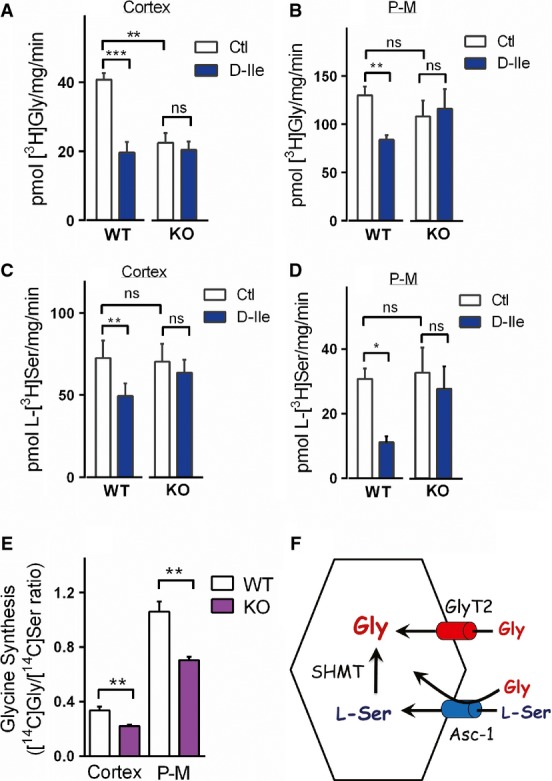
- A, B Glycine uptake by purified synaptosomes from cerebral cortex (A) and the pons–medulla (P-M) (B) of WT and Asc-1-KO mice, either in the absence or in the presence of 1 mM D-Ile.
- C, D L-serine uptake in cortical synaptosomes (C) and P-M synaptosomes (D) of WT and Asc-1-KO mice.
- E Impaired synthesis of glycine from L-serine in Asc-1-KO mice in vivo. Synthesis of [14C]glycine in the cortex and P-M is expressed as the ratio of the recovered radioactivity in [14C]Gly to L-[14C]Ser.
- F Schematic model of the dual action of Asc-1 on glycine homeostasis. Direct transport of glycine by Asc-1 may contribute to glycine accumulation in concert with GlyT2 at the spinal cord and brainstem. Asc-1 also appears to transport L-serine that is subsequently converted into glycine by SHMT, providing a source of glycine. Disruption of the later process seems to underlie the global decrease in the levels of glycine in the nervous system of Asc-1-KO mice.
Data information: (A–D) The results are average ± SEM of 5 (A, B) or 4 (C, D) independent experiments with different preparations of P14–18 mice. (E) The data are average ± SEM of 4 (cortex) and 5 (P-M) mice at P14 (cortex) or P11, P14, and P20 (P-M). *P < 0.05, **P < 0.01, ***P < 0.001; repeated-measures ANOVA and Bonferroni's post hoc test (A–D) or two-tailed unpaired Student's t-test (E). Source data are available online for this article.
Surprisingly, we found that the rates of glycine transport in synaptosomal preparations from the pons–medulla of WT and Asc-1-KO mice are approximately the same (Fig4B), which may be due to compensatory mechanisms in glycine transport as a result of the non-conditional deletion of Asc-1 gene during development. To overcome this limitation, we used D-Ile as a tool to selectively inhibit Asc-1 and found that it decreases the uptake of [3H]Gly into WT synaptosomes by 35%, but has no effect on Asc-1-KO preparations (Fig4B). This observation indicates that Asc-1 is required for glycine transport in the pons–medulla as well.
We found that Asc-1 also transports L-serine in the cortex and pons–medulla. The use of an Asc-1 inhibitor to overcome compensatory changes in Asc-1-KO mice was a key to reveal Asc-1 as an L-serine transporter. D-Ile inhibits the L-[3H]Ser uptake in WT cortical synaptosomes by 30%, but has no effect on Asc-1-KO preparations (Fig4C). Remarkably, D-Ile inhibits by 70% the uptake of L-[3H]Ser in pons–medulla synaptosomes from WT, without any effect on Asc-1-KO mice (Fig4D). Likewise, we found that D-Ala, another transportable inhibitor of Asc-1 23, promotes a 70% inhibition of L-serine uptake in the pons–medulla of WT mice, with little or no effect on Asc-1-KO mice (Supplementary Fig S4). Altogether, the data indicate that Asc-1 is a major transporter for L-serine in addition to glycine, but Asc-1-KO synaptosomal preparations display adaptive increases in L-serine transport.
