

Abstract

Peptide aptamers are small combinatorial proteins that are selected to bind to specific sites on their target molecules. Peptide aptamers consist of short, 5-20 amino acid residues long sequences, typically embedded as a loop within a stable protein scaffold. Various peptide aptamer scaffolds and in vitro and in vivo selection techniques are reviewed with emphasis on specific biomedical, bioimaging, and bioanalytical applications.
Keywords: Affibodies, affilins, anticalins, armadillo repeat, atrimers, avimers, cell display, combinatorial libraries, darpins, directed evolution, DNA display, fynomers, knottins, kunitz domain, monobodies, mRNA display, OBodies, phage display, peptide aptamers, protein scaffold, ribosome display, thioredoxin, yeast two hybrid
1. Introduction
Designing molecules that modulate cellular processes through selective high affinity binding to discrete sites on biological molecules still remains a “Holy Grail” of biomedicine [1]. Protein recognition of and interaction with other cell components in the dynamic context of complex signaling and metabolic networks provide the basis of life. Harnessing the power of these interactions constitutes one of the biggest challenges of modern biological science.
The concept of Antikörper (Antibodies), that was introduced by Paul Ehrlich more than 120 years ago [2], and his idea of a magic bullet [3] culminated in the development of hybridoma technology and monoclonal antibodies, now an indispensable part of contemporary research, diagnostics and therapy. The numerous achievements of modern antibody technologies are indisputable and are covered in a variety of recent reviews [4-14]. The numbers of antibodies used in research and diagnostics is measured in the thousands, hundreds are in drug discovery company pipelines, and ∼30 are already used in clinical applications [15]. Still, a few inherent characteristics related to antibody properties and production limits their usefulness and clinical efficacy [16]. For example, the generation of antibodies depends on animal immunization, which rules out toxic, low-immunogenic or otherwise incompatible targets. Due to the considerable size (150 kDa for IgG) of antibodies, applications for most intracellular therapeutic targets are restricted, delivery must be accomplished by injection or infusion, and tissue penetration and accumulation can be an issue as well. Antibodies are temperature sensitive, undergo irreversible denaturation and have a limited shelf life. Diagnostic applications are generally limited to physiological conditions, and in spite of considerable efforts for antibody humanization [17], Fcmediated complement-dependent cytotoxicity (CDC) and antibody-dependent cellular cytotoxicity (ADCC) can be a serious problem. Selection of the binding surface of an antigen is determined by the immune system, which prefers a planar binding interface, as a result, binding to other topologies such as folds, cavities and the clefts of catalytic sites is generally problematic [18, 19]. Finally, the complex molecular architecture of antibodies includes a multitude of glycosylation sites and disulfide bonds that requires a eukaryotic system to manufacture. This process is laborious, expensive and suffers from batch to batch variations in activity.
The emergence of Aptamers created an attractive alternative to antibodies [1]. In 1990 two revolutionary studies presented the method of in vitro generation of high-affinity molecules against selected targets when Tuerk and Gold succeeded in selecting RNA ligands against T4 DNA polymerase [20]; and Ellington and Szostak against organic dyes [21]. The first group coined the moniker SELEX (for Systematic Evolution of Ligands by EXponential enrichment), while the second introduced the term Aptamer (from the Latin aptus - fit, and Greek meros - part), now defined as single-stranded nucleic acids (NA), RNA or DNA molecules of 20-100 bases long capable of spontaneously folding into 3-D structures and selectively binding to their cognate targets. SELEX technology allowed for rapid interrogation of large synthetic libraries (1014-1016 molecules) and drastically broadened the spectrum of targets, which now includes not only toxic and non-immunogenic molecules, but also many synthetic and natural materials, and small compounds [22]. Similar to antibodies in binding affinity (nanomolar to picomolar range binding constants), aptamers are less immunogenic, smaller (10-50 kDa), and can be used in a variety of environmental conditions. Aptamers can be engineered and produced completely in a test tube, and can be readily modified during and after chemical synthesis to increase the stability and variability of the library [23]. But the greatest advantage is the robustness and speed of in vitro generation, selection and evolution of aptamers [24]. Owing to these advantages, aptamers gained a lot of interest over the past decade and today are widely used in therapy and diagnostics [1, 25-27], targeted drug delivery [28-30], in the area of molecular imaging [31, 32] and biosensors [33-36].
Yet another alternative to antibodies developed around 1996 were Peptide Aptamers (PA). The concept, originally introduced by Roger Brent [37], proposed a short amino acid sequence embedded (“double constrained”) within the context of a small and very stable protein backbone (“scaffold”). Conformational constraint is important, since it stabilizes the insert loop and makes it more likely to fold and recognize cognate surfaces. PAs can be viewed as scaled down versions of immunoglobulin T-cell receptors, they are extremely small and simple molecules characterized by high stability, high solubility, fast folding kinetics and available in large quantities through chemical synthesis or bacterial expression [38]. PAs are essentially a “loop on a frame” design, where the 5-20 residue peptide loop grafted onto a neutral scaffold is the source of variability for selecting high affinity binders to a target protein or small molecule from combinatorial libraries. The binding affinity of constrained aptamers can be as much as 1000 times higher than the free peptide, a fact generally attributed to the lower conformational entropy of the restricted peptide loop [39, 40].
PAs can be produced and selected in vivo through yeast two hybrid and similar techniques, making PAs ideal candidates for interrogating intracellular targets in a physiological environment [41]. Whereas in vitro selected NA aptamers might encounter problems getting into live cells in sufficient amounts due inefficient cellular uptake and to intracellular processing of endosome-targeted RNAs [42]. Contact surfaces implicated in protein-protein interactions tend to be flat and large (1,500-3,000 Å2) relative to those involved in protein-small-molecule interactions (300-1,000 Å2) [43]. NA aptamers bind more precisely and strongly because they extend the surface contact with their cognate targets through adaptive conformational changes, resulting in the creation of specific binding sites [1, 44]. This results in large interaction surfaces compared to immunoglobulins (i.e. ∼2600 Å2 of the enzyme surface is masked by one NA aptamer against HIV reverse transcriptase [44, 45]). PAs generally exhibit a smaller binding footprint that permits a more precise interrogation of the target than that afforded by NA-based probes, which makes PAs an excellent research tool [46, 46]. Nevertheless, both NA aptamers and PAs share a great deal of similarity. Representing two classes of biomolecules, NA aptamers and PAs provide a large degree of variability in functional groups (i.e. different pH-dependence on the formation of hydrogen and electrostatic bonds) and in conformational flexibility [38], which eventually translates into a plethora of binding molecules against various chemical surfaces, thus improving the chances of finding an ideal interactor.
Advances in the technology of scaffold design, represented by antibody-based [48] single-domain (dAbs), single-chain variable fragment (scFv), antigen-binding fragment (Fab), Avibody, minibody, CH2D domain, Fcab, and bispecific T-cell engager (BiTE) molecules [15], and non-immunoglobin protein structures, circumvent some of the limitations of classical immunoglobulins as biomedical tools. These scaffolds represent smaller and more rigid entities with improved stability and the capability of directed evolution through rounds of combinatorial screening [49-51]. Antibody fragments designed on the basis of naturally occurring heavy-chain-only antibodies, like nanobodies derived from Camelidae [52, 53] and VNARs from sharks [54, 55], look especially promising. It will be interesting to watch which approach will eventually gain the upper hand, but most likely we will see some degree of technological convergence [16, 56] and segregation into the fields where these technologies will prove to be most effective.
2. Scaffolds
The first step in creating a combinatorial peptide aptamer library is to select a proper scaffold, i.e. a rigid, compact, preferably monomeric, stable protein core capable of displaying variable target interaction surfaces in a manner analogous to the immunoglobulin complementarity determining region (CDR). The scaffold has to be soluble, non-toxic and capable of high-level expression in prokaryotic systems. The cloned scaffold gene should be engineered to accommodate functional protein elements such as localization signals, and epitope and purification tags [57]. It is highly desirable that permutations introduced into variable regions do not adversely affect solubility, folding and the aggregating properties of the resulting combinatorial product.
The current outlook in the field of developing novel bioaffinity materials is based on the inherent limitations plaguing the classic antibody approach. Consequently, in addition to small antibody-based scaffolds, there emerged a second group of scaffolds based on naturally tough non-immunoglobulin protein structures that can be genetically manipulated into forming high-affinity patches without compromising the overall stability of the molecule. And while nature has been working for many million years polishing its antibody project, it took just two decades for researchers to develop more than fifty different alternative protein scaffold designs, summarized in a number of comprehensive reviews [16, 56-61].
2.1. “Loop on a Frame” Scaffolds
The original PA concept of Roger Brent [37], which proposed a “loop on a frame” model imitating antibody paratope, was later expanded to include scaffolds that contain multiple variable loops and incorporate more complex combinatorial proteins. These improvements create target binding surfaces consisting of variable non-continuous surface-exposed side chains spread over several structural frame elements or across several variable loops [41, 62], and are capable of tolerating significant structural changes [38]. These new platforms are called engineered (non-antibody) scaffolds, (double-)constrained combinatorial proteins or antibody mimics (mimetics) [16, 38, 59]. Both loop- and rigid structure-mediated binding are suitable for generating high affinity surfaces. We discuss several scaffold designs that are particularly effective due to the ability to yield high affinity binders and to function as alternatives to conventional antibodies [63].
2.1.1. Thioredoxins
Bacterial Thioredoxin A (TrxA) was the original scaffold design pioneered by Roger Brent lab [37] for display of conformationally constrained peptides. Thioredoxin is a highly soluble, small and structurally rigid 12 kD oxidoreductase enzyme involved in maintaining the E.coli cytosolic thiol/disulfide equilibrium, it is non-toxic even when expressed at high levels in bacteria. Thioredoxin has an active site composed of a Cys-Gly-Pro-Cys stretch that is capable of accommodating long peptide insertions albeit with a concomitant loss of enzymatic function [64]. Thioredoxin is a well-folded single domain protein consisting of four α -helices that flank a central five-stranded β-sheet and an active site disulfide group protruding from the protein surface [65]. Comparative studies demonstrated that Thioredoxin possesses superior stability over other scaffolds [66]. The solvent accessible loop where peptide aptamers are presented at the surface of the molecule is essential for recognizing and binding to target compounds (see Fig. 1). Employing the yeast two-hybrid method a number of peptide aptamers, such as PA against human cyclin-dependent kinase 2 (Cdk2) [37] and E2F transcription factor, were identified from corresponding random loop libraries, as well as several other targets, mostly inhibitors of signaling pathways involved in cancer development and progression. Owing to its remarkable characteristics, TrxA was the first widely used peptide aptamer scaffold (see Table 1 in review [57]). In 2012 Groner et al. [67] described a PA against Stat3 and Survivin, selected using human Thioredoxin (hTrx) that was modified to provide high levels of expression and facilitate cellular delivery; in 2013 the same group developed a PA against Stat5 [68].
Fig. (1).
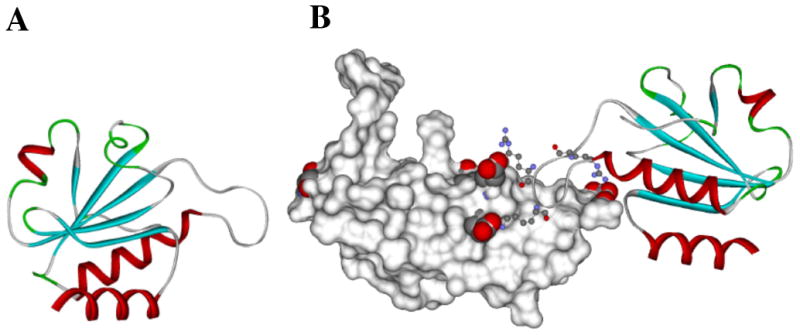
Computer simulation of TrxA-based peptide aptamer docking with the C2 domain of RAGE based on contact residues determined by using NMR spectroscopy (a) TrxA based PA is shown in ribbons Secondary structural elements turns helices and strands are colored in green red and cyan respectively. (b) Surface rendering of the C2 domain of RAGE with glutamic acid residues 243 245 and 322 (shown as space fills) forming negatively charged contact points that interact with positively charged Arginines 41 43 and 45 (shown as ball-and-stick models) of the aptamer loop The PA is presented as a solid ribbon with coloring indicative of the secondary structure Adapted from [47].
2.1.1.1. Combinatorial Library of Improved Peptide Aptamers (CLIPs)
In 2013, Reverdatto et al. [47] used a Thioredoxin scaffold to develop an efficient design for constructing a Combinatorial Library of Improved Peptide aptamers (CLIPs) and used that library to identify PAs that alter signal transduction initiated by the binding of the physiological ligand S100B to the cell Receptor for Advanced Glycated End products (RAGE) [69]. Signal transduction of RAGE is implicated in the etiology of many diseases, including diabetes, neurode-generation, cancer, and inflammation [69-72]. At the core of CLIPs technology is efficient directional cloning of randomized oligonucleotides within the context of the E.coli thiore-doxin gene, coupled with amplification of a ligated library, which preserves its complexity (Fig. 2). The scheme is robust and facilitated the creation of a library of 3×1010 clones, a small portion of which was tested by yeast two hybrid selection, resulting in a number of binders to RAGE. No limitations exist in expanding the random octapeptide library to full theoretical complexity, 1.1×1012 clones and beyond [47].
Fig. (2).
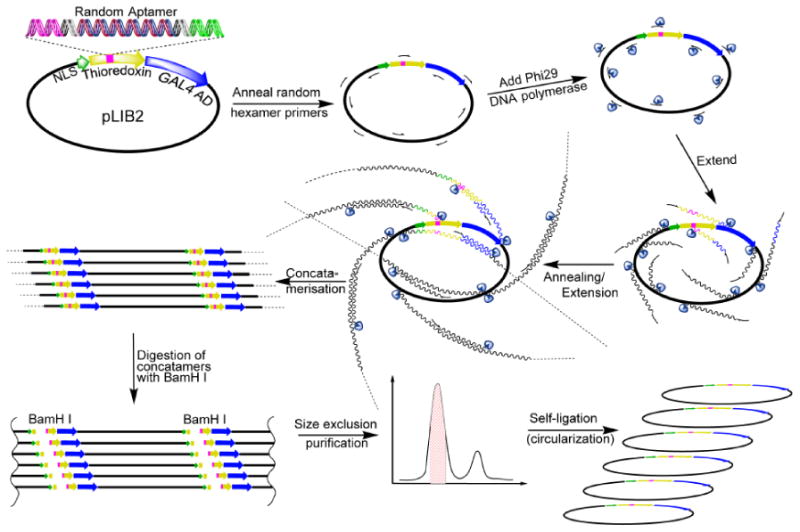
General scheme for Phi 29 amplification of CLIPs. Random hexamer primers are annealed to pLib2. Plasmids are amplified by using Phi29 DNA polymerase forming concatameric DNA strands. The concatamers are digested with BamHI and purifed by using size exclusion chromatography. Self-ligation provides up to a 1000-fold amplification of CLIPs. Adapted from [47].
Inserting random aptamer loop sequences into the wild type Thioredoxin structure often destabilizes the Thioredoxin fold, causing PAs to oligomerize. To overcome this problem a mutation was introduced within the Thioredoxin platform to produce soluble monomeric PAs. The D26A mutation was critical since it eliminated a buried negative charge in the wild type Thioredoxin that became a destabilizing factor in the engineered platform. The small size of the PAs facilitated the characterization of the interactions of PAs with target molecules at atomic resolution by using NMR spectroscopy and allowed the PAs to be considered as drug-like molecules rather than large particles that bind non-specifically [73].
By using RAGE domains as targets in a yeast two-hybrid screen, several PAs were isolated that recognize all three RAGE immunoglobulin domains [47]. The interaction surfaces on the VC1 and C2 domains resolved by NMR clearly showed that the PAs bind to distinctly different sites with affinities ranging from tens of nanomolar to micromolar. NMR analyses of the interaction surfaces between PAs and the C2 domain of RAGE resulted in a solution model of the complex (Fig. 1A).
The effect of PAs on S100B-induced RAGE signal transduction was examined in mammalian cells and found to dramatically decrease the phosphorylation of RAGE effectors [47]. A few PAs bind to sites proximal to the S100B binding site [74, 75], suggesting that these PAs inhibit signaling by directly blocking the S100B-RAGE interaction. The binding sites for other PAs were located in the membrane-proximal part of RAGE far from the S100B binding site, implying that they may allosterically affect RAGE signaling. Thus, monomeric PAs selected from CLIPs block specific sites on the target receptor without affecting other physiological interactions of the receptor.
In summary, the availability of specific binders that recognize different sites on a target combined with structural biology technology and functional assays provides an opportunity to dissect complex biological pathways.
2.1.2. Monobodies
Monobodies, also known as Adnectins, were introduced as a platform for combinatorial peptide library design in 2000 [76, 77], and are among the most well-studied and used in clinical studies [78]. The scaffold is based on the structure of the tenth extracellular type III domain of human fibronectin (10Fn3) which consists of seven β-sheets that form a barrel and three solvent-accessible loops on each side that closely resemble the complementarity determining regions (CDRs) of antibody variable domains [79] (Fig. 3A). The 10Fn3 domain is a small 10 kDa structure that lacks any disulfide bridges or free cysteines, but still retains a remarkable rigidity and thermostability up to 80 °C even under reducing conditions, and can be produced in bacteria with high yields [78]. For the purpose of engineering novel binding entities based on 10Fn3 architecture, the loops (green areas in Fig. 3A) are the obvious choice for diversification.
