

SUMMARY
Escherichia coli FtsN is a bitopic membrane protein that is essential for triggering active cell constriction. A small periplasmic subdomain (EFtsN) is required and sufficient for function, but its mechanism of action is unclear. We isolated extragenic EFtsN*-suppressing mutations that restore division in cells producing otherwise non-functional variants of FtsN. These mapped to the IC domain of FtsA in the cytoplasm and to small subdomains of the FtsB and FtsL proteins in the periplasm. All FtsB and FtsL variants allowed survival without EFtsN, but many then imposed a new requirement for interaction between the cytoplasmic domain of FtsN (NFtsN) with FtsA. Alternatively, variants of FtsA, FtsB or FtsL acted synergistically to allow cell division in the complete absence of FtsN. Strikingly, moreover, substitution of a single residue in FtsB (E56) proved sufficient to rescue ΔftsN cells as well. In FtsN+ cells, EFtsN*-suppressing mutations promoted cell fission at an abnormally small cell size, and caused cell shape and integrity defects under certain conditions. This and additional evidence support a model in which FtsN acts on either side of the membrane to induce a conformational switch in both FtsA and the FtsBLQ subcomplex to derepress septal peptidoglycan synthesis and membrane invagination.
Keywords: Division, FtsB, FtsL, FtsQ, PBP3
INTRODUCTION
Bacterial cell fission (cytokinesis, constriction, septation) is mediated by a trans-envelope ring-shaped organelle called the septal ring (SR) or divisome. The E.coli SR is a complex apparatus with over 30 distinct protein components. Ten (FtsA, B, I, K, L, N, Q, W, Z, and ZipA) are essential and cells that lack any of these core components form smooth multi-nucleoid filaments that eventually die. Many of the other, non-core, SR proteins also play important roles in the fission process, but are individually not essential for cell survival (de Boer, 2010, Lutkenhaus et al., 2012, Egan & Vollmer, 2013).
SR assembly starts with assembly of the FtsZ ring (Z-ring, ZR) on the cytoplasmic face of the inner membrane (IM), well before actual constriction at this site. The ZR consists of polymers of the tubulin-like FtsZ protein that are decorated by various non-core FtsZ associated proteins (Zap’s), and anchored to the membrane via interactions between the C-terminus of FtsZ with either FtsA or ZipA (de Boer, 2010, Lutkenhaus et al., 2012, Egan & Vollmer, 2013). ZipA is a type Ib (N-out) bitopic integral membrane protein (Hale & de Boer, 1997), and FtsA binds membrane peripherally via a C-terminal amphiphatic helix (Pichoff & Lutkenhaus, 2005). FtsA is also a remarkable domain variant of the actin family that instead of a typical IB domain flanked by the IA and 2B domains, possesses a unique IC domain positioned between the IA and 2A domains (van Den Ent & Löwe, 2000). Despite this profound deviation of archetypical actin structure, FtsA can form homo-polymeric filaments, and it may be short oligomers/polymers of FtsA that help tether longer FtsZ polymers to the IM (Lara et al., 2005, Szwedziak et al., 2012). Though both FtsA and ZipA are normally essential SR components, mutant variants of FtsA that bypass the requirement for ZipA can be readily isolated (Geissler et al., 2003, Bernard et al., 2007, Goehring et al., 2007, Pichoff et al., 2012). Most of these variants also display reduced self-interaction, indicating that the polymerization state of FtsA controls some critical aspect of SR assembly and/or function (Pichoff et al., 2012).
ZR assembly is followed by ordered recruitment of the remaining seven core-proteins (ZR<FtsK<FtsBLQ<FtsIW<FtsN), which are all integral IM species. FtsB, FtsI (=PBP3), FtsL, FtsN and FtsQ are all type II bitopic (N-in) membrane proteins with relatively large periplasmic domains. FtsK and FtsW are polytopic. Strong accumulation of FtsN at the SR marks the successful incorporation of all core proteins and maturation to a constriction-competent organelle. Most of the non-core SR proteins subsequently join the assembly at/after active constriction has started (Goehring & Beckwith, 2005, de Boer, 2010, Lutkenhaus et al., 2012, Egan & Vollmer, 2013).
The complexity of the SR reflects the variety of intricate tasks it must execute and coordinate. In E.coli and other Gram-negative bacteria, these include: i) invagination of the IM, ii) synthesis of an inward growing layer of septal peptidoglycan (sPG), iii) precise splitting of this growing sPG layer from the periplasmic side to form the two new polar caps, iv) invagination of the outer-membrane (OM) in the space created by sPG splitting, and v) closure of septal pores in both membranes. Interestingly, only IM invagination/closure and sPG synthesis are essential processes for survival of E.coli and the subsequent steps are mostly executed by non-core SR components (Gerding et al., 2007, Peters et al., 2011, Yang et al., 2011). Cells that are blocked at the sPG splitting and/or OM invagination steps form long, but viable, chains of cell compartments held together by bridges of unseparated sPG and/or OM (Meury & Devilliers, 1999, Heidrich et al., 2001, Gerding et al., 2007, Uehara et al., 2009). Disintegration of internal cell units limits the length of such chains (Priyadarshini et al., 2007), which probably aids in their survival and propagation.
IM invagination may be driven by both active contraction of the ZR and ingrowth of the sPG layer. In vitro experiments indicate that as FtsA tethers FtsZ filaments to the membrane, it can also destabilize the filaments (Beuria et al., 2009, Loose & Mitchison, 2014). In the presence of GTP and ATP, moreover, purified FtsZ and FtsA are sufficient to spontaneously form membrane-associated contractile rings in liposomes (Osawa & Erickson, 2013). How the SR controls and executes sPG synthesis is relatively poorly understood. Of the core SR proteins, only the monofunctional murein transpeptidase PBP3 (FtsI) has a clearly defined and direct role in sPG synthesis (Adam et al., 1997, Pisabarro et al., 1986). Beside its own synthetic activity, PBP3 anchors a larger sPG synthase subcomplex in the SR (Höltje, 1998, Vollmer & Bertsche, 2008). This includes its direct partners FtsW (Mercer & Weiss, 2002, Fraipont et al., 2011), which may (Mohammadi et al., 2011) or may not (Sham et al., 2014) be a membrane flippase for the lipidII murein precursor, and the bifunctional murein synthase PBP1B (Bertsche et al., 2006), which likely contributes both glycosyltransferase and transpeptidase activities to the SR (Bertsche et al., 2005). PBP1B itself is not essential because the redundant synthase PBP1A compensates when PBP1B function is compromised (Yousif et al., 1985, Born et al., 2006).
Why the remaining core SR proteins FtsB, K, L, N and Q are essential for cell fission and viability is not clear. FtsK plays well defined roles in proper chromosome resolution and segregation, but these activities are not essential for cell fission and survival per se (Sherratt et al., 2010). Prior recruitment of FtsK by the ZR is normally required to recruit subsequent core components (Chen & Beckwith, 2001). This role in SR assembly might be its only essential function as normal expression of certain mutant FtsA variants, or overexpression of each of several SR proteins, can restore fission in cells that lack FtsK completely (Draper et al., 1998, Geissler & Margolin, 2005, Bernard et al., 2007, Goehring et al., 2007, Dubarry et al., 2010). FtsB, L and Q form a phylogenetically well-conserved subcomplex that joins the maturing SR after FtsK (Buddelmeijer & Beckwith, 2004, Goehring & Beckwith, 2005). The subcomplex directly recruits FtsW and its PBP3 partner, and is thought to primarily function as a scaffold for maturation of the SR (Gonzalez & Beckwith, 2009, Gonzalez et al., 2010).
FtsN is the last of the core proteins to accumulate sharply at the SR (Addinall et al., 1997, Chen & Beckwith, 2001, Wissel & Weiss, 2004). It is a fairly abundant protein (Ursinus et al., 2004), and is proposed to both stabilize the SR (Chen & Beckwith, 2001, Rico et al., 2010), and help trigger the active constriction phase of cell fission (Moll & Thanbichler, 2009, Gerding et al., 2009). Various evidence suggests that FtsN can interact with many other SR proteins, including components of the ZR (FtsA, ZapA), the FtsBLQ subcomplex, and the sPG synthase subcomplex (FtsW, PBP3, PBP1B, MtgA) (Di Lallo et al., 2003, Karimova et al., 2005, Muller et al., 2007, Derouaux et al., 2008, Alexeeva et al., 2010, Busiek et al., 2012). The functional significance of most of these interactions is still unclear.
FtsN consists of a small cytoplasmic (FtsN1–30, NFtsN), a trans-membrane (FtsN31–54, TMFtsN), and a large periplasmic (FtsN55–319) domain (Fig. 1A) (Dai et al., 1996). The latter consists of juxta-membrane α-helices H1 (~FtsN62–67), H2 (~FtsN80–93) and H3 (~FtsN117–123,), a long linker peptide (FtsN124–242), and a C-terminal globular SPOR domain (FtsN243–319, SFtsN) (Yang et al., 2004). The isolated SFtsN domain binds peptidoglycan in vitro (Ursinus et al., 2004, Moll & Thanbichler, 2009, Duncan et al., 2013). Like SPOR domains from other proteins, however, SFtsN accumulates sharply at sites of active constriction in vivo, indicating it specifically prefers a form of PG that is transiently enriched at these sites (Moll & Thanbichler, 2009, Gerding et al., 2009, Arends et al., 2010). Interestingly, NFtsN interacts directly with the IC domain of FtsA (Busiek et al., 2012), and overexpression of an FtsA variant with a substitution in this domain (FtsAE124A) can rescue cells that lack FtsN completely (Bernard et al., 2007, Gerding et al., 2009). Thus, FtsN is well positioned to communicate progress in sPG synthesis to the ZR in the cytoplasm, and may help license the latter to contract. If so, however, this function of FtsN is dispensible, because neither NFtsN nor SFtsN are required for the essential activity of the protein (Dai et al., 1996, Ursinus et al., 2004).
Figure 1. Domain structure of E.coli FtsN, properties of genetic constructs, and critical residues in the essential domain, EFtsN.

(A) Depicted are the full-length protein (FtsN1–319) and an expanded view of an N-terminal portion (FtsN1–128), immediately below. The transmembrane domain (TMFtsN, light grey), helices H1, H2, and H3 (black) in the periplasmic juxtamembrane region, and the C-terminal SPOR domain (SFtsN, dark grey) are indicated. The small periplasmic peptide that is required and sufficient for FtsN’s essential function in cell division (EFtsN) is indicated with the double-headed arrow in the expanded view.
Also shown are inserts present on plasmids that produce fusions of various portions of FtsN to RFP, GFP or TTGFP under control of the ara (pBL142 and pLP160) or lac (all other constructs) regulatory region. TTGFP-fusions contain the TorA signal peptide (hatched box) that is cleaved upon export to the periplasm via the twin arginine transport (Tat) system. Grey lines represent non-FtsN residues encoded by deletion-substitution constructs. TM1 represents the first transmembrane domain of MalF. Some fusions end with the non-FtsN Leu-Glu dipeptide (LE), as indicated. Columns indicate the FtsN residues present in each fusion, and whether the fusion could (+) or could not (−) compensate for the absence of native FtsN in CH31 [PBAD::ftsN] and/or CH34 [ΔftsN].
(B) Spot titer analyses of strain CH34 [ΔftsN] harboring plasmids pCH201 [Plac::gfp-ftsN], pBL205 [Plac::gfp-ftsNΔ(59–73)<>6], pBL211 [Plac::gfp-ftsNΔ(59–73)<>15], or pBL210 [Plac::gfp-ftsNΔ(59–73)<>95]. The number of residues between the end of TMFtsN and the beginning of EFtsN (TM-E) and the difference with the native protein (Δ, in parentheses) in each encoded fusion protein is indicated to the right of each row. Cells were grown overnight in LB with 25 μM IPTG, diluted in LB to OD600=4×10X, and 5 μl of each dilution was spotted on LB agar with or without 500 μM IPTG, as indicated. Plates were incubated at 30°C for 18 hr.
(C) Spot titer analyses of strain CH34 [ΔftsN] harboring plasmids encoding periplasmic TTGFP-FtsNX-Y fusions under control of Plac. The contiguous stretch of FtsN residues present in each fusion (X–Y) is indicated on the right of the rows. Cells were grown overnight in M9-maltose with 250 μM IPTG, diluted in M9-maltose medium to OD600=4×10X, and 5 μl of each dilution was spotted on LB (upper panels) or M9-maltose (lower panels) agar with or without 250 μM IPTG, as indicated. Plates were incubated at 30°C for 18 (LB) or 24 (M9) hr.
(D) FtsN residues 75–93, corresponding to EFtsN, are colored according to phylogenetic conservation (% identity in OMA group 145686, release 16 (Altenhoff et al., 2011)); red (100–95%), blue (95–90%), green (90–85%), and black (<85%). Permissible (P) and non-permissible (NP) residue substitutions are indicated above and below the EFtsN sequence, respectively. NP substitutions encircled in orange were used as query in screens for extragenic EFtsN*-suppressors.
Rather, the essential domain of FtsN (EFtsN) is confined to a small periplasmic peptide of at most 35 residues centered about helix H2 (Gerding et al., 2009). The EFtsN peptide by itself shows no obvious affinity for the SR, and needs to be overproduced to restore normal division of ΔftsN cells. On the other hand, generation of the SFtsN-target at the SR requires the activity of EFtsN, as well as that of PBP3 and at least one of the murein amidases responsible for splitting sPG (Gerding et al., 2009). Hence, we proposed that FtsN is integral to a positive feedback mechanism that helps trigger and sustain the active constriction phase. In the model, EFtsN allosterically stimulates sPG synthesis and splitting of new sPG by murein hydrolases generates the substrate for SFtsN, which then recruits more FtsN to the SR, increasing the local concentration of EFtsN, et cetera (Gerding et al., 2009).
Here, we addressed the mechanisms of action of FtsN in more depth. Consistent with the idea that EFtsN is required for PBP3 activity, we show that reduced EFtsN activity is very poorly tolerated in cells that lack PBP1B, and causes cell lysis rather than chaining or filamentation. In principle, EFtsN could regulate PBP3 directly, or via a more circuitous route that involves one or more of the other essential SR components. We took a genetic approach to search for the proximal target of EFtsN. First, we narrowed the domain down to a 19-residue peptide (FtsN75–93), and established that single substitutions at one of three FtsN residues (W83, Y85, and L89) abrogate it’s essential function. We then screened for extragenic suppressors that restore viability to cells producing non-functional FtsN variants as the sole source of the protein. Notably, this yielded suppressing mutations affecting either the IC domain of FtsA, or a small periplasmic subdomain of either FtsB or FtsL. All suppressing variants of FtsB or FtsL allowed cells to survive in the absence of EFtsN, but the majority then imposed a requirement for the normally non-essential interaction between NFtsN and FtsA in the cytoplasm. This new requirement could be overcome by combining suppressing mutations in ftsB or ftsL with each other, or with those in ftsA, resulting in strains that survive and divide in the complete absence of FtsN. Remarkably, moreover, some FtsB variants rescued ΔftsN cells without the need for additional mutations, and all were affected in a single periplasmic residue (E56). Under normal growth conditions, the suppressing mutations stimulated premature cell fission in otherwise wt cells (FtsN+). At 42°C on low osmotic medium, however, they also caused cell shape and integrity defects, and associated lethality was suppressed by removal of ftsN.
The results imply that FtsN stimulates cell fission by acting on both sides of the membrane and that the FtsBLQ subcomplex is not a mere scaffold for maturation of the SR, but also regulates its activity in an FtsN-dependent manner. They further support a model in which EFtsN allosterically induces a conformational state of FtsBLQ that is required to allow sPG synthesis by PBP3 and associated synthases. The interaction between NFtsN and FtsA may promote this same state of FtsBLQ via direct or indirect interactions of the subcomplex with FtsA.
