

Summary
The N-acetylmuramoyl-l-alanine amidases of Escherichia coli (AmiA, B and C) are periplasmic enzymes that remove murein cross-links by cleaving the peptide moiety from N-acetylmuramic acid. Ami− cells form chains, indicating that the amidases help to split the septal murein. Interestingly, cells defective in the twin-arginine protein transport (Tat) pathway show a similar division defect. We find that both AmiA and AmiC are routed to the periplasm via Tat, providing an explanation for the Tat− division phenotype. Taking advantage of the ability of Tat to export prefolded (fluorescent) green fluorescent protein (GFP) to the periplasm, we sublocalized AmiA and AmiC in live cells using functional fusions to GFP. Interestingly, the periplasmic localization of the fusions differed markedly. AmiA–GFP appeared to be dispersed throughout the periplasm in all cells. AmiC–GFP similarly appeared throughout the periplasm in small cells, but was concentrated almost exclusively at the septal ring in constricting cells. Recruitment of AmiC to the ring was mediated by an N-terminal non-amidase targeting domain and required the septal ring component FtsN. AmiC therefore replaces FtsN as the latest known recruit to the septal ring and is the first entirely periplasmic component to be localized.
Introduction
During cytokinesis in Escherichia coli, the cytoplasmic (inner) membrane, murein (peptidoglycan) layer and outer membrane are co-ordinately constricted at mid-cell to form the new daughter cell poles. Ingrowth of the envelope layers is thought to be driven and co-ordinated by the septal ring, a dynamic membrane-associated organelle that assembles at the division site well before constriction and constricts in concert with the ingrowing septum (Lutkenhaus and Addinall, 1997; Rothfield et al., 1999; Margolin, 2000). With the possible exception of the homologue of Bacillus subtilis ZapA (YgfE) (Gueiros-Filho and Losick, 2002), the known septal ring components are essential division proteins, which are either cytoplasmic (FtsA, FtsZ, YgfE) or integral inner membrane species (FtsI, FtsK, FtsL, FtsN, FtsQ, FtsW, YgbQ and ZipA). These proteins assemble in a specific order to form a functional division apparatus. The earliest known step is polymerization of the tubulin-like FtsZ protein into a ring (Z ring) just underneath the cytoplasmic membrane (Bi and Lutkenhaus, 1991). Formation of the Z ring requires at least one of the FtsZ-binding proteins, FtsA or ZipA, which join FtsZ independently of one another (Hale and de Boer, 1999; Liu et al., 1999; Pichoff and Lutkenhaus, 2002). After assembly of FtsZ, FtsA and ZipA, the remaining division proteins join the ring in the order: FtsK, FtsQ, FtsL + YgbQ, FtsW, FtsI and, finally, FtsN (Chen and Beckwith, 2001; Buddelmeijer et al., 2002; Hale and de Boer, 2002).
To date, the septal ring is loosely defined as an assemblage of proteins serving a common function (cell division) and showing a common and interdependent subcellular localization pattern (a ring-like accumulation at the division site). The actual molecular architecture of the organelle is still poorly understood. So far, the only direct contacts known to occur within the ring are between FtsZ and its binding partners FtsA, ZipA and YgfE (Hale and de Boer, 1997; Wang et al., 1997; Gueiros-Filho and Losick, 2002), but many more interactions between its various components are expected to take place. Some of these interactions may be indirect, involving molecules (be they proteins, lipids, amino sugars or other types) that still remain to be identified. Important goals are to identify all division factors associated with the septal ring and to understand how they co-operate to form a functional apparatus capable of accomplishing cell constriction.
Constriction of the cell envelope is coupled to a highly localized synthesis of septal murein (Wientjes and Nanninga, 1989; de Pedro et al., 1997; 2001; Nanninga, 1998). So far, the d,d-transpeptidase FtsI (penicillin-binding protein 3) is the only E. coli septal ring component directly associated with murein synthesis (Ishino and Matsuhashi, 1981; Adam et al., 1997). It is clear, however, that other murein synthetic, as well as degrading, activities must be present at the septal ring to generate septal murein and shape the new polar caps (Höltje, 1998; Pedersen et al., 1999; Höltje and Heidrich, 2001; Vollmer and Höltje, 2001).