Conceivably, transport of L-serine by Asc-1 is required for the subsequent synthesis of glycine by SHMT, the main pathway for glycine production in the nervous system 15. To evaluate this possibility, we injected WT and Asc-1-KO mice with L-[14C]serine and monitored the synthesis of [14C]glycine by analyzing the ratio of radioactive serine to newly synthesized glycine in the brain using HPLC analysis. We found a significant decrease in the synthesis of [14C]glycine both in the cortex and in the pons–medulla of Asc-1-KO mice (Fig4E), indicating that Asc-1 is critical for the synthesis of glycine in vivo.
Although other L-serine transport pathways are observed in synaptosomes of Asc-1-KO mice (Fig4C and D), they are not enough to compensate for the deficient glycine synthesis in these mice (Fig4E). The preparation of purified synaptosomes contains a mixture of nerve terminals from different neuronal types as well as contaminant glial particles 24. Therefore, it is possible that other L-serine transporters present in Asc-1-KO mice are not expressed in the same cells or cellular locations that normally contain Asc-1. Our data imply that these putative compensatory L-serine transporter mechanisms are not directly coupled to glycine synthesis process.
It is unlikely that Asc-1 mediates vesicular transport of glycine or L-serine. L-Serine is not transported into synaptic vesicles and does not compete with glycine uptake into synaptic vesicles 25, indicating that the vesicular glycine transporter is distinct from Asc-1.
Our data indicate that Asc-1 may regulate glycine homeostasis by two mechanisms (see scheme in Fig4F). First, Asc-1 may be directly involved in glycine accumulation by glycinergic neurons along with GlyT2. This is supported by our observation that Asc-1 works as a glycine transporter in synaptosomes (Fig4A and B). Furthermore, Asc-1 is expressed by neurons at pre-synaptic areas 12 and has a high affinity for glycine (Km = 8 μM) 11. The second mechanism encompasses the transport of L-serine into neurons for the subsequent synthesis of glycine by SHMT, an enzyme highly expressed in neurons 26 (Fig4F). L-Serine is known to shuttle from astrocytes to neurons, but the identity of neuronal L-serine transporter has been elusive 27. Our data indicate that Asc-1 might be crucial for L-serine import into neurons, and this is supported by our experiments showing that Asc-1 is a major L-serine transporter in synaptosomes (Fig4C and D). Most importantly, we found that deletion of Asc-1 decreases the synthesis of glycine from L-serine in vivo both in the cortex and in pons–medulla (Fig4E). Thus, disruption of L-serine transport and diminished conversion of L-serine into glycine are likely to contribute to the global reduction in glycine we observed in Asc-1-KO mice. However, the participation of some glial populations expressing Asc-1 cannot be completely discarded, since Asc-1 RNA has been described in brainstem astrocytes 28. Furthermore, our data do not exclude other roles for Asc-1, such as regulation of NMDA receptor function via release of D-serine 12.
In summary, our observations indicate that Asc-1 is essential for glycine metabolism and is required for glycinergic inhibitory transmission. Asc-1 is a novel candidate for hyperekplexia in patients where mutations in the primary genes (GLRA1, GLRB, and SLC6A5) have not been detected 7.
On the other hand, mutations in the glycine cleavage system cause non-ketotic hyperglycinemia (OMIM 605899), characterized by massive accumulation of glycine in the CNS, intractable seizures, mental retardation, hypotonia, and frequently neonatal death 29. So far, no effective treatments can prevent the severe neurological impairment in this disease. Selective Asc-1 inhibitors can potentially decrease glycine accumulation in the CNS of patients with non-ketotic hyperglycinemia.
Materials and Methods
Mice breeding and genotyping
Non-conditional heterozygote Asc-1 (Slc7a10)-KO mice were generated by Deltagen Inc. (San Mateo, CA) and maintained in C57Bl/6J 129sv mixed background by back-crossing with C57Bl/6 for 5 generations. The mice were genotyped from tail biopsies using primers to detect WT and Asc-1-KO alleles as previously described 17. All the procedures used in this study were approved by the Committee of Animal Experimentation of the Technion, Israel Institute of Technology.