Fig. (3).
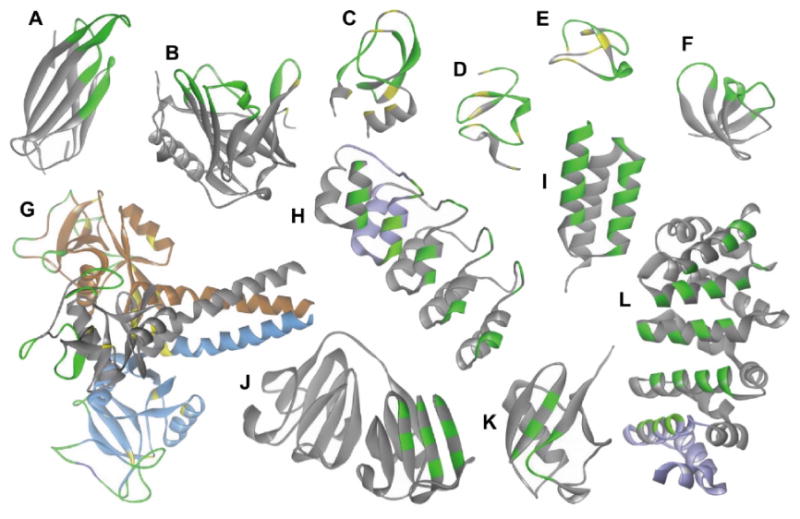
Ribbon representation of the scaffold structures discussed in text. Amino acids patches involved in binding are indicated in green; the frameworks in grey and disulfide bridges are indicated in yellow. For structures with repeated elements one unit repeat is indicated in violet. A: Adnectin scaffold (pdb code: 3QWQ). B: Anticalin scaffold (pdb code: 3BX7). C: Kunitz domain scaffold (pdb code: 1KTH). D: Avimer scaffold (pdb code: 1AJJ). E: Knottin scaffold (pdb code: 1CLV). F: Fynomer scaffold (pdb code: 4AFQ). G: Atrimer scaffold (pdb code: 1HTN, the trimeric assembly is indicated in grey, blue, and brown). H: Darpin (pdb code: 4DX5). I: Affibody (pdb code: 1LP1). J and K: Affilins, either modified γ-B crystallin proteins (J; pdb code, 2JDG) or based on the ubiquitin protein (K; pdb code, 1UBQ). L: Armadillo repeats (pdb code: 1JDH). Adapted from [63]
Initial library designs included variations of 4-10 residues in all or some of the loops, and produced binders with affinities ranging from low micromolar for Ubiquitin to subnanomolar for TNF-α [76, 77]. The thermostability and solubility of 10Fn3 can degrade when the loops are randomized to create diversity [80, 81]. Creating biased arrangements of amino acid residues in each diversified position and varying the loop length resulted in interactors with low nanomolar to picomolar affinity [80, 82]. The structures of co-crystals of Adnectins with their biological targets revealed that besides the interactions of diversified loops, non-loop residues from the N-terminus and β-strands also participate in binding thus expanding the available interaction footprint [77, 83]. The relative autonomy of fibronectin domains provides an opportunity to generate multi-functional binding chimeras, which was demonstrated by creating a bispecific molecule based on two individual adnectins with affinities for epidermal growth factor receptor (EGFR) and for insulin-like growth factor receptor-1 (IGF1R) (see Applications).
2.1.3. Anticalins
Anticalins as an alternative architecture for peptidic combinatorial library design appeared about the same time as monobodies [84, 85]. They are based on the diverse family of small (∼20 kDa, 150-160 residues) single-chain extracellular proteins called Lipocalins, which are abundant in both prokaryotes and eukaryotes. Human species contain twelve different Lipocalins circulating in blood and extracellular fluids. Lipocalins bind and store or transport hydrophobic or chemically active molecules, such as vitamins, lipids and various secondary metabolites [86]. Lipocalins share a conserved structural fold comprised of eight antiparallel β-strands, forming a cup-shaped β-barrel core connected in a pair-wise fashion with four structurally variable loops, which together with the adjacent residues of the β-barrel core form the ligand pocket. Different Lipocalins have a high degree of structural similarity in the β-barrel core while displaying a broad diversity in the four-loop region that resembles the organization of antibody antigen binding sites (Fig. 3B). Unlike the usually flat interface of antibody CDRs, Lipocalins possess a characteristic deep ligand pocket making them particularly suitable for tightly binding small, hapten-type compounds or peptides [87]. Anticalins are structurally very stable, with melting temperatures exceeding 70ºC, and are not affected by loop permutations, they do not have disulfide bridges and are not glycosylated, which makes them suitable for production in yeast or bacteria. A range of 16-24 randomized residues per loop appears to be optimal for the design of Anticalin libraries [84, 88].
2.1.4. Kunitz Domains
A Kunitz domain is a common structural fold found in a family of reversible serine protease inhibitors. Examples of Kunitz-type protease inhibitors are bovine pancreatic trypsin inhibitor (BPTI), human pancreatic secretory trypsin inhibitor (PSTI), Alzheimer's amyloid β-protein precursor inhibitor (APPI), the leech-derived trypsin inhibitor (LTDI), the mustard trypsin inhibitor II (MTI II), and the periplasmic E. coli protease inhibitor Ecotin [60]. The Kunitz domain is a representative of alpha/beta proteins with irregular secondary structure stabilized by three disulfide bonds and presenting three peptide loops that can be varied without introducing much destabilization to the scaffold (Fig. 3C). Protease inhibitors meet the scaffold criteria in that they are small (60 residues), stable and capable of evolving the binding activity of exposed peptide loops through targeted randomization to construct combinatorial libraries [58]. Kunitz domain-based scaffolds have been successfully utilized to construct and select a library of protease inhibitors [89] with the potential for therapeutic applications (see below).
2.1.5. Avimers
“Avidity multimers” or Avimers as scaffolds were proposed by Silverman et al. in 2005 [90]. Avimers are derived from the family of human receptor domains known as Adomains, which are present in some cell-surface receptors including low-density lipoprotein receptor (LDLR). Adomains recognize over 100 different targets including small molecules, proteins and viruses, with each ligand often contacting multiple domains [90]. Individual A-domains are very small, ∼4 kDa (35 residues), and form uniform stable structures stabilized by three disulfide bridges and calcium binding (Fig. 3D). Only 12 residues are required for scaffold maintenance, making Avimers ideal candidates for diversification. Avimers are remarkably heat stable, withstanding prolonged incubation at 50-80 ºC. Avimers containing up to eight A-domain modules, with total of 48 cysteines, were produced in E.coli with yields exceeding 1 g/L and were reported to be properly folded upon air oxidation [90]. Employing the nature-inspired strategy of combining sequentially selected A-domains generated each time against the new epitope in one Avimer, Silverman et al. [90] developed Avimers with picomolar affinity for Interleukin 6.
2.1.6. Knottins
Knottins, also known as cystine knot miniproteins, share the same cystine knot motif that is commonly found in some plant proteases and in toxins of several invertebrates (such as spiders, scorpions and cone snails). The small, ∼3 kDa (25-35 residues), protein forms a very rigid structure (Fig. 3E) in which three antiparallel β-sheets connected by interspersed variable peptide loops are stitched together with conserved interlocking disulfide bridges in a characteristic knotted topology [60]. Excellent structural and thermal stability and remarkable tolerance to introducing sequence diversification within the loop regions, often without the loss of structural integrity and bioactivity [91], led to the development of several combinatorial libraries based on the Knottin scaffold [56, 60, 63, 92-94]. In two cases, libraries generated on the basis of diversification of a six-residue surface loop of the agouti-related protein (AgRP) scaffold with only two disulfide bridges [95], and a squash (Ecballium elaterium) trypsin-inhibitor based scaffold [96] yielded Knottins with nanomolar affinity against tumor vasculature-related Integrins, such as αvβ3, αvβ5 and αvβ1. The small size of the Knottin scaffold makes chemical synthesis of the molecule practical, as well as incorporating unnatural and modified residues to increase diversity and functionality [91].
2.1.7. Fynomers
Fynomers as a scaffold concept dates to 2007 when several libraries based on this architecture were generated [97, 98]. Fynomers are based on the src homology 3 (SH3) domain of the human proto-oncogene tyrosine-protein kinase Fyn called FynSH3 (amino acid residues 83-145). The SH3 domain binds to proline rich peptides containing a PXXP binding motif, its sequence is fully conserved between humans, mouse, rat and gibbons. SH3 is composed of two antiparallel β-sheets connected through two flexible loops (called RT-Src and n-Src-loop) that are positioned to interact with other proteins (Fig. 3F). Up to six residues in each loop can be varied without significantly reducing the solubility and folding properties, and the length of the n-Src-loop can be changed from four to six residues [99]. These substitutions generally abolish the original PXXP binding activity. Grabulovski et al. [98] describe their Fynomer design as a particularly attractive scaffold for the generation of binding proteins, since it “(1) can be expressed in bacteria in soluble form in high amounts, (2) is monomeric and does not aggregate when stored in solution, (3) is very stable (Tm ∼70 °C), (4) lacks cysteine residues and (5) is of human origin featuring an amino acid sequence completely conserved from mouse to man and, hence, is considered to be non-immunogenic” [98]. The claims are supported by the recent successful screening of Fynomer libraries that produced potent inhibitors of the serine protease Chymase (expressed in the secretory granules of mast cells) with subnanomolar affinities [99].
2.1.8. Atrimers
Atrimers are another example of a scaffold that capitalizes on an oligomeric structure and multiple contacts to form interaction surfaces with high avidities. The platform was introduced in 2009, and is based on the human homotrimeric protein Tetranectin, a member of the lectin family characterized by a C-type lectin domain (CTLD), found in blood plasma and tissue [100]. Tetranectin is a 60 kDa protein made up of three identical structural units in which the CTLD elements are positioned C-terminally to a trimerizing coil-coil region (Fig. 3G). CTLD retain their structural integrity as separate protein domains and trimerization leads to an apparent 100-fold increase in binding affinity for a Tetranectin ligand, plasminogen kringle-4. Each CTLD fragment has five loop regions 6-9 residues long that provide binding specificity. The diversification of loop residues can be performed in iterative or sequential fashion without compromising the structural integrity of the domain [100]. The molecular weight of 60-70 kDa and the lack of glycosylation improves the likelihood of overexpression of Atrimers in E.coli. The origin of the platform may shield it from the human immune system and its smaller size should favor better tissue penetration [101]. The modular nature of Atrimers makes them particularly suited to engage with trimeric targets, many of which include proteins of therapeutic interest.
2.2. Scaffolds Containing Rigid Combinatorial Motifs
In contrast to the “loop on a frame” design another major class of scaffold motifs utilizes permutations of residues embedded into rigid, regular secondary structural elements at positions that can tolerate side chain replacements without significantly destabilizing the structure. The introduction of binding sites into relatively rigid secondary structure elements is considered advantageous because of the reduced entropy cost for structural rearrangement of the binding domain upon target interaction relative to those that may occur with the flexible binding structures generated in hypervariable or single loops [102]. The following examples provide the most recent developments of this scaffold concept.
2.2.1. Darpins
DARPins (Designed Ankyrin Repeat Proteins) were introduced as a scaffold design by the Plückthun group in 2003 [103-107]. The Ankyrin repeat proteins are one of the most abundant binding proteins in the human proteome. The natural function of Ankyrin repeat is to engage its target prompting enzyme inhibition, or simply anchoring proteins together. Ankyrin repeat (AR) proteins are assembled from repeats of usually 33 residues with no cysteines, each forming a structural unit comprised of two antiparallel α-helices joined by a short β-turn (Fig. 3H). DARPins are an ensemble of repetitive structural modules (including several randomized design AR units) that forms a stable protein framework with a large potential target interaction surface. AR modules are held together by a hydrophobic interface, with the first (N-cap) and last (C-cap) repeat being different to present a hydrophilic outer surface, which would otherwise negatively affect folding and aggregating properties [103]. With cap ping modules, DARPins of 4, 5 and 6 repeats have molecular weights of 14, 18 and 22 kDa correspondingly, about one tenth the size of an antibody. They express very well in E. coli as soluble monomers (up to several g/L in fermentation), and because their stability increases with length, some of them become resistant even to denaturation by boiling or guanidine hydrochloride [108]. Deuterium exchange experiments on “full-consensus” DARPins indicate that this high stability is due to strong coupling between repeats. Some amide protons require more than a year to exchange at 37 °C [109], highlighting the extraordinary stability of the proteins. The location of these very slowly exchanging protons indicates a very stable core structure resulting from a combination of hydrophobic shielding coupled with favorable electrostatic interactions [107].
Following the same modular assembly principle as those established for Avimers, a single DARPin can be directed against different epitopes on the same target or against several targets; up to six positions can be diversified in each module as is the case with bispecific DARPins [110]. A number of randomized DARPin libraries have been established, with binder selection being performed through ribosome or phage display technologies. Biomedical applications utilizing DARPins, discussed in detail below, have proven to be very successful [63, 107, 111, 112]. Further extending the concept of DARPin architecture, the Plückthun group proposed the idea of Loop DARPins, with continuous protracted loops mimicking the antibody long CDR-H3 region grafted onto the scaffold [107, 113]. This expands the scope of possible epitopes that can be targeted without sacrificing the stability of the scaffold. Novel designs yielded binders against BCL-2, an important regulator of the apoptosis pathway, with 30 pM affinity after only one round of Loop DARPins library screening.
2.2.2. Affibodies
Affibody molecules, presented in 1997 by Nord et al. [114, 115], are another example of a protein interaction surface within a structural framework. The scaffold design is based on the Z-domain (the IgG binding domain) of Staphylococcus aureus protein A. The Z-domain is a small, ∼6 kDa (58 amino acids), three α-helix bundle protein, consisting of a single polypeptide chain (Fig. 3I). The domain does not contain cysteine residues, folds rapidly, resists proteolysis, and can be overexpressed in soluble form in various host cells [116]. The small size makes chemical synthesis of the molecule feasible, and modifications introduced into Z domains allow for directional head-to-tail polymerization, facilitating connection of multiple domains for increased avidity. Typically, randomization of 13 solvent accessible amino acids on the surface of helices 1 and 2 is used to generate novel combinatorial libraries.
2.2.3. Affilins
The Affilin scaffold design was announced in 2005 [117]. It comes in two forms: The first platform is based on γ-B Crystallin, a 20 kDa protein (176 amino acids) from a family of structural proteins found in the eye lens and cornea of vertebrates, and the second on Ubiquitin, a highly conserved abundant 8.5 kDa (76 amino acids) eukaryotic protein used by cells for posttranslational modification. γ-B Crystalline consists of two identical domains with mainly β-sheet structure (Fig. 3J). Ubiquitin consists of three and a half α-turns and a five stranded β-sheet (Fig. 3K). β-sheets within both structures provide the binding surfaces, of which eight near-surface residues in γ-B Crystallin and six in Ubiquitin are suitable for modification [63]. Reevaluation of the platform revealed the benefits of using dimeric, head-to-tail, Ubiquitin units in the library, and a more extensive randomization of residues, five in the ²-sheet and four in the ²-turn [102]. Changes introduced by diversifying those positions do not cause any significant loss of stability. In fact, some of the binders identified in library screenings demonstrated remarkable stability to pH changes, thermal denaturation and high concentrations of denaturing agents [102, 117-121].
2.2.4. Armadillo Repeat
Armadillo repeat proteins (ArmRPs) have been in the works for a while (see [122] for review), but the first library based on this scaffold appeared in 2012 [123]. The name derives from the appearance of a Drosophila mutant defective in a segment polarity protein later identified as Drosophila β-Catenin. ArmRPs are abundant in eukaryotes and take part in signaling, nucleocytoplasmic transport and cell adhesion. Unlike most of the platforms discussed above, ArmRP is a modular peptide-binding scaffold developed for the purpose of binding to folded proteins, in which the unstructured regions can act as linear peptide targets. Such transient and generally low-affinity but highly specific interactions between a globular protein and short linear peptide regions have been found in many highly dynamic cellular networks involved in signaling, regulation, and protein trafficking, and may constitute 15-40% of all cellular interactions [122].
The structure of natural ArmRPs is a right-handed superhelix or solenoid, formed by 4-12 stacked tandem Armadillo repeat motifs, each consisting of approximately 42 amino acids folded into three α-helices, H1, H2, and H3 (Fig. 3L). The binding mode is highly conserved. A polypeptide in extended conformation is bound in an antiparallel orientation relative to the ArmRP, forming an asymmetric double helix. This antiparallel binding mode is maintained by an array of conserved asparagine residues that hydrogen bond to the backbone of the extended peptide [122]. The affinities of ArmRPs for their ligands extend to the subnanomolar range [123]. Similar to the DARPin design, specially constructed N- and C-terminal capping repeats were introduced to shield the hydrophobic core of the ArmRPs assembly, dramatically increasing in vivo folding and preventing aggregation [124]. Randomizable positions for a combinatorial library were identified by analyzing natural complexes of Armadillo proteins with their targets. Four residues in H3, one in H1 and one in the loop following H3 were targeted. Two nonrandomized internal repeats flanking the three randomized repeats were introduced into the library to improve stability and folding characteristics [123]. Model screening of the library based on the ArmRPs scaffold was directed against Neurotensin, a 13 amino acid neuropeptide ligand of the G-protein coupled Neurotensin receptor. Specific peptide binders were selected after four rounds using ribosome display. All were highly overexpressed in E.coli, monomeric in solution, and exhibited affinities of 7 μM [123].