The N-acetylmuramoyl-l-alanine amidases (AmiA, AmiB and AmiC) are periplasmic enzymes capable of destroying cell wall cross-links by cleaving the peptide moiety from N-acetylmuramic acid. It was shown recently that Ami− cells form chains, indicating that the amidases contribute to daughter cell separation by helping to split the septal murein (Heidrich et al., 2001; 2002; Höltje and Heidrich, 2001). To investigate whether amidases are specific components of the septal ring complex, we studied their subcellular localization in live cells using fusions to green fluorescent protein (GFP).
GFP exported to the periplasm by the general secretory (Sec) system folds incorrectly and is not fluorescent (Feilmeier et al., 2000). However, it was demonstrated recently that prefolded (fluorescent) GFP can be exported to the periplasm by the twin-arginine transport (Tat) system when the signal peptide of the Tat substrate TorA is appended to its N-terminus (Santini et al., 2001; Thomas et al., 2001). Interestingly, Tat− cells have a cell-chaining phenotype very similar to that of Ami− mutants (Stanley et al., 2001). Moreover, we noted that the predicted signal peptides of both AmiA and AmiC contain motifs resembling the Tat-targeting consensus (S/T)RRXFLK (Berks et al., 2000; DeLisa et al., 2002).
These observations suggested that the amidases might reach the periplasm via Tat, allowing the use of GFP fusions to study their subcellular localization. In this report, we show that this is indeed the case, providing an explanation for the division defect of Tat− cells. In addition, we found that the periplasmic localization patterns of AmiA and AmiC differed markedly. AmiA–GFP appeared to be distributed throughout the periplasm in all cells. In contrast, whereas AmiC–GFP was similarly present throughout the periplasm in small cells, the fusion localized almost exclusively to a ring at the site of constriction in dividing cells. This result indicated that AmiC is a periplasmic component of the septal ring organelle. Accordingly, we found that the accumulation of AmiC–GFP at the site of constriction requires the prior assembly of the septal ring component FtsN, placing AmiC as the latest known recruit to the division apparatus. Using deletion derivatives, we also established that an N-terminal domain of AmiC is dispensable for amidase activity, but both required and sufficient for septal targeting.
Thus, we have localized the first exported, entirely periplasmic, septal ring component in live bacteria, as well as identified the first autolysin-targeting domain in a Gram-negative organism.
Results
Tat-dependent translocation of functional AmiA–GFP to the periplasm
Inspection of the predicted signal sequences (Fig. 1A) of the periplasmic amidases suggested that AmiA and AmiC might be exported to the periplasm by Tat (DeLisa et al., 2002). This possibility is attractive for it might explain the cell-chaining phenotype of Tat− cells (Stanley et al., 2001), as well as provide an opportunity to sublocalize the amidases in live cells using GFP fusions (Santini et al., 2001; Thomas et al., 2001). We explored this possibility using constructs in which amiA and amiC sequences were fused to gfpmut2 (Cormack et al., 1996) and placed under control of the lac promoter of plasmid pMLB1113 (Fig. 1B and C).
Fig. 1.

Amidase domains and plasmid and phage inserts.
A. Signal sequences of E. coli periplasmic murein amidases AmiA, B and C (SWISSPROT accession P36548, P26365 and Q46929), as predicted by signalp version 1.1 (Nielsen et al., 1997). Potential Tat-targeting motifs (Berks et al., 2000; DeLisa et al., 2002) in AmiA and AmiC are underlined.