Metabolic profiling and HPLC analysis
Homozygote Asc-1-KO and WT littermates (eight P7 mice in each group) were anesthetized with isoflurane and decapitated. Their brains were dissected, frozen on dry ice, and shipped to Metabolon Inc. (North Carolina) for metabolic profiling. The samples were analyzed using LC/MS and GC/MS techniques as previously described 30. Statistical analysis was carried out with Student's t-test and Welch correction. To adjust for multiple comparisons, the P-values were corrected for the false discovery rate, as described previously 31. HPLC analysis for amino acids was carried out as previously described 16.
Behavioral analysis
Acoustic startle reflex was evaluated as previously described 32, using a sound proof and ventilated chamber (Kinder Scientific, CA, USA). The values were corrected for the mice weights (WT, 7.4 g ± 0.9 vs Asc-1-KO, 5.9 ± 0.4) and expressed as Newtons/g. In order to evaluate tremors, activity from the sensor was recorded without the auditory stimuli. Activity in the open field was analyzed for 5 min as described 33.
Pharmacological treatments
All compounds used for i.p. injections were dissolved in 0.9% NaCl. To determine brain amino acids, mice were anesthetized with isoflurane, cardiacally perfused for 2 min with ice-cold PBS to remove the blood, and the tissues were subsequently processed for HPLC analysis 16.
Ex vivo electrophysiology
Mice were decapitated after isoflurane anesthesia, and the brainstem was dissected. Transversal slices containing the hypoglossal nucleus (280 μm) were cut with a vibrating microtome 7000 (Campden Instruments). Then, the slices were equilibrated for 30 min in oxygenated aCSF consisting of (in mM) 125 NaCl, 25 NaHCO3, 25 glucose, 3 KCl, 1.25 NaH2PO4, 2 CaCl2, and 1 MgCl2, pH 7.4 (at room temperature). Whole-cell patch-clamp recordings of glycinergic spontaneous inhibitory post-synaptic currents (sIPSCs) were monitored in voltage clamp mode essentially as described 2, 22. Briefly, for the isolation of glycinergic sIPSCs, the aCSF was supplemented with 20 μM 6-cyano-7-nitroquinoxaline-2,3-dione (CNQX, Sigma), 50 μM 2-amino-5-phosphopentanoate (D-AP5, Sigma), and 20 μM bicuculline methobromide (Sigma). Slices were transferred to a recording chamber equipped with an upright BX61WI Olympus microscope and a 60× infrared–differential interference contrast optics, and perfused with oxygenated aCSF at 28°C. After the identification of hypoglossal neurons by their size, shape, and location, whole-cell patch-clamp recordings were obtained using Multi-Clamp 700A (Molecular Devices, Union City, CA). The recording patch electrodes were filled with an intracellular solution consisting of (in mM) 110 CsCl, 30 TEA-Cl, 1 CaCl2, 2 MgCl2, 4 Na2ATP, 10 Hepes, 10 EGTA (adjusted to pH 7.2 with KOH), and 5 5-lidocaine-N-ethyl bromide, giving a resistance of 3–6 MΏ. The hypoglossal neurons were voltage-clamped at −70 mV. Series resistance ranged from 8 to 25 MΏ. Signals were sampled at 5-10 kHz, and the data were analyzed using Clampfit-10.3 (Molecular Devices).
Amino acid uptake
Purified synaptosomes 24 were incubated at 30°C for 1–3 min in the presence of HBSS supplemented with 5 μM of either [3H]glycine or L-[3H]serine in HBSS buffer, in the presence or in the absence of an Asc-1 inhibitor. The uptake was terminated by filtration through 0.45-μm nitrocellulose filters (Millipore) in a filtration apparatus, followed by 4 quick washes with 9 ml of ice-cold HBSS. Blanks were performed by incubating the synaptosomes on ice. The radioactivity retained in the filters was monitored by scintillation counting.