RESULTS
Further definition of the essential domain of FtsN (EFtsN)
To define the essential domain of FtsN (EFtsN), we previously used complementation assays with various portions of FtsN fused to the C-terminus of either cytoplasmic GFP or Tat-targeted periplasmic TTGFP. These showed that ΔftsN cells can be rescued by trans-membrane GFP-FtsN1–90 or periplasmic TTGFP-FtsN71–105, but not by GFP-FtsN1–81 or TTGFP-FtsN71–90 (Fig. 1A). Rescue by the former two fusions suggested that EFtsN resides in the T71-E90 interval, though the failure of the periplasmic TTGFP-FtsN71–90 fusion to support cell division in ΔftsN cells raised the possibility that truncation of FtsN near/at the C-terminal boundary (e.g. in functional GFP-FtsN1–90) imposes a requirement for additional N-terminal residues in the 1–70 interval (Gerding et al., 2009).
We tested additional fusions to define the boundaries of EFtsN more precisely (Fig. 1A). Western analyses indicated that none of the studied fusion proteins were subject to excessive degradation (Fig. S1, S2). Periplasmic fusions TTGFP-FtsN75–105 and TTGFP-FtsN75–99 were as effective as TTGFP-FtsN71–105 in stimulating cell division, and ΔftsN cells producing any of these fusions propagated readily on both rich (LB) and minimal (M9) medium (Fig. 1A and C, and data not shown). In contrast, a TTGFP-FtsN80–105 fusion was not functional (Fig. 1A), suggesting EFtsN starts between L75–P79. Further trimming of TTGFP-FtsN75–99 at the C-terminal end yielded TTGFP-FtsN75–93, the smallest functional fusion we identified. Though TTGFP-FtsN75–93 could still rescue ΔftsN cells, it was significantly less effective in doing so than the other functional fusions. Consequently, cells of strain CH34/pLP218 [ΔftsN/Plac::ttgfp-ftsN75–93] could grow on M9 in the presence of IPTG, but suffered a lethal division defect on LB, even at high concentrations of inducer (Fig. 1C, and data not shown). Thus, EFtsN does not extend past Q93, but residues in the P94–P99 interval promote the activity or stability of the domain, when produced as a fusion to periplasmic TTGFP.
As alluded to above, the C-terminal boundary of EFtsN is slightly different when the domain is expressed as a trans-membrane fusion (Fig. 1A). The finding that fusion GFP-FtsN1–90 is functional while TTGFP-FtsN71–90 is not suggested some additional requirement for the first 70 residues in EFtsN function, when the latter is truncated at E90. Additional fusions were studied to further explore this possibility. Like TTGFP-FtsN71–90, a larger periplasmic fusion containing all periplasmic residues up to E90 (TTGFP-FtsN55–90) was still not capable of rescuing ΔftsN cells (Fig. 1A). This implied that the presence of residues in the 55–71 interval are not sufficient to explain the functionality of GFP-FtsN1–90, and raised the possibility that specific residues in the cytoplasmic (NFtsN) or transmembrane (TMFtsN) domains might be important instead. This was tested with a variant of a functional trans-membrane RFP-FtsN1–90 fusion in which both NFtsN and TMFtsN were replaced with corresponding domains of the MalF protein. As summarized in Fig. 1A, the RFP-MalF2–39-FtsN55–90 variant was still capable of supporting cell division in ΔftsN cells. Altogether, the complementation results indicate that the functionality of transmembrane XFP-FtsN1–90 fusions on one hand, and the non-functionality of periplasmic TTGFP-FtsN71–90 or TTGFP-FtsN55–90 fusions on the other, is not determined by any specific FtsN residues in the 1–71 interval. Rather, being membrane tethered per se may help to position EFtsN at an optimal distance from the membrane and/or hold it in an active conformation.
We also explored the importance of the periplasmic residues between TMFtsN and EFtsN more specifically by studying derivatives of a full length GFP-FtsN fusion (encoded by pCH201) in which 15 of these (FtsN59–73, including H1) are replaced with unrelated peptides of 6 (pBL205), 15 (pBL211) or 95 (pBL210) residues, with the latter two mostly derived from the unstructured P/Q-rich domain of the ZipA protein (Hale & de Boer, 1997, Ohashi et al., 2002). As summarized in Fig. 1A, all three substitution derivatives supported viability of ΔftsN cells. Notably, however, substitution of FtsN59–73 with only 6 residues (a deficit of 9 residues) severely compromised the ability of the protein to support cell division, while substitution with an equal number (15) or with 80 additional residues had relatively little effect (Fig. 1B, S2, and data not shown). We infer that restriction of the maximal distance of EFtsN to the outer leaflet of the inner membrane is far more detrimental to function than expansion of this distance. Hence, the residues between TMFtsN and EFtsN serve as a linker that allows EFtsN to reach a sufficient distance from the IM, and this may be their only role.
Identification of critical residues in EFtsN
To identify individual residues critical to FtsN function, each in the FtsN75–93 interval was next subjected to site-scanning (residues 80–93) and/or site-directed mutagenesis, and resulting mutants were tested for their ability to rescue growth of the FtsN-depletion strain CH31 [PBAD::ftsN] in the absence of arabinose. Specific site-directed mutants were generated in the context of pCH201 [Plac::gfp-ftsN], encoding full length GFP-FtsN. Random site-scanning mutants were obtained in the context of pBL116 [Plac::gfp-ftsNT64G, S67G, +A102]. This plasmid is similar to pCH201, except that ftsN codons 68–101 are flanked by two unique SfiI sites that were introduced to facilitate the generation of site-scanning libraries. It encodes a fully functional variant of GFP-FtsN in which FtsN residues T64 and S67 are each replaced with glycine, and an alanine residue is inserted between FtsN residues 101 and 102. We analyzed 114 ftsN alleles with mutations of a single codon, yielding a total of 57 distinct residue substitutions within the FtsN75–93 interval (Table S1). Of the latter, 26 had no effect on the ability of GFP-FtsN to rescue FtsN-depleted cells, and such permissible substitutions were identified for each of the 19 residues in the targeted interval (Fig. 1D). The remaining 31 substitutions abrogated the essential function of FtsN. Interestingly, all affected one of only three residues; W83, Y85, or L89. With the exception of Y85G, permissible substitutions in these three residues generally conserved their aromatic and/or hydrophobic character, though many substitutions were not tolerated. For example, Phe was permissible at each one of the three positions, but Ala was not and Y85 could also not be substituted with I, L, V, or W (Fig. 1D, Table S1). The fact that site-scanning mutagenesis did not yield non-permissible substitutions in any other residue within the 80–93 interval is notable, and suggests that W83, Y85, and L89 may be the only FtsN residues that are truly critical for division and viability.
PBP1B-dependent phenotypes of cells with reduced FtsN activity
Given that EFtsN is essential, periplasmic, and small, we hypothesize that it stimulates cell constriction via allosteric regulation of a periplasmic domain of one of the other essential SR proteins. FtsI (=PBP3) and FtsW are directly involved in sPG synthesis and are attractive candidates for regulation by EFtsN. Whereas treatment of wt or ponA cells (lacking PBP1A) with PBP3-specific β-lactams typically results in the formation of long filaments, treatment of ponB cells (lacking PBP1B) results in rapid lysis instead (Schmidt et al., 1981, Garcia del Portillo & de Pedro, 1990, Denome et al., 1999).
We reasoned that if EFtsN were required for normal PBP3 activity it’s depletion phenotype might similarly depend on PBP1B, and this was indeed the case (Fig. 2). The chromosomal ftsNslm117 (ftsN::EZTnKan-2) allele encodes a SPOR-less version of the protein, and confers a mild cell chaining phenotype due to diminished EFtsN activity at the SR (Gerding et al., 2009). The allele could readily be combined with ΔponA, resulting in strain BL105 [ΔponA ftsNslm117] that grew as well as wt or single mutant strains (Fig. 2A) and showed the typical ftsNslm117 chaining phenotype (Fig. 2B). In contrast, ftsNslm117 could not be combined with ΔponB, unless cells carried complementing constructs encoding PBP1B or EFtsN (Fig. 2, and data not shown). For example, strain BL84(iMG62) [ΔponB ftsNslm117(Plac::gfp-ftsN1–123)] carries a chromosomal CRIM construct integrated at attHK022 (Haldimann & Wanner, 2001) that encodes SPOR-less GFP-FtsN1–123 under control of the lac promotor. The strain only grew well in the presence of high concentrations of inducer (≥ 500 μM IPTG) and its division phenotype was then close to normal, due to suppression of cell chaining by GFP-FtsN1–123 (Fig. 2A,D). When inoculated in medium without inducer, however, cells lysed after a few mass doublings, with many cells showing signs of initial lysis at septation sites (Fig. 2A,C). Thus, ftsNslm117 is synthetically lethal with ΔponB, but not with ΔponA, and EFtsN-depletion in ΔponB cells induces lysis rather than chain and/or filament formation. These PBP1B-dependent phenotypes parallel those induced by PBP3-specific drugs and support the idea that EFtsN is needed for full PBP3 activity.
Figure 2. Reduced FtsN function in the absence of PBP1B causes massive cell lysis.

(A) Growth curves in LB at 30°C of strains TB28 [wt] (black), TB77 [ftsNslm117] (dark grey), CH82 [ΔponA] (grey), BL105 [ΔponA ftsNslm117] (light grey), BL24 [ΔponB] (blue), and BL84(iMG62) [ΔponB ftsNslm117(Plac::gfp-ftsN1–123)] (bracketed curves) growing with 500 μM IPTG (green), 100 μM IPTG (magenta), 0.1 % glucose (red), or nothing (orange) supplemented to the medium. Note that the TB28, TB77, CH82, and BL105 curves essentially superimpose. Also note the distinct drop around 180 min in optical densities of the BL84(iMG62) cultures without IPTG. Cultures were grown to density overnight in LB, or in LB with 500 μM IPTG (strain BL84(iMG62) only), and diluted 200-fold in the same, or supplemented as indicated for BL84(iMG62). Growth was continued at 30°C and OD600 values were determined every 20 minutes.
(B–D) Cells of strains BL105 [ΔponA ftsNslm117] (B), and BL84(iMG62) [ΔponB ftsNslm117(Plac::gfp-ftsN1–123)] growing in the presence of 0.1 % glucose (C) or 500 μM IPTG (D). Cultures were grown in parallel as in panel A. Cells in B and D were imaged at OD600=~0.6, and cells in C were sampled at the same time as those in D. Note septal bulge (arrow) and ‘rabbit ear’ (arrow head), indicative of septal lysis, in panel C. Bar equals 4 μm.
Screen for extragenic suppressors of non-functional ftsN alleles
In principle, EFtsN could regulate PBP3 directly, or its allosteric ‘signal’ could be relayed by additional SR components via more circuitous routes before affecting the sPG synthase. To help elucidate the function of EFtsN more precisely, we screened for spontaneous extragenic suppressors of specific lethal mutations in one of the three essential EFtsN residues identified above (Fig. 1D). Forcing cells to survive with such a non-functional FtsN variant would ideally select for compensatory mutations in the gene for the immediate target of EFtsN that restore a productive EFtsN-target interaction.
Strain BL86 [ΔftsN ΔrecA ΔlacIZYA leu::Tn10] carrying plasmid pBL200 [aadA repAts Psyn135::ftsN I-SceI cI857 PλR::i-sceI] was constructed to serve as host in these screens. At 30°C, BL86/pBL200 cells express wt ftsN exclusively from a constitutive promotor on the plasmid. At elevated temperatures (above ~35°C), however, ftsN expression ceases because pBL200 both fails to replicate, and self-destructs due to de-repression of the plasmid-borne gene for the I-SceI meganuclease, which cuts the plasmid at an I-SceI site that is unique in the strain. Incubation of BL86/pBL200 in LB at 38–42°C caused severe cell filamentation and reduced the number of colony forming units over 108-fold after 24 hr, indicating effective elimination of pBL200 (data not shown). For screens, the strain was transformed with an unstably inherited mini-F plasmid [bla lacI Plac::gfp-ftsN*] encoding a mutant GFP-FtsN* variant under control of the lac promotor, and cells were incubated at 38–42°C for 24–48hr in liquid LB with IPTG. In case cultures showed an appreciable density, survivors were colony-purified on solid medium with IPTG, and then subjected to several tests to ensure that they indeed propagated without native FtsN (SpecS) but still required FtsN* (retention of gfp-ftsN* allele on mini-F plasmid, and IPTG-dependent cell division).
Most essential SR proteins (save FtsB, FtsK, FtsN itself, and ZipA) are encoded by a gene cluster at 2′ on the chromosome, near the leu::Tn10 marker in strain BL86. To map a suppressor mutation, therefore, we first determined if it was co-transducible with leu::Tn10. If so, the nucleotide sequence of (parts of) the 2′ cluster of the suppressed strain was determined by conventional means. If not, the entire genome sequence of the suppressed strain was determined, and compared to that of unsuppressed BL86.
Screens were performed with BL86/pBL200 producing non-functional GFP-FtsN variants with one of seven distinct substitutions in one of the three critical residues of EFtsN (W83L, W83M, W83T, Y85S, Y85W, L89H or L89S). Only screens with GFP-FtsNW83L and GFP-FtsNY85W yielded verifiable extragenic suppressor mutations (collectively called EFtsN*-suppressors), and these are discussed below.
Suppression of ftsNY85W by ftsAI143L
Screening for survivors of strain BL86/pBL200 carrying pBL215 [Plac::gfp-ftsNY85W] yielded two promising clones, BL86-KK1/pBL215 and BL86-AK1/pBL215, that had lost pBL200 and relied on pBL215 for survival. Co-transduction experiments indicated that both harbored leu::Tn10-linked suppressing mutations. The co-transduction frequency for BL86-KK1/pBL215, was ~58%, and sequencing of the chromosomal ftsQAZ region revealed a transversion in codon 143 of ftsA (ATC>CTC), causing an I143L substitution in the 1C domain of the FtsA protein (FtsAI143L). Notably, this same allele was previously isolated as a suppressor of a hypomorphic variant of the FtsQ protein (FtsQV92D) (Goehring et al., 2007). To verify that the ftsAI143L mutation is sufficient for survival of cells producing GFP-FtsNY85W as the sole source of FtsN, we first cloned the ftsAI143L allele on plasmid pBL236 [repAts ftsAI143L] and used it for gene replacement (Hamilton et al., 1989) in wt strain TB28. The resulting strain, BL114 [ftsAI143L], was then transformed with pBL215 [Plac::gfp-ftsNY85W] or a vector control (pRC7 [Plac::lacZ]), and transformants were tested for their ability to tolerate the introduction of ΔftsN<>aph via transduction in the presence of IPTG.
As expected, strain TB28 [wt] harboring pRC7, or any plasmid encoding a non-functional variant of GFP-FtsN, could not be transduced to ΔftsN<>aph (Table 1). Strain BL114 [ftsAI143L] devoid of a complementing plasmid also failed to yield viable transductants, indicating that a chromosomal ftsAI143L allele is not sufficient to bypass the need for FtsN altogether. In contrast, BL114/pBL215 cells readily tolerated removal of chromosomal ftsN, yielding strain BL120/pBL215 [ftsAI143L ΔftsN/ Plac::gfp-ftsNY85W] (Table 1). As expected, the division phenotype of BL120/pBL215 cells depended on the presence of IPTG. In its absence, cells formed long filaments with few constrictions in LB medium and shorter filaments in M9, but constriction frequencies increased significantly in both media in the presence of inducer (Fig. S3 A–D).
Table 1.
EFtsN*-suppressing mutations in ftsA, ftsB or ftsL.
| Mini-F plasmid | a Strain viable without chromosomal ftsN | |||||
|---|---|---|---|---|---|---|
| Name | Protein under Plac control | TB28 [wt] | BL18 [ftsAE124A] | BL114 [ftsAI143L] | BL140 [ftsBD59H] | BL154 [ftsLD93G] |
| pRC7 | LacZ | − | − | − | − | − |
| pBL209 | GFP-FtsN | + | + | + | + | + |
| pBL216 | GFP-FtsNW83L | − | + | + | + | + |
| pBL226 | GFP-FtsNW83T | − | + | + | + | + |
| pBL215 | GFP-FtsNY85W | − | + | + | + | + |
| pBL225 | GFP-FtsNY85S | − | + | + | + | + |
| pBL217 | GFP-FtsNL89S | − | + | + | + | + |
Each strain harboring the indicated plasmid was subjected to transduction of a chromosomal ΔftsN<>aph allele using an equal amount of P1 lysate prepared on strain CH34/pCH201 [ΔftsN<>aph/Plac::gfp-ftsN]. Cells were incubated at 30°C on M9-maltose agar containing Amp, Kan, and 100 μM IPTG. −, no transductants; +, viable transductants (15–101 per plate).