B–D. Domain organization of AmiA (B), AmiC (C) and AmiB (D), plasmid and phage inserts and summary of functionality and sublocalization studies. Predicted signal sequences (SS), septal targeting (T) and catalytic (C) domains, relevant residue numbers and the approximate position of a native amiC promoter are indicated in the diagrams. Plasmid and phage inserts (solid lines), the lac promoter (arrows), gfpmut2 (boxes) and the presence of a non-native ribosome binding site (*) are indicated below the diagrams. The amidase domains were drawn based on the Pfam database (Bateman et al., 2002). Columns on the right-hand side summarize the functionality and sublocalization studies. TB53 [ΔamiA ΔamiC] cells carrying the indicated construct were grown in the presence of 0, 50, 100, 250 and 500 μM IPTG, and their division phenotypes (Phen.) were examined. ccc, severe chaining phenotype, over 80% of cells in chains of 4–14 units; c, mild chaining phenotype, 10–30% of cells in chains of 4–6 units; wt, wild type, less than 5% of cells in chains. Minimum IPTG concentrations (in μM) required to attain the indicated phenotype were: 0 (pTB27, λTB27, pTB39), 50 (pTB28, pTB41), 100 (λTB28), 250 (pTB32) and 500 (pTB33). Plasmids pTB34 and pTB37 failed to suppress chaining at all concentrations. The localization patterns (Loc.) of the fusion proteins are given in the second column: P, peripheral in all cells; P/R, accumulated at the septal ring in constricting cells, peripheral otherwise; ?, cytoplasmic fluorescence but localization unreliable because of excessive degradation of the fusion protein.
PreAmiA is predicted to consist of a signal peptide (residues 1–34), a domain of unknown function (residues 35–117) and an amidase domain (residues 118–289) (Fig. 1B). We constructed plasmid pTB32 [Plac∷amiA–gfp], encoding full-length preAmiA fused to GFP, and tested its ability to direct the production of functional amidase. Overproduction of any one of the periplasmic amidases can suppress the chaining phenotype of mutants lacking all three enzymes, indicating that their activities (partially) overlap (Heidrich et al., 2001).
To test the functionality of amiA–gfp, we tested the ability of pTB32 to suppress chaining in the double mutant strain TB53 [ΔamiA ΔamiC]. Cells were grown to density in the absence or presence of IPTG and examined by microscopy. TB53 cells carrying the pMLB1113 vector showed a distinct division defect with over 80% of cells present in chains of 4–14 units (Figs 1 and 2A). In comparison, pTB32 [Plac∷amiA–gfp] markedly suppressed chaining in an IPTG-dependent manner, indicating that AmiA–GFP retained amidase activity (Figs 1 and 2B).
Fig. 2.
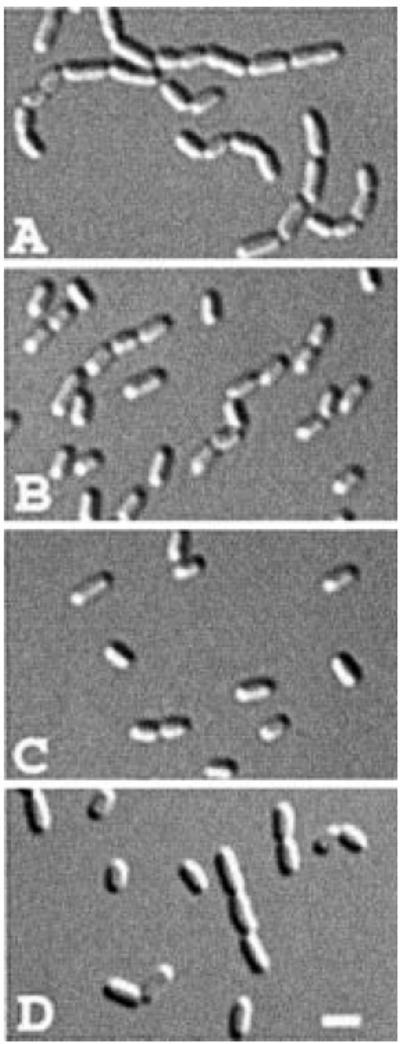
Functionality of the amidase fusion proteins. Differential interference contrast (DIC) micrographs show representative fields of TB53/pMLB1113 [ΔamiA ΔamiC/vector] (A), TB53/pTB32 [ΔamiA ΔamiC/Plac∷amiA–gfp] (B), TB53(λTB27) [ΔamiA ΔamiC (Plac∷PamiC∷amiC–gfp)] (C) and TB53/pTB33 [ΔamiA ΔamiC/Plac∷amiB–gfp] cells grown overnight in LB with no (C), 250 μM (A and B) or 500 μM (D) IPTG. Bar equals 2 μm.