2.2.5. OBodies
The oligonucleotide binding (OB)-fold anticodon recognition domain from Pyrobaculum aerophilum Aspartyl tRNA Synthetase is utilized in OBodies. The OB-fold is a small, single domain binding element, generally not containing disulfide linkages and comprising a 5-stranded β-barrel that binds ligands via a concave interaction surface. OB-folds exist in many organisms, including archaea, bacteria, yeast and mammals, and exhibit no sequence conservation. A common feature of the OB-folds is a rather characteristic structure formed from the combination of concave β-sheets and loops, with the ability to accommodate very diverse sequences and modifications. OB-folds are capable of binding to a broad range of ligands, including oligonucleotides, oli-gosaccharides, proteins, and small molecules. Construction and screening of a combinatorial library based on this scaffold design using phage display yielded an OBody with 3 nM affinity for hen egg white lysozyme [125].
3. Selection Techniques
It is a daunting task to identify and isolate high affinity ligands from combinatorial libraries. A large and continually expanding arsenal of powerful screening methods and techniques have been developed to assist researchers in this endeavor [126].
In the SELEX method, strongly binding NA aptamers can be easily amplified by PCR for subsequent rounds of selection, and identified through DNA sequencing. Similarly, in order to allow for the repeated cycles of selection-amplification, phenotypically selected combinatorial peptide molecules must be identifiable and amplifiable through the linked genetic information. This can be accomplished either by directly linking a related nucleic acid with a peptide molecule, or by confining both in the closed environment of a cell, virus particle or artificial microvesicle (Fig. 4). Selection technologies can be divided into two classes. The first and most widely used approach is selection through extracellular display, such as the surface of cells, filamentous phages, or in vitro. The second is in vivo screening represented by a plethora of in-cell protein complementation assays, including two-hybrid systems, in which the target and ligand are both produced and interact inside a living cell.
Fig. (4).
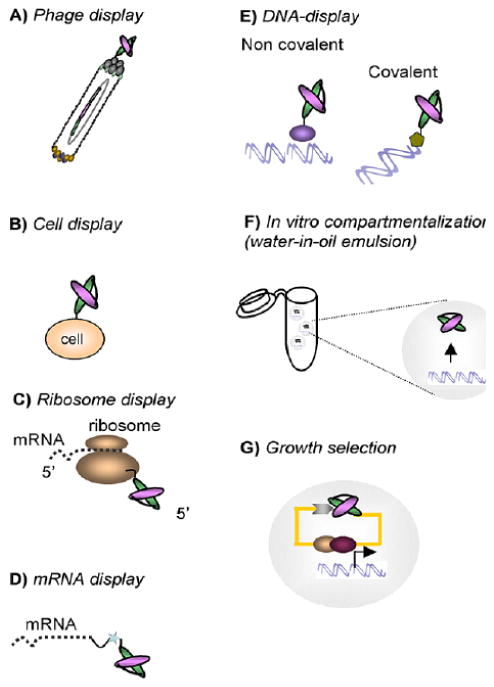
Schematic representation of PA library screening platforms providing the phenotype-genotype linking. A) PA fragment is displayed at the surface of the phage particle via fusion with a phage coat protein, usually pIII, or B) on the surface of bacterial, yeast and mammalian cells. C) Ribosome provides a non-covalent link between the nascent protein and mRNA in vitro. D) More stable puromycine covalent linker directly binds novel protein with its encoding mRNA. E) In DNA-display, the DNA-protein linkage is established either non-covalently through the bacterial RepA protein, or covalently with a bacteriophage P2 protein which binds to its own DNA sequence. F) In vitro compartmentalization ensures that each DNA molecule gets is own confined space within a microdroplet linking to synthesized proteins through affinity tags. G) In-cell growth selection systems are based on the protein fragment complementation, with Yeast-two Hybrid technology most often used. In vivo selection procedures rely on a phenotypical change (growth advantage and/or color readout) induced by interaction of target protein with a library member. Adapted from (126).
3.1. In vitro Displays
3.1.1. Phage Display
Phage display is a method of presenting polypeptides on the surface of bacteriophage to select for the ability of the polypeptide to bind preselected targets (Fig. 4A). This technique was introduced by Smith in 1985 [127] to map epitope binding sites of immunoglobulins by panning phage libraries of random peptides onto immobilized antibodies. It was shown that bacteriophage genomes can be engineered to accommodate designed combinatorial libraries. The genome can tolerate fairly large inserts into coat proteins which then present novel polypeptide chains on the phage surface. The most commonly used vectors for phage display are filamentous phage M13 and the closely related strains fd and f1. The single stranded circular DNA genome of M13 is covered by ∼2700 copies (for a wild type) of the major coat protein pVIII and capped with five copies each of minor coat proteins pIII, pVI and pVII, and pIX at opposite ends of the capsid [128]. All of the coat proteins were tested for the ability to display foreign polypeptides on the phage surface [129, 130], with pIII and pVIII receiving the most attention. The smaller size of coat protein pVIII (50 amino acids) limits inserts to 6-8 residues, but because they are displayed in thousands of copies on the surface the potential avidity of the interactions are greatly increased. The larger coat protein pIII (406 amino acids) can accommodate inserts of 100 residues and more that are displayed in limited numbers on the ends of the phage particle, favoring selection of stronger binders.
Libraries of diversified antibody fragments or peptidic combinatorial libraries built on artificial scaffolds can be introduced directly into the genome of M13 phage (6.4 kb) by fusing to the gene of the appropriate coat protein, usually at the 5' end. The resulting construct is transferred to E.coli where it is amplified, translated, replicated, and packed into newly assembled M13 phage particles, which escape the cell. During assembly the fusion proteins are displayed on the phage surface while the encoding ssDNA is packed within the same phage particle. Next the phage population is presented against the target of interest (biopanning), non-bound particles are washed away. After eluting the bound phage it is re-amplified in E.coli for the next round of selection; strong binding candidates are identified in 3-10 cycles. The high transformation efficiencies of E.coli allow the construction of large phage libraries of 1010-1012 unique clones. But the biggest advantage of the phage display method, accounting for its ever-growing acceptance, is the efficiency and convenience of library screening combined with the inherent flexibility of the selection process [131]. An assortment of combinatorial phage display libraries based on variable 6-30 residues pep-tide loops embedded directly into coat proteins [129], on antibody scFv- and Fab-fragments [132], and on protein scaffolds, such as Kunitz domain, Lipocalins, Anticalins and Affibodies [16, 59, 63, 84, 132], have been designed and successfully screened. An interesting extension of this technique was introduced by the Liu group [133-135]. The which is called Phage Assisted Continuous Evolution (PACE), allows for nonstop rounds of selection with very little operator involvement and adjustable selection stringency (via anhydrotetracycline). The was tested for evolution of RNA polymerases to different promoter specificities, and successfully produced an enzyme with a 10000-fold net change in sequence preferences.
The limitations of phage displays result from producing large or structurally incompatible fusions with the coat protein, which results in compromised assembly of the phage particles and an impeded growth cycle. The development of the phagemid system [136], a hybrid of M13 and plasmid DNA packaged as single stranded DNA in viral particles with the presence of a helper phage, allows the display of a mosaic of both recombinant and wild type coat proteins thus attenuating possible defects in phage function. Alternative display systems that use lytic phages, e.g. T4 [137-139] and T7 [140, 141], do not depend on the ability of fusion proteins to be translocated across the bacterial membrane for phage assembly. The phagemid system allows expressed protein to be fused at the carboxy terminus rather than the amino terminus, but its usage is limited thus far to displaying small peptides and cDNA libraries. Lambda display has been suggested as an alternative for low solubility proteins that tend to form in inclusion bodies when expressed in bacteria [142], and is mostly used for protein display from cDNA libraries [142-145] Advantages of the lambda phage display are the ability to utilize multimeric proteins and no need to secrete fusion proteins. Efforts to adapt phage display technology to eukaryotic viruses are surveyed in [131].
One of the interesting applications of phage display technology is panning live cell cultures, or even whole organs. The successful selection of peptides targeting vascular endothelial cells in gastric cancer [146] and of a VEGF-mimic [147] illustrates the validity of this approach. In vivo selection is used to identify tissue- or organ-specific ligands, which results in the segregation of the phage particles from the intravenously administered library. When recovered from the organ of choice, specific ligands tend to cluster in particular tissues while the non-selective phage will be spread evenly throughout the organism. Such organ or tissue specific ligands have great potential as diagnostic tools or as components of drug delivery platforms [129].
3.1.2. Cell Surface Display
Cell surface display screening is similar to phage display, but uses the whole cell as a vehicle to display diversified antibodies or combinatorial peptide libraries (Fig. 4B). The method finds its application in affinity maturation of preselected binders, and screening is usually coupled with magnetic- or fluorescence-activated cell sorting [148]. The idea of using the bacterial surface for the library display has been around since 1997 [149]. Both gram negative (mostly E.coli [150]) and gram positive hosts (from Staphylococcus) have been used. A number of outer membrane proteins including OmpA, OmpC, Porins, and autotransporters, were tested as display anchors, with some of them being able to tolerate inserts exceeding 100 residues [131]. A fusion construct is delivered to the cell on a plasmid and, due to the high efficiency of E.coli transformation, library sizes of 109-1011 can be achieved [129]. The development of periplasmic bacterial display [151] facilitated the assembly and display of full length antibodies in the E.coli periplasm, allowing for spheroplast screening through thew binding of fluorescently labeled ligand. Compared to bacterial surface display, yeast have fewer problems regarding proper protein folding, post-translational processing and correct assembly on the cell surface. Pioneered by Boder and Wittrup in 1997 [152], yeast display works through the fusion of a foreign DNA fragment with the sequence of a cell wall mannoprotein, in which expressed protein is delivered to the cell wall and secured to the cell surface through its C-terminal glycosylphosphatidylinositol (GPI) anchor. A few cell wall proteins have proven capable of displaying foreign proteins on the cell surface, including a-agglutinin [152], Flo1p [153, 154], and α -agglutinin [155]. A-agglutinin is one of the two mating factor-specific agglutinins that facilitate intercellular contacts during mating, it is composed of two subunits, Aga1p, which is covalently anchored to the cell wall matrix through GPI, and the smaller Aga2p subunit, which is linked to the core Aga1p by two disulfide bridges. Driven by the Gal1 promoter, engineered Aga2p can express 104-105 copies of recombinant protein on the surface of each yeast cell. Although the somewhat lower transformation efficiency in yeast, compared to E. coli, makes construction of larger naïve libraries difficult, and the protein glycosylation pattern can differ from that of mammals, the use of yeast display for affinity maturation has proven to be a powerful tool [148]. A comparative evaluation of yeast and phage display performed in 2007 [156] revealed that the sampling of a given small-size library by yeast display was more comprehensive and provided more high affinity (nanomolar) binders than by phage display, which is particularly important for isolating rare combinatorial molecules from large libraries.
Features important for the production of proteins with complex folds, reduced aggregation and correct glycosylation can only be implemented using a higher eukaryotic system. Successful display of single-chain antibodies on the surface of HEK293T cells demonstrated in 2006 by Ho et al. [157], established an important breakthrough in mammalian display technology and is more extensively discussed in [148, 158, 159].
It should be mentioned that all the cell-based display platforms described above are limited by the library size, which is determined by DNA transformation efficiency. The toxicity of the combinatorial library proteins to the host cell can also be a problem. These limitations are alleviated in cell free display systems that can support larger libraries and eliminate the toxicity problem. The most well-developed methods use ribosomal, mRNA, and covalent DNA cell-free systems.
3.1.3. Ribosome Display
The idea of ribosome display capitalizes on the formation of a stable ternary complex between mRNA, ribosome and the novel polypeptide chain during translation, when the lack of a terminal stop codon prevent release factors from binding and triggering the disassembly of the translational complex (Fig. 4C). It was first introduced by Mattheakis et al. in 1994 [160] and subsequently adapted for selection by Hanes and Plückthun in 1997 [161]. Usually, a mixture of mRNAs encoding the combinatorial peptide library with a ribosome binding site at 5' end and the spacer without stop codon at 3' end, is prepared and then translated in vitro. Spacer sequence stays attached to the peptidyl tRNA and occupies the ribosomal tunnel, effectively stalling off translation and providing a non-covalent complex of mRNA, nascent polypeptide and the ribosome. The complex is panned against the desired target in a manner similar to phage display selection; alternatively, the combination of fluorescently labeled target and in vitro compartmentalization permits the selection by cell sorting (see below). Selected ternary complexes are dissociated by adding ethylenediaminetetraacetic acid, EDTA, and the recovered binders are reverse-transcribed and amplified by PCR for analysis or further selection. Because the ternary complexes are highly labile, the selections are performed at low temperatures and at high concentrations of magnesium. The vulnerability of RNA and polypeptides to nucleases and proteases present in E. coli or eukaryotic cell extracts used for in vitro translation prompted the creation of a minimal system containing a bare bones set of recombinant E. coli protein factors and purified 70S ribosomes [50]. This system increased the stability and yield of the ribosomal display. Since cell-free synthesis creates the possibility of generating large pools of polypeptides, with a diversity of 1012-1014 clones, ribosomal display has become a valuable method to search for high affinity compounds from diverse libraries. One example is the discovery of high-affinity binders to MAP-kinases selected through screening of the combinatorial library, embedded within a DARPin scaffold, by means of a ribosome-display [162].
3.1.4. mRNA Display
Another cell-free display system introduced in 1997 is the mRNA display, in which mRNA is covalently attached to a cognate polypeptide by a linker containing the antibiotic puromycin (Fig. 4D). The result is a high stability complex [163, 164]. A diversified pool of mRNAs is first modified by attaching a short DNA linker conjugated with puromycin at the 3' end. When the ribosome stalls at the junction during translation, puromycin enters the ribosome A site and forms a covalent bond with the nascent peptide. Very similar to ribosome display in terms of selection and amplification, mRNA display exhibits increased stability of the formed complex, which translates into improved handling and speed. The size of the library is limited by the number of ribosomes present in the extract, and can reach 1012-1014 unique molecules. In vitro techniques allow rapid and convenient monitoring of the selection process through deep sequencing [165], and introduce additional diversity by using error-prone PCR amplification or by incorporating non-naturally occurring amino acids during the translation step [166]. Due to the inherent limitations of in vitro translation, cell free display methods are not very efficient in presenting polypeptides of more than 300 amino acids or multiple chains. Nonetheless, the methods are still an excellent choice for screening large libraries of single chain antibodies and designs based on non-antibody scaffolds, i.e. peptide aptamers. Successful generation of libraries based on Adnectins [167], DARPins [168], Fynomers [97, 165], Armadillo repeat proteins [123] and other scaffolds have been reported [169-172]. One of the recent modifications of mRNA display technology is the ‘TRAP display’, in which the puromycin linker is attached via base pairing, instead of covalent modification, shortening a round of selection to just 2.5 hours compared to the usual 2-3 days [173].
3.1.5. DNA Display
In a further development of the mRNA display method, conversion of mRNA to cDNA was introduced to avoid problems with nuclease-driven RNA degradation, thus creating DNA display technology [174, 175], which was successfully applied to the evolution of disulfide-rich peptide ap-tamers [176] (Fig. 4E). In modern practice mRNA gets reverse transcribed after translation, forming an RNA-DNA complex attached to a polypeptide, which then goes through selection and PCR amplification directly (mRNA display) or is digested with RNAse H before selection (cDNA display) [177].
An interesting twist in methodology aimed at increasing the chemical stability of the panning complexes is the proprietary in vitro library display platform named covalent display technology [178]. It exploits the unusual properties of a replication initiator protein from E. coli bacteriophage P2, which is also an endonuclease. The gene coding for this protein (P2A) is placed downstream from the randomized library construct. After transcription and translation, the newly formed P2A fused at the C-terminus of polypeptide chain nicks its own gene and covalently attaches to the exposed 5' phosphate through the tyrosine residue in the active site of P2A. Once this link to the genotype is established, screening and selection of binding candidates is accomplished in a manner similar to the other in vitro display methods. This platform was successfully used to screen a model singlechain antibody library with 107 complexity [179], but it did not get as much attention as other display methods. A similar technology named CIS display utilizes the properties of a DNA replication initiator protein, RepA, to bind non-covalently to a unique template DNA from which it has been expressed [180]. It was shown that the resulting complexes are stable enough for high affinity ligand selection and that libraries encoding >1012 random 18-mer peptides can be con structed and used to isolate specific binders.