The ΔftsN<>aph lesion could also be readily introduced in BL114 [ftsAI143L] cells carrying extra copies of ftsAI143L on plasmid pBL236 [repAts ftsAI143L]. Cells of the resulting strain BL120/pBL236 [ftsAI143L ΔftsN/ repAts ftsAI143L] did not show a normal division phenotype but formed filaments of various lengths at 30°C (Fig. S3 E and F), and failed to propagate when replication of pBL236 ceased at 42°C (data not shown). Thus, an elevated level of FtsAI143L can bypass the absolute essentiality of FtsN, but compensates for its absence only incompletely. These properties of FtsAI143L are similar to those of the FtsAE124A variant described previously (Fig. S3 G and H)(Bernard et al., 2007, Gerding et al., 2009). It is interesting to note that both variants carry substitutions in the 1C domain of FtsA, though they affect residues that reside at opposite ends of this domain (van Den Ent & Löwe, 2000, Bernard et al., 2007, Goehring et al., 2007).
Suppression of ftsNY85W by ftsLD93G
The co-transduction frequency of the suppressing mutation in clone BL86-AK1/pBL215 with leu::Tn10 was ~75%. Nucleotide sequencing of the chromosomal ftsL-Z region revealed a transition in codon 93 of ftsL (GAC>GGC), causing a D93G substitution in the periplasmic domain of the FtsL protein (Fig. 3C). To test if this substitution is indeed sufficient for survival of cells producing GFP-FtsNY85W as the sole source of FtsN, we first recombineered the ftsLD93G mutation onto the chromosome of a leu::Tn10 derivative of TB28. Strain BL154 [leu::Tn10 ftsLD93G] was then transformed with pBL215 [Plac::gfp-ftsNY85W], and tested for its ability to tolerate the introduction of ΔftsN<>aph in the presence of IPTG (Table 1). BL154 carrying the vector control could not be transduced to ΔftsN<>aph, indicating that the ftsLD93G mutation did not compensate for the complete absence of FtsN. The presence of pBL215, by contrast, readily allowed the creation of strain BL157/pBL215 [leu::Tn10 ftsLD93G ΔftsN/ Plac::gfp-ftsNY85W], implying that the FtsLD93G protein indeed imparted sufficient functionality to GFP-FtsNY85W. Accordingly, BL157/pBL215 required IPTG for good growth (Fig. S4D, and data not shown).
Figure 3. Features of E.coli FtsN, FtsB, FtsL, and mutant variants.
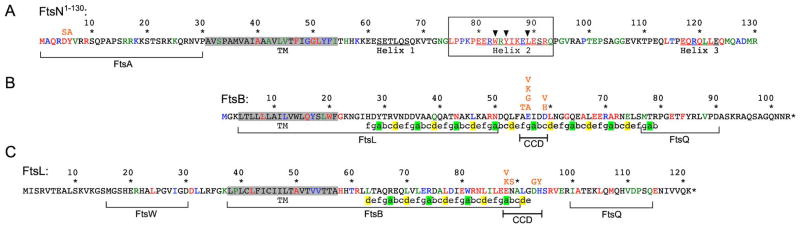
Amino acid residues of FtsN1–130 (A), FtsB (B), and FtsL (C) are colored as in figure 1D according to % identity in OMA groups 145686, 368384, and 412816, respectively (Altenhoff et al., 2011). Pertinent substitutions (in orange) are indicated immediately above affected residues. All three proteins are type II bitopic (N-in) inner membrane species and the panels are arranged to align their trans-membrane domains (TM), highlighted in grey. Portions of the proteins previously implicated in interactions are bracketed with the protein partner indicated underneath (Gonzalez & Beckwith, 2009, Gonzalez et al., 2010, Busiek et al., 2012, van den Berg van Saparoea et al., 2013).
In panel A, the essential domain of FtsN (EFtsN) is boxed and the three critical residues are indicated by arrowheads. The three proposed juxtamembrane helices (Yang et al., 2004) are underlined.
In Panels B and C, predicted heptad repeats in the periplasmic domains of FtsB and FtsL are indicated with A and D positions highlighted in green and yellow, respectively. Predictions were generated with Paircoil2 with a window length of 21 and P-score cutoff <0.08 (McDonnell et al., 2006). The proposed constriction control subdomain (CCD) in each protein (see text) is indicated. If the coiled-coil of FtsB were perpendicular to the membrane, its CCD would reach about 5.0–5.5 nm into the periplasm (LaPointe et al., 2013).
Suppression of ftsNW83L by ftsBD59H
Screening for survivors of strain BL86/pBL200 carrying pBL216 [Plac::gfp-ftsNW83L] yielded clones BL86-AK11/pBL216 and BL86-AK12/pBL216 that had lost pBL200 and relied on pBL216 for survival. The suppressing mutation in neither clone was co-transducible with leu::Tn10, indicating they mapped outside the 2′ cluster. Genomic sequencing revealed the same transversion (GAT>CAT) in both clones, suggesting they were siblings. The mutation maps to ftsB, and leads to a D59H substitution in the periplasmic domain of the protein (Fig. 3B). To verify that this mutation is sufficient for survival of cells producing GFP-FtsNW83L as the sole source of FtsN, we first again used gene replacement (Hamilton et al., 1989) to convert wt strain TB28 to ftsBD59H. The resulting strain, BL140 [ftsBD59H], was then transformed with pBL216 [Plac::gfp-ftsNW83L], and tested for its ability to tolerate the introduction of ΔftsN<>aph via transduction in the presence of IPTG. Indeed, transductants were readily obtained (Table 1) and the resulting strain, BL141/pBL216 [ftsBD59H ΔftsN/ Plac::gfp-ftsNW83L], displayed IPTG-dependent growth and division (Fig. S4C, and data not shown). In contrast, BL140 cells carrying pRC7 [Plac::lacZ] yielded no transductants (Table 1). Thus, production of normally non-functional GFP-FtsNW83L was critical for survival of ftsBD59H ΔftsN cells.
ftsBD59H or ftsLD93G cells no longer require EFtsN for viability, but depend on NFtsN in its absence
The viability of strains BL140 [ftsBD59H] and BL154 [ftsLD93G] indicated that neither suppression of ftsNW83L by ftsBD59H, nor of ftsNY85W by ftsLD93G, occurred in a strictly allele-specific fashion, and this was confirmed by additional observations. Production of periplasmic TTGFP-FtsN75–99 was still sufficient to rescue ΔftsN ftsBD59H or ΔftsN ftsLD93G cells, showing that neither FtsBD59H nor FtsLD93G prevented the wild type EFtsN peptide from executing its essential activity (Table 2). Moreover, either ftsBD59H or ftsLD93G was sufficient to rescue ΔftsN cells producing any of the tested GFP-FtsN* variants with otherwise lethal substitutions of W83, Y85, or L89 in their EFtsN domain (Table 1, Fig. S4, C and D).
Table 2.
NFtsN contributes to septal ring localization, and supports division of ftsBD59H or ftsLD93G cells in the absence of EFtsN.
| Plasmid | a At SR | b Strain viable without chromosomal ftsN | |||||
|---|---|---|---|---|---|---|---|
| Name | Protein under Plac control | TB28 [wt] | BL18 [ftsAE124A] | BL114 [ftsAI143L] | BL140 [ftsBD59H] | BL154 [ftsLD93G] | |
| vector | none | NA | − | − | − | − | − |
| pCH201 | GFP-FtsN | +++ | + | + | + | + | + |
| pBL330 | GFP-FtsND5S,Y6A | +++ | + | + | + | + | + |
| pLP219 | TTGFP-FtsN75–99 | − − − | + | + | + | + | + |
| pBL136 | GFP-FtsNΔ(64–101)<>22 | +++ | − | − | − | + | + |
| pLP171 | GFP-FtsN1–71 | − −+ | − | − | − | + | + |
| pMG13 | GFP-FtsN1–81 | − −+ | − | − | − | + | + |
| pBL335 | GFP-FtsN1–81; D5S,Y6A | − − − | − | − | − | − | − |
| pLP170 | GFP-FtsN1–31-MalF17–39-FtsN55–81 | − −+ | − | − | − | + | + |
| pBL312 | GFP-MalF2–14-FtsN27–81 | − − − | − | − | − | − | − |
Strain TB28 carrying the indicated plasmid was grown in M9-glucose with 5 μM IPTG to OD600~0.4, and live cells were observed by fluorescence microscopy. Indicated is if a fusion protein accumulated at constriction sites sharply (+++) or weakly (−−+), or if it appeared evenly distributed along the cell periphery (−−−). NA, not applicable.
Each strain harboring the indicated plasmid was subjected to transduction of a chromosomal ΔftsN<>aph allele using an equal amount of P1 lysate prepared on strain CH34/pCH201 [ΔftsN<>aph/Plac::gfp-ftsN]. Cells were incubated at 30°C on M9 maltose agar containing Amp, Kan and 500 μM IPTG. −, no transductants; +, viable transductants (15–106 per plate). Vector used was pMLB1113ΔH [Plac::].
Because neither ftsBD59H nor ftsLD93G was sufficient to bypass the need for FtsN altogether (Tables 1 and 2), this lack of allele specificity was unexpected. It also raised the possibility that the EFtsN domain might no longer be essential in ftsBD59H or ftsLD93G cells, but that they become critically dependent on another part of the FtsN protein when EFtsN is non-functional or absent. This possibility was confirmed by the observation that production of GFP-FtsNΔ(64–101)<>22, an otherwise non-functional fusion in which the EFtsN domain and flanking residues are replaced with an arbitrary peptide (Fig. 1A), was sufficient to rescue ΔftsN ftsBD59H or ΔftsN ftsLD93G cells (Table 2).
To elucidate which of the non-EFtsN domains of FtsN gained a critical function in ftsBD59H or ftsLD93G cells, GFP-FtsN derivatives that lacked EFtsN and one or more additional domains were tested for their ability to support division of ΔftsN ftsBD59H or ΔftsN ftsLD93G cells. As summarized in Table 2, fusions GFP-FtsN1–81 and GFP-FtsN1–71 could rescue these cells, implying that most of periplasmic FtsN, including its SPOR domain, was dispensable for this function. A variant of GFP-FtsN1–81 in which the transmembrane domain of FtsN is replaced with TM1 of MalF (GFP-FtsN1–31-MalF17–39-FtsN55–81) also rescued, showing that TMFtsN was also not specifically required. In contrast, a variant of GFP-FtsN1–81 in which the cytoplasmic domain of FtsN is replaced with cytoplasmic MalF residues (GFP-MalF2–14-FtsN27–81) failed to correct a ΔftsN lesion in ftsBD59H or ftsLD93G cells. Hence, in contrast to wt cells, ftsBD59H or ftsLD93G cells can survive without the essential periplasmic domain of FtsN, provided that the cytoplasmic domain of FtsN is still present (Table 2).
NFtsN is not sufficient to rescue ftsAI143L or ftsAE124A cells that lack EFtsN
Like the EFtsN*-suppressors in ftsB or ftsL, the suppressing mutations in ftsA showed little allele specificity. Thus, production of any of the tested plasmid-encoded GFP-FtsN* variants with different (and normally lethal) substitutions in EFtsN (Table 1, Fig. S4, A and B), or of any trans-membrane or periplasmic fusion with an intact EFtsN peptide (Table 2, and data not shown), allowed survival of both ΔftsN ftsAI143L and ΔftsN ftsAE124A cells. In contrast to ftsBD59H and ftsLD93G, however, ftsAE124A and ftsAI143L did not support viability of ΔftsN cells producing fusions that lack the EFtsN peptide altogether, including those with an intact NFtsN domain such as GFP-FtsNΔ(64–101)<>22 or GFP-FtsN1–81 (Table 2). Hence, suppression of EFtsN* by the mutations in the IC domain of FtsA differs significantly from that by FtsBD59H or FtsLD93G. While the former still critically depend on the presence of native or mutant EFtsN, the latter can bypass this need as long as NFtsN is present.
NFtsN residues required for viability of ftsBD59H or ftsLD93G cells lacking EFtsN
The N-terminal cytoplasmic domain of E.coli FtsN (NFtsN) consists of only ~30 residues, and sequence comparisons indicate that the most N-terminal ones (FtsN1–9) are the best conserved (Fig. 3A). To test if this region is important for viability of ftsBD59H or ftsLD93G cells that lack EFtsN, we compared the complementing properties of GFP-FtsN and GFP-FtsN1–81 fusions with those of derivatives bearing the double substitution D5S and Y6A (DY>SA) in two adjacent conserved FtsN residues (Fig. 3A, Table 2). Consistent with the notion that ftsBD59H or ftsLD93G cells gained the ability to survive in the presence of either the NFtsN or EFtsN domain (see above), production of GFP-FtsN (NFtsN+, EFtsN+), GFP-FtsNDY>SA (EFtsN+), or GFP-FtsN1–81 (NFtsN+) supported cell division and viability of ΔftsN ftsBD59H and ΔftsN ftsLD93G cells. In contrast, the GFP-FtsN1–81,DY>SA fusion failed to rescue ΔftsN ftsBD59H or ΔftsN ftsLD93G cells (Table 2), and this was not due to excessive instability of the protein (Fig. S5). Thus, residue D5 and/or Y6 in the N-terminal cytoplasmic domain of FtsN play a critical role in the EFtsN-independent division process that can occur in ftsBD59H or ftsLD93G cells.
The N-terminus of FtsN promotes localization to the SR, and interaction with FtsA in BACTH assays
The NFtsN domain can interact directly with the FtsA protein (Busiek et al., 2012), raising the possibility that this interaction is an important step during EFtsN-independent division of ftsBD59H or ftsLD93G cells. Support for this idea came from BACTH assays in which we used SPOR-less versions of FtsN (FtsN1–105) to help increase the specificity of the assay for interactions involving the N-terminal portion of the protein. Notably, T18-FtsA interacted robustly with T25-FtsN1–105 in this assay, but failed to do so with a derivative bearing the DY>SA substitution. In contrast, both T25-FtsN1–105 and T25-FtsN1–105,DY>SA interacted weakly, but comparably, with T18-FtsQ (fig. 4A).
Figure 4. NFtsN promotes interaction with FtsA and localization to constriction sites.

(A) Bacterial two-hybrid assays. Strain BTH101 [cya] was co-transformed with plasmid pairs as indicated, and plated on LB agar containing Amp, Kan, and 0.2 % glucose. Purified colonies were subsequently striped on M9-glucose agar supplemented with Amp, Kan, 40 μg/ml X-gal, and 250 μM IPTG. Plates were incubated at 30°C for 18 hr and then at RT for 30 hr.
(B) Fluorescence micrographs of live cells. Strain TB28 [wt] carrying pCH201 [Plac::gfp-ftsN] (left), pMG13 [Plac::gfp-ftsN1–81] (middle), or pBL335 [Plac::gfp-ftsN1–81, DY>SA] (right) was grown at 30°C in M9-glucose medium with 5 μM IPTG to OD600=~0.5, and imaged immediately. Note the even peripheral distribution of GFP-FtsN1–81, DY>SA in contrast to the weak (red arrows) or sharp (yellow arrowheads) accumulations of GFP-FtsN1–81 and GFP-FtsN, respectively, at constriction sites. Bar equals 4 μm.
Even though the periplasmic SPOR domain of FtsN is required and sufficient for the strong accumulation of the protein at SR’s, we previously noted that SPOR-less versions of GFP-FtsN still weakly accumulate at SR’s (Gerding et al., 2009). The interaction of NFtsN with FtsA suggested an involvement of NFtsN in this weak accumulation. Accordingly, fusions GFP-FtsN1–81 and GFP-FtsN1–31-MalF17–39-FtsN55–81 (both NFtsN+) showed weak accumulation at constriction sites of wt cells, while GFP-MalF2–14-FtsN27–81 (NFtsN−) did not (Fig. 4B, Table 2). Importantly, the GFP-FtsN1–81,DY>SA fusion also failed to accumulate, emphasizing the role of residues D5 and/or Y6 in proper NFtsN function (Fig. 4B, Table 2).