To test whether AmiA–GFP required Tat for export, pTB32 was introduced in strain MC4100 and its ΔtatC derivative B1LKO (Bogsch et al., 1998). Cells were grown in the presence of IPTG, and AmiA–GFP was identified by Western analyses using anti-GFP antibodies. Whereas the MC4100/pTB32 extract contained two closely migrating forms of AmiA–GFP, the B1LKO/pTB32 extract contained only the slower migrating one (Fig. 3A, lanes 1 and 2). This result indicated that the signal peptide of AmiA–GFP failed to be processed in B1LKO, supporting a role for Tat in the export of AmiA to the periplasm.
Fig. 3.

Immunoblot analyses of GFP fusions. Blots probed with anti-GFP antibodies.
Lanes in (A) contained whole-cell extracts of MC4100/pTB32 [wt/Plac∷amiA–gfp] (1) and B1LKO/pTB32 [ΔtatC/Plac∷amiA–gfp] (2). Cells were grown with 250 μM IPTG. The arrow indicates the positions of processed and unprocessed forms of AmiA–GFP (a).
Lanes in (B) contained extracts of MC4100/pTB28 [wt/Plac∷amiC–gfp] (1), B1LKO/pTB28 [ΔtatC/Plac∷amiC–gfp] (2), MC4100/pTB34 [wt/Plac∷TamiC–gfp] (3), B1LKO/pTB34 [ΔtatC/Plac∷TamiC–gfp] (4) and TB54/pTB28 [PBAD∷ftsN/Plac∷amiC–gfp] (5 and 6). Cells were grown with 50 μM (1–4) or 100 μM (5 and 6) IPTG, and without (1–4 and 6) or with (5) 0.2% arabinose. Arrows indicate the positions of processed and unprocessed forms of AmiC–GFP (a) and TAmiC–GFP (b).
Lanes in (C) contained extracts of TB28/pTB39 [wt/Plac∷amiC–gfp] (1) and TB28/pTB41 [wt/Plac∷CamiC–gfp] (2). Cells were grown with 10 μM IPTG. Arrows indicate the positions of processed and unprocessed forms of AmiC–GFP (a) and CAmiC–GFP (b). Note that the processed and unprocessed forms of the fusions to full-length AmiC migrated too closely to be distinguishable on these blots. For, this, as well as for Figs 5-8, cells were grown in M9-based medium to OD600 = 0.4–0.7.
Consistent with this interpretation, conversion of cells of the Tat+ strain TB28/pTB32 to spheroplasts led to the preferential release of the faster migrating, presumably processed, form of AmiA–GFP into the surrounding buffer (Fig. 4A). Release of AmiA–GFP was markedly inefficient compared with the other periplasmic proteins MalE (Fig. 4A) and TAmiC–GFP (see below), but could be enhanced by the addition of 250 mM NaCl to the spheroplasts (Fig. 4A; data not shown). Perhaps AmiA–GFP is electrostatically bound to a membrane component or is poorly soluble in the spheroplast buffer.
Fig. 4.
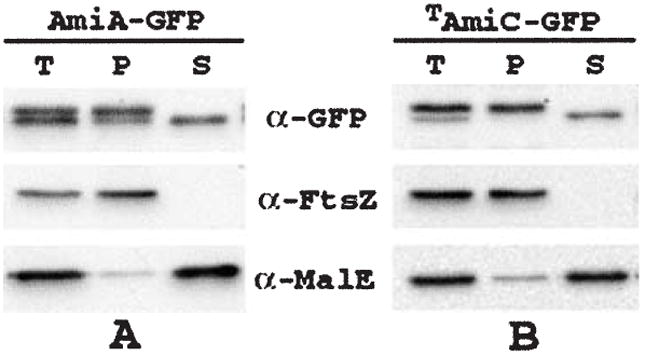
Release of AmiA–GFP and TAmiC–GFP from spheroplasts. Strains TB28/pTB32 [wt/Plac∷amiA–gfp] (A) and TB28/pTB34 [wt/Plac∷TamiC–gfp] (B) were grown in LB supplemented with 0.2% maltose and 250 (A) or 50 (B) μM IPTG. One aliquot of cells was used to prepare a total-cell extract. The remaining cells were converted to spheroplasts and pelleted by centrifugation (see Experimental procedures). The resulting pellet (P) and supernatant (S) fractions, along with the total-cell extract (T), were analysed by SDS-PAGE and immunoblotting for GFP, FtsZ or MalE as indicated. FtsZ and MalE served as markers for the cytoplasm and periplasm respectively. Note that cells grown in rich medium, as used here for efficient spheroplast formation, show a ratio of unprocessed (non-periplasmic) to processed (periplasmic) forms of either fusion that is significantly greater than the ratio observed in cells grown in M9-based medium (see Fig. 3; results not shown).