In vivo synthesis of glycine
Aliquots of L-[U-14C]serine (8 μCi/mice) were dried in a speed vacuum, dissolved in 0.9% NaCl, and injected i.p. into Asc-1-KO and WT littermates. Thirty minutes after injection, the mice were anesthetized with isoflurane, perfused with ice-cold PBS to remove blood, and the different brain areas were dissected on ice and frozen in dry ice. To monitor glycine synthesis, the regions of interest were homogenized in 4 volumes of trichloroacetic acid (TCA) 5% and the samples were subjected to HPLC analysis as previously described 16. The peaks corresponding to serine and glycine were collected, and the radioactivity was monitored by scintillation counting. The results were expressed as the ratio of [14C]glycine to L-[14C]serine in the different brain regions.
Acknowledgments
We thank Oded Bodner for expert assistance with movie editing, Alessandro Usiello for helpful advice, Maya Sandler for help in the electrophysiology setup, and Irena Reiter for technical assistance. This work was supported by Israel Science Foundation (HW and SE), Legacy Heritage Fund (HW), and the Allen and Jewel Prince Center for Neurodegenerative Processes of the Brain (HW, SE, and JS).
Author contributions
HS measured amino acid contents by HPLC, and HS and HW did transport assays and in vivo glycine synthesis assays. SZ, HS, and SN did behavioral tests. HS, SN, and IR carried out Western blot quantification. YS did electrophysio-logy and analyzed the data. DR and HS prepared the samples for metabolomics. AA supervised the behavioral tests and their statistical analysis. SE analyzed the biochemical data. SH and JS supervised and analyzed the electrophysiology data. HW is responsible for the execution of the project, data analysis, and manuscript preparation.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting Information
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Movie S1
Supplementary Movie S2
Supplementary Information
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
References
- Becker CM, Hoch W, Betz H. Glycine receptor heterogeneity in rat spinal cord during postnatal development. EMBO J. 1988;7:3717–3726. doi: 10.1002/j.1460-2075.1988.tb03255.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomeza J, Ohno K, Hulsmann S, Armsen W, Eulenburg V, Richter DW, Laube B, Betz H. Deletion of the mouse glycine transporter 2 results in a hyperekplexia phenotype and postnatal lethality. Neuron. 2003;40:797–806. doi: 10.1016/s0896-6273(03)00673-1. [DOI] [PubMed] [Google Scholar]
- Shiang R, Ryan SG, Zhu YZ, Hahn AF, O'Connell P, Wasmuth JJ. Mutations in the alpha 1 subunit of the inhibitory glycine receptor cause the dominant neurologic disorder, hyperekplexia. Nat Genet. 1993;5:351–358. doi: 10.1038/ng1293-351. [DOI] [PubMed] [Google Scholar]
- Rees MI, Lewis TM, Kwok JB, Mortier GR, Govaert P, Snell RG, Schofield PR, Owen MJ. Hyperekplexia associated with compound hetero-zygote mutations in the beta-subunit of the human inhibitory glycine receptor (GLRB) Hum Mol Genet. 2002;11:853–860. doi: 10.1093/hmg/11.7.853. [DOI] [PubMed] [Google Scholar]
- Rees MI, Harvey K, Pearce BR, Chung SK, Duguid IC, Thomas P, Beatty S, Graham GE, Armstrong L, Shiang R, et al. Mutations in the gene encoding GlyT2 (SLC6A5) define a presynaptic component of human startle disease. Nat Genet. 2006;38:801–806. doi: 10.1038/ng1814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eulenburg V, Becker K, Gomeza J, Schmitt B, Becker CM, Betz H. Mutations within the human GLYT2 (SLC6A5) gene associated with hyperekplexia. Biochem Biophys Res Commun. 2006;348:400–405. doi: 10.1016/j.bbrc.2006.07.080. [DOI] [PubMed] [Google Scholar]