3.1.6. In vitro Compartmentalization
Another approach that maintains a genotype-phenotype link during selection rounds and provides more flexibility to translated proteins is in vitro compartmentalization (IVC) (Fig. 4F). Water-oil emulsion microdroplets 2 μm in diameter are used as artificial cell-mimics. Such constructs can be filled with a single copy of library DNA and components of transcription and translation machinery. These model systems can synthesize up to 1010 copies of individually compartmentalized proteins in a 50 μL reaction volume [181-184]. Several methods are employed to maintain the connection between protein and its encoding DNA.
Since the requirement of immediate linkage of a nascent polypeptide to its mRNA or DNA is alleviated, conventional affinity pairs like biotin-streptavidin can be used (STABLE display [185, 186]). Alternatively, a covalent link can be established by fusing a polypeptide with the constant domain of DNA methyltransferase, M. HaeIII, which attaches to the end of the coding DNA through a suicide inhibitor (5-fluorodeoxycytidine) [97], or through a fusion with human DNA repair protein O6-alkylguanine-DNA alkyltransferase (hAGT), which binds to benzylguanine (BG)-modified DNA strand [182, 187]. Subsequent selection is performed when the emulsion is disrupted, followed by amplification, encapsulation and more screening rounds if needed. The technique can be further modified by introducing microbeads into the emulsion microdroplets [188, 189]. By means of emulsion PCR (ePCR) the DNA in the microdroplets can be amplified from single to multiple copies [190-192] with subsequent attachment to the bead surface. Breaking and recreating emulsions for purposes of buffer exchange and adding new reaction components, without allowing different library members and their products to intermix, now becomes possible. Fluorescently-labeled targets [187] permit IVC technology to employ custom microfluidic chips or cell sorters [193]) to rank affinity binders. While conventional panning requires labor intensive biophysical analysis, flow cytometry provides the opportunity for fast (up to 1.5×104 droplets per second) analysis of the sample and allows for direct selection and ranking of the binders based on their relative affinities [187]. Since IVC technology allows for disengagement of protein from negatively charged RNA/DNA molecules and bulky ribosomes, which might interfere with binding and/or catalytic functions, it is believed to be particularly well-suited for enzyme selection and evolution through rapid cycles of selection and recovery (see refs [166, 181, 184, 192, 194, 195] for discussion). Among the current problems of IVC technology are droplet size, uniformity of DNA loading, and the possibility of droplet fusion [194].
3.1.7. Advantages and Disadvantages of In vitro Selection
There are several reviews discussing the advantages and limitations of in vitro display methods [166, 170, 181, 196], of which the large library size and independence from cellular physiology are the main benefits. Restrictions in the size of the investigated protein, the inability to deal with protein complexes and membrane components, as well as difficulties of working in an RNAse-free environment [197] are the main limitations. Some problems are intrinsic to all extracellular display methods that employ multiple amplification/selection rounds. Thus, binding to components of the screening system other than the target molecule, which generates Target Unrelated Peptides (TUPs) [198, 199], or the accumulation of phage clones with propagation advantages can severely affect library diversity [200-202]. In vitro selection usually requires a substantial amount of purified target to be obtained ahead of the screening, which is not always easily accomplished. Selection is carried out outside the cell environment, which might lead to the absence of proper folding or post-translational modifications, but probably the biggest concern is the potential loss of peptide binders arising from intermolecular competition between many potential ligands for a limited number of binding sites on the target [203]. In vitro competition between molecules leads to the “the survival of the fittest”, in which only the few highest affinity binders remain after final selection [204], and a number of weaker binders with potentially superior characteristics might be missed. Some of these issues were addressed by using single round high stringency selection with subsequent examination of selected clones by high throughput sequencing and population level statistical analysis [165, 166, 205], but some issues still require attention.
3.2. In-cell (Growth) Selection
In-cell selection, represented by various forms of protein fragment complementation assays like Yeast two Hybrid (Y2H), provides several advantages over screens based on extracellular interactions (Fig. 4G). Yeast are eukaryotes, which allows for N-linked glycosylation and oxidative protein folding, ensuring proper formation of disulfide bonds Within the intracellular setting proteins have a higher chance of proper assembly and interaction, with no need for previously prepared and purified targets. An important feature of in-cell selection is the lack of competition for each peptide aptamer expressed within a given cell to bind to a target protein [203], which makes possible isolation of combinatorial peptides with different affinities and distinct binding sites within the same target. Several methods designed for evaluating protein-protein interaction within the environment of living cells are employed for screening peptide aptamer libraries.
The central idea behind protein complementation is to identify a protein that does not function when split apart, but can reconstitute its function when the parts are brought back together. The concept of the Yeast two Hybrid (Y2H) screen, originally introduced by Fields and Song [206], is based on reconstituting the modular transcription factor GAL4, which drives the expression of marker genes in the nucleus of Saccharomyces cerevisiae [206]. The binding domain (BD) of Gal4 (amino acids 1 to 147) recognizes the upstream activating sequence (UAS) of the GAL1 promoter, while the C-terminal activation domain (AD) (residues 768 to 881) activates the transcriptional machinery. Although the domains are functionally independent, only together they can initiate transcription. Two known interacting yeast proteins Snf1 and Snf4 were used in the study to create fusions with the Gal4 BD and Gal4 AD, correspondingly termed the ‘bait’ and the ‘prey’. When transformed on separate plasmids into a yeast strain harboring a lacZ reporter gene under the control of the Gal1 promoter, the physical interaction between the baitprey pair reconstructs the functional transcription factor, resulting in reporter gene expression and a color readout. This general principle was employed by Brent [37] in designing the first peptide aptamer library using fusions of Thioredoxin-constrained peptides with Gal4 AD as a prey and Cdk2-fused BD as bait. This was later followed by construction and Y2H screening of more peptide aptamer libraries with scaffold-constrained and linear peptides [57].
Improvements in methodology brought new reporter genes like MEL1, gusA, lacA3, EGFP, HIS3, LEU2, URA3, ADE2, LYS2, and Aureobasidin A resistance gene; more prey AD options, such as Herpes simplex virus VP16 activating region, E.coli B42 polypeptide, and more options for bait BDs, E.coli repressor LexA BD, human estrogen receptor BD, bacteriophage λ cI repressor and Tet repressor (summarized in [207]). During library construction both the target protein-BD fusion and PA library-BD fusions are modified with nuclear localization sequences at the N-termini and placed under the constitutively active ADH1 promoter. Plasmids are maintained with different prototrophic markers, like LEU2 and TRP1, to track retention by the yeast cell. The resulting vectors are delivered either by simultaneous transformation of the cells, or through separate transformation of different haploid strains followed by mating [208] and plating on corresponding selection medium. Bait and prey fusions, expressed from plasmids inside the cell, are translocated into the cell nucleus where interaction of the target protein with the library member is manifested through growth advantage (colony formation) or color development. Analysis and confirmation of interactors is accomplished by using standard laboratory techniques [208, 209]. In addition to Y2H, a number of other methods for studying proteinprotein interactions are based on protein complementation techniques (reviewed in [207, 210]).
The modest transformation rates of most yeast strains (∼106 cfu/μg of input DNA) and high rates of false positives [210-212] are the predominant drawbacks of Y2H library screening. The problem of false positives can be partially relieved by placing multiple reporter readouts into the yeast to detect bait-prey interactions, and by introducing galactoseinducible prey expression, like in the LexA interaction trap system [57, 208]. The need to increase the library size can be addressed by improving yeast transformation methods [213], or by increasing the scale of the transformation. In our lab we obtained the necessary amounts of input DNA (Fig. 2) by first amplifying initial ligation mixes in vitro by using Phi29 DNA polymerase to produce hundreds of micrograms of plasmid DNA, then further amplifying the DNA by propagation in E. coli [47].
The discovery of interactors after extensive library screening is just the first step on a long road. Comprehensive biophysical and biochemical characterization of the binding molecules, confirmation of their target recognition abilities, and ideally the detailed atomic resolution of the binding interface is required [214] Molecules with moderate affinity to the target or with unfavorable characteristics (low solubility, aggregation, toxicity, etc) are subjected to several rounds of affinity maturation under conditions of increased stringency with special emphasis on selecting high stability compounds. This is usually carried out through random mutagenesis of variable regions (sometimes involving the scaffold), or by applying more focused approaches guided by rational computer aided design based on structural data and the results of comprehensive mutational scanning, generating heatmaps of antigen-binding enrichment ratios [166, 214-220]. Quite often maturation rounds are performed in different library screening settings and display methods used during primary selection rounds can be complemented by Y2H for affinity evolution. Besides random mutagenesis, non-naturally occurring amino acids and various chemical modifications can be used to increase the interaction space and library complexity [23, 166, 221]. Although requiring sufficient resources and time investment, affinity maturation generally appears to provide the desired improvements in protein characteristics, sometimes producing extraordinary results. Only then are the evolved high affinity products ready for testing in biomedical or industrial settings.
4. Applications
The advance of monoclonal antibodies ignited a growing interest in creating new molecules that retain the high affinity and specificity of antibodies together with low toxicity and low immunogenicity, and are faster, easier and less expensive to discover and manufacture. The flexibility of PA platforms permits a variety of binding surfaces capable of accommodating large flat protein-protein interfaces as well as the clefts and pockets targeted by small molecules [220]. This fast growing class of antibody mimetics is finding its way into the field of biomedical and bioanalytical applications dominated by antibodies and nucleic acids aptamers. Most of the efforts directed at developing alternative scaffold designs are concentrated in the area of therapeutic and diagnostic applications [15, 63, 222, 223]. Below are a few examples.
4.1. Biomedical Applications
4.1.1. Adnectins
A bispecific molecule based on two individual Adnectins with affinities of 10 nM for epidermal growth factor receptor (EGFR) and 1 nM for insulin-like growth factor receptor-1 (IGF1R) was designed to inhibit cross-talk between receptors in cancer cells [224]. Fragments were connected through a short linker and PEGylated. The hybrid molecule successfully inhibited phosphorylation and downstream signaling of both receptors, induced receptor degradation and decreased proliferation of several human cancer cell lines. Another molecule built on an Adnectin scaffold, Angiocept1 (CT-322), is the first Adnectin tested in humans, paving the way for a potential treatment for glioblastoma, pancreatic cancer and other oncological conditions. It targets the VEGFR2 pathway by strongly binding to receptor (Kd=11 nM) and effectively inhibiting its signaling in human umbilical vein endothelial cells. The growth of a broad range of human tumor xenografts was inhibited by CT-322 when tested in mouse models [225], CT-322 demonstrated marked antitumor activity and low toxicity in patients with advanced stage solid tumors when administered at 2 mg/kg [15].
A unique scaffold based on the 14th extracellular domain of human fibronectin III is a platform for the proprietary Pronectin design. The most advanced Pronectins, directed against various cellular antigens, including AXL (metastatic cancer), VEGF-R2 (macular degeneration, various cancers), and Frizzled receptors (stem cell differentiation and cancer) are in preclinical development [15].
4.1.2. Anticalins
Great potential for the Anticalin platform was revealed after screening the human lcn2-based library produced Anticalins with subnanomolar affinities against the extracellular domain of cytotoxic T-lymphocyte associated antigen 4 (CTLA-4, CD152). Selected compounds demonstrated potent blocking activity and showed an immunostimulatory effect on the T-cell response in an animal model [226]. The crystal structure of the extracellular domains of CTLA4 and the Anticalin binder demonstrated that although all four randomized loops contributed to the binding interface, three of them exhibited a induced fit relative to the uncomplexed form [226]. Other Anticalin drug candidates are Angiocel (PRS- 050), targeted against the vascular endothelial growth factor (VEGF-A), and PRS-110 directed against the hepatocyte growth factor receptor (HGFR; c-Met proto-oncogene), both have binding affinities in the nanomolar to picomolar range. Angiocel as a potential treatment for a number of solid tumors has been recommended to enter clinical Phase II [87].
The unique ability of Anticalins to target small molecules like the cardiac steroid digoxigenin with high affinity and selectivity [85] can be utilized, for example, in developing antidotes for neutralization of digitalis and other intoxications, or as cargo vehicles for delivering various payloads to specific targets. Another potential application involves selectively blocking receptors by scavenging their small molecule ligands or inhibiting access to the binding sites [63]. There are a few other Anticalin projects in the discovery phase, with one of them, administered by inhalation, targeting Interleukin-4 receptor (IL4α) for the treatment of asthma [222].
4.1.3. Kunitz PAs
Screening of a phage display library based on the human lipoprotein-associated coagulation inhibitor (LACI) of the Kunitz domain family yielded the compound DX-88, which was approved by the FDA in 2009 as the drug Kalbitor (Ecallantide), the only product based on a non-Ig scaffold that has been successfully commercialized to date. DX-88 is an inhibitor of plasma protease Kallikrein, and is used for the treatment of a life-threatening disorder, hereditary angioedema (HAE), and the prevention of blood loss in cardiothoracic surgery. In the development of this drug, the principle of iterative library screening was employed, where the high affinity interactors selected from the library with mutated primary loops were the foundation for the next library created by varying a second region. In the last round residues that were strongly selected in the initial library received limited variability, while the conserved positions received the majority of diversification. This eventually resulted in the development of an inhibitor to plasma Kallikrein with high specificity and potency (Ki= 25 pM) [132].
4.1.4. DARPins
The most advanced development using DARPin resulted in a highly potent VEGF-A inhibitor (IC50 < 10 pM) as a possible treatment for various ophthalmologic diseases including age-related macular degeneration and diabetic macular edema [227]. The molecule was evaluated in clinical trials and shown to be safe and well tolerated by patients [222]. Another example is the development of a DARPin targeted against human epidermal growth factor 2 (HER2), which after several rounds of affinity maturation showed a remarkable strong binding constant of 90 pM [227]. To create a potent antagonist for epidermal growth-factor receptor (EGFR), commonly overexpressed on surface of many human cancer cells, two pairs of DARPins with low nanomolar affinities were joined to form a tetravalent, bispecific molecule which proved to be more effective in inhibiting A431 tumor cell proliferation than the antibody drug Cetuximab [112]. In developing a model for an affinity-mediated drug delivery system, DARPins directed against epithelial cellular adhesion molecules (EpCAM) was attached to a truncated version of Pseudomonas exotoxin [228]. During in vitro experiments inhibition IC50 ranged between 0.005 pM and 0.7 pM for EpCAM-positive tumor cells, while 10,000-fold higher values for EpCAM-negative tumor cells were observed [63].
4.1.5. Avimers
The most advanced Avimer product candidate AMG-220 (C326) neutralizes the proinflammatory cytokine interleukin-6 as a possible treatment for Crohn's disease. AMG-220 demonstrated picomolar binding affinity for IL-6 [90] and subpicomolar potency in an IL-6-stimulated TF-1 leukemia cell proliferation assay. Although phase 1 testing was successfully completed, clinical development of the molecule has stopped [15].
4.1.6. Atrimers
An example of a successful implementation of an Atrimer platform is the discovery of inhibitor of human tumor necrosis factor (TNFα), which was selected from a phage display library where loops 1 and 4 of Atrimer scaffold were randomized. Subsequent maturation steps ,which included more randomization in loops 1,4 and 3 plus selection strategy favoring the clones with low off-rates, led to nanomolar binders and a 200-fold improvement in the biological activity of Tetranectin trimers over monomers after the third round of maturation [100].
4.1.7. Affilins
The Affilin scaffold-based design allowed for the successful selection of binding molecules generated against the fibronectin Extradomain B (ED-B). This is a domain of a Fibronectin isoform normally absent in most adult tissues, but specifically expressed during wound healing, inflammation and in most cancers. Primary screening of a phage display library composed of Ubiquitin dimers was followed by ribosome display affinity maturation, with evolved versions displaying affinities in the range of 30-200 pM [102].
4.2. Bioimaging Applications
4.2.1. Affibodies
Affibodies are considered to be the protein scaffold design most fit for in vivo imaging [223]. Derived from protein A, they tend to generate an immune response after being repeatedly administered to patients, making them poor therapeutic agents. As imaging tools they benefit from their small size and fast renal clearance. Screening phage display libraries produced several examples of high-affinity binders, like Affibodies targeting HER2 receptor (22 pM) [229], three different anti-epidermal growth factor receptor (EGFR) Affibodies (0.9-50 nM), and an Affibody engineered to 0.5 nM affinity for insulin-like growth factor type I receptor (IGF1R) [11]. In mice ovarian carcinoma xenografts demonstrated great selectivity of Affibody molecules for HER2 over healthy tissue both in vivo and in vitro. High tumor uptake and fast renal clearance allowed high contrast images to be taken as early as 1 hour after injection. After subsequent maturation rounds and more tests in mouse models, Affibody became the first non-antibody design for live cancer imaging submitted for clinical trials [230].
4.2.2. Knottins
Small cystine knot peptides, or Knottins is another design with great value for molecular imaging. Employing combinatorial techniques and directed evolution, two ligands based on this platform were selected against a series of Integrin domains which are overexpressed on the cell surfaces in cancerous tissues and are shown to facilitate angiogenesis and metastasis. The first is derived from the cystine knot trypsin inhibitor from Ecballium elaterium, and the second, based on the agouti-related protein Knottin, exhibited corresponding affinities of <5 nM and 44 nM against cultured cancer cells [96, 231]. More applications of Adnectins and DARPins designs in molecular imaging are reviewed in [232].