Strong support for a role for Tat in export of AmiA came from fluorescence microscopy. Consistent with a periplasmic location for AmiA–GFP (Santini et al., 2001; Thomas et al., 2001), wild-type cells expressing the fusion appeared as if surrounded by a fluorescent halo with some accumulation of signal at the cell poles (Figs 5A and 6A and B). All cells in the population showed this distribution, indicating that AmiA remains dispersed in the periplasm throughout the division cycle. This distribution is identical to that seen with Tat-targeted, but otherwise unfused, GFP (Santini et al., 2001; Thomas et al., 2001) (Fig. 8B; see below). The apparent polar bias of the GFP signal indicates that the periplasmic volume surrounding the poles is relatively large. This could either reflect a natural state of affairs or be caused by some plasmolysis during microscopy. In contrast to the peripheral distribution of AmiA–GFP in wild-type cells, the fusion appeared to be dispersed throughout the cytoplasm in ΔtatC cells (Fig. 5B). This discrepancy in fluorescence distribution was not caused by any enhanced non-specific degradation of the fusion in Tat−cells, as judged from the Western analyses (Fig. 3A, compare lanes 1 and 2).
Fig. 5.

Aberrant distribution of AmiA–GFP and AmiC–GFP in a ΔtatC mutant. Fluorescence (A–E) and DIC (A′–E′) micrographs show live cells of strains MC4100/pTB32 [wt/Plac∷amiA–gfp] (A), B1LKO/pTB32 [ΔtatC/Plac∷amiA–gfp] (B), MC4100/pTB28 [wt/Plac∷amiC–gfp] (C and D) and B1LKO/pTB28 [ΔtatC/Plac∷amiC–gfp] (E) grown with 250 μM (A and B) or 50 μM (C–E) IPTG. Note the chaining phenotype of Tat− cells (B and E). We infer that the deeply constricted cell in (D) was about to separate, whereas the one in (C) was still finishing constriction (see text). Bar equals 1 μm.
Fig. 6.

Localization of AmiA–GFP and AmiC–GFP. Fluorescence (A–G) and DIC (A′–G′) micrographs show the distribution of AmiA–GFP (A and B) and AmiC–GFP (C–G) in live cells. Strains TB28/pTB32 [wt/Plac∷amiA–gfp] (A and B), TB36(λTB27) [ΔamiC(Plac∷PamiC∷amiC–gfp)] (C and D) and TB28/pTB28 [wt/Plac∷amiC–gfp] (E and G) were grown with 250 μM (A and B), no (C and D) or 50 μM (E–G) IPTG. The arrow in (G) points to a faint ring-like accumulation of AmiC–GFP in a cell at an early stage of constriction. Bar equals 1 μm.
Fig. 8.
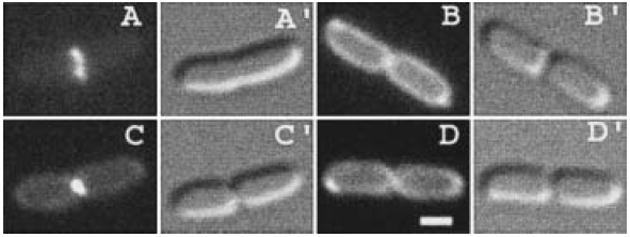
Localization of AmiC–GFP deletion derivatives. Fluorescence (A–D) and DIC (A′–D′) micrographs show live cells of strain TB28[wt] carrying pTB34 [Plac∷TamiC–gfp] (A), pTB37 [Plac∷SSamiC–gfp] (B), pTB39 [Plac∷amiC–gfp] (C) or pTB41 [Plac∷CamiC–gfp] (D) and grown with 10 μM (B–D) or 50 μM (A) IPTG. Bar equals 1 μm.