- Harvey RJ, Topf M, Harvey K, Rees MI. The genetics of hyperekplexia: more than startle! Trends Genet. 2008;24:439–447. doi: 10.1016/j.tig.2008.06.005. [DOI] [PubMed] [Google Scholar]
- Simon ES. Phenotypic heterogeneity and disease course in three murine strains with mutations in genes encoding for alpha 1 and beta glycine receptor subunits. Mov Disord. 1997;12:221–228. doi: 10.1002/mds.870120213. [DOI] [PubMed] [Google Scholar]
- Becker CM. Disorders of the inhibitory glycine receptor: the spastic mouse. FASEB J. 1990;4:2767–2774. doi: 10.1096/fasebj.4.10.2165011. [DOI] [PubMed] [Google Scholar]
- Xiong W, Chen SR, He L, Cheng K, Zhao YL, Chen H, Li DP, Homanics GE, Peever J, Rice KC, et al. Presynaptic glycine receptors as a potential therapeutic target for hyperekplexia disease. Nat Neurosci. 2014;17:232–239. doi: 10.1038/nn.3615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fukasawa Y, Segawa H, Kim JY, Chairoungdua A, Kim DK, Matsuo H, Cha SH, Endou H, Kanai Y. Identification and characterization of a Na(+)-independent neutral amino acid transporter that associates with the 4F2 heavy chain and exhibits substrate selectivity for small neutral D- and L-amino acids. J Biol Chem. 2000;275:9690–9698. doi: 10.1074/jbc.275.13.9690. [DOI] [PubMed] [Google Scholar]
- Helboe L, Egebjerg J, Moller M, Thomsen C. Distribution and pharmacology of alanine-serine-cysteine transporter 1 (asc-1) in rodent brain. Eur J Neurosci. 2003;18:2227–2238. doi: 10.1046/j.1460-9568.2003.02966.x. [DOI] [PubMed] [Google Scholar]
- Xie X, Dumas T, Tang L, Brennan T, Reeder T, Thomas W, Klein RD, Flores J, O'Hara BF, Heller HC, et al. Lack of the alanine-serine-cysteine transporter 1 causes tremors, seizures, and early postnatal death in mice. Brain Res. 2005;1052:212–221. doi: 10.1016/j.brainres.2005.06.039. [DOI] [PubMed] [Google Scholar]
- Wang XF, Cynader MS. Astrocytes provide cysteine to neurons by releasing glutathione. J Neurochem. 2000;74:1434–1442. doi: 10.1046/j.1471-4159.2000.0741434.x. [DOI] [PubMed] [Google Scholar]
- Shank RP, Aprison MH. The metabolism in vivo of glycine and serine in eight areas of the rat central nervous system. J Neurochem. 1970;17:1461–1475. doi: 10.1111/j.1471-4159.1970.tb00513.x. [DOI] [PubMed] [Google Scholar]
- Rosenberg D, Artoul S, Segal AC, Kolodney G, Radzishevsky I, Dikopoltsev E, Foltyn VN, Inoue R, Mori H, Billard JM, et al. Neuronal D-serine and glycine release via the Asc-1 transporter regulates NMDA receptor-dependent synaptic activity. J Neurosci. 2013;33:3533–3544. doi: 10.1523/JNEUROSCI.3836-12.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutter AR, Fradley RL, Garrett EM, Chapman KL, Lawrence JM, Rosahl TW, Patel S. Evidence from gene knockout studies implicates Asc-1 as the primary transporter mediating d-serine reuptake in the mouse CNS. Eur J Neurosci. 2007;25:1757–1766. doi: 10.1111/j.1460-9568.2007.05446.x. [DOI] [PubMed] [Google Scholar]
- Yee BK, Chang DL, Feldon J. The Effects of dizocilpine and phencyclidine on prepulse inhibition of the acoustic startle reflex and on prepulse-elicited reactivity in C57BL6 mice. Neuropsychopharmacology. 2004;29:1865–1877. doi: 10.1038/sj.npp.1300480. [DOI] [PubMed] [Google Scholar]
- Lapin IP. Antagonism of L-glycine to seizures induced by L-kynurenine, quinolinic acid and strychnine in mice. Eur J Pharmacol. 1981;71:495–498. doi: 10.1016/0014-2999(81)90195-3. [DOI] [PubMed] [Google Scholar]
- Kawai N, Sakai N, Okuro M, Karakawa S, Tsuneyoshi Y, Kawasaki N, Takeda T, Bannai M, Nishino S. The Sleep-Promoting and Hypothermic Effects of Glycine are Mediated by NMDA Receptors in the Suprachiasmatic Nucleus. Neuropsychopharmacology. 2015;40:1405–1416. doi: 10.1038/npp.2014.326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouagazzal AM, Jenck F, Moreau JL. Drug-induced potentiation of prepulse inhibition of acoustic startle reflex in mice: a model for detecting antipsychotic activity? Psychopharmacology. 2001;156:273–283. doi: 10.1007/s002130100763. [DOI] [PubMed] [Google Scholar]