These results agree with a recent report that a fraction of FtsN molecules in the cell can be recruited to the SR in an SFtsN-independent fashion via an interaction between NFtsN and FtsA (Busiek & Margolin, 2014). Moreover, our results indicate that the same cytoplasmic NFtsN-FtsA interaction is critical for the survival of ΔftsN ftsBD59H or ΔftsN ftsLD93G cells that lack the periplasmic EFtsN peptide.
Synergy of FtsAI143L with FtsBD59H or FtsLD93G in compensating for the absence of FtsN
Why the E124A or I143L substitutions in the IC domain of FtsA promote survival of cells with compromised FtsN function is unclear, but one straightforward scenario is that NFtsN promotes a conformational state of FtsA that is also favored by the residue substitutions. Because ftsBD59H or ftsLD93G cells can survive with NFtsN as the sole portion of FtsN present in the cell, this scenario predicted that introduction of a IC domain substitution allele in ftsBD59H or ftsLD93G cells might allow their survival in the complete absence of FtsN. Hence, we created strains BL149 [ftsAI143L ftsBD59H], BL151 [ftsAE124A ftsBD59H], and BL164 [ftsAI143L ftsLD93G], and tested their ability to survive complete removal of ftsN by transduction of the ΔftsN<>aph allele. Indeed, while parent strains bearing only one of the mutant ftsA, B or L genes failed to yield viable ΔftsN transductants, the three new strains readily tolerated removal of ftsN (Table 3, Fig. 5).
Table 3.
Synergy between EFtsN*-suppressing mutations.
| Strain | Genotype | a Permits ΔftsN | bΔftsN derivative |
|---|---|---|---|
| TB28 | wt | − | CH34* |
| BL18 | ftsAE124A | − | BL20* |
| BL114 | ftsAI143L | − | BL120* |
| BL140 | ftsBD59H | − | BL141* |
| BL154 | ftsLD93G | − | BL157* |
| BL149 | ftsAI143L ftsBD59H | + | BL150 |
| BL151 | ftsAE124A ftsBD59H | + | BL152 |
| BL164 | ftsAI143L ftsLD93G | + | BL165 |
| BL159 | ftsBD59H ftsLD93G | + | BL163 |
| BL167 | ftsBE56A | + | BL173 |
| BL172 | ftsBE56K | + | BL175 |
The indicated strains were subjected to transduction of a chromosomal ΔftsN<>aph allele using an equal amount of P1 lysate prepared on strain CH34/pCH201 [ΔftsN<>aph/Plac::gfp-ftsN]. Cells were incubated at 30°C on M9-maltose agar containing Kan. −, no transductants; +, viable transductants (27–81 per plate).
Asterisk (*) indicates that strain requires a complementing or suppressing genetic construct for survival.
Figure 5. Cell fission in the complete absence of FtsN.

Differential interference contrast images of live cells of the indicated FtsN+ strains (A–J) and their ΔftsN offspring (K–R), grown exponentially at 30°C to OD600=0.5–0.6 in LB (A–E, and K–N) or M9-glucose (F–J, and O–R) medium. Arrows in panels F–I point at some of the FtsN+ cells with cell shape defects in minimal medium. Bar equals 8 (K–N) or 4 (all other panels) μm.
Synergy of FtsBD59H with FtsLD93G in compensating for the absence of FtsN
Why D59H or D93G substitutions in the periplasmic domains of FtsB or FtsL, respectively, can compensate for absence of the otherwise essential domain of FtsN (EFtsN) is also unclear. Similar to the proposed role of NFtsN in directly promoting a conformation of FtsA that promotes constriction of the SR, one parsimonious possibility is that EFtsN directly or indirectly promotes a conformation of the FtsBLQ subcomplex that is also favored by the FtsBD59H and FtsLD93G substitutions. However, the FtsBD59H or FtsLD93G substitutions do not fully compensate for absence of EFtsN, as ftsBD59H or ftsLD93G cells still require NFtsN in lieu of EFtsN for survival (see above). We constructed strain BL159 [ftsBD59H ftsLD93G] to test if the mutations in the two genes act synergistically in compensating for a lack of FtsN activity. Interestingly, the ΔftsN<>aph allele could indeed be readily introduced into BL159, yielding strain BL163 [ftsBD59H ftsLD93G ΔftsN] (Table 3, Fig. 5).
Random mutagenesis identifies constriction control domains (CCD) in FtsB and FtsL that determine the essentiality of EFtsN
Plasmids pBL304 [Psyn135::ftsBD59H] and pBL305 [Psyn135::ftsLD93G] are low copynumber plasmids that direct constitutive production of FtsBD59H and FtsLD93G, respectively. Strain TB28 [wt] carrying either one of these plasmids readily tolerated removal of chromosomal ftsN, provided that it also harbored a second plasmid directing production of either NFtsN, EFtsN, or both (results not shown). Thus, ftsBD59H and ftsLD93G are both dominant over wt ftsB and ftsL, respectively, in allowing viability in the absence of EFtsN.
We took advantage of this in plasmid-based screens for additional ftsB and ftsL alleles that overcome the lethality that is normally associated with loss of EFtsN function. Error-prone PCR was used to prepare libraries of randomly mutated ftsB and ftsL in the context of plasmids pBL336 [Psyn135::ftsB] and pJH2 [Psyn135::ftsL], respectively. Libraries were then introduced into strain JH1 [ΔftsN ΔrecA ΔlacIZYA] already harboring pBL200 [aadA repAts Psyn135::ftsN I-SceI cI857 PλR::i-sceI], and one of several mini-F derivatives encoding variants of GFP-FtsN that lack functional EFtsN (Table S2). Transformants were then plated at 37°C or 42°C to promote loss of pBL200 and, hence, select for plasmid-borne ftsB or ftsL alleles that allow survival of EFtsN cells. These screens yielded a total of 15 clones from the pBL336 [Psyn135::ftsB] library (~7000 screened), and 13 from the pJH2 [Psyn135::ftsL] one (~9000 screened). Subsequent sequence analyses and subcloning, to resolve clones with multiple silent and/or missense mutations, then allowed us to identify a total of 6 and 5 relevant single residue substitutions in FtsB and FtsL, respectively. Interestingly, substitutions in the proteins were limited to 3 (FtsB A55, E56, and D59) or 4 (FtsL E88, N89, D93, and H94) neighboring residues (Fig. 3B,C and Table S2).
Clustering of these mutations is notable, and suggests that the affected residues comprise small periplasmic subdomains in FtsB and FtsL that are important in determining the essentiality of EFtsN. In FtsB, the affected residues reside in the fourth (A55 and E56) or fifth (D59) complete heptad repeat of the periplasmic coiled-coil domain of the protein (Fig. 3B) for which a partial crystal structure is available (LaPointe et al., 2013). In FtsL, the affected residues reside just C-terminal to the third complete heptad repeat of the predicted periplasmic coiled-coil domain of the protein (Fig. 3C). Whether this region forms a fourth heptad or a turn in the protein structure is presently not clear (Villanelo et al., 2011). For brevity, we refer to the proposed subdomains as CCD (constriction control domain, Fig. 3B and C).
Substitution of FtsB E56 allows cell fission and viability in the complete absence of FtsN
The plasmid-borne EFtsN*-suppressing alleles of ftsB and ftsL isolated above (Fig. 3B and C, Table S2) were next introduced in TB28 [wt] and tested for retention of their essential division functions by their ability to allow removal of chromosomal ftsB or ftsL via transduction. Transductants were readily obtained in all cases, yielding the strains listed in Table 4. None of these showed overt division problems under standard growth conditions (data not shown), indicating that, like ftsBD59H and ftsLD93G, none of the isolated EFtsN*-suppressing mutations in the two genes significantly interfered with their essential function. The strains were next transformed with a compatible plasmid encoding GFP-FtsN1–81 (NFtsN+, EFtsN−), or a vector control, and tested for tolerance to removal of chromosomal ftsN via transduction. The properties of two of the new ftsB alleles (A55T and D59V) and all four of the ftsL ones (E88K, E88V, N89S, and H94Y) were similar to those of ftsBD59H and ftsLD93G in that they all rendered EFtsN non-essential, provided GFP-FtsN1–81 was produced (Table 4).
Table 4.
Viability of ftsB or ftsL mutant cells that lack EFtsN or all of FtsN.
| Straina | genotype | pMG13 [Plac::gfp-ftsN1–81] | pMLB1113ΔH [Plac::-] |
|---|---|---|---|
| BL155/pBL336 | ΔftsB/Psyn135::ftsB | − | − |
| BL155/pBL340 | ΔftsB/Psyn135::ftsBA55T | + | − |
| BL155/pBL339 | ΔftsB/Psyn135::ftsBE56A | + | + |
| BL155/pBL342 | ΔftsB/Psyn135::ftsBE56G | + | + |
| BL155/pBL341 | ΔftsB/Psyn135::ftsBE56K | + | + |
| BL155/pBL338 | ΔftsB/Psyn135::ftsBE56V | + | + |
| BL155/pBL343 | ΔftsB/Psyn135::ftsBD59V | + | −b |
| BL156/pJH2 | ΔftsL/Psyn135::ftsL | − | − |
| BL156/pBL333 | ΔftsL/Psyn135::ftsLE88K | + | −b |
| BL156/pBL332 | ΔftsL/Psyn135::ftsLE88V | + | − |
| BL156/pBL334 | ΔftsL/Psyn135::ftsLN89S | + | −b |
| BL156/pBL331 | ΔftsL/Psyn135::ftsLH94Y | + | −b |
Each strain harboring either pMG13 [Plac::gfp-ftsN1–81] or the vector control pMLB1113ΔH [Plac::] was subjected to transduction of a chromosomal ΔftsN<>cat allele using an equal amount of P1 lysate prepared on strain BL71/pCH288 [ΔftsN<>cat/ Plac::sstorA-gfp-ftsN55–123]. Cells were incubated at 30°C on M9-maltose agar containing Cam and 100 μM IPTG. −, no transductants; +, viable transductants (74–116 per plate).
Tiny colonies appeared on the transduction plate, but these failed to propagate upon restreaking.
Remarkably, however, the four ftsB alleles affected in Glu56 (E56A, E56G, E56K, and E56V) sustained cell viability and fission in the absence of any part of FtsN (Table 4). To further verify this result, two of these alleles (ftsBE56A and ftsBE56K) were recombined with the chromosome of wt strain TB28 and the resulting strains were tested for tolerance to removal of ftsN in the absence of any (complementing) plasmid. As summarized in Table 3, both BL167 [ftsBE56A] and BL172 [ftsBE56K] readily tolerated removal of ftsN, yielding strains BL173 [ftsBE56A ΔftsN] and BL175 [ftsBE56K ΔftsN], respectively. Thus, a single amino acid substitution in the periplasmic CCD subdomain of the FtsB protein is indeed sufficient to render FtsN completely non-essential (Table 3, Fig. 5).
EFtsN*-suppressing mutations promote cell division in both FtsN− and FtsN+ cells
Cells of the extragenically suppressed ΔftsN strains listed in Table 3 displayed a filamentous/chaining phenotype that was only mild in minimal medium, but more pronounced in rich medium (Fig. 5, and data not shown). Hence, none of the (combinations of) EFtsN*-suppressing mutations in ftsA, ftsB, and/or ftsL tested so far conferred a normal division frequency to ΔftsN cells. Still, the fact that these cells divide at all in the complete absence of FtsN, and fairly well in minimal medium (Fig. 5, compare O-R to J), is remarkable and indicates that the EFtsN*-suppressing mutations in ftsA, ftsB, and ftsL have significant stimulatory effects on the division machinery, at least when FtsN is absent.
To assess if EFtsN*-suppressing mutations also stimulate cell fission when FtsN is present, we measured cell dimensions of corresponding FtsN+ strains during exponential growth in LB at 30°C (Table 5, Fig. S6). Interestingly, while the mass doubling times of all tested FtsN+ strains bearing EFtsN*-suppressing mutations were essentially equivalent to that of TB28 [wt], their average cell sizes were significantly (by ~11–34%) below normal. Small size was mainly due to reduced average cell lengths (by ~10–29%), though cells of some strains were also slightly thinner or wider than normal (Table 5).
Table 5.
EFtsN*-suppressing mutations promote early cell fission in otherwise wt cells.
| Strain | Genotype | b Td | c N | a Avg. Length | a Avg. Width | a Avg. Volume | |||
|---|---|---|---|---|---|---|---|---|---|
| μm (SD) | % WT | μm (SD) | % WT | μm3 (SD) | % WT | ||||
| LB | |||||||||
| TB28 | wt | 41 | 331 | 4.41 (0.93) | 100 | 0.95 (0.07) | 100 | 2.90 (0.72) | 100 |
| BL18 | ftsAE124A | 41 | 366 | 3.96 (1.06) | 90 | 0.91 (0.09) | 96 | 2.42 (0.84) | 83 |
| BL114 | ftsAI143L | 40 | 346 | 3.46 (0.89) | 79 | 0.87 (0.11) | 92 | 1.92 (0.75) | 66 |
| BL140 | ftsBD59H | 42 | 359 | 3.63 (0.87) | 82 | 0.97 (0.07) | 102 | 2.44 (0.67) | 84 |
| BL154 | ftsLD93G | 39 | 369 | 3.81 (0.93) | 86 | 0.93 (0.08) | 98 | 2.38 (0.69) | 82 |
| BL149 | ftsAI143L ftsBD59H | 41 | 362 | 3.24 (0.81) | 73 | 0.91 (0.09) | 96 | 1.92 (0.63) | 66 |
| BL164 | ftsAI143L ftsLD93G | 42 | 345 | 3.24 (0.74) | 73 | 0.92 (0.08) | 97 | 1.94 (0.53) | 67 |
| BL159 | ftsBD59H ftsLD93G | 44 | 353 | 3.66 (0.95) | 83 | 0.99 (0.09) | 104 | 2.58 (0.77) | 89 |
| BL167 | ftsBE56A | 43 | 362 | 3.12 (0.72) | 71 | 0.97 (0.14) | 102 | 2.11 (0.77) | 73 |
| BL172 | ftsBE56K | 43 | 399 | 3.34 (0.73) | 76 | 0.99 (0.09) | 104 | 2.32 (0.65) | 80 |
| M9-glucose | |||||||||
| TB28 | wt | 92 | 354 | 2.43 (0.49) | 100 | 0.95 (0.08) | 100 | 1.45 (0.33) | 100 |
| BL114 | ftsAI143L | 99 | 377 | 2.16 (0.46) | 89 | 0.95 (0.09) | 100 | 1.26 (0.32) | 87 |
| BL167 | ftsBE56A | 101 | 360 | 1.93 (0.38) | 79 | 1.02 (0.09) | 107 | 1.24 (0.25) | 86 |
Overnight cultures in LB were diluted to OD600=0.02 in LB, or to OD600=0.05 in M9-glucose, and further incubated at 30°C to OD600=0.5–0.6. Cells were chemically fixed before microscopy. SD, standard deviation of the mean. Average cell lengths and volumes of the mutants were smaller than those of TB28 as tested by one-way ANOVA followed by Tukey’s test (α=0.005).
Mass doubling time in minutes.
Number of cells measured.
The effects of separate EFtsN*-suppressing mutations on the average size of FtsN+ cells were not additive. For example, BL140 [ftsBD59H] cells were on average 16% smaller than TB28 [wt] cells, but both BL114 [ftsAI143L] and BL149 [ftsAI143L ftsBD59H] cells were 34% smaller. Intrinsic constraints, such as nucleoid volume and production rates of division proteins, are likely to impose a lower limit to cell size, and reduction by a third may be the limit under these growth conditions. Regardless, these results indicate that the EFtsN*-suppressing mutations promote cell fission in the absence or presence of FtsN, and lead to premature division in otherwise wt (FtsN+) cells.