4.3. Bioanalytical Applications
Peptide aptamers find many applications in the fields of biomedical research, bioanalytics and biotechnology, and have already been discussed in several reviews [38, 56, 61, 87, 107, 233, 234]. FAffinity matrices based on Affibody and Affilin designs have been used to isolate several biologically important proteins [235], while PAs derived from Affibody and Anticalin scaffolds were tested as antibody alternatives in immunochemical assays [56]. Affibodies [233] and Adnectins [6] were tried in protein array formats, and the FRET system for detecting analytes in solution was developed on an Affibody platform [236]. A potential role in environmental monitoring was reported for Anticalins as they can bind small molecules with affinities up to 80 nM in biosensor prototypes [237]. Additionally, the specific binding of affinity ligands can often stabilize the target protein and facilitate high-resolution structure determination. In this regard small affinity binders based on DARPin and Affibody scaffolds have a clear advantage over bulky antibodies, and a number of complexes have been solved by X-ray crystallography and NMR technology [61].
Conclusion
Peptide aptamers represent a new class of biologically active molecules that have the potential to bind a given target in solution as well as under extracellular and intracellular conditions. A wide variety of PA scaffolds and selection schemes are available to satisfy the demanding applications required in the biomedical, bioimaging and bioanalytical fields.
Acknowledgments
This work was supported by NIH grant R01GM08500605.
Footnotes
Conflict of Interest: The authors confirm that this article content has no conflict of interest.
References
- 1.Nimjee SM, Rusconi CP, Sullenger BA. Aptamers: an emerging class of therapeutics. Annu Rev Med. 2005;56:555–583. doi: 10.1146/annurev.med.56.062904.144915. [DOI] [PubMed] [Google Scholar]
- 2.Silverstein A. Paul Ehrlich's Receptor Immunology: The Magnificent Obsession. Academic Press; London, UK: 2002. [Google Scholar]
- 3.Bosch F, Rosich L. The contributions of Paul Ehrlich to pharmacology: a tribute on the occasion of the centenary of his Nobel Prize. Pharmacology. 2008;82:171–179. doi: 10.1159/000149583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Buss NA, Henderson SJ, McFarlane M, Shenton JM, de Haan L. Monoclonal antibody therapeutics: history and future. Curr Opin Pharmacol. 2012;12:615–622. doi: 10.1016/j.coph.2012.08.001. [DOI] [PubMed] [Google Scholar]
- 5.Smith AJ. New Horizons in Therapeutic Antibody Discovery: Opportunities and Challenges versus Small-Molecule Therapeutics. J Biomol Screen. 2014 doi: 10.1177/1087057114562544. [DOI] [PubMed] [Google Scholar]
- 6.Shirai H, Prades C, Vita R, Marcatili P, Popovic B, Xu J, Overington JP, Hirayama K, Soga S, Tsunoyama K, Clark D, Lefranc MP, Ikeda K. Antibody informatics for drug discovery. Biochim Biophys Acta. 2014;1844:2002–2015. doi: 10.1016/j.bbapap.2014.07.006. [DOI] [PubMed] [Google Scholar]
- 7.Solier C, Langen H. Antibody-based proteomics and biomarker research - current status and limitations. Proteomics. 2014;14:774–783. doi: 10.1002/pmic.201300334. [DOI] [PubMed] [Google Scholar]
- 8.Vedi A, Ziegler DS. Antibody therapy for pediatric leukemia. Front Oncol. 2014;4:82. doi: 10.3389/fonc.2014.00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Winnall WR, Beasley MD, Center RJ, Parsons MS, Kiefel BR, Kent SJ. The maturation of antibody technology for the HIV epidemic. Immunol Cell Biol. 2014;92:570–577. doi: 10.1038/icb.2014.35. [DOI] [PubMed] [Google Scholar]
- 10.Byrne H, Conroy PJ, Whisstock JC, O'Kennedy RJ. A tale of two specificities: bispecific antibodies for therapeutic and diagnostic applications. Trends Biotechnol. 2013;31:621–632. doi: 10.1016/j.tibtech.2013.08.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Murad JP, Lin OA, Espinosa EV, Khasawneh FT. Current and experimental antibody-based therapeutics: insights, breakthroughs, setbacks and future directions. Curr Mol Med. 2013;13:165–178. [PubMed] [Google Scholar]
- 12.Sliwkowski MX, Mellman I. Antibody therapeutics in cancer. Science. 2013;341:1192–1198. doi: 10.1126/science.1241145. [DOI] [PubMed] [Google Scholar]
- 13.Adams JJ, Sidhu SS. Synthetic antibody technologies. Curr Opin Struct Biol. 2014;24:1–9. doi: 10.1016/j.sbi.2013.11.003. [DOI] [PubMed] [Google Scholar]
- 14.Panowksi S, Bhakta S, Raab H, Polakis P, Junutula JR. Site-specific antibody drug conjugates for cancer therapy. MAbs. 2014;6:34–45. doi: 10.4161/mabs.27022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Mintz CS, Crea R. Protein Scaffolds. The Next Generation of Protein Therapeutics? BioProcess International. 2013;11:40–48. [Google Scholar]
- 16.Gebauer M, Skerra A. Engineered protein scaffolds as next-generation antibody therapeutics. Curr Opin Chem Biol. 2009;13:245–255. doi: 10.1016/j.cbpa.2009.04.627. [DOI] [PubMed] [Google Scholar]
- 17.Ahmadzadeh V, Farajnia S, Feizi MA, Nejad RA. Antibody humanization methods for development of therapeutic applications. Monoclon Antib Immunodiagn Immunother. 2014;33:67–73. doi: 10.1089/mab.2013.0080. [DOI] [PubMed] [Google Scholar]
- 18.Streltsov VA, Varghese JN, Carmichael JA, Irving RA, Hudson PJ, Nuttall SD. Structural evidence for evolution of shark Ig new antigen receptor variable domain antibodies from a cell-surface receptor. Proc Natl Acad Sci U S A. 2004;101:12444–12449. doi: 10.1073/pnas.0403509101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Saerens D, Ghassabeh GH, Muyldermans S. Single-domain antibodies as building blocks for novel therapeutics. Curr Opin Pharmacol. 2008;8:600–608. doi: 10.1016/j.coph.2008.07.006. [DOI] [PubMed] [Google Scholar]
- 20.Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment: RNA ligands to bacteriophage T4 DNA polymerase. Science. 1990;249:505–510. doi: 10.1126/science.2200121. [DOI] [PubMed] [Google Scholar]
- 21.Ellington AD, Szostak JW. In vitro selection of RNA molecules that bind specific ligands. Nature. 1990;346:818–822. doi: 10.1038/346818a0. [DOI] [PubMed] [Google Scholar]
- 22.Song KM, Lee S, Ban C. Aptamers and Their Biological Applications. Sensors. 2012;12:612–631. doi: 10.3390/s120100612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Uzawa T, Tada S, Wang W, Ito Y. Expansion of the aptamer library from a “natural soup” to an “unnatural soup”. Chem Commun (Camb) 2013;49:1786–1795. doi: 10.1039/c2cc36348h. [DOI] [PubMed] [Google Scholar]
- 24.Szeitner Z, Andras J, Gyurcsanyi RE, Meszaros T. Is less more? Lessons from aptamer selection strategies. J Pharm Biomed Anal. 2014;101C:58–65. doi: 10.1016/j.jpba.2014.04.018. [DOI] [PubMed] [Google Scholar]
- 25.Kanwar JR, Shankaranarayanan JS, Gurudevan S, Kanwar RK. Aptamer-based therapeutics of the past, present and future: from the perspective of eye-related diseases. Drug Discov Today. 2014;19:1309–1321. doi: 10.1016/j.drudis.2014.02.009. [DOI] [PubMed] [Google Scholar]
- 26.Santosh B, Yadava PK. Nucleic acid aptamers: research tools in disease diagnostics and therapeutics. Biomed Res Int. 2014;2014:540451. doi: 10.1155/2014/540451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Xing H, Hwang K, Li J, Torabi SF, Lu Y. DNA Aptamer Technology for Personalized Medicine. Curr Opin Chem Eng. 2014;4:79–87. doi: 10.1016/j.coche.2014.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang C, Liu B, Lu J, Zhang G, Lu A. Strategies for combination of aptamer and targeted drug delivery. J Nanosci Nanotechnol. 2014;14:501–512. doi: 10.1166/jnn.2014.8746. [DOI] [PubMed] [Google Scholar]
- 29.Zhou J, Rossi JJ. Cell-type-specific, Aptamerfunctionalized Agents for Targeted Disease Therapy. Mol Ther Nucleic Acids. 2014;3:e169. doi: 10.1038/mtna.2014.21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Xiang D, Shigdar S, Qiao G, Wang T, Kouzani AZ, Zhou S, Kong L, Li Y, Pu C, Duan W. Nucleic Acid Aptamer-Guided Cancer Therapeutics and Diagnostics: the Next Generation of Cancer Medicine. Theranostics. 2015;5:23–42. doi: 10.7150/thno.10202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Chen ZY, Wang YX, Lin Y, Zhang JS, Yang F, Zhou QL, Liao YY. Advance of molecular imaging technology and targeted imaging agent in imaging and therapy. Biomed Res Int. 2014;2014:819324. doi: 10.1155/2014/819324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang AZ, Farokhzad OC. Current progress of aptamer-based molecular imaging. J Nucl Med. 2014;55:353–356. doi: 10.2967/jnumed.113.126144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Kim YS, Gu MB. Advances in aptamer screening and small molecule aptasensors. Adv Biochem Eng Biotechnol. 2014;140:29–67. doi: 10.1007/10_2013_225. [DOI] [PubMed] [Google Scholar]
- 34.MacKay S, Wishart D, Xing JZ, Chen J. Developing trends in aptamer-based biosensor devices and their applications. IEEE Trans Biomed Circuits Syst. 2014;8:4–14. doi: 10.1109/TBCAS.2014.2304718. [DOI] [PubMed] [Google Scholar]
- 35.Yoshida W, Abe K, Ikebukuro K. Emerging techniques employed in aptamer-based diagnostic tests. Expert Rev Mol Diagn. 2014;14:143–151. doi: 10.1586/14737159.2014.868307. [DOI] [PubMed] [Google Scholar]
- 36.Zhou W, Huang PJ, Ding J, Liu J. Aptamer-based biosensors for biomedical diagnostics. Analyst. 2014;139:2627–2640. doi: 10.1039/c4an00132j. [DOI] [PubMed] [Google Scholar]
- 37.Colas P, Cohen B, Jessen T, Grishina I, McCoy J, Brent R. Genetic selection of peptide aptamers that recognize and inhibit cyclin-dependent kinase 2. Nature. 1996;380:548–550. doi: 10.1038/380548a0. [DOI] [PubMed] [Google Scholar]
- 38.Mascini M, Palchetti I, Tombelli S. Nucleic acid and peptide aptamers: fundamentals and bioanalytical aspects. Angew Chem Int Ed Engl. 2012;51:1316–1332. doi: 10.1002/anie.201006630. [DOI] [PubMed] [Google Scholar]
- 39.Cohen BA, Colas P, Brent R. An artificial cell-cycle inhibitor isolated from a combinatorial library. Proc Natl Acad Sci U S A. 1998;95:14272–14277. doi: 10.1073/pnas.95.24.14272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ladner RC. Constrained peptides as binding entities. Trends in Biotechnology. 1995;13:426–430. doi: 10.1016/S0167-7799(00)88997-0. [DOI] [PubMed] [Google Scholar]
- 41.Baines IC, Colas P. Peptide aptamers as guides for small-molecule drug discovery. Drug Discov Today. 2006;11:334–341. doi: 10.1016/j.drudis.2006.02.007. [DOI] [PubMed] [Google Scholar]
- 42.Dassie JP, Giangrande PH. Current progress on aptamer-targeted oligonucleotide therapeutics. Ther Deliv. 2013;4:1527–1546. doi: 10.4155/tde.13.118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wells JA, McClendon CL. Reaching for highhanging fruit in drug discovery at protein-protein interfaces. Nature. 2007;450:1001–1009. doi: 10.1038/nature06526. [DOI] [PubMed] [Google Scholar]
- 44.Bunka DH, Stockley PG. Aptamers come of age - at last. Nat Rev Microbiol. 2006;4:588–596. doi: 10.1038/nrmicro1458. [DOI] [PubMed] [Google Scholar]
- 45.Jaeger J, Restle T, Steitz TA. The structure of HIV-1 reverse transcriptase complexed with an RNA pseudoknot inhibitor. Embo j. 1998;17:4535–4542. doi: 10.1093/emboj/17.15.4535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hamdi A, Colas P. Yeast two-hybrid methods and their applications in drug discovery. Trends Pharmacol Sci. 2012;33:109–118. doi: 10.1016/j.tips.2011.10.008. [DOI] [PubMed] [Google Scholar]
- 47.Reverdatto S, Rai V, Xue J, Burz DS, Schmidt AM, Shekhtman A. Combinatorial library of improved peptide aptamers, CLIPs to inhibit RAGE signal transduction in mammalian cells. PLoS One. 2013;8:e65180. doi: 10.1371/journal.pone.0065180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Holliger P, Hudson PJ. Engineered antibody fragments and the rise of single domains. Nat Biotechnol. 2005;23:1126–1136. doi: 10.1038/nbt1142. [DOI] [PubMed] [Google Scholar]
- 49.Hammers CM, Stanley JR. Antibody phage display: technique and applications. J Invest Dermatol. 2014;134:e17. doi: 10.1038/jid.2013.521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Kanamori T, Fujino Y, Ueda T. PURE ribosome display and its application in antibody technology. Biochim Biophys Acta. 2014;1844:1925–1932. doi: 10.1016/j.bbapap.2014.04.007. [DOI] [PubMed] [Google Scholar]
- 51.Shukra AM, Sridevi NV, Dev C, Kapil M. Production of recombinant antibodies using bacteriophages. Eur J Microbiol Immunol (Bp) 2014;4:91–98. doi: 10.1556/EuJMI.4.2014.2.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Chakravarty R, Goel S, Cai W. Nanobody: the “magic bullet” for molecular imaging? Theranostics. 2014;4:386–398. doi: 10.7150/thno.8006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.De Meyer T, Muyldermans S, Depicker A. Nanobody-based products as research and diagnostic tools. Trends Biotechnol. 2014;32:263–270. doi: 10.1016/j.tibtech.2014.03.001. [DOI] [PubMed] [Google Scholar]
- 54.Kovaleva M, Ferguson L, Steven J, Porter A, Barelle C. Shark variable new antigen receptor biologics - a novel technology platform for therapeutic drug development. Expert Opin Biol Ther. 2014;14:1527–1539. doi: 10.1517/14712598.2014.937701. [DOI] [PubMed] [Google Scholar]
- 55.Liu JL, Zabetakis D, Brown JC, Anderson GP, Goldman ER. Thermal stability and refolding capability of shark derived single domain antibodies. Mol Immunol. 2014;59:194–199. doi: 10.1016/j.molimm.2014.02.014. [DOI] [PubMed] [Google Scholar]
- 56.Skerra A. Alternative non-antibody scaffolds for molecular recognition. Curr Opin Biotechnol. 2007;18:295–304. doi: 10.1016/j.copbio.2007.04.010. [DOI] [PubMed] [Google Scholar]
- 57.Borghouts C, Kunz C, Groner B. Peptide aptamer libraries. Comb Chem High Throughput Screen. 2008;11:135–145. doi: 10.2174/138620708783744462. [DOI] [PubMed] [Google Scholar]
- 58.Nygren PA, Skerra A. Binding proteins from alternative scaffolds. J Immunol Methods. 2004;290:3–28. doi: 10.1016/j.jim.2004.04.006. [DOI] [PubMed] [Google Scholar]
- 59.Binz HK, Amstutz P, Pluckthun A. Engineering novel binding proteins from nonimmunoglobulin domains. Nat Biotechnol. 2005;23:1257–1268. doi: 10.1038/nbt1127. [DOI] [PubMed] [Google Scholar]
- 60.Hosse RJ, Rothe A, Power BE. A new generation of protein display scaffolds for molecular recognition. Protein Sci. 2006;15:14–27. doi: 10.1110/ps.051817606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Gronwall C, Stahl S. Engineered affinity proteins--generation and applications. J Biotechnol. 2009;140:254–269. doi: 10.1016/j.jbiotec.2009.01.014. [DOI] [PubMed] [Google Scholar]
- 62.Hoppe-Seyler F, Butz K. Peptide aptamers: powerful new tools for molecular medicine. J Mol Med (Berl) 2000;78:426–430. doi: 10.1007/s001090000140. [DOI] [PubMed] [Google Scholar]
- 63.Weidle UH, Auer J, Brinkmann U, Georges G, Tiefenthaler G. The emerging role of new protein scaffold-based agents for treatment of cancer. Cancer Genomics Proteomics. 2013;10:155–168. [PubMed] [Google Scholar]