We conclude that AmiA indeed relies on the Tat system for transport to the periplasm.
Tat-dependent translocation of functional AmiC–GFP to the periplasm
PreAmiC is predicted to consist of three domains. Residues 1–31 are likely to make up the signal peptide, residues 32–249 a domain of unknown function and residues 250–417 the catalytic amidase domain. We prepared constructs encoding fusions of GFP to full-length preAmiC (AmiC–GFP), as well as to deletion derivatives lacking residues 187–417 (TAmiC–GFP), 32–126 (CAmiC–GFP) or 32–417 (SSAmiC–GFP) (Fig. 1A and C).
Plasmids pTB27, pTB28 and pTB39, as well as the lysogenic phages λTB27 and λTB28, encode the same fusion of full-length preAmiC to GFP, and each was able to completely suppress the division defect of TB53 (Figs 1C and 2C). Plasmid pTB27 and λTB27 differ from pTB28 and λTB28, respectively, only in that the former contain 149 bp of chromosomal DNA upstream of the predicted start codon of AmiC, whereas the latter contain only 78 bp. In contrast to cells containing pTB28 or λTB28, those harbouring pTB27 or λTB27 expressed a significant amount of AmiC–GFP (not shown) and readily suppressed the phenotype of TB53 (Figs 1C and 2C), even in the absence of inducer. This probably results from the presence of a native amiC promoter in pTB27 and λTB27, which is absent in the other constructs (Fig. 1). Plasmid pTB39 is identical to pTB28 except that the amiC ribosome binding site was changed to that of T7 gene10, and silent mutations created an NheI restriction site that was used in the construction of pTB37, pTB39 and pTB41 (see Experimental procedures).
As expected, pTB37 [Plac∷SSamiC–gfp], encoding only the AmiC signal peptide fused to GFP, did not correct chaining in TB53. Neither did pTB34 [Plac∷TamiC–gfp], which encodes a fusion to the signal peptide and a portion of the N-terminal domain. Interestingly, however, pTB41 [Plac∷CamiC–gfp] did correct chaining in the presence of IPTG. This ability of pTB41 showed that residues 32–126 are dispensable for amidase activity, which is consistent with the proposal that catalytic activity resides in the C-terminal domain of AmiC (Fig. 1C).
Strains MC4100 and B1LKO [ΔtatC] were used as above to test whether AmiC is also a substrate for Tat. Microscopic examination showed that expression of any of the AmiC fusions in wild-type cells resulted in pronounced peripheral fluorescence patterns (Fig. 5C and D), which are discussed in detail below. In contrast, as is shown for B1LKO/pTB28 [ΔtatC/Plac∷amiC–gfp] in Fig. 5E, expression in Tat− cells resulted in homogeneous cytoplasmic fluorescence. Western analyses on cells expressing AmiC–GFP and TAmiC–GFP showed that the fusions were largely intact in both wild-type and ΔtatC cells, indicating that the differences in fluorescence distributions were not the result of protein degradation (Fig. 3B, lanes 1–4). Moreover, two closely migrating forms were readily detectable in extracts of wild-type cells expressing TAmiC–GFP, whereas in the Tat− cells, only the larger form was present (Fig. 3B, lanes 3 and 4). Spheroplast formation of Tat+ cells caused the efficient release of the smaller form of TAmiC–GFP, whereas the larger, presumably unprocessed, form remained associated with the spheroplasts (Fig. 4B).
We conclude that AmiC, like AmiA, relies on the Tat system for export to the periplasm.