- Latal AT, Kremer T, Gomeza J, Eulenburg V, Hulsmann S. Development of synaptic inhibition in glycine transporter 2 deficient mice. Mol Cell Neurosci. 2010;44:342–352. doi: 10.1016/j.mcn.2010.04.005. [DOI] [PubMed] [Google Scholar]
- Rosenberg D, Kartvelishvily E, Shleper M, Klinker CM, Bowser MT, Wolosker H. Neuronal release of D-serine: a physiological pathway controlling extracellular D-serine concentration. FASEB J. 2010;24:2951–2961. doi: 10.1096/fj.09-147967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunkley PR, Jarvie PE, Robinson PJ. A rapid Percoll gradient procedure for preparation of synaptosomes. Nat Protoc. 2008;3:1718–1728. doi: 10.1038/nprot.2008.171. [DOI] [PubMed] [Google Scholar]
- Christensen H, Fykse EM, Fonnum F. Inhibition of gamma-aminobutyrate and glycine uptake into synaptic vesicles. Eur J Pharmacol. 1991;207:73–79. doi: 10.1016/s0922-4106(05)80040-9. [DOI] [PubMed] [Google Scholar]
- Abarinov EV, Beaudin AE, Field MS, Perry CA, Allen RH, Stabler SP, Stover PJ. Disruption of shmt1 impairs hippocampal neurogenesis and mnemonic function in mice. J Nutr. 2013;143:1028–1035. doi: 10.3945/jn.113.174417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolosker H, Radzishevsky I. The serine shuttle between glia and neurons: implications for neurotransmission and neurodegeneration. Biochem Soc Trans. 2013;41:1546–1550. doi: 10.1042/BST20130220. [DOI] [PubMed] [Google Scholar]
- Schnell C, Shahmoradi A, Wichert SP, Mayerl S, Hagos Y, Heuer H, Rossner MJ, Hulsmann S. The multispecific thyroid hormone transporter OATP1C1 mediates cell-specific sulforhodamine 101-labeling of hippocampal astrocytes. Brain Struct Funct. 2015;220:193–203. doi: 10.1007/s00429-013-0645-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hennermann JB, Berger JM, Grieben U, Scharer G, Van Hove JL. Prediction of long-term outcome in glycine encephalopathy: a clinical survey. J Inherit Metab Dis. 2012;35:253–261. doi: 10.1007/s10545-011-9398-1. [DOI] [PubMed] [Google Scholar]
- Evans AM, DeHaven CD, Barrett T, Mitchell M, Milgram E. Integrated, nontargeted ultrahigh performance liquid chromatography/electrospray ionization tandem mass spectrometry platform for the identification and relative quantification of the small-molecule complement of biological systems. Anal Chem. 2009;81:6656–6667. doi: 10.1021/ac901536h. [DOI] [PubMed] [Google Scholar]
- Storey JD, Tibshirani R. Statistical significance for genomewide studies. Proc Natl Acad Sci USA. 2003;100:9440–9445. doi: 10.1073/pnas.1530509100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avital A, Dolev T, Aga-Mizrachi S, Zubedat S. Environmental enrichment preceding early adulthood methylphenidate treatment leads to long term increase of corticosterone and testosterone in the rat. PLoS ONE. 2011;6:e22059. doi: 10.1371/journal.pone.0022059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aga-Mizrachi S, Cymerblit-Sabba A, Gurman O, Balan A, Shwam G, Deshe R, Miller L, Gorodetsky N, Heinrich N, Tzezana O, et al. Methylphenidate and desipramine combined treatment improves PTSD symptomatology in a rat model. Transl Psychiat. 2014;4:e447. doi: 10.1038/tp.2014.82. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure S1
Supplementary Figure S2
Supplementary Figure S3
Supplementary Figure S4
Supplementary Table S1
Supplementary Table S2
Supplementary Movie S1
Supplementary Movie S2
Supplementary Information
Review Process File
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4