- 64.LaVallie ER, DiBlasio EA, Kovacic S, Grant KL, Schendel PF, McCoy JM. A thioredoxin gene fusion expression system that circumvents inclusion body formation in the E. coli cytoplasm. Biotechnology (N Y) 1993;11:187–193. doi: 10.1038/nbt0293-187. [DOI] [PubMed] [Google Scholar]
- 65.Martin JL. Thioredoxin--a fold for all reasons. Structure. 1995;3:245–250. doi: 10.1016/s0969-2126(01)00154-x. [DOI] [PubMed] [Google Scholar]
- 66.Klevenz B, Butz K, Hoppe-Seyler F. Peptide aptamers: exchange of the thioredoxin-A scaffold by alternative platform proteins and its influence on target protein binding. Cell Mol Life Sci. 2002;59:1993–1998. doi: 10.1007/PL00012521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Groner B, Weber A, Mack L. Increasing the range of drug targets: interacting peptides provide leads for the development of oncoprotein inhibitors. Bioengineered. 2012;3:320–325. doi: 10.4161/bioe.21272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Weber A, Borghouts C, Brendel C, Moriggl R, Delis N, Brill B, Vafaizadeh V, Groner B. The inhibition of stat5 by a Peptide aptamer ligand specific for the DNA binding domain prevents target gene transactivation and the growth of breast and prostate tumor cells. Pharmaceuticals (Basel) 2013;6:960–987. doi: 10.3390/ph6080960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hofmann MA, Drury S, Fu C, Qu W, Taguchi A, Lu Y, Avila C, Kambham N, Bierhaus A, Nawroth P, Neurath MF, Slattery T, Beach D, McClary J, Nagashima M, Morser J, Stern D, Schmidt AM. RAGE mediates a novel proinflammatory axis: a central cell surface receptor for S100/calgranulin polypeptides. Cell. 1999;97:889–901. doi: 10.1016/s0092-8674(00)80801-6. [DOI] [PubMed] [Google Scholar]
- 70.Taguchi A, Blood DC, del Toro G, Canet A, Lee DC, Qu W, Tanji N, Lu Y, Lalla E, Fu C, Hofmann MA, Kislinger T, Ingram M, Lu A, Tanaka H, Hori O, Ogawa S, Stern DM, Schmidt AM. Blockade of RAGE-amphoterin signalling suppresses tumour growth and metastases. Nature. 2000;405:354–360. doi: 10.1038/35012626. [DOI] [PubMed] [Google Scholar]
- 71.Kislinger T, Tanji N, Wendt T, Qu W, Lu Y, Ferran LJ, Jr, Taguchi A, Olson K, Bucciarelli L, Goova M, Hofmann MA, Cataldegirmen G, D'Agati V, Pischetsrieder M, Stern DM, Schmidt AM. Receptor for advanced glycation end products mediates inflammation and enhanced expression of tissue factor in vasculature of diabetic apolipoprotein E-null mice. Arterioscler Thromb Vasc Biol. 2001;21:905–910. doi: 10.1161/01.atv.21.6.905. [DOI] [PubMed] [Google Scholar]
- 72.Yan SD, Chen X, Fu J, Chen M, Zhu H, Roher A, Slattery T, Zhao L, Nagashima M, Morser J, Migheli A, Nawroth P, Stern D, Schmidt AM. RAGE and amyloid-beta pep-tide neurotoxicity in Alzheimer's disease. Nature. 1996;382:685–691. doi: 10.1038/382685a0. [DOI] [PubMed] [Google Scholar]
- 73.Cobbert JD, DeMott C, Majumder S, Smith EA, Burz DS, McDonough KA, Shekhtman A. Caught in Action: Selecting Peptide Aptamers Against Intrinsically Disordered Proteins in Live Cells. Scientific Reports. 2015;5:9402. doi: 10.1038/srep09402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Koch M, Chitayat S, Dattilo BM, Schiefner A, Diez J, Chazin WJ, Fritz G. Structural basis for ligand recognition and activation of RAGE. Structure. 2010;18:1342–1352. doi: 10.1016/j.str.2010.05.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Fritz G. RAGE: a single receptor fits multiple ligands. Trends Biochem Sci. 2011;36:625–632. doi: 10.1016/j.tibs.2011.08.008. [DOI] [PubMed] [Google Scholar]
- 76.Koide A, Bailey CW, Huang X, Koide S. The fibronectin type III domain as a scaffold for novel binding proteins. J Mol Biol. 1998;284:1141–1151. doi: 10.1006/jmbi.1998.2238. [DOI] [PubMed] [Google Scholar]
- 77.Xu L, Aha P, Gu K, Kuimelis RG, Kurz M, Lam T, Lim AC, Liu H, Lohse PA, Sun L, Weng S, Wagner RW, Lipovsek D. Directed evolution of high-affinity antibody mimics using mRNA display. Chem Biol. 2002;9:933–942. doi: 10.1016/s1074-5521(02)00187-4. [DOI] [PubMed] [Google Scholar]
- 78.Lipovsek D. Adnectins: engineered target-binding protein therapeutics. Protein Eng Des Sel. 2011;24:3–9. doi: 10.1093/protein/gzq097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Koide A, Koide S. Monobodies: antibody mimics based on the scaffold of the fibronectin type III domain. Methods Mol Biol. 2007;352:95–109. doi: 10.1385/1-59745-187-8:95. [DOI] [PubMed] [Google Scholar]
- 80.Hackel BJ, Kapila A, Wittrup KD. Picomolar affinity fibronectin domains engineered utilizing loop length diversity, recursive mutagenesis, and loop shuffling. J Mol Biol. 2008;381:1238–1252. doi: 10.1016/j.jmb.2008.06.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Hackel BJ, Wittrup KD. The full amino acid repertoire is superior to serine/tyrosine for selection of high affinity immunoglobulin G binders from the fibronectin scaffold. Protein Eng Des Sel. 2010;23:211–219. doi: 10.1093/protein/gzp083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Koldobskaya Y, Duguid EM, Shechner DM, Suslov NB, Ye J, Sidhu SS, Bartel DP, Koide S, Kossiakoff AA, Piccirilli JA. A portable RNA sequence whose recognition by a synthetic antibody facilitates structural determination. Nat Struct Mol Biol. 2011;18:100–106. doi: 10.1038/nsmb.1945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Ramamurthy V, Krystek SR, Jr, Bush A, Wei A, Emanuel SL, Das Gupta R, Janjua A, Cheng L, Murdock M, Abramczyk B, Cohen D, Lin Z, Morin P, Davis JH, Dabritz M, McLaughlin DC, Russo KA, Chao G, Wright MC, Jenny VA, Engle LJ, Furfine E, Sheriff S. Structures of adnectin/protein complexes reveal an expanded binding footprint. Structure. 2012;20:259–269. doi: 10.1016/j.str.2011.11.016. [DOI] [PubMed] [Google Scholar]
- 84.Beste G, Schmidt FS, Stibora T, Skerra A. Small antibody-like proteins with prescribed ligand specificities derived from the lipocalin fold. Proc Natl Acad Sci U S A. 1999;96:1898–1903. doi: 10.1073/pnas.96.5.1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Schlehuber S, Beste G, Skerra A. A novel type of receptor protein, based on the lipocalin scaffold, with specificity for digoxigenin. J Mol Biol. 2000;297:1105–1120. doi: 10.1006/jmbi.2000.3646. [DOI] [PubMed] [Google Scholar]
- 86.Skerra A. Alternative binding proteins: anticalins - harnessing the structural plasticity of the lipocalin ligand pocket to engineer novel binding activities. Febs j. 2008;275:2677–2683. doi: 10.1111/j.1742-4658.2008.06439.x. [DOI] [PubMed] [Google Scholar]
- 87.Richter A, Eggenstein E, Skerra A. Anticalins: exploiting a non-Ig scaffold with hypervariable loops for the engineering of binding proteins. FEBS Lett. 2014;588:213–218. doi: 10.1016/j.febslet.2013.11.006. [DOI] [PubMed] [Google Scholar]
- 88.Gebauer M, Skerra A. Anticalins small engineered binding proteins based on the lipocalin scaffold. Methods Enzymol. 2012;503:157–188. doi: 10.1016/B978-0-12-396962-0.00007-0. [DOI] [PubMed] [Google Scholar]
- 89.Williams A, Baird LG. DX-88 and HAE: a developmental perspective. Transfus Apher Sci. 2003;29:255–258. doi: 10.1016/S1473-0502(03)00170-8. [DOI] [PubMed] [Google Scholar]
- 90.Silverman J, Liu Q, Bakker A, To W, Duguay A, Alba BM, Smith R, Rivas A, Li P, Le H, Whitehorn E, Moore KW, Swimmer C, Perlroth V, Vogt M, Kolkman J, Stemmer WP. Multivalent avimer proteins evolved by exon shuffling of a family of human receptor domains. Nat Biotechnol. 2005;23:1556–1561. doi: 10.1038/nbt1166. [DOI] [PubMed] [Google Scholar]
- 91.Kolmar H. Biological diversity and therapeutic potential of natural and engineered cystine knot miniproteins. Curr Opin Pharmacol. 2009;9:608–614. doi: 10.1016/j.coph.2009.05.004. [DOI] [PubMed] [Google Scholar]
- 92.Reinwarth M, Nasu D, Kolmar H, Avrutina O. Chemical synthesis, backbone cyclization and oxidative folding of cystine-knot peptides: promising scaffolds for applications in drug design. Molecules. 2012;17:12533–12552. doi: 10.3390/molecules171112533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Austin J, Wang W, Puttamadappa S, Shekhtman A, Camarero JA. Biosynthesis and biological screening of a genetically encoded library based on the cyclotide MCoTI-I. Chembiochem. 2009;10:2663–2670. doi: 10.1002/cbic.200900534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Sancheti H, Camarero JA. “Splicing up” drug discovery Cell-based expression and screening of genetically-encoded libraries of backbone-cyclized polypeptides. Adv Drug Deliv Rev. 2009;61:908–917. doi: 10.1016/j.addr.2009.07.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Silverman AP, Levin AM, Lahti JL, Cochran JR. Engineered cystine-knot peptides that bind alpha(v)beta(3) integrin with antibody-like affinities. J Mol Biol. 2009;385:1064–1075. doi: 10.1016/j.jmb.2008.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Kimura RH, Cheng Z, Gambhir SS, Cochran JR. Engineered knottin peptides: a new class of agents for imaging integrin expression in living subjects. Cancer Res. 2009;69:2435–2442. doi: 10.1158/0008-5472.CAN-08-2495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bertschinger J, Grabulovski D, Neri D. Selection of single domain binding proteins by covalent DNA display. Protein Eng Des Sel. 2007;20:57–68. doi: 10.1093/protein/gzl055. [DOI] [PubMed] [Google Scholar]
- 98.Grabulovski D, Kaspar M, Neri D. A novel, non-immunogenic Fyn SH3-derived binding protein with tumor vascular targeting properties. J Biol Chem. 2007;282:3196–3204. doi: 10.1074/jbc.M609211200. [DOI] [PubMed] [Google Scholar]
- 99.Schlatter D, Brack S, Banner DW, Batey S, Benz J, Bertschinger J, Huber W, Joseph C, Rufer A, van der Klooster A, Weber M, Grabulovski D, Hennig M. Generation, characterization and structural data of chymase binding proteins based on the human Fyn kinase SH3 domain. MAbs. 2012;4:497–508. doi: 10.4161/mabs.20452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Byla P, Andersen MH, Holtet TL, Jacobsen H, Munch M, Gad HH, Thogersen HC, Hartmann R. Selection of a novel and highly specific tumor necrosis factor alpha (TNFal-pha) antagonist: insight from the crystal structure of the antagonist-TNFalpha complex. J Biol Chem. 2010;285:12096–12100. doi: 10.1074/jbc.M109.063305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Rohn J. Newsmaker: anaphore. Nat Biotechnol. 2010;28:1143. doi: 10.1038/nbt1110-1143. [DOI] [PubMed] [Google Scholar]
- 102.Lorey S, Fiedler E, Kunert A, Nerkamp J, Lange C, Fiedler M, Bosse-Doenecke E, Meysing M, Gloser M, Rundfeldt C, Rauchhaus U, Hanssgen I, Gottler T, Steuernagel A, Fiedler U, Haupts U. Novel ubiquitin-derived high affinity binding proteins with tumor targeting properties. J Biol Chem. 2014;289:8493–8507. doi: 10.1074/jbc.M113.519884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Binz HK, Stumpp MT, Forrer P, Amstutz P, Pluckthun A. Designing repeat proteins: well-expressed, soluble and stable proteins from combinatorial libraries of consensus ankyrin repeat proteins. J Mol Biol. 2003;332:489–503. doi: 10.1016/s0022-2836(03)00896-9. [DOI] [PubMed] [Google Scholar]
- 104.Forrer P, Stumpp MT, Binz HK, Pluckthun A. A novel strategy to design binding molecules harnessing the modular nature of repeat proteins. FEBS Lett. 2003;539:2–6. doi: 10.1016/s0014-5793(03)00177-7. [DOI] [PubMed] [Google Scholar]
- 105.Stumpp MT, Amstutz P. DARPins: a true alternative to antibodies. Curr Opin Drug Discov Devel. 2007;10:153–159. [PubMed] [Google Scholar]
- 106.Zahnd C, Amstutz P, Pluckthun A. Ribosome display: selecting and evolving proteins in vitro that specifically bind to a target. Nat Methods. 2007;4:269–279. doi: 10.1038/nmeth1003. [DOI] [PubMed] [Google Scholar]
- 107.Pluckthun A. Designed Ankyrin Repeat Proteins (DARPins): Binding Proteins for Research, Diagnostics, and Therapy. Annu Rev Pharmacol Toxicol. 2015;55:489–511. doi: 10.1146/annurev-pharmtox-010611-134654. [DOI] [PubMed] [Google Scholar]
- 108.Wetzel SK, Settanni G, Kenig M, Binz HK, Pluckthun A. Folding and unfolding mechanism of highly stable full-consensus ankyrin repeat proteins. J Mol Biol. 2008;376:241–257. doi: 10.1016/j.jmb.2007.11.046. [DOI] [PubMed] [Google Scholar]
- 109.Wetzel SK, Ewald C, Settanni G, Jurt S, Pluckthun A, Zerbe O. Residue-resolved stability of full-consensus ankyrin repeat proteins probed by NMR. J Mol Biol. 2010;402:241–258. doi: 10.1016/j.jmb.2010.07.031. [DOI] [PubMed] [Google Scholar]
- 110.Eggel A, Baumann MJ, Amstutz P, Stadler BM, Vogel M. DARPins as bispecific receptor antagonists analyzed for immunoglobulin E receptor blockage. J Mol Biol. 2009;393:598–607. doi: 10.1016/j.jmb.2009.08.014. [DOI] [PubMed] [Google Scholar]
- 111.Stumpp MT, Binz HK, Amstutz P. DARPins: a new generation of protein therapeutics. Drug Discov Today. 2008;13:695–701. doi: 10.1016/j.drudis.2008.04.013. [DOI] [PubMed] [Google Scholar]
- 112.Boersma YL, Pluckthun A. DARPins and other repeat protein scaffolds: advances in engineering and applications. Curr Opin Biotechnol. 2011;22:849–857. doi: 10.1016/j.copbio.2011.06.004. [DOI] [PubMed] [Google Scholar]
- 113.Schilling J, Schoppe J, Pluckthun A. From DARPins to LoopDARPins: novel LoopDARPin design allows the selection of low picomolar binders in a single round of ribosome display. J Mol Biol. 2014;426:691–721. doi: 10.1016/j.jmb.2013.10.026. [DOI] [PubMed] [Google Scholar]
- 114.Nord K, Nilsson J, Nilsson B, Uhlen M, Nygren PA. A combinatorial library of an alpha-helical bacterial receptor domain. Protein Eng. 1995;8:601–608. doi: 10.1093/protein/8.6.601. [DOI] [PubMed] [Google Scholar]
- 115.Nord K, Gunneriusson E, Ringdahl J, Stahl S, Uhlen M, Nygren PA. Binding proteins selected from combinatorial libraries of an alpha-helical bacterial receptor domain. Nat Bio-technol. 1997;15:772–777. doi: 10.1038/nbt0897-772. [DOI] [PubMed] [Google Scholar]
- 116.Nygren PA. Alternative binding proteins: affibody binding proteins developed from a small three-helix bundle scaffold. Febs j. 2008;275:2668–2676. doi: 10.1111/j.1742-4658.2008.06438.x. [DOI] [PubMed] [Google Scholar]
- 117.Hey T, Fiedler E, Rudolph R, Fiedler M. Artificial, non-antibody binding proteins for pharmaceutical and industrial applications. Trends Biotechnol. 2005;23:514–522. doi: 10.1016/j.tibtech.2005.07.007. [DOI] [PubMed] [Google Scholar]
- 118.Fiedler E, Fiedler M, Proetzel G, Scheuermann T, Fiedler U, Rudolph R. Affilin™ Molecules: Novel Ligands for Bioseparation. Food and Bioproducts Processing. 2006;84:3–8. [Google Scholar]
- 119.Ebersbach H, Fiedler E, Scheuermann T, Fiedler M, Stubbs MT, Reimann C, Proetzel G, Rudolph R, Fiedler U. Affilin-novel binding molecules based on human gamma-B-crystallin, an all beta-sheet protein. J Mol Biol. 2007;372:172–185. doi: 10.1016/j.jmb.2007.06.045. [DOI] [PubMed] [Google Scholar]