FtsB and FtsL protect each other from proteolysis in both E.coli (Buddelmeijer et al., 2002, Gonzalez & Beckwith, 2009) and B.subtilis (Daniel et al., 1998, Daniel & Errington, 2000, Wadenpohl & Bramkamp, 2010). In B.subtilis, moreover, absence of the FtsL protease RasP (YluC) leads to stabilization of both FtsL and DivIC (FtsB) as well as to a premature division phenotype (Bramkamp et al., 2006). We performed Western analyses to assess possible effects of EFtsN*-suppressing mutations on FtsB and FtsL levels, but observed no marked effects (Fig. S7). We infer that stimulation of cell fission by the EFtsN*-suppressing mutations is not simply the result of increased FtsB and/or FtsL levels. This is consistent with the observations that modest overexpression of the native proteins is insufficient to compensate for the absence of EFtsN (Table 4, and data not shown), and has little, if any, effect on the division phenotype of wt cells (Guzman et al., 1992).
Fission at a reduced cell mass could result from premature initiation of cell constriction, an acceleration of the constriction process, or a combination of the two. To address these possibilities, we chose BL167 [ftsBE56A] as representative of a prematurely dividing strain, and compared the periods of sPG synthesis during exponential growth in cells of this strain with wt cells. To this end, the fluorescent D-amino acid HADA (hydroxy coumarin-carbonyl-amino-D-alanine) was used to covalently pulse-label newly synthesized PG (Kuru et al., 2012). Pulse-labeled cells were imaged and those with a clear accumulation of fluorescence in a ‘HADA-ring’ at/near midcell were taken to have started the cell fission process (Fig. 6). Sites of visible constriction were scored as a secondary marker, and the average ages of cells showing a constriction and/or ‘HADA-ring’ was calculated (Den Blaauwen et al., 1999). As before (Table 5), TB28 and BL167 grew with identical mass doubling times, and the average size of BL167 cells was significantly below that of wt cells (by 23%, Table S3). Even so, sPG synthesis and visible constriction started at about the same times in the division cycles of both strains, and lasted for about as long before cell separation (Fig. 6C). We conclude that BL167 [ftsBE56A] cells initiate constriction at an abnormally low cell mass and that this is the primary reason for its small cell size.
Figure 6. Premature initiation of cell constriction in ftsBE56A cells.

(A, B) Fluorescence and corresponding DIC images of TB28 [wt] (A) and BL167 [ftsBE56A] (B) cells grown exponentially at 30°C in LB to OD600=0.5–0.6, and pulse-labeled with HADA for 1 min immediately before fixation in ethanol. Arrows in the DIC images point at cells in which a clear ‘HADA ring’ is absent, and in which sPG synthesis had presumably not yet started. Note the difference in cell sizes between the two strains. Bar equals 4 μm.
(C) Inferred division cycles of the two strains based on measurement of relevant parameters of about 300 of the cells shown in panels A and B (see also Table S3). Periods of sPG synthesis (i.e. presence of ‘HADA-ring’) and of cell constriction, are shown in red and black, respectively. Average cell volumes are indicated on the right.
EFtsN*-suppressing mutations cause conditional and FtsN-dependent cell shape defects and lethality
Small cell sizes were also observed when FtsN+ strains carrying EFtsN*-suppressing mutations grew at lower rates in M9 minimal medium (Fig. 5F–J), and quantitative analyses of BL114 [ftsAI143L] and BL167 [ftsBE56A] cells revealed that they were about 14% smaller in volume than TB28 [wt] cells (Table 5). Interestingly, while BL114 [ftsAI143L] and BL18 [ftsAE124A] cells still resembled normal (though short) rods (Table 5, and data not shown), BL167 [ftsBE56A] as well as other FtsN+ strains with (combinations of) EFtsN*-suppressing mutations in ftsB and/or ftsL displayed shape defects in this medium. Thus, many cells were both shorter and wider than usual and resembled fat rods, ovals, or even spheres (Fig. 5F–J, Table 5, and data not shown).
Notably, one of the ftsL alleles (ftsLE88K) we recovered in the plasmid-based EFtsN*-suppression screen was previously reported to cause cell lysis at 42°C in low-osmotic LB (Ishino et al., 1989, Ueki et al., 1992), and is also the subject of an independent and complementary study in this issue (Tsang & Bernhardt, 2015). To assess if conditional lethality is a more common property of EFtsN*-suppressing mutations, we used spot-titer analyses to compare plating efficiencies of strains listed in Table 3 on regular LB (containing 0.5% NaCl) or half-strength LB without NaCl (0.5LBNS) at 30°C and 42°C. None of the strains showed obvious growth or plating defects at either temperature on regular LB agar (Fig. 7A, Fig. S8A). On 0.5LBNS agar, however, several strains indeed showed severe plating defects at 42°C. Interestingly, most sensitive to this condition were the six FtsN+ strains (BL149, BL151, BL164, BL159, BL167, and BL172) bearing those (combinations of) EFtsN*-suppressing alleles that allow for survival in the complete absence of FtsN (see Table 3). These strains also grew poorly in liquid 0.5LBNS at 42°C (not shown), and inspection of cells by microscopy revealed pronounced cell shape and integrity defects in all cases. In addition to fat, spherical or otherwise mis-shapen cells, cultures also contained adundant cell ‘ghosts’ and debris. Membrane-blebbing, indicative of breaches of the murein layer, was occasionally observed, but whether cell death was primarily caused by cell wall lysis or some other catastrophy is unclear (Fig. 7E, G, Fig. S8D, F, I, K).
Figure 7. Conditional lethality of EFtsN*-suppressing mutations in FtsN+ cells.

(A) Cells of the indicated strains were grown overnight in LB at 30°C. Cultures were serially diluted in LB to OD600=4.10X as indicated, and 5 μl of each dilution was spotted on LB or 0.5LBNS agar. Plates were incubated for 16 hr at 42°C or 20 hr at 30°C.
(B–H) Cells of the indicated strains were grown overnight in LB at 30°C, diluted 400-fold in 0.5LBNS, grown for 3.5 hr at 42°C, and imaged live. Arrows point at examples of cells with shape defects (black) or of cell ghosts/debris (white). See Fig. S8 for images of additional strains.
(I) Strain BL173 [ftsBE56A ΔftsN] carrying plasmid pMLB1113ΔH (vector control), pCH201 [Plac::gfp-ftsN], pBL136 [Plac::gfp-ftsN Δ(64–101)<>22], or pBL330 [Plac::gfp-ftsNDY>SA] was grown overnight at 30°C in LB with 0.2% glucose. Cultures were serially diluted in LB to OD600=4×10X as indicated, and 5 μl of each dilution was spotted on 0.5LBNS agar. The plate was incubated for 16 hr at 42°C. Note that basal production of GFP-FtsN or GFP-FtsNDY>SA was sufficient to observe their toxicity under these conditions, and no IPTG was added to the medium.
Strikingly, cell death on 0.5LBNS at 42°C was almost completely alleviated in the ΔftsN derivatives of these strains (BL150, BL152, BL165, BL163, BL173, and BL175), and this was accompanied by a significant restoration of rod-shape as well (Fig. 7F, H, Fig. S8E, G, J, L). In fact, the morphology of these ΔftsN strains was about as close to WT under these conditions as when grown in M9 medium at 30°C (Fig. 5O–R, and data not shown).
Put differently, to cells that can survive without the protein, FtsN becomes toxic under these growth conditions (Fig. 7, Fig. S8). Accordingly, plasmid-encoded GFP-FtsN induced lethality in BL173 [ftsBE56A ΔftsN] cells on 0.5LBNS agar at 42°C (Fig. 7I). In addition, the GFP-FtsNDY>SA fusion (lacking intact NFtsN) similarly induced lethality under these conditions, but the GFP-FtsNΔ(64–101)<>22 fusion (lacking EFtsN) did not. It thus appears that the EFtsN activity that is normally essential for cell division is also responsible for the toxicity observed under these circumstances (Fig. 7I).
DISCUSSION
As a core component of the septal ring, FtsN is normally essential for cell constriction and viability. We previously showed that a small periplasmic domain of at most 35 residues is both required and sufficient for FtsN function, and that elevated levels of just this essential peptide (EFtsN) in the periplasm can fully rescue ΔftsN cells (Gerding et al., 2009). Here, we narrowed EFtsN to 19 residues, and found that single substitutions at 3 of these abbrogate FtsN function. We hypothesize that the EFtsN peptide allosterically activates septal PG synthesis and that these residues (W83, Y85, and L89) are particularly important for productive interaction of EFtsN with the periplasmic portion of another essential SR component. The likeliest ultimate target for EFtsN regulation is the PBP3-FtsW subcomplex. Like EFtsN, both PBP3 and FtsW are essential SR components and the subcomplex is both required for, and directly participates in, sPG synthesis. The fact that ponB cells lyse, rather than form filaments, upon depletion of EFtsN (Fig. 2) is also fully compatible with the assumption that EFtsN is normally needed for proper PBP3 activity. In the simplest scenario, EFtsN interacts with the PBP3-FtsW subcomplex directly to stimulate sPG synthesis. Alternatively, EFtsN regulates the subcomplex via one or more intermediary SR components. To help understand the mechanism of EFtsN action we identified extragenic suppressors of otherwise lethal substitutions of the critical residues in the peptide, and studied their properties.
Activity of EFtsN in the periplasm
As summarized in figure 8A, our results show that the normal requirement for the FtsN protein can be overcome in a variety of ways. Importantly, they also implicate the FtsBLQ subcomplex as a regulator of constriction initiation and a likely intermediary between EFtsN and the sPG synthase complex. The discovery that substitution of a single glutamic acid residue (E56) in the periplasmic portion of FtsB is sufficient to restore cell division and viability in the complete absence of FtsN is particularly striking. The recovery of ‘weaker’ EFtsN*-suppressing alleles in both ftsB and ftsL, and the synergy between at least two such alleles (ftsBD59H and ftsLD93G) in similarly rescuing ΔftsN cells, further indicates that it is a complex containing both proteins that determines the essentiality of FtsN. Taken together with the observation that EFtsN*-suppressing variants of FtsB and FtsL also cause premature initiation of sPG synthesis and cell constriction in otherwise wildtype cells, we conclude that the FtsBLQ subcomplex is not merely a scaffold for maturation of the SR, but regulates its activity as well.
Figure 8. Model for FtsN action in stimulation of cell constriction in E.coli.

(A) Summary of how cells lacking EFtsN or both NFtsN and EFtsN (ΔftsN) can be rescued by alterations to the SR on either or both sides of the membrane. The first row represents the wild type situation. Rescue by overexpression (⇧) of the FtsA subdomain IC variant FtsAE124A in the cytoplasm, or of the EFtsN peptide in the periplasm was described before (Bernard et al., 2007, Gerding et al., 2009). The other rows (in blue) summarize the new rescue conditions reported here. FtsB* and FtsL* variants harbor residue substitutions in their periplasmic CCD subdomains that render EFtsN non-essential. NFtsN refers to a membrane-associated form of the cytoplasmic subdomain of FtsN.
(B and C) Model for FtsN action in stimulation of cell constriction in E.coli. Relevant SR components are depicted. For clarity, the peptidoglycan layer, outer-membrane, FtsK, ZipA, and all non-essential SR proteins are omitted.
(B) The concentration of FtsN at the SR is low. The FtsBLQ subcomplex (BQL) is in a conformation (off) that suppresses septal PG synthesis by PBP3, FtsW (W), and associated proteins. FtsA (A) tethers FtsZ (Z) polymers to the membrane, and is also in an off conformation. The FtsA-off state may help to stabilize the FtsBLQ-off state, and vice versa, via direct or indirect interactions (stippled double headed arrow). The FtsA-off state may also suppress contraction of the Z-ring on the cytoplasmic face of the membrane.
(C) The concentration of FtsN (N) at the SR has increased. EFtsN (red) directly or indirectly stimulates a switch in FtsBLQ conformation to a state (on) that no longer suppresses, and may now stimulate, sPG synthesis (star in PBP3). NFtsN (magenta) directly binds the IC domain of FtsA and stimulates a conformation (on) of the protein that stabilizes the FtsBLQ-on state, and vice versa, via direct or indirect interactions (double headed arrow). FtsA-on may simultaneously stimulate active Z-ring contraction. The possibility that Z-ring components can (also) control the sPG synthase in a manner independent of FtsBLQ (green stippled arrow) is not excluded.
We propose that when the concentration of FtsN at the SR is below a certain treshold, FtsBLQ mostly exists in a conformation or state (off) that actively suppresses sPG synthesis by the PBP3-FtsW subcomplex and associated enzymes (Fig. 8B). Suppression is likely imposed via direct physical contact, as a variety of evidence support direct interaction between the two subcomplexes (Karimova et al., 2005, Goehring et al., 2006, D’Ulisse et al., 2007, Gonzalez et al., 2010, Noirclerc-Savoye et al., 2013). When FtsN accumulates at the SR, the EFtsN peptide induces a conformational switch in FtsBLQ (on) that relieves the suppression of PBP3-FtsW activity, thus promoting synthesis of sPG (Fig. 8C). Processing of this sPG by murein hydrolases then generates substrate for SFtsN, leading to additional accumulation of FtsN at the SR, to complete the self-enhancing cycle of the protein in stimulating cell constriction (Gerding et al., 2009). In this scenario, the EFtsN*-suppressing residue substitutions in FtsB and FtsL promote the on-state of FtsBLQ to a degree that is sufficient to bypass the need for EFtsN or all of FtsN. Though the switch in FtsBLQ conformation may merely relieve repression of sPG synthesis, the possibility that FtsBLQ in the on-state (FtsBLQ-on) actively stimulates the sPG synthase complex and/or has other additional functions during the constriction process is attractive as well.
The EFtsN*-suppressing residue substitutions in FtsB and FtsL define a new subdomain (CCD) in each of these small proteins that is flanked by subdomains previously shown to be involved in forming the FtsBLQ subcomplex (Fig. 3). The CCD’s at least partially correspond to phylogenetically conserved zones (‘negative patches’) in the coiled-coil folds of both FtsB (residues ~50–70) and FtsL (residues ~85–95) with predominant negative charge (Villanelo et al., 2011). It is also notable that all EFtsN*-suppressing substitutions are either of an acidic residue, or one immediately adjacent, and that the charged residues in each case are replaced with ones that carry no or opposite charge. Electrostatic interactions involving the affected residues in FtsB (E56, D59) and FtsL (E88, D93, H94) may be instrumental in maintaining the proposed FtsBLQ-off conformation when EFtsN is lacking or non-functional (Fig. 8B).
In the simplest variant of the model, EFtsN interacts with the FtsBLQ subcomplex directly to induce the conformational switch. In this regard, it may be relevant that EFtsN and the CCD subdomains in FtsB and FtsL that determine its essentiality are spaced roughly the same number of residues from the membrane (Fig. 3), and that reducing this distance for EFtsN significantly abrogates its activity (Fig. 1B). However, the possibilities that EFtsN acts on FtsBLQ via FtsK or the PBP3-FtsW subcomplex itself, cannot not be excluded. Regardless of how the off/on switch is accomplished precisely, it presumes some degree of structural flexibility of the FtsBLQ subcomplex. Support for this has come from molecular dynamics modeling, in vivo cross-linking, and in vitro reconstitution experiments (Villanelo et al., 2011, van den Berg van Saparoea et al., 2013, Noirclerc-Savoye et al., 2013). Clearly, more biochemical, physical and/or structural work is needed to fully understand how EFtsN or mutations in the CCD subdomains of FtsB and FtsL affect the FtsBLQ subcomplex and how this is transmitted to the PBP3-FtsW subcomplex and FtsA (see below).