AmiC is a periplasmic septal ring component
In contrast to AmiA–GFP, the periplasmic distribution of AmiC–GFP in Tat+ cells changed strikingly during the cell cycle (Fig. 6C–G). In non-dividing cells, the location of AmiC–GFP appeared to be identical to that of AmiA–GFP (Fig. 6C and E). In dividing cells, however, the protein accumulated in a ring at the site of constriction (Fig. 6D, F and G). This accumulation appeared to coincide with a depletion of fluorescence in the remainder of the cell’s periphery, including the polar regions, suggesting that the protein is recruited from throughout the periplasm to concentrate in the ring. Because of higher signal intensities, AmiC–GFP was most easily visualized in cells expressing the fusion from a plasmid (pTB27, 28 or 39). However, the same localization patterns were readily evident when amiC–gfp was present as a single copy integrated in the chromosome. Figure 6C and D, for example, shows cells of the lysogen TB36(λTB27) [ΔamiC (Plac∷PamiC∷amiC–gfp)] grown under conditions (no IPTG) in which transcription of amiC–gfp is presumably driven by a native amiC promoter (Fig. 1C).
To correlate recruitment of AmiC–GFP to septation sites more quantitatively, we examined 224 TB28/pTB28 [wt/Plac∷amiC–gfp] cells grown in the presence of 50 μM IPTG by differential interference contrast (DIC) and fluorescence microscopy. The majority of cells (60%) showed no sign of constriction, and the remainder were slightly (14%), moderately (8%) or deeply (18%) constricted. The population of cells showing homogeneous peripheral fluorescence (63% of the total) consisted of almost all the unconstricted cells (96% of class) and a minority of the deeply constricted ones (27% of class). We assume that the latter were very close to the point of complete separation. All other cells (37% of the total) showed AmiC–GFP concentrated almost exclusively at the site of septation. These results indicate that AmiC recruitment to the division site virtually coincides with the initiation of cell wall constriction, and that the enzyme redisperses throughout the periplasm at, or just before, cell separation.
AmiC requires FtsN for recruitment to the septal ring
The results above indicated that, compared with other septal ring proteins, AmiC joins the structure relatively late. In the mostly linear dependency pathway for the recruitment of septal ring components, the FtsN protein is the latest recruit known to date (Addinall et al., 1997; Chen and Beckwith, 2001; Buddelmeijer et al., 2002; Hale and de Boer, 2002). To place AmiC within this pathway, we studied the localization of AmiC–GFP in FtsN-depleted filaments. To this end, we constructed strain TB54/pTB28 [araC PBAD∷ftsN/Plac∷amiC–gfp], in which the native ftsN promoter on the chromosome is replaced with the araBAD promoter. Both cell division and septal recruitment of AmiC–GFP proceeded normally when this strain grew in the presence of 0.2% arabinose and 100 μM IPTG (Fig. 7A). In the absence of arabinose, however, cells failed to divide and formed long non-septate filaments. Moreover, ring-like accumulations of AmiC–GFP appeared to be completely absent from these filaments (Fig. 7B). Western analyses showed that this was not caused by possible degradation of the fusion in the periplasm (Fig. 3B, lanes 5 and 6). As expected, GFP fusions to the earlier septal ring recruits ZipA and FtsL accumulated in multiple regularly spaced rings in TB54 filaments (not shown). We conclude that AmiC requires FtsN for its recruitment to the septal ring. Consequently, AmiC replaces FtsN as the last known component to join the ring complex.
Fig. 7.
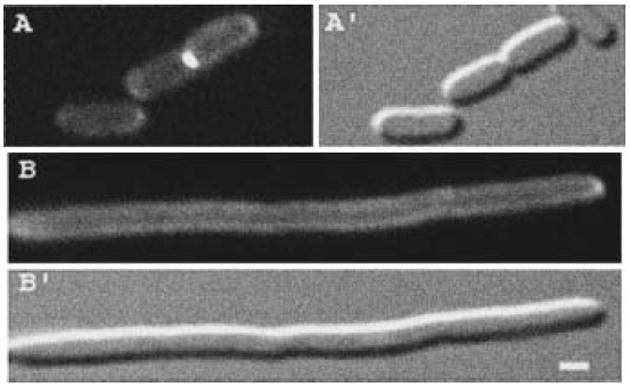
Localization of AmiC–GFP in FtsN-depleted filaments. Fluorescence (A and B) and DIC (A′ and B′) micrographs show live cells of strain TB54/pTB28 [PBAD∷ftsN/Plac∷amiC–gfp] grown with 100 μM IPTG in the presence (A) or absence (B) of 0.2% arabinose. Bar equals 1 μm.