- 120.Mirecka EA, Hey T, Fiedler U, Rudolph R, Hatzfeld M. Affilin molecules selected against the human papillomavirus E7 protein inhibit the proliferation of target cells. J Mol Biol. 2009;390:710–721. doi: 10.1016/j.jmb.2009.05.027. [DOI] [PubMed] [Google Scholar]
- 121.Hoffmann A, Kovermann M, Lilie H, Fiedler M, Balbach J, Rudolph R, Pfeifer S. New binding mode to TNF-alpha revealed by ubiquitin-based artificial binding protein. PLoS One. 2012;7:e31298. doi: 10.1371/journal.pone.0031298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Reichen C, Hansen S, Pluckthun A. Modular peptide binding: from a comparison of natural binders to designed armadillo repeat proteins. J Struct Biol. 2014;185:147–162. doi: 10.1016/j.jsb.2013.07.012. [DOI] [PubMed] [Google Scholar]
- 123.Varadamsetty G, Tremmel D, Hansen S, Parmeggiani F, Pluckthun A. Designed Armadillo repeat proteins: library generation, characterization and selection of peptide binders with high specificity. J Mol Biol. 2012;424:68–87. doi: 10.1016/j.jmb.2012.08.029. [DOI] [PubMed] [Google Scholar]
- 124.Parmeggiani F, Pellarin R, Larsen AP, Varadamsetty G, Stumpp MT, Zerbe O, Caflisch A, Pluckthun A. Designed armadillo repeat proteins as general peptide-binding scaffolds: consensus design and computational optimization of the hydrophobic core. J Mol Biol. 2008;376:1282–1304. doi: 10.1016/j.jmb.2007.12.014. [DOI] [PubMed] [Google Scholar]
- 125.Steemson JD, Baake M, Rakonjac J, Arcus VL, Liddament MT. Tracking molecular recognition at the atomic level with a new protein scaffold based on the OB-fold. PLoS One. 2014;9:e86050. doi: 10.1371/journal.pone.0086050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Mondon P, Dubreuil O, Bouayadi K, Kharrat H. Human antibody libraries: a race to engineer and explore a larger diversity. Front Biosci. 2008;13:1117–1129. doi: 10.2741/2749. [DOI] [PubMed] [Google Scholar]
- 127.Smith GP. Filamentous fusion phage: novel expression vectors that display cloned antigens on the virion surface. Science. 1985;228:1315–1317. doi: 10.1126/science.4001944. [DOI] [PubMed] [Google Scholar]
- 128.Marvin DA. Filamentous phage structure, infection and assembly. Curr Opin Struct Biol. 1998;8:150–158. doi: 10.1016/s0959-440x(98)80032-8. [DOI] [PubMed] [Google Scholar]
- 129.Hamzeh-Mivehroud M, Alizadeh AA, Morris MB, Church WB, Dastmalchi S. Phage display as a technology delivering on the promise of peptide drug discovery. Drug Discov Today. 2013;18:1144–1157. doi: 10.1016/j.drudis.2013.09.001. [DOI] [PubMed] [Google Scholar]
- 130.Loset GA, Sandlie I. Next generation phage display by use of pVII and pIX as display scaffolds. Methods. 2012;58:40–46. doi: 10.1016/j.ymeth.2012.07.005. [DOI] [PubMed] [Google Scholar]
- 131.Sergeeva A, Kolonin MG, Molldrem JJ, Pasqualini R, Arap W. Display technologies: application for the discovery of drug and gene delivery agents. Adv Drug Deliv Rev. 2006;58:1622–1654. doi: 10.1016/j.addr.2006.09.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Nixon AE, Sexton DJ, Ladner RC. Drugs derived from phage display: from candidate identification to clinical practice. MAbs. 2014;6:73–85. doi: 10.4161/mabs.27240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Esvelt KM, Carlson JC, Liu DR. A system for the continuous directed evolution of biomolecules. Nature. 2011;472:499–503. doi: 10.1038/nature09929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Dickinson BC, Leconte AM, Allen B, Esvelt KM, Liu DR. Experimental interrogation of the path dependence and stochasticity of protein evolution using phage-assisted continuous evolution. Proc Natl Acad Sci U S A. 2013;110:9007–9012. doi: 10.1073/pnas.1220670110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Carlson JC, Badran AH, Guggiana-Nilo DA, Liu DR. Negative selection and stringency modulation in phage-assisted continuous evolution. Nat Chem Biol. 2014;10:216–222. doi: 10.1038/nchembio.1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Qi H, Lu H, Qiu HJ, Petrenko V, Liu A. Phagemid Vectors for Phage Display: Properties, Characteristics and Construction. Journal of Molecular Biology. 2012;417:129–143. doi: 10.1016/j.jmb.2012.01.038. [DOI] [PubMed] [Google Scholar]
- 137.Efimov VP, Nepluev IV, Mesyanzhinov VV. Bacteriophage T4 as a surface display vector. Virus Genes. 1995;10:173–177. doi: 10.1007/BF01702598. [DOI] [PubMed] [Google Scholar]
- 138.Jiang J, Abu-Shilbayeh L, Rao VB. Display of a PorA peptide from Neisseria meningitidis on the bacteriophage T4 capsid surface. Infect Immun. 1997;65:4770–4777. doi: 10.1128/iai.65.11.4770-4777.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Gamkrelidze M, Dabrowska K. T4 bacteriophage as a phage display platform. Arch Microbiol. 2014;196:473–479. doi: 10.1007/s00203-014-0989-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Krumpe LR, Atkinson AJ, Smythers GW, Kandel A, Schumacher KM, McMahon JB, Makowski L, Mori T. T7 lytic phage-displayed peptide libraries exhibit less sequence bias than M13 filamentous phage-displayed peptide libraries. Proteomics. 2006;6:4210–4222. doi: 10.1002/pmic.200500606. [DOI] [PubMed] [Google Scholar]
- 141.Sharma SC, Memic A, Rupasinghe CN, Duc AC, Spaller MR. T7 phage display as a method of peptide ligand discovery for PDZ domain proteins. Biopolymers. 2009;92:183–193. doi: 10.1002/bip.21172. [DOI] [PubMed] [Google Scholar]
- 142.Kong B, Ma WJ. Display of aggregation-prone ligand binding domain of human PPAR gamma on surface of bacteriophage lambda. Acta Pharmacol Sin. 2006;27:91–99. doi: 10.1111/j.1745-7254.2006.00253.x. [DOI] [PubMed] [Google Scholar]
- 143.Santi E, Capone S, Mennuni C, Lahm A, Tramontano A, Luzzago A, Nicosia A. Bacteriophage lambda display of complex cDNA libraries: a new approach to functional genomics. J Mol Biol. 2000;296:497–508. doi: 10.1006/jmbi.1999.3471. [DOI] [PubMed] [Google Scholar]
- 144.Niwa M, Fukuoka K, Fujimoto T, Maruyama IN. Efficient isolation of cDNA clones encoding rheumatoid arthritis autoantigens by lambda phage surface display. J Biotechnol. 2004;114:55–58. doi: 10.1016/j.jbiotec.2004.05.010. [DOI] [PubMed] [Google Scholar]
- 145.Beghetto E, Gargano N. Lambda-display: a powerful tool for antigen discovery. Molecules. 2011;16:3089–3105. doi: 10.3390/molecules16043089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Liang S, Lin T, Ding J, Pan Y, Dang D, Guo C, Zhi M, Zhao P, Sun L, Hong L, Shi Y, Yao L, Liu J, Wu K, Fan D. Screening and identification of vascularendothelial-cell-specific binding peptide in gastric cancer. J Mol Med (Berl) 2006;84:764–773. doi: 10.1007/s00109-006-0064-2. [DOI] [PubMed] [Google Scholar]
- 147.Giordano RJ, Anobom CD, Cardo-Vila M, Kalil J, Valente AP, Pasqualini R, Almeida FC, Arap W. Structural basis for the interaction of a vascular endothelial growth factor mimic peptide motif and its corresponding receptors. Chem Biol. 2005;12:1075–1083. doi: 10.1016/j.chembiol.2005.07.008. [DOI] [PubMed] [Google Scholar]
- 148.Doerner A, Rhiel L, Zielonka S, Kolmar H. Therapeutic antibody engineering by high efficiency cell screening. FEBS Lett. 2014;588:278–287. doi: 10.1016/j.febslet.2013.11.025. [DOI] [PubMed] [Google Scholar]
- 149.Georgiou G, Stathopoulos C, Daugherty PS, Nayak AR, Iverson BL, Curtiss R., 3rd Display of heterologous proteins on the surface of microorganisms: from the screening of combinatorial libraries to live recombinant vaccines. Nat Biotechnol. 1997;15:29–34. doi: 10.1038/nbt0197-29. [DOI] [PubMed] [Google Scholar]
- 150.Lofblom J. Bacterial display in combinatorial protein engineering. Biotechnol J. 2011;6:1115–1129. doi: 10.1002/biot.201100129. [DOI] [PubMed] [Google Scholar]
- 151.Mazor Y, Van Blarcom T, Mabry R, Iverson BL, Georgiou G. Isolation of engineered, full-length antibodies from libraries expressed in Escherichia coli. Nat Biotechnol. 2007;25:563–565. doi: 10.1038/nbt1296. [DOI] [PubMed] [Google Scholar]
- 152.Boder ET, Wittrup KD. Yeast surface display for screening combinatorial polypeptide libraries. Nat Biotechnol. 1997;15:553–557. doi: 10.1038/nbt0697-553. [DOI] [PubMed] [Google Scholar]
- 153.Schreuder MP, Mooren AT, Toschka HY, Verrips CT, Klis FM. Immobilizing proteins on the surface of yeast cells. Trends Biotechnol. 1996;14:115–120. doi: 10.1016/0167-7799(96)10017-2. [DOI] [PubMed] [Google Scholar]
- 154.Sato N, Matsumoto T, Ueda M, Tanaka A, Fukuda H, Kondo A. Long anchor using Flo1 protein enhances reactivity of cell surface-displayed glucoamylase to polymer substrates. Appl Microbiol Biotechnol. 2002;60:469–474. doi: 10.1007/s00253-002-1121-6. [DOI] [PubMed] [Google Scholar]
- 155.Ueda M, Tanaka A. Genetic immobilization of proteins on the yeast cell surface. Biotechnol Adv. 2000;18:121–140. doi: 10.1016/s0734-9750(00)00031-8. [DOI] [PubMed] [Google Scholar]
- 156.Bowley DR, Labrijn AF, Zwick MB, Burton DR. Antigen selection from an HIV-1 immune antibody library displayed on yeast yields many novel antibodies compared to selection from the same library displayed on phage. Protein Eng Des Sel. 2007;20:81–90. doi: 10.1093/protein/gzl057. [DOI] [PubMed] [Google Scholar]
- 157.Ho M, Nagata S, Pastan I. Isolation of anti-CD22 Fv with high affinity by Fv display on human cells. Proc Natl Acad Sci U S A. 2006;103:9637–9642. doi: 10.1073/pnas.0603653103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Bowers PM, Horlick RA, Kehry MR, Neben TY, Tomlinson GL, Altobell L, Zhang X, Macomber JL, Krapf IP, Wu BF, McConnell AD, Chau B, Berkebile AD, Hare E, Verdino P, King DJ. Mammalian cell display for the discovery and optimization of antibody therapeutics. Methods. 2014;65:44–56. doi: 10.1016/j.ymeth.2013.06.010. [DOI] [PubMed] [Google Scholar]
- 159.King DJ, Bowers PM, Kehry MR, Horlick RA. Mammalian cell display and somatic hypermutation in vitro for human antibody discovery. Curr Drug Discov Technol. 2014;11:56–64. doi: 10.2174/15701638113109990037. [DOI] [PubMed] [Google Scholar]
- 160.Mattheakis LC, Bhatt RR, Dower WJ. An in vitro polysome display system for identifying ligands from very large peptide libraries. Proc Natl Acad Sci U S A. 1994;91:9022–9026. doi: 10.1073/pnas.91.19.9022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Hanes J, Pluckthun A. In vitro selection and evolution of functional proteins by using ribosome display. Proc Natl Acad Sci U S A. 1997;94:4937–4942. doi: 10.1073/pnas.94.10.4937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Amstutz P, Koch H, Binz HK, Deuber SA, Pluckthun A. Rapid selection of specific MAP kinase-binders from designed ankyrin repeat protein libraries. Protein Eng Des Sel. 2006;19:219–229. doi: 10.1093/protein/gzl004. [DOI] [PubMed] [Google Scholar]
- 163.Nemoto N, Miyamoto-Sato E, Husimi Y, Yanagawa H. In vitro virus: Bonding of mRNA bearing puromycin at the 3′-terminal end to the C-terminal end of its encoded protein on the ribosomein vitro. FEBS Letters. 1997;414:405–408. doi: 10.1016/s0014-5793(97)01026-0. [DOI] [PubMed] [Google Scholar]
- 164.Roberts RW, Szostak JW. RNA-peptide fusions for the in vitro selection of peptides and proteins. Proc Natl Acad Sci U S A. 1997;94:12297–12302. doi: 10.1073/pnas.94.23.12297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Olson CA, Nie J, Diep J, Al-Shyoukh I, Takahashi TT, Al-Mawsawi LQ, Bolin JM, Elwell AL, Swanson S, Stewart R, Thomson JA, Soh HT, Roberts RW, Sun R. Single-round, multiplexed antibody mimetic design through mRNA display. Angew Chem Int Ed Engl. 2012;51:12449–12453. doi: 10.1002/anie.201207005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Lane MD, Seelig B. Advances in the directed evolution of proteins. Curr Opin Chem Biol. 2014;22C:129–136. doi: 10.1016/j.cbpa.2014.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Parker MH, Chen Y, Danehy F, Dufu K, Ekstrom J, Getmanova E, Gokemeijer J, Xu L, Lipovsek D. Antibody mimics based on human fibronectin type three domain engineered for thermostability and high-affinity binding to vascular endothelial growth factor receptor two. Protein Eng Des Sel. 2005;18:435–444. doi: 10.1093/protein/gzi050. [DOI] [PubMed] [Google Scholar]
- 168.Barendt PA, Ng DTW, McQuade CN, Sarkar CA. Streamlined Protocol for mRNA Display. Acs Combinatorial Science. 2013;15:77–81. doi: 10.1021/co300135r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Golynskiy MV, Haugner JC, 3rd, Morelli A, Morrone D, Seelig B. In vitro evolution of enzymes. Methods Mol Biol. 2013;978:73–92. doi: 10.1007/978-1-62703-293-3_6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Valencia CA, Zou J, Liu R. In vitro selection of proteins with desired characteristics using mRNA-display. Methods. 2013;60:55–69. doi: 10.1016/j.ymeth.2012.11.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Wada A. Development of Next-Generation Peptide Binders Using In vitro Display Technologies and Their Potential Applications. Front Immunol. 2013;4:224. doi: 10.3389/fimmu.2013.00224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Naimuddin M, Kobayashi S, Tsutsui C, Machida M, Nemoto N, Sakai T, Kubo T. Directed evolution of a three-finger neurotoxin by using cDNA display yields antagonists as well as agonists of interleukin-6 receptor signaling. Mol Brain. 2011;4:2. doi: 10.1186/1756-6606-4-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Ishizawa T, Kawakami T, Reid PC, Murakami H. TRAP display: a high-speed selection method for the generation of functional polypeptides. J Am Chem Soc. 2013;135:5433–5440. doi: 10.1021/ja312579u. [DOI] [PubMed] [Google Scholar]
- 174.Yamaguchi J, Naimuddin M, Biyani M, Sasaki T, Machida M, Kubo T, Funatsu T, Husimi Y, Nemoto N. cDNA display: a novel screening method for functional disulfide-rich peptides by solid-phase synthesis and stabilization of mRNA-protein fusions. Nucleic Acids Res. 2009;37:e108. doi: 10.1093/nar/gkp514. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Ueno S, Nemoto N. cDNA display: rapid stabilization of mRNA display. Methods Mol Biol. 2012;805:113–135. doi: 10.1007/978-1-61779-379-0_8. [DOI] [PubMed] [Google Scholar]
- 176.Mochizuki Y, Nemoto N. Evolution of disulfide-rich Peptide aptamers using cDNA display. Methods Mol Biol. 2012;805:237–250. doi: 10.1007/978-1-61779-379-0_13. [DOI] [PubMed] [Google Scholar]
- 177.Murray CJ, Baliga R. Cell-free translation of peptides and proteins:from high throughput screening to clinical production. Current Opinion in Chemical Biology. 2013;17:420–426. doi: 10.1016/j.cbpa.2013.02.014. [DOI] [PubMed] [Google Scholar]
- 178.FitzGerald K. In vitro display technologies - new tools for drug discovery. Drug Discov Today. 2000;5:253–258. doi: 10.1016/s1359-6446(00)01501-4. [DOI] [PubMed] [Google Scholar]