Activity of NFtsN in the cytoplasm
It was previously shown that elevated levels of FtsAE124A in the cytoplasm can also rescue ΔftsN cells (Bernard et al., 2007, Gerding et al., 2009). We identified I143L as a second residue substitution in the IC domain of FtsA that similarly promotes survival without FtsN (Fig. 8A). Like FtsAE124A, FtsAI143L rescues ΔftsN cells when overexpressed (Fig. S3). However, cells bearing ftsAE124A or ftsAI143L as the sole chromosomal copy only tolerate the absence of native FtsN if one of two additional conditions are met. One is that cells produce mutant variants (FtsN*) with otherwise lethal substitutions within EFtsN (Tables 1 and 2). The fact that FtsAE124A or FtsAI143L can impart sufficient functionality to these FtsN* variants indicates that the latter still retain some EFtsN activity that is either masked or simply insufficient to drive cell constriction in cells producing native FtsA. The other, more revealing, condition is that ftsAE124A or ftsAI143L cells carry an additional EFtsN*-suppressing mutation in ftsB or ftsL (Fig. 8A). This synergy implies that (mutant) FtsA can collaborate with (mutant) FtsBLQ in an FtsN-independent manner to drive constriction. This could involve direct FtsA-FtsBLQ interactions in the cytoplasm and/or mediation by other SR components, including the PBP3-FtsW subcomplex (Di Lallo et al., 2003, Corbin et al., 2004, Karimova et al., 2005). It is particularly striking in this regard that cells bearing any of the EFtsN*-suppressing mutations in ftsB or ftsL together with native ftsA can also tolerate the absence of EFtsN, but only when NFtsN is still intact (Fig. 8A). Furthermore, we showed that residues at the N-terminal end of NFtsN are required for both this activity (Table 2) and its interaction with FtsA (Fig. 4). In other words, to strains with EFtsN*-suppressing mutations in ftsB or ftsL, the interaction between native FtsA and intact NFtsN appears equivalent to expression of FtsAE124A or FtsAI143L.
These results support further elaborations of the model. Thus, we propose that FtsA also exists in either an off or on conformation, and that the latter is promoted by the interaction of NFtsN with the IC domain, as well as by the E124A or I143L substitutions in this domain. We also suggest that either state of FtsA can reinforce the corresponding state of the FtsBLQ complex, and vice versa, and that this mutual reinforcement can occur in an FtsN-independent manner (Fig. 8B, C). Such functional coupling between FtsA and FtsBLQ is supported by: i) The observed synergies between the EFtsN*-suppressing mutations in ftsA and ftsB or ftsL in rescuing ΔftsN cells (Fig. 8A), ii) The fact that the same rescue can be accomplished solely by overexpression of FtsAE124A or FtsAI143L in the cytoplasm, or by substitution of FtsBE56 or overexpression of native EFtsN in the periplasm (Fig. 8A), iii) The previous finding that ftsAI143L rescues cells with otherwise lethally low levels of FtsQ (Goehring et al., 2007) and iv) The new finding that ftsLE88K rescues cells with otherwise lethally low levels of FtsA (Tsang & Bernhardt, 2015). In the model, FtsA spends most time in the off state prior to constriction initiation, when the concentration of FtsN at the SR is still low (Fig. 8B). As the concentration of FtsN at the SR rises, the interaction of FtsA with NFtsN causes the concentration of FtsA-on to rise as well and this helps to reinforce the FtsBLQ-on state that is simultaneously induced via EFtsN in the periplasm (Fig. 8C). It is attractive to speculate that the same conformational states of FtsA might also suppress (off) or promote (on) Z-ring contraction as it would provide a means to coordinate active IM invagination with sPG synthesis via concerted regulation by FtsA, the FtsBLQ subcomplex, and FtsN.
The IC domain participates in forming the subunit interface of polymeric FtsA, and it seems likely that NFtsN binding will affect polymer properties somehow (Krupka et al., 2012, Pichoff et al., 2012, Szwedziak et al., 2012, Hsin et al., 2013). Hence, the off/on states of FtsA could correspond to different states of oligomerization. On the other hand, the E124A or I143L substitutions did not strongly affect FtsA self-interaction in yeast two-hybrid assays (Pichoff et al., 2012). Subtle differences in self-interaction may be sufficient to favor the on or off state of FtsA. Alternatively, differences in interactions of FtsA with other SR components may be more decisive. Again, additional work is needed to understand how NFtsN or mutations in its IC subdomain affect the state of FtsA and how this is communicated to the FtsBLQ subcomplex and other SR components. It will also be interesting to learn if EFtsN*-suppressing mutations affecting additional SR components (e.g. FtsQ or the sPG synthase subcomplex) can be recovered and, if so, what their phenotypes are.
Conditional lethality of EFtsN*-suppressing mutations
The presence of EFtsN*-suppressing mutations in otherwise wildtype cells had several interesting morphological effects that depended on growth conditions. In regular LB or M9 medium (Table 5, Fig. 5, 6, S6), they caused septation at a reduced cell mass, leading to a small-cell phenotype. In M9 medium, several (combinations) of the mutations also caused a modest loss of rod-shape. A far more dramatic loss of normal cell shape was seen when these cells were grown at high temperature in low osmotic medium (Fig. 7, S8). In addition, this was accompanied by cell death, especially in the strains with the potential to survive without FtsN altogether. In fact, FtsN proved toxic to these strains under these growth conditions, and EFtsN was responsible for this toxicity in at least one of these strains (Fig. 7, S8). These results, and those reported by Tsang and Bernhardt (Tsang & Bernhardt, 2015), strongly indicate that overstimulation of the cell constriction machinery is, at least partially, responsible for the observed loss of cell shape and integrity.
In the context of our model, we imagine that an abnormally high FtsBLQ on:off ratio leads to hyperactive sPG synthase complexes. Increased competition for PG precursors and/or protein components with the PG synthase complexes responsible for proper elongation of the murein sacculus (Vollmer & Bertsche, 2008), could then explain the loss of rod-shape. Hyperactivity of sPG synthases and/or the high on-state of FtsBLQ more directly, might also lead to disregulation of murein hydrolase activities (Uehara & Bernhardt, 2011), contributing to cell death. Both low osmolarity and high temperature were required to observe the severe cell shape and viability defects in these strains (Fig. 7, S8, and data not shown). Both conditions are stressful and have a variety of physiological consequences (Wood, 1999, Guisbert et al., 2008). More work is needed to understand how each condition contributes to the lethality of these mutants. Regardless, the results suggest that both blocking and overstimulation of the cell constriction machinery can be lethal to E.coli, at least under certain growth conditions.
EXPERIMENTAL PROCEDURES
Strains, genetic constructs, and growth conditions
Strains and plasmids used in this study are listed in Tables S4 and S5, respectively, and their construction is detailed in the supplement as well. Unless stated otherwise, cells were grown at 30°C in LB (1% tryptone, 0.5% yeast extract, 0.5% NaCl, pH=7.0) or in M9 minimal medium (47.7 mM Na2HPO4, 22.0 mM KH2PO4, 8.6 mM NaCl, 18.7 mM NH4Cl, 2.0 mM MgSO4, 0.1 mM CaCl) (Roskams & Rodgers, 2002) supplemented with 0.2% casamino acids, 50 μg/ml L-tryptophan, 50 μM thiamine, and 0.2% of either glucose (M9-glucose) or maltose (M9-maltose). When appropriate, medium was supplemented with 50 μg/ml ampicillin (Amp), 25 μg/ml chloramphenicol (Cam), 25 μg/ml kanamycin (Kan), 50 μg/ml spectinomycin (Spec), or 12.5 μg/ml tetracycline (Tet). Amp or Cam concentrations were reduced to 15 and 10 μg/ml, respectively, when cells carried bla or cat integrated in the chromosome. For the transduction experiments in Tables 1–4, cells were incubated with the equivalent of 0.1 μl undiluted P1 lysate grown on either strain CH34/pCH201 [ΔftsN<>aph/ Plac::gfp-ftsN] (Tables 1–3) or BL71/pCH288 [ΔftsN<>cat/ Plac::sstorA-gfp-ftsN55–123] (Table 4). Other details are specified in the text.
ftsN mutagenesis and functionality screens
FtsN residues D5 and Y6, and each of the 14 residues of EFtsN (FtsN75–93), were substituted by site-directed and/or site-scanning mutagenesis in the context of a full-length GFP-FtsN fusion. Site-directed mutagenesis was done by the Quickchange (Stratagene) method or by asymmetric amplification and overlap extension (Xiao & Pei, 2011), and yielded mutant derivatives of pCH201 [Plac::gfp-ftsN].
For site scanning mutagenesis of residues 80–93, DNA2.0 (Menlo Park, CA) prepared 14 collections of 132 bp fragments that encode FtsN68–101, and in which the codon of one of the targeted residues is randomized to allow substitution with any other residue. The fragments also contain a unique SfiI site at each end, allowing directional insertion in pBL143 [Plac::gfp-ftsNΔ(64–101)<>(SphI_XhoI-stuffer)] to generate libraries of pBL116 [Plac::gfp-ftsN T64G, S67G, +A102] mutants. For a mixed library, equimolar amounts of DNA from the 14 collections were mixed, and the mixture was either used directly for cloning, or as template for amplification with primers 5′-GTGGCCTGCAAGGCCAGAAAGTGACCGGAAACGG-3′ and 5′-CAGGCCGAAGGGGCCTCTGTGGGCGCACGCACTCCCGG-3′ to increase the amount of material. Fragments were treated with SfiI, and the released 113 bp fragments were used to replace the 122 bp SfiI-SfiI stuffer fragment of pBL143. FtsNY85- and FtsNL89-specific libraries were prepared similarly, except that the corresponding un-mixed fragment collection was used.
Unmutated pCH201 [Plac::gfp-ftsN] or pBL116 [Plac::gfp-ftsN T64G, S67G, +A102] fully rescued strain CH34 [ΔftsN] and the cells displayed an almost normal division phenotype in LB, even without IPTG. Hence, to test the functionality of mutant derivatives of pCH201 or pBL116, they were moved into the FtsN-depletion strain CH31 [PBAD::ftsN] in the presence of 0.5% arabinose (Ara), and transformants were tested for growth on LB agar with or without Ara.
From the mixed pBL116 site-scanning libraries, 90 out of 216 transformants failed to grow without Ara, and the ftsN sequences of 49 non-functional and of 40 functional pBL116 derivatives was determined. To increase the diversity of substitutions in Y85 and L89, we additionally determined the ftsN sequences of pBL116 derivatives from the FtsNY85-specific (55 non-functional, 5 functional) and FtsNL89-specific (26 non-functional, 14 functional) libraries, respectively. The results are summarized in Fig. 1D and Table S1.
Isolation and mapping of spontaneous extragenic EFtsN*-suppressors
Strain BL86/pBL200 [ΔftsN<>aph ΔrecA ΔlacIZYA leu::Tn10 / aadA repAts Psyn135::ftsN I-SceI cI857 PλR::i-sceI] was transformed with an unstably inherited mini-F plasmid [bla lacIq Plac::gfp-ftsN*:: GlacZYA] encoding a non-functional GFP-FtsN* fusion with FtsN substitution W83L (pBL216), W83M (pBL218) W83T (pBL226) Y85S (pBL225), Y85W (pBL215), L89H (pBL219), or L89S (pBL217), as well as lacZYA, under control of the lac regulatory region. Transformants were grown overnight at 30°C in LB with Amp and 100 μM IPTG to OD600~4.0, and aliquots of cultures (~4.108 cells) were diluted 500-fold in 50 ml of fresh medium. Diluted cultures were incubated at 38°C or 42°C for 24 hr, diluted again 500-fold in 5.0 ml fresh medium lacking Amp (in duplicate for some screens), and incubated for another 24 hr at 38°C or 42°C. Cultures showing an appreciable density were streaked out on LB-IX agar (containing 100 μM IPTG and 60 μg/ml X-gal, but no Amp) at 30°C to distinguish colonies that retained the mini-F plasmid (blue) from those that did not (white). The latter were relatively rare and, when examined, were spectinomycin resistant (AadA+), suggesting they retained (at least part of) pBL200 via an unclear mechanism. Blue colonies were retained and tested for growth on LB agar with or without 100 μM IPTG, or with both 100 μM IPTG and Spec. Clones that showed IPTG-dependent growth and sensitivity to Spec were retained and ftsN on the resident mini-F plasmid was sequenced to ensure it still carried the initial query allele. Screens with each query allele were done at least twice. Only screens with W83L (pBL216), Y85S (pBL225), and Y85W (pBL215) yielded survivors that had lost pBL200, showed IPTG-dependent growth, and had retained the initial query allele.
Phage P1 was grown on these survivors, and the lysate was used to transduce leu::Tn10 to strain CH34/pBL200 [ΔftsN<>aph ΔlacIZYA / aadA repAts Psyn135::ftsN I-SceI cI857 PλR::i-sceI] carrying the same mini-F plasmid that was used to select for the survivor. Selection was at 30°C on LB agar containing Tet, Amp, Spec, and 100 μM IPTG. Transductants (36 of each) were then purified in duplicate on the same medium lacking Spec and incubated at 30°C or 42°C. The number of temperature-resistant transductants was used to calculate the co-transduction frequency of the EFtsN*-suppressing mutation with leu::Tn10. The suppressing mutations in BL86-AK1/pBL215 and BL86-KK1/pBL215 showed 75% and 58% linkage with leu::Tn10, respectively. Chromosomal fragments were amplified with primer pairs 5′-GGTCTAGAGGTGGCTGAGAACCCTCGTGCC-3′ and 5′-TCGTCGACCTACTACAGCTACAAAGAGATC-3′ (yielding a 2337bp ftsLI fragment), 5′-CGTCTAGAGCGTAATTGGCTCACGCAAAGTG-3′ and 5′-CGGTCGACGCCAAAGCTGACGCAGCGTTC-3′ (yielding a 2464 bp mraY fragment), 5′-ATTCTAGATATGGTTCTGCTCTCCCCAGCC-3′ and 5′-CGGTCGACGCATTTCGGGCACGATGGAACG-3′ (yielding a 2526 bp ftsW murG fragment), and 5′-GTAGTACGAATTCTGGAACTGGCGGAC-3′ and 5′-GAGGCCGTAATCATCGTCGGCCTC-3′ (yielding a 2152 bp ftsQA fragment). Sequence analyses of these fragments revealed the presence of ftsLD93G and ftsAI143L in BL86-AK1 and BL86-KK1, respectively.
EFtsN*-suppressing mutations in survivors of screens with W83L (pBL216) or Y85S (pBL225) did not co-transduce with leu::Tn10. The entire genome of two survivors from each screen was determined and compared to that of unsuppressed strain BL86/pBL209 at the CWRU Genomics Sequencing Core. Total DNA was extracted using the Epicenter MasterPure DNA Purification kit, libraries were prepared using the Epicentre Nextera DNA Sample Prep Kit, and sequences were obtained with the Illumina HiScan system. Illumina reads were mapped to the reference genome (MG1655) using the Bowtie program (Langmead et al., 2009) and SNPs were called using the SAMtools package (Li et al., 2009). This revealed the same ftsBD59H allele in suppressed strains BL86-AK11/pBL216 and BL86-AK12/pBL216. The two survivors of Y85S (pBL225) did not reveal any SNP’s in/near genes known to affect cell division or elongation, and more work will be required to understand the genetic basis for suppression in these strains.
EFtsN*-suppression by randomly mutagenized ftsB or ftsL
Libraries of randomly mutagenized ftsB and ftsL were prepared in the context of plasmids pBL336 [Psyn135::ftsB] and pJH2 [Psyn135::ftsL], respectively, using Error-prone amplification of fragments with Taq polymerase essentially as described (Cadwell & Joyce, 1992). For the pBL336 library, ftsB was amplified from pBL294 [Plac::ftsB] with primers 5′-CCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATAC-3′ and 5′-GCCAAGCTTGTCGACTGGCCGAGGCGGCC-3′, and the 374 bp XbaI-HindIII fragments of the product were used to replace the 414bp XbaI-HindIII fragment of plasmid pJH2 [Psyn135::ftsL]. For the pJH2 library, ftsL was amplified from plasmid pJH1 [Psyn135::ftsL] with primers 5′-CCTCTAGAAATAATTTTGTTTAACTTTAAGAAGGAGATATAC-3′ and 5′-CATGATTACGCCAAGCTTGTCGAC-3′, and the 414 bp XbaI-HindIII fragments of the product were used to replace the equivalent fragment of plasmid pJH2 [Psyn135::ftsL]. Ligation mixtures were transformed to strain DH5α for amplification of the libraries.