The N-terminal domain of AmiC is a septal targeting domain
The observations that a large portion of the N-terminal domain of AmiC is dispensable for its amidase activity, and that a comparably long N-terminal extension is lacking in AmiA (Fig. 1), suggested that the N-terminus of AmiC might have a role in targeting the enzyme to the septal ring. Direct evidence for such a role came from analyses of TB28/pTB34 [wt/Plac∷TamiC–gfp] cells, which showed that the localization patterns of TAmiC were identical to that of the full-length protein (Fig. 8A and C). In contrast, cells carrying pTB37 [Plac∷SSamiC–gfp] or pTB41 [Plac∷CamiC–gfp] showed homogeneous peripheral fluorescence throughout their division cycles, indicating that neither the signal peptide nor the catalytic domain of AmiC is capable of directing GFP to the site of division (Fig. 8B and D). As judged by Western analyses, the failure of CAmiC–GFP to localize to rings is not due to enhanced degradation of the fusion (Fig. 3C, lanes 1 and 2).
We conclude that TAmiC is both required and sufficient for targeting to the septal ring.
AmiB is not a Tat substrate
In addition to AmiA and AmiC, E. coli encodes one other known periplasmic amidase, AmiB. This enzyme is unlikely to play a significant role in cell separation under normal circumstances because a ΔamiB mutation has no apparent phenotype on its own and also contributes little to the chaining phenotype of multiple amidase deletion mutants (Heidrich et al., 2001). Residues 1–22 of preAmiB are likely to comprise the signal sequence, residues 23–253 a domain of unknown function and residues 254–445 the amidase domain (Fig. 1D). The predicted signal peptide lacks a Tat-targeting motif, suggesting that AmiB is exported by the Sec system (Fig. 1A). Even so, as part of this study, we prepared and studied plasmid pTB33 [Plac∷amiB–gfp], encoding a fusion of GFP to preAmiB. Perhaps not surprisingly, AmiB could not be reliably sublocalized using this fusion. Cells expressing AmiB–GFP showed fluorescence throughout the cell, and Western analyses indicated that virtually all the fusion was degraded to smaller fragments, some of which could still have been fluorescent (not shown). Even though AmiB–GFP was subject to processing, pTB33 was still able to significantly reduce the severity of the TB53 [ΔamiA ΔamiC] chaining phenotype (Figs 1 and 2D), indicating that it directed the production of a significant amount of functional amidase (Heidrich et al., 2001). Interestingly, pTB33 also significantly suppressed the chaining phenotype of strain B1LKO [ΔtatC], whereas pTB32 [Plac∷amiA–gfp] and pTB28 [Plac∷amiC–gfp] completely failed to do so (Fig. 9). This result is consistent with the prediction that, unlike AmiA and AmiC, AmiB does not require the Tat system for export.
Fig. 9.
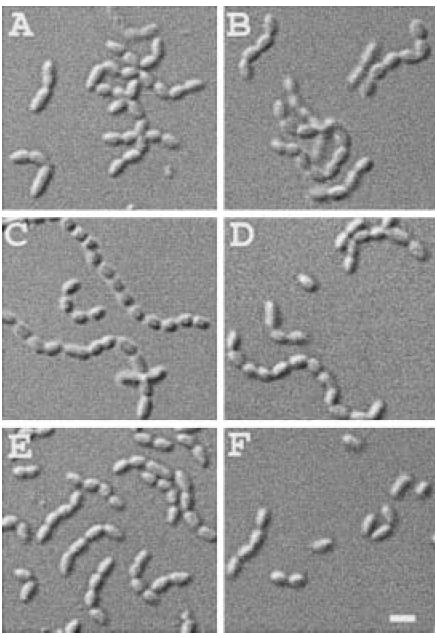
Overexpression of amiB–gfp suppresses the chaining phenotype of Tat− cells. DIC micrographs show representative fields of B1LKO/pTB32 [ΔtatC/Plac∷amiA–gfp] (A and B), B1LKO/pTB28 [ΔtatC/Plac∷amiC–gfp] (C and D) and B1LKO/pTB33 [ΔtatC/Plac∷amiB–gfp] (E and F) cells grown overnight in LB with no (A, C and E), 0.5 mM (F) or 1 mM (B