- 179.Reiersen H, Lobersli I, Loset GA, Hvattum E, Simonsen B, Stacy JE, McGregor D, Fitzgerald K, Welschof M, Brekke OH, Marvik OJ. Covalent antibody display--an in vitro antibody-DNA library selection system. Nucleic Acids Res. 2005;33:e10. doi: 10.1093/nar/gni010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Odegrip R, Coomber D, Eldridge B, Hederer R, Kuhlman PA, Ullman C, FitzGerald K, McGregor D. CIS display: In vitro selection of peptides from libraries of protein-DNA complexes. Proc Natl Acad Sci U S A. 2004;101:2806–2810. doi: 10.1073/pnas.0400219101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Leemhuis H, Stein V, Griffiths AD, Hollfelder F. New genotype-phenotype linkages for directed evolution of functional proteins. Curr Opin Struct Biol. 2005;15:472–478. doi: 10.1016/j.sbi.2005.07.006. [DOI] [PubMed] [Google Scholar]
- 182.Keppler A, Gendreizig S, Gronemeyer T, Pick H, Vogel H, Johnsson K. A general method for the covalent labeling of fusion proteins with small molecules in vivo. Nat Biotechnol. 2003;21:86–89. doi: 10.1038/nbt765. [DOI] [PubMed] [Google Scholar]
- 183.Kaltenbach M, Hollfelder F. SNAP display: in vitro protein evolution in microdroplets. Methods Mol Biol. 2012;805:101–111. doi: 10.1007/978-1-61779-379-0_7. [DOI] [PubMed] [Google Scholar]
- 184.Tawfik DS, Griffiths AD. Man-made cell-like compartments for molecular evolution. Nat Biotechnol. 1998;16:652–656. doi: 10.1038/nbt0798-652. [DOI] [PubMed] [Google Scholar]
- 185.Doi N, Yanagawa H. STABLE: protein-DNA fusion system for screening of combinatorial protein libraries in vitro. FEBS Lett. 1999;457:227–230. doi: 10.1016/s0014-5793(99)01041-8. [DOI] [PubMed] [Google Scholar]
- 186.Yonezawa M, Doi N, Kawahashi Y, Higashinakagawa T, Yanagawa H. DNA display for in vitro selection of diverse peptide libraries. Nucleic Acids Res. 2003;31:e118. doi: 10.1093/nar/gng119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Diamante L, Gatti-Lafranconi P, Schaerli Y, Hollfelder F. In vitro affinity screening of protein and peptide binders by megavalent bead surface display. Protein Eng Des Sel. 2013;26:713–724. doi: 10.1093/protein/gzt039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 188.Sepp A, Tawfik DS, Griffiths AD. Microbead display by in vitro compartmentalisation: selection for binding using flow cytometry. FEBS Lett. 2002;532:455–458. doi: 10.1016/s0014-5793(02)03740-7. [DOI] [PubMed] [Google Scholar]
- 189.Griffiths AD, Tawfik DS. Directed evolution of an extremely fast phosphotriesterase by in vitro compartmentalization. Embo j. 2003;22:24–35. doi: 10.1093/emboj/cdg014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 190.Gan R, Yamanaka Y, Kojima T, Nakano H. Microbeads display of proteins using emulsion PCR and cell-free protein synthesis. Biotechnol Prog. 2008;24:1107–1114. doi: 10.1002/btpr.43. [DOI] [PubMed] [Google Scholar]
- 191.Gan R, Furuzawa S, Kojima T, Kanie K, Kato R, Okochi M, Honda H, Nakano H. Directed evolution of angiotensin II-inhibiting peptides using a microbead display. J Biosci Bioeng. 2010;109:411–417. doi: 10.1016/j.jbiosc.2009.10.009. [DOI] [PubMed] [Google Scholar]
- 192.Paul S, Stang A, Lennartz K, Tenbusch M, Uberla K. Selection of a T7 promoter mutant with enhanced in vitro activity by a novel multi-copy bead display approach for in vitro evolution. Nucleic Acids Res. 2013;41:e29. doi: 10.1093/nar/gks940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 193.Bernath K, Hai M, Mastrobattista E, Griffiths AD, Magdassi S, Tawfik DS. In vitro compartmentalization by double emulsions: sorting and gene enrichment by fluorescence activated cell sorting. Anal Biochem. 2004;325:151–157. doi: 10.1016/j.ab.2003.10.005. [DOI] [PubMed] [Google Scholar]
- 194.Rothe A, Surjadi RN, Power BE. Novel proteins in emulsions using in vitro compartmentalization. Trends Biotechnol. 2006;24:587–592. doi: 10.1016/j.tibtech.2006.10.007. [DOI] [PubMed] [Google Scholar]
- 195.Skirgaila R, Pudzaitis V, Paliksa S, Vaitkevicius M, Janulaitis A. Compartmentalization of destabilized enzymemRNA-ribosome complexes generated by ribosome display: a novel tool for the directed evolution of enzymes. Protein Engineering Design & Selection. 2013;26:453–461. doi: 10.1093/protein/gzt017. [DOI] [PubMed] [Google Scholar]
- 196.Wang H, Liu R. Advantages of mRNA display selections over other selection techniques for investigation of proteinprotein interactions. Expert Rev Proteomics. 2011;8:335–346. doi: 10.1586/epr.11.15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Cotten SW, Zou J, Valencia CA, Liu R. Selection of proteins with desired properties from natural proteome libraries using mRNA display. Nat Protoc. 2011;6:1163–1182. doi: 10.1038/nprot.2011.354. [DOI] [PubMed] [Google Scholar]
- 198.Menendez A, Scott JK. The nature of target-unrelated peptides recovered in the screening of phage-displayed random peptide libraries with antibodies. Anal Biochem. 2005;336:145–157. doi: 10.1016/j.ab.2004.09.048. [DOI] [PubMed] [Google Scholar]
- 199.Vodnik M, Zager U, Strukelj B, Lunder M. Phage display: selecting straws instead of a needle from a haystack. Molecules. 2011;16:790–817. doi: 10.3390/molecules16010790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Thomas WD, Golomb M, Smith GP. Corruption of phage display libraries by target-unrelated clones: diagnosis and countermeasures. Anal Biochem. 2010;407:237–240. doi: 10.1016/j.ab.2010.07.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Newton-Northup JR, Deutscher SL. Contending With Target Unrelated Peptides from Phage Display. J Mol Imaging Dynam. 2012;2:1–3. [Google Scholar]
- 202.Derda R, Tang SK, Li SC, Ng S, Matochko W, Jafari MR. Diversity of phage-displayed libraries of peptides during panning and amplification. Molecules. 2011;16:1776–1803. doi: 10.3390/molecules16021776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Crawford M, Woodman R, Ko Ferrigno P. Peptide aptamers: tools for biology and drug discovery. Brief Funct Genomic Proteomic. 2003;2:72–79. doi: 10.1093/bfgp/2.1.72. [DOI] [PubMed] [Google Scholar]
- 204.Shi H, Fan X, Sevilimedu A, Lis JT. RNA aptamers directed to discrete functional sites on a single protein structural domain. Proc Natl Acad Sci U S A. 2007;104:3742–3746. doi: 10.1073/pnas.0607805104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.'t Hoen PAC, Jirka SMG, tsen Broeke BR, Schultes EA, Aguilera B, Pang KH, Heemskerk H, Aartsma-Rus A, van Ommen GJ, den Dunnen JT. Phage display screening without repetitious selection rounds. Anal Biochem. 2012;421:622–631. doi: 10.1016/j.ab.2011.11.005. [DOI] [PubMed] [Google Scholar]
- 206.Fields S, Song O. A novel genetic system to detect protein-protein interactions. Nature. 1989;340:245–246. doi: 10.1038/340245a0. [DOI] [PubMed] [Google Scholar]
- 207.Stynen B, Tournu H, Tavernier J, Van Dijck P. Diversity in genetic in vivo methods for protein-protein interaction studies: from the yeast two-hybrid system to the mammalian splitluciferase system. Microbiol Mol Biol Rev. 2012;76:331–382. doi: 10.1128/MMBR.05021-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Bickle MB, Dusserre E, Moncorge O, Bottin H, Colas P. Selection and characterization of large collections of peptide aptamers through optimized yeast two-hybrid procedures. Nat Protoc. 2006;1:1066–1091. doi: 10.1038/nprot.2006.32. [DOI] [PubMed] [Google Scholar]
- 209.Causier B, Davies B. Analysing protein-protein interactions with the yeast two-hybrid system. Plant Mol Biol. 2002;50:855–870. doi: 10.1023/a:1021214007897. [DOI] [PubMed] [Google Scholar]
- 210.Koegl M, Uetz P. Improving yeast two-hybrid screening systems. Brief Funct Genomic Proteomic. 2007;6:302–312. doi: 10.1093/bfgp/elm035. [DOI] [PubMed] [Google Scholar]
- 211.Suter B, Kittanakom S, Stagljar I. Two-hybrid technologies in proteomics research. Curr Opin Biotechnol. 2008;19:316–323. doi: 10.1016/j.copbio.2008.06.005. [DOI] [PubMed] [Google Scholar]
- 212.Ferro E, Trabalzini L. The yeast two-hybrid and related methods as powerful tools to study plant cell signalling. Plant Mol Biol. 2013;83:287–301. doi: 10.1007/s11103-013-0094-4. [DOI] [PubMed] [Google Scholar]
- 213.Benatuil L, Perez JM, Belk J, Hsieh CM. An improved yeast transformation method for the generation of very large human antibody libraries. Protein Eng Des Sel. 2010;23:155–159. doi: 10.1093/protein/gzq002. [DOI] [PubMed] [Google Scholar]
- 214.Sudha G, Nussinov R, Srinivasan N. An overview of recent advances in structural bioinformatics of protein-protein interactions and a guide to their principles. Prog Biophys Mol Biol. 2014;116:141–150. doi: 10.1016/j.pbiomolbio.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 215.Eijsink VG, Gaseidnes S, Borchert TV, van den Burg B. Directed evolution of enzyme stability. Biomol Eng. 2005;22:21–30. doi: 10.1016/j.bioeng.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 216.Romero PA, Arnold FH. Exploring protein fitness landscapes by directed evolution. Nat Rev Mol Cell Biol. 2009;10:866–876. doi: 10.1038/nrm2805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 217.Schmid FX. Lessons about protein stability from in vitro selections. Chembiochem. 2011;12:1501–1507. doi: 10.1002/cbic.201100018. [DOI] [PubMed] [Google Scholar]
- 218.Golynskiy MV, Haugner JC, 3rd, Seelig B. Highly diverse protein library based on the ubiquitous (beta/alpha)(8) enzyme fold yields well-structured proteins through in vitro folding selection. Chembiochem. 2013;14:1553–1563. doi: 10.1002/cbic.201300326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 219.Socha RD, Tokuriki N. Modulating protein stability - directed evolution strategies for improved protein function. Febs j. 2013;280:5582–5595. doi: 10.1111/febs.12354. [DOI] [PubMed] [Google Scholar]
- 220.Vanhee P, van der Sloot AM, Verschueren E, Serrano L, Rousseau F, Schymkowitz J. Computational design of peptide ligands. Trends Biotechnol. 2011;29:231–239. doi: 10.1016/j.tibtech.2011.01.004. [DOI] [PubMed] [Google Scholar]
- 221.Brustad EM, Arnold FH. Optimizing non-natural protein function with directed evolution. Curr Opin Chem Biol. 2011;15:201–210. doi: 10.1016/j.cbpa.2010.11.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 222.Wurch T, Pierre A, Depil S. Novel protein scaffolds as emerging therapeutic proteins: from discovery to clinical proof-of-concept. Trends Biotechnol. 2012;30:575–582. doi: 10.1016/j.tibtech.2012.07.006. [DOI] [PubMed] [Google Scholar]
- 223.Banta S, Dooley K, Shur O. Replacing antibodies: engineering new binding proteins. Annu Rev Biomed Eng. 2013;15:93–113. doi: 10.1146/annurev-bioeng-071812-152412. [DOI] [PubMed] [Google Scholar]
- 224.Emanuel SL, Engle LJ, Chao G, Zhu RR, Cao C, Lin Z, Yamniuk AP, Hosbach J, Brown J, Fitzpatrick E, Gokemei-jer J, Morin P, Morse BA, Carvajal IM, Fabrizio D, Wright MC, Das Gupta R, Gosselin M, Cataldo D, Ryseck RP, Doyle ML, Wong TW, Camphausen RT, Cload ST, Marsh HN, Gottardis MM, Furfine ES. A fibronectin scaffold approach to bispecific inhibitors of epidermal growth factor receptor and insulin-like growth factor-I receptor. MAbs. 2011;3:38–48. doi: 10.4161/mabs.3.1.14168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Tolcher AW, Sweeney CJ, Papadopoulos K, Patnaik A, Chiorean EG, Mita AC, Sankhala K, Furfine E, Gokemeijer J, Iacono L, Eaton C, Silver BA, Mita M. Phase I and pharmacokinetic study of CT-322 (BMS-844203), a targeted Adnectin inhibitor of VEGFR-2 based on a domain of human fibronectin. Clin Cancer Res. 2011;17:363–371. doi: 10.1158/1078-0432.CCR-10-1411. [DOI] [PubMed] [Google Scholar]
- 226.Schonfeld D, Matschiner G, Chatwell L, Trentmann S, Gille H, Hulsmeyer M, Brown N, Kaye PM, Schlehuber S, Hohlbaum AM, Skerra A. An engineered lipocalin specific for CTLA-4 reveals a combining site with structural and conformational features similar to antibodies. Proc Natl Acad Sci U S A. 2009;106:8198–8203. doi: 10.1073/pnas.0813399106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 227.Zahnd C, Kawe M, Stumpp MT, de Pasquale C, Tamaskovic R, Nagy-Davidescu G, Dreier B, Schibli R, Binz HK, Waibel R, Pluckthun A. Efficient tumor targeting with high-affinity designed ankyrin repeat proteins: effects of affinity and molecular size. Cancer Res. 2010;70:1595–1605. doi: 10.1158/0008-5472.CAN-09-2724. [DOI] [PubMed] [Google Scholar]
- 228.Martin-Killias P, Stefan N, Rothschild S, Pluckthun A, Zangemeister-Wittke U. A novel fusion toxin derived from an EpCAM-specific designed ankyrin repeat protein has potent antitumor activity. Clin Cancer Res. 2011;17:100–110. doi: 10.1158/1078-0432.CCR-10-1303. [DOI] [PubMed] [Google Scholar]
- 229.Friedman M, Nordberg E, Hoiden-Guthenberg I, Brismar H, Adams GP, Nilsson FY, Carlsson J, Stahl S. Phage display selection of Affibody molecules with specific binding to the extracellular domain of the epidermal growth factor receptor. Protein Eng Des Sel. 2007;20:189–199. doi: 10.1093/protein/gzm011. [DOI] [PubMed] [Google Scholar]
- 230.Baum RP, Prasad V, Muller D, Schuchardt C, Orlova A, Wennborg A, Tolmachev V, Feldwisch J. Molecular imaging of HER2-expressing malignant tumors in breast cancer patients using synthetic 111In- or 68Ga-labeled affibody molecules. J Nucl Med. 2010;51:892–897. doi: 10.2967/jnumed.109.073239. [DOI] [PubMed] [Google Scholar]
- 231.Ackerman SE, Currier NV, Bergen JM, Cochran JR. Cystine-knot peptides: emerging tools for cancer imaging and therapy. Expert Rev Proteomics. 2014;11:561–572. doi: 10.1586/14789450.2014.932251. [DOI] [PubMed] [Google Scholar]
- 232.Stern LA, Case BA, Hackel BJ. Alternative Non-Antibody Protein Scaffolds for Molecular Imaging of Cancer. Curr Opin Chem Eng. 2013;2 doi: 10.1016/j.coche.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 233.Lofblom J, Feldwisch J, Tolmachev V, Carlsson J, Stahl S, Frejd FY. Affibody molecules: engineered proteins for therapeutic, diagnostic and biotechnological applications. FEBS Lett. 2010;584:2670–2680. doi: 10.1016/j.febslet.2010.04.014. [DOI] [PubMed] [Google Scholar]
- 234.Borghouts C, Kunz C, Groner B. Peptide aptamers: recent developments for cancer therapy. Expert Opin Biol Ther. 2005;5:783–797. doi: 10.1517/14712598.5.6.783. [DOI] [PubMed] [Google Scholar]
- 235.Clonis YD. Affinity chromatography matures as bioin-formatic and combinatorial tools develop. J Chromatogr A. 2006;1101:1–24. doi: 10.1016/j.chroma.2005.09.073. [DOI] [PubMed] [Google Scholar]
- 236.Engfeldt T, Renberg B, Brumer H, Nygren PA, Karlstrom AE. Chemical synthesis of triple-labelled three-helix bundle binding proteins for specific fluorescent detection of unlabelled protein. Chembiochem. 2005;6:1043–1050. doi: 10.1002/cbic.200400388. [DOI] [PubMed] [Google Scholar]
- 237.Ramoni R, Bellucci S, Grycznyski I, Grycznyski Z, Grolli S, Staiano M, Bellis GD, Micciulla F, Pastore R, Tiberia A, Conti V, Merli E, Varriale A, Rossi M, D'Auria S. The protein scaffold of the lipocalin odorant-binding protein is suitable for the design of new biosensors for the detection of explosive components. Journal of Physics: Condensed Matter. 2007;19:395012. [Google Scholar]