For the screens, libraries were introduced into strain JH1 [ΔftsN ΔrecA ΔlacIZYA] harboring both pBL200 [aadA repAts Psyn135::ftsN I-SceI cI857 PλR::i-sceI] and one of the mini-F derivatives encoding mutant GFP-FtsN* variants. Transformants were plated on LB agar with 200 μM IPTG at 37°C or 42°C to induce loss of pBL200 and, hence, select for plasmid-borne ftsB or ftsL alleles that allow survival of EFtsN cells. Survivors were tested to ensure sensitivity to spectinomycin before the ftsB or ftsL alleles were sequenced. Plasmid clones with multiple silent and/or missense mutations in ftsB or ftsL were used for subcloning to resolve the actual EFtsN*-suppressing mutation. The results are summarized in Fig. 3 and Table S2.
Microscopy, HADA labeling, and image analyses
Imaging was performed on a Zeiss Axioplan-2 microscope system as previously described (Johnson et al., 2002). Live cells were imaged on pads of 1.2% agarose in M9 salts. For the cell dimension measurements shown in Table 5 and Fig. S6, cells were chemically fixed (Bendezu & de Boer, 2008) and imaged on poly-L-lysine-coated coverslips using phase contrast optics. Cell length and diameter were measured in automated mode using the ObjectJ 1.03a plug-in (van der Ploeg et al., 2013) of imageJ 1.48a (Schneider et al., 2012).
For the pulse-labeling experiments with HADA (Kuru et al., 2012) shown in Fig. 6 and Table S3, overnight cultures (LB, 30°C) were diluted to OD600=0.02 in fresh LB and growth was continued at 30°C to OD600=0.6. One hundredth of a 100 mM stock solution of hydroxy coumarin-carbonyl-amino-D-alanine (HADA, kindly supplied by Erkin Kuru and Michael VanNieuwenhze) in DMSO was added to aliquots of each culture. After incubation at 37°C for one min, ice-cold ethanol was added to 70% and mixtures were incubated on ice for 15 min. The ethanol-fixed cells were washed five times in ice-cold PBS (10.1 mM Na2HPO4, 1.8 mM KH2PO4, 137 mM NaCl, 2.7 mM KCl, pH 7.4), and then imaged on pads of 1.2% agarose in M9 salts using fluorescence (DAPI) and DIC optics. Cell length and diameter, and the presence of a constriction and/or fluorescent ring were determined manually using ObjectJ. The periods of septal peptidoglycan (sPG) synthesis and of visible cell constriction during the division cycle were determined essentially as described (Den Blaauwen et al., 1999), assuming HADA accumulation at midcell coincides with synthesis of sPG. The average time after cell birth when sPG synthesis (tpg) or constriction (tc) starts was calculated using: , where Td is the mass doubling time and F(x) is the fraction of cells with no visible constriction (tx=tc), or with neither a constriction nor a ‘HADA ring’ (tx=tpg). The average time before cell separation when sPG synthesis is completed was calculated using: , where F(x) is the fraction of cells with a (deep) constriction but without associated ‘HADA ring’. Cell volume was calculated using V=4/3πr3+πr2h with r representing radius and h cylinder length. Data analysis was performed in Microsoft Excel and Origin 9 (OriginLab Corporation).
Immunoblotting
Western analyses with anti-GFP antibodies (Rockland) were done essentially as before (Bernhardt & de Boer, 2003). The procedure was similar for detection of FtsB and FtsL, with the following exceptions. Cells were resuspended in Tricine sample buffer (450 mM Tris.Cl, 12% glycerol, 4% SDS, 0.0025% Coomassie Blue G, pH=8.5), and broken by incubation at 65°C for 15 min followed by brief sonication. Whole cell lysates were fractionated in Tricine running buffer (100 mM Tris.OH, 100 mM Tricine, 0.1% SDS, pH=8.3) on Novex 10–20% Tricine gradient gels (Life Technologies). Proteins were transferred to 0.2 μm nitrocellulose filter in transfer buffer (25 mM Tris.Cl, 192 mM Glycine, 10% Methanol) for 2 hr in a Genie Blotter (Idea Scientific Company). Blots were heated in boiling PBS for 5 min for heat-induced antigen retrieval (Swerdlow et al., 1986) immediately before subsequent routine blocking and detection steps. Rabbit polyclonal α-FtsB and α-FtsL antisera were kindly provided by Morgan Feeney and Jon Beckwith.
Other methods
Bacterial two hybrid (BACTH) assays (Karimova et al., 1998) were performed as before (Bendezu et al., 2009). For the growth curves in Fig. 2, 0.2 ml cultures were grown shaking at 30°C in a 96-well Falcon 353072 plate in a BioTek Synergy HT microplate reader.
Supplementary Material
Acknowledgments
We thank Jillian Houtz, Cynthia Hale, Matthew Gerding, Anita Boyapati, and Thomas Bernhardt for help in plasmid and/or strain construction, Michael VanNieuwenhze, David Lee, Erkin Kuru, Morgan Feeney and Jon Beckwith for strains, plasmids, antisera or other reagents, and Mary-Jane Tsang and Thomas Bernhardt for advice and sharing results before publication. Supported by NIH GM57059 (to PdB).
References
- Adam M, Fraipont C, Rhazi N, Nguyen-Disteche M, Lakaye B, Frere JM, Devreese B, Van Beeumen J, van Heijenoort Y, van Heijenoort J, Ghuysen JM. The bimodular G57–V577 polypeptide chain of the class B penicillin-binding protein 3 of Escherichia coli catalyzes peptide bond formation from thiolesters and does not catalyze glycan chain polymerization from the lipid II intermediate. J Bacteriol. 1997;179:6005–6009. doi: 10.1128/jb.179.19.6005-6009.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol. 1997;25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- Alexeeva S, Gadella TW, Jr, Verheul J, Verhoeven GS, den Blaauwen T. Direct interactions of early and late assembling division proteins in Escherichia coli cells resolved by FRET. Mol Microbiol. 2010;77:384–398. doi: 10.1111/j.1365-2958.2010.07211.x. [DOI] [PubMed] [Google Scholar]
- Altenhoff AM, Schneider A, Gonnet GH, Dessimoz C. OMA 2011: orthology inference among 1000 complete genomes. Nucleic Acids Res. 2011;39:D289–294. doi: 10.1093/nar/gkq1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arends SJ, Williams K, Scott RJ, Rolong S, Popham DL, Weiss DS. Discovery and characterization of three new Escherichia coli septal ring proteins that contain a SPOR domain: DamX, DedD, and RlpA. J Bacteriol. 2010;192:242–255. doi: 10.1128/JB.01244-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendezu FO, de Boer PA. Conditional lethality, division defects, membrane involution, and endocytosis in mre and mrd shape mutants of Escherichia coli. J Bacteriol. 2008;190:1792–1811. doi: 10.1128/JB.01322-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. RodZ (YfgA) is required for proper assembly of the MreB actin cytoskeleton and cell shape in E.coli. EMBO J. 2009;28:193–204. doi: 10.1038/emboj.2008.264. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernard CS, Sadasivam M, Shiomi D, Margolin W. An altered FtsA can compensate for the loss of essential cell division protein FtsN in Escherichia coli. Mol Microbiol. 2007;64:1289–1305. doi: 10.1111/j.1365-2958.2007.05738.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernhardt TG, de Boer PAJ. The Escherichia coli amidase AmiC is a periplasmic septal ring component exported via the twin-arginine transport pathway. Mol Microbiol. 2003;48:1171–1182. doi: 10.1046/j.1365-2958.2003.03511.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertsche U, Breukink E, Kast T, Vollmer W. In vitro murein peptidoglycan synthesis by dimers of the bifunctional transglycosylase-transpeptidase PBP1B from Escherichia coli. J Biol Chem. 2005;280:38096–38101. doi: 10.1074/jbc.M508646200. [DOI] [PubMed] [Google Scholar]
- Bertsche U, Kast T, Wolf B, Fraipont C, Aarsman ME, Kannenberg K, von Rechenberg M, Nguyen-Disteche M, den Blaauwen T, Holtje JV, Vollmer W. Interaction between two murein (peptidoglycan) synthases, PBP3 and PBP1B, in Escherichia coli. Mol Microbiol. 2006;61:675–690. doi: 10.1111/j.1365-2958.2006.05280.x. [DOI] [PubMed] [Google Scholar]
- Beuria TK, Mullapudi S, Mileykovskaya E, Sadasivam M, Dowhan W, Margolin W. Adenine nucleotide-dependent regulation of assembly of bacterial tubulin-like FtsZ by a hypermorph of bacterial actin-like FtsA. J Biol Chem. 2009;284:14079–14086. doi: 10.1074/jbc.M808872200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Born P, Breukink E, Vollmer W. In vitro synthesis of cross-linked murein and its attachment to sacculi by PBP1A from Escherichia coli. J Biol Chem. 2006;281:26985–26993. doi: 10.1074/jbc.M604083200. [DOI] [PubMed] [Google Scholar]
- Bramkamp M, Weston L, Daniel RA, Errington J. Regulated intramembrane proteolysis of FtsL protein and the control of cell division in Bacillus subtilis. Mol Microbiol. 2006;62:580–591. doi: 10.1111/j.1365-2958.2006.05402.x. [DOI] [PubMed] [Google Scholar]
- Buddelmeijer N, Beckwith J. A complex of the Escherichia coli cell division proteins FtsL, FtsB and FtsQ forms independently of its localization to the septal region. Mol Microbiol. 2004;52:1315–1327. doi: 10.1111/j.1365-2958.2004.04044.x. [DOI] [PubMed] [Google Scholar]
- Buddelmeijer N, Judson N, Boyd D, Mekalanos JJ, Beckwith J. YgbQ, a cell division protein in Escherichia coli and Vibrio cholerae, localizes in codependent fashion with FtsL to the division site. Proc Natl Acad Sci U S A. 2002;99:6316–6321. doi: 10.1073/pnas.092128499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busiek KK, Eraso JM, Wang Y, Margolin W. The early divisome protein FtsA interacts directly through its 1c subdomain with the cytoplasmic domain of the late divisome protein FtsN. J Bacteriol. 2012;194:1989–2000. doi: 10.1128/JB.06683-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Busiek KK, Margolin W. A role for FtsA in SPOR-independent localization of the essential Escherichia coli cell division protein FtsN. Mol Microbiol. 2014;92:1212–1226. doi: 10.1111/mmi.12623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cadwell RC, Joyce GF. Randomization of genes by PCR mutagenesis. PCR Methods Appl. 1992;2:28–33. doi: 10.1101/gr.2.1.28. [DOI] [PubMed] [Google Scholar]
- Chen JC, Beckwith J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol Microbiol. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- Corbin BD, Geissler B, Sadasivam M, Margolin W. Z-Ring-Independent Interaction between a Subdomain of FtsA and Late Septation Proteins as Revealed by a Polar Recruitment Assay. J Bacteriol. 2004;186:7736–7744. doi: 10.1128/JB.186.22.7736-7744.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Ulisse V, Fagioli M, Ghelardini P, Paolozzi L. Three functional subdomains of the Escherichia coli FtsQ protein are involved in its interaction with the other division proteins. Microbiology. 2007;153:124–138. doi: 10.1099/mic.0.2006/000265-0. [DOI] [PubMed] [Google Scholar]
- Dai K, Xu Y, Lutkenhaus J. Topological characterization of the essential Escherichia coli cell division protein FtsN. J Bacteriol. 1996;178:1328–1334. doi: 10.1128/jb.178.5.1328-1334.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daniel RA, Errington J. Intrinsic instability of the essential cell division protein FtsL of Bacillus subtilis and a role for DivIB protein in FtsL turnover. Mol Microbiol. 2000;36:278–289. doi: 10.1046/j.1365-2958.2000.01857.x. [DOI] [PubMed] [Google Scholar]
- Daniel RA, Harry EJ, Katis VL, Wake RG, Errington J. Characterization of the essential cell division gene ftsL (yllD) of Bacillus subtilis and its role in the assembly of the division apparatus. Mol Microbiol. 1998;29:593–604. doi: 10.1046/j.1365-2958.1998.00954.x. [DOI] [PubMed] [Google Scholar]
- de Boer PAJ. Advances in understanding E.coli cell fission. Curr Opin Microbiol. 2010;13:730–737. doi: 10.1016/j.mib.2010.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Blaauwen T, Buddelmeijer N, Aarsman ME, Hameete CM, Nanninga N. Timing of FtsZ assembly in Escherichia coli. J Bacteriol. 1999;181:5167–5175. doi: 10.1128/jb.181.17.5167-5175.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Denome SA, Elf PK, Henderson TA, Nelson DE, Young KD. Escherichia coli mutants lacking all possible combinations of eight penicillin binding proteins: viability, characteristics, and implications for peptidoglycan synthesis. J Bacteriol. 1999;181:3981–3993. doi: 10.1128/jb.181.13.3981-3993.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Derouaux A, Wolf B, Fraipont C, Breukink E, Nguyen-Disteche M, Terrak M. The monofunctional glycosyltransferase of Escherichia coli localizes to the cell division site and interacts with penicillin-binding protein 3, FtsW, and FtsN. J Bacteriol. 2008;190:1831–1834. doi: 10.1128/JB.01377-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Di Lallo G, Fagioli M, Barionovi D, Ghelardini P, Paolozzi L. Use of a two-hybrid assay to study the assembly of a complex multicomponent protein machinery: bacterial septosome differentiation. Microbiology. 2003;149:3353–3359. doi: 10.1099/mic.0.26580-0. [DOI] [PubMed] [Google Scholar]
- Draper GC, McLennan N, Begg K, Masters M, Donachie WD. Only the N-terminal domain of FtsK functions in cell division. J Bacteriol. 1998;180:4621–4627. doi: 10.1128/jb.180.17.4621-4627.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dubarry N, Possoz C, Barre FX. Multiple regions along the Escherichia coli FtsK protein are implicated in cell division. Mol Microbiol. 2010;78:1088–1100. doi: 10.1111/j.1365-2958.2010.07412.x. [DOI] [PubMed] [Google Scholar]
- Duncan TR, Yahashiri A, Arends SJ, Popham DL, Weiss DS. Identification of SPOR domain amino acids important for septal localization, peptidoglycan binding, and a disulfide bond in the cell division protein FtsN. J Bacteriol. 2013;195:5308–5315. doi: 10.1128/JB.00911-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Egan AJ, Vollmer W. The physiology of bacterial cell division. Ann N Y Acad Sci. 2013;1277:8–28. doi: 10.1111/j.1749-6632.2012.06818.x. [DOI] [PubMed] [Google Scholar]
- Fraipont C, Alexeeva S, Wolf B, van der Ploeg R, Schloesser M, den Blaauwen T, Nguyen-Disteche M. The integral membrane FtsW protein and peptidoglycan synthase PBP3 form a subcomplex in Escherichia coli. Microbiology. 2011;157:251–259. doi: 10.1099/mic.0.040071-0. [DOI] [PubMed] [Google Scholar]
- Garcia del Portillo F, de Pedro MA. Differential effect of mutational impairment of penicillin-binding proteins 1A and 1B on Escherichia coli strains harboring thermosensitive mutations in the cell division genes ftsA, ftsQ, ftsZ, and pbpB. J Bacteriol. 1990;172:5863–5870. doi: 10.1128/jb.172.10.5863-5870.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Elraheb D, Margolin W. A gain-of-function mutation in ftsA bypasses the requirement for the essential cell division gene zipA in Escherichia coli. Proc Natl Acad Sci U S A. 2003;100:4197–4202. doi: 10.1073/pnas.0635003100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Geissler B, Margolin W. Evidence for functional overlap among multiple bacterial cell division proteins: compensating for the loss of FtsK. Mol Microbiol. 2005;58:596–612. doi: 10.1111/j.1365-2958.2005.04858.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Liu B, Bendezu FO, Hale CA, Bernhardt TG, de Boer PA. Self-enhanced accumulation of FtsN at division sites, and roles for other proteins with a SPOR domain (DamX, DedD, and RlpA) in Escherichia coli cell constriction. J Bacteriol. 2009;191:7383–7401. doi: 10.1128/JB.00811-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gerding MA, Ogata Y, Pecora ND, Niki H, de Boer PA. The trans-envelope Tol-Pal complex is part of the cell division machinery and required for proper outer-membrane invagination during cell constriction in E. coli. Mol Microbiol. 2007;63:1008–1025. doi: 10.1111/j.1365-2958.2006.05571.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goehring NW, Beckwith J. Diverse paths to midcell: assembly of the bacterial cell division machinery. Curr Biol. 2005;15:R514–526. doi: 10.1016/j.cub.2005.06.038. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Gonzalez MD, Beckwith J. Premature targeting of cell division proteins to midcell reveals hierarchies of protein interactions involved in divisome assembly. Mol Microbiol. 2006;61:33–45. doi: 10.1111/j.1365-2958.2006.05206.x. [DOI] [PubMed] [Google Scholar]
- Goehring NW, Petrovska I, Boyd D, Beckwith J. Mutants, suppressors, and wrinkled colonies: mutant alleles of the cell division gene ftsQ point to functional domains in FtsQ and a role for domain 1C of FtsA in divisome assembly. J Bacteriol. 2007;189:633–645. doi: 10.1128/JB.00991-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MD, Akbay EA, Boyd D, Beckwith J. Multiple interaction domains in FtsL, a protein component of the widely conserved bacterial FtsLBQ cell division complex. J Bacteriol. 2010;192:2757–2768. doi: 10.1128/JB.01609-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gonzalez MD, Beckwith J. Divisome under construction: distinct domains of the small membrane protein FtsB are necessary for interaction with multiple cell division proteins. J Bacteriol. 2009;191:2815–2825. doi: 10.1128/JB.01597-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guisbert E, Yura T, Rhodius VA, Gross CA. Convergence of molecular, modeling, and systems approaches for an understanding of the Escherichia coli heat shock response. Microbiol Mol Biol Rev. 2008;72:545–554. doi: 10.1128/MMBR.00007-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guzman L-M, Barondess JJ, Beckwith J. FtsL, an essential cytoplasmic membrane protein involved in cell division in Escherichia coli. J Bacteriol. 1992;174:7716–7728. [PMC free article] [PubMed] [Google Scholar]
- Haldimann A, Wanner BL. Conditional-replication, integration, excision, and retrieval plasmid-host systems for gene structure-function studies of bacteria. J Bacteriol. 2001;183:6384–6393. doi: 10.1128/JB.183.21.6384-6393.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E.coli. Cell. 1997;88:175–185. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- Hamilton CM, Aldea M, Washburn BK, Babitzke P, Kushner SR. New method for generating deletions and gene replacements in Escherichia coli. J Bacteriol. 1989;171:4617–4622. doi: 10.1128/jb.171.9.4617-4622.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, de Pedro MA, Höltje JV. Involvement of N-acetylmuramyl-L-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hsin J, Fu R, Huang KC. Dimer dynamics and filament organization of the bacterial cell division protein FtsA. J Mol Biol. 2013;425:4415–4426. doi: 10.1016/j.jmb.2013.07.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ishino F, Jung HK, Ikeda M, Doi M, Wachi M, Matsuhashi M. New mutations fts-36, lts-33, and ftsW clustered in the mra region of the Escherichia coli chromosome induce thermosensitive cell growth and division. J Bacteriol. 1989;171:5523–5530. doi: 10.1128/jb.171.10.5523-5530.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Johnson JE, Lackner LL, de Boer PAJ. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ-rings. J Bacteriol. 2002;184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Dautin N, Ladant D. Interaction network among Escherichia coli membrane proteins involved in cell division as revealed by bacterial two-hybrid. analysis. J Bacteriol. 2005;187:2233–2243. doi: 10.1128/JB.187.7.2233-2243.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karimova G, Pidoux J, Ullmann A, Ladant D. A bacterial two-hybrid system based on a reconstituted signal transduction pathway. Proc Natl Acad Sci U S A. 1998;95:5752–5756. doi: 10.1073/pnas.95.10.5752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krupka M, Rivas G, Rico AI, Vicente M. Key role of two terminal domains in the bidirectional polymerization of FtsA protein. J Biol Chem. 2012;287:7756–7765. doi: 10.1074/jbc.M111.311563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuru E, Hughes HV, Brown PJ, Hall E, Tekkam S, Cava F, de Pedro MA, Brun YV, VanNieuwenhze MS. In Situ probing of newly synthesized peptidoglycan in live bacteria with fluorescent D-amino acids. Angew Chem Int Ed Engl. 2012;51:12519–12523. doi: 10.1002/anie.201206749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, Trapnell C, Pop M, Salzberg SL. Ultrafast and memory-efficient alignment of short DNA sequences to the human genome. Genome Biol. 2009;10:R25. doi: 10.1186/gb-2009-10-3-r25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaPointe LM, Taylor KC, Subramaniam S, Khadria A, Rayment I, Senes A. Structural organization of FtsB, a transmembrane protein of the bacterial divisome. Biochemistry. 2013;52:2574–2585. doi: 10.1021/bi400222r. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lara B, Rico AI, Petruzzelli S, Santona A, Dumas J, Biton J, Vicente M, Mingorance J, Massidda O. Cell division in cocci: localization and properties of the Streptococcus pneumoniae FtsA protein. Mol Microbiol. 2005;55:699–711. doi: 10.1111/j.1365-2958.2004.04432.x. [DOI] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics. 2009;25:2078–2079. doi: 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Loose M, Mitchison TJ. The bacterial cell division proteins FtsA and FtsZ self-organize into dynamic cytoskeletal patterns. Nat Cell Biol. 2014;16:38–46. doi: 10.1038/ncb2885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutkenhaus J, Pichoff S, Du S. Bacterial cytokinesis: From Z ring to divisome. Cytoskeleton (Hoboken) 2012;69:778–790. doi: 10.1002/cm.21054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonnell AV, Jiang T, Keating AE, Berger B. Paircoil2: improved prediction of coiled coils from sequence. Bioinformatics. 2006;22:356–358. doi: 10.1093/bioinformatics/bti797. [DOI] [PubMed] [Google Scholar]
- Mercer KL, Weiss DS. The Escherichia coli cell division protein FtsW is required to recruit its cognate transpeptidase, FtsI (PBP3), to the division site. J Bacteriol. 2002;184:904–912. doi: 10.1128/jb.184.4.904-912.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Meury J, Devilliers G. Impairment of cell division in tolA mutants of Escherichia coli at low and high medium osmolarities. Biol Cell. 1999;91:67–75. [PubMed] [Google Scholar]
- Mohammadi T, van Dam V, Sijbrandi R, Vernet T, Zapun A, Bouhss A, Diepeveen-de Bruin M, Nguyen-Disteche M, de Kruijff B, Breukink E. Identification of FtsW as a transporter of lipid-linked cell wall precursors across the membrane. EMBO J. 2011;30:1425–1432. doi: 10.1038/emboj.2011.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moll A, Thanbichler M. FtsN-like proteins are conserved components of the cell division machinery in proteobacteria. Mol Microbiol. 2009;72:1037–1053. doi: 10.1111/j.1365-2958.2009.06706.x. [DOI] [PubMed] [Google Scholar]
- Muller P, Ewers C, Bertsche U, Anstett M, Kallis T, Breukink E, Fraipont C, Terrak M, Nguyen-Disteche M, Vollmer W. The essential cell division protein FtsN interacts with the murein (peptidoglycan) synthase PBP1B in Escherichia coli. J Biol Chem. 2007;282:36394–36402. doi: 10.1074/jbc.M706390200. [DOI] [PubMed] [Google Scholar]
- Noirclerc-Savoye M, Lantez V, Signor L, Philippe J, Vernet T, Zapun A. Reconstitution of membrane protein complexes involved in pneumococcal septal cell wall assembly. PloS one. 2013;8:e75522. doi: 10.1371/journal.pone.0075522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ohashi T, Hale CA, De Boer PA, Erickson HP. Structural Evidence that the P/Q Domain of ZipA Is an Unstructured, Flexible Tether between the Membrane and the C-Terminal FtsZ-Binding Domain. J Bacteriol. 2002;184:4313–4315. doi: 10.1128/JB.184.15.4313-4315.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osawa M, Erickson HP. Liposome division by a simple bacterial division machinery. Proc Natl Acad Sci USA. 2013;110:11000–11004. doi: 10.1073/pnas.1222254110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters NT, Dinh T, Bernhardt TG. A fail-safe mechanism in the septal ring assembly pathway generated by the sequential recruitment of cell separation amidases and their activators. J Bacteriol. 2011;193:4973–4983. doi: 10.1128/JB.00316-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Tethering the Z ring to the membrane through a conserved membrane targeting sequence in FtsA. Mol Microbiol. 2005;55:1722–1734. doi: 10.1111/j.1365-2958.2005.04522.x. [DOI] [PubMed] [Google Scholar]
- Pichoff S, Shen B, Sullivan B, Lutkenhaus J. FtsA mutants impaired for self-interaction bypass ZipA suggesting a model in which FtsA’s self-interaction competes with its ability to recruit downstream division proteins. Mol Microbiol. 2012;83:151–167. doi: 10.1111/j.1365-2958.2011.07923.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pisabarro AG, Prats R, Vázquez D, Rodríguez-Tébar A. Activity of penicillin-binding protein 3 from Escherichia coli. J Bacteriol. 1986;168:199–206. doi: 10.1128/jb.168.1.199-206.1986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priyadarshini R, de Pedro MA, Young KD. Role of peptidoglycan amidases in the development and morphology of the division septum in Escherichia coli. J Bacteriol. 2007;189:5334–3347. doi: 10.1128/JB.00415-07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rico AI, Garcia-Ovalle M, Palacios P, Casanova M, Vicente M. Role of Escherichia coli FtsN protein in the assembly and stability of the cell division ring. Mol Microbiol. 2010;76:760–771. doi: 10.1111/j.1365-2958.2010.07134.x. [DOI] [PubMed] [Google Scholar]
- Roskams J, Rodgers L. Lab Ref: A Handbook of Recipes, Reagents, and Other Reference Tools for Use at the Bench. Cold Spring Harbor Laboratory Press; Cold Spring Harbor, New York: 2002. [Google Scholar]
- Schmidt LS, Botta G, Park JT. Effects of furazlocillin, a beta-lactam antibiotic which binds selectively to penicillin-binding protein 3, on Escherichia coli mutants deficient in other penicillin-binding proteins. J Bacteriol. 1981;145:632–637. doi: 10.1128/jb.145.1.632-637.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods. 2012;9:671–675. doi: 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sham LT, Butler EK, Lebar MD, Kahne D, Bernhardt TG, Ruiz N. Bacterial cell wall. MurJ is the flippase of lipid-linked precursors for peptidoglycan biogenesis. Science. 2014;345:220–222. doi: 10.1126/science.1254522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sherratt DJ, Arciszewska LK, Crozat E, Graham JE, Grainge I. The Escherichia coli DNA translocase FtsK. Biochem Soc Trans. 2010;38:395–398. doi: 10.1042/BST0380395. [DOI] [PubMed] [Google Scholar]
- Swerdlow PS, Finley D, Varshavsky A. Enhancement of immunoblot sensitivity by heating of hydrated filters. Anal Biochem. 1986;156:147–153. doi: 10.1016/0003-2697(86)90166-1. [DOI] [PubMed] [Google Scholar]
- Szwedziak P, Wang Q, Freund SM, Lowe J. FtsA forms actin-like protofilaments. EMBO J. 2012;31:2249–2260. doi: 10.1038/emboj.2012.76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsang M-J, Bernhardt TG. An ftsL allele that bypasses defects in essential divisome components. Mol Microbiol. 2015 doi: 10.1111/mmi.12905. in press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Bernhardt TG. More than just lysins: peptidoglycan hydrolases tailor the cell wall. Curr Opin Microbiol. 2011;14:698–703. doi: 10.1016/j.mib.2011.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uehara T, Dinh T, Bernhardt TG. LytM-domain factors are required for daughter cell separation and rapid ampicillin-induced lysis in Escherichia coli. J Bacteriol. 2009;191:5094–5107. doi: 10.1128/JB.00505-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ueki M, Wachi M, Jung H-K, Ishino F, Matsuhashi M. Escherichia coli mraR gene involved in cell growth and division. J Bacteriol. 1992;174:7841–7843. doi: 10.1128/jb.174.23.7841-7843.1992. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ursinus A, van den Ent F, Brechtel S, de Pedro M, Holtje JV, Lowe J, Vollmer W. Murein (peptidoglycan) binding property of the essential cell division protein FtsN from Escherichia coli. J Bacteriol. 2004;186:6728–6737. doi: 10.1128/JB.186.20.6728-6737.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Berg van Saparoea HB, Glas M, Vernooij IG, Bitter W, den Blaauwen T, Luirink J. Fine-mapping the contact sites of the Escherichia coli cell division proteins FtsB and FtsL on the FtsQ protein. J Biol Chem. 2013;288:24340–24350. doi: 10.1074/jbc.M113.485888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Den Ent F, Löwe J. Crystal structure of the cell division protein FtsA from Thermotoga maritima. EMBO J. 2000;19:5300–5307. doi: 10.1093/emboj/19.20.5300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van der Ploeg R, Verheul J, Vischer NO, Alexeeva S, Hoogendoorn E, Postma M, Banzhaf M, Vollmer W, den Blaauwen T. Colocalization and interaction between elongasome and divisome during a preparative cell division phase in Escherichia coli. Mol Microbiol. 2013;87:1074–1087. doi: 10.1111/mmi.12150. [DOI] [PubMed] [Google Scholar]
- Villanelo F, Ordenes A, Brunet J, Lagos R, Monasterio O. A model for the Escherichia coli FtsB/FtsL/FtsQ cell division complex. BMC Struct Biol. 2011;11:28. doi: 10.1186/1472-6807-11-28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vollmer W, Bertsche U. Murein (peptidoglycan) structure, architecture and biosynthesis in Escherichia coli. Biochim Biophys Acta. 2008;1778:1714–1734. doi: 10.1016/j.bbamem.2007.06.007. [DOI] [PubMed] [Google Scholar]
- Wadenpohl I, Bramkamp M. DivIC stabilizes FtsL against RasP cleavage. J Bacteriol. 2010;192:5260–5263. doi: 10.1128/JB.00287-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wissel MC, Weiss DS. Genetic analysis of the cell division protein FtsI (PBP3): amino acid substitutions that impair septal localization of FtsI and recruitment of FtsN. J Bacteriol. 2004;186:490–502. doi: 10.1128/JB.186.2.490-502.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood JM. Osmosensing by bacteria: signals and membrane-based sensors. Microbiol Mol Biol Rev. 1999;63:230–262. doi: 10.1128/mmbr.63.1.230-262.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao YH, Pei Y. Asymmetric overlap extension PCR method for site-directed mutagenesis. Methods Mol Biol. 2011;687:277–282. doi: 10.1007/978-1-60761-944-4_20. [DOI] [PubMed] [Google Scholar]
- Yang DC, Peters NT, Parzych KR, Uehara T, Markovski M, Bernhardt TG. An ATP-binding cassette transporter-like complex governs cell-wall hydrolysis at the bacterial cytokinetic ring. Proc Natl Acad Sci U S A. 2011;108:E1052–1060. doi: 10.1073/pnas.1107780108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang JC, Van Den Ent F, Neuhaus D, Brevier J, Lowe J. Solution structure and domain architecture of the divisome protein FtsN. Mol Microbiol. 2004;52:651–660. doi: 10.1111/j.1365-2958.2004.03991.x. [DOI] [PubMed] [Google Scholar]
- Yousif SY, Broome-Smith JK, Spratt BG. Lysis of Escherichia coli by beta-lactam antibiotics: deletion analysis of the role of penicillin-binding proteins 1A and 1B. J Gen Microbiol. 1985;131:2839–2845. doi: 10.1099/00221287-131-10-2839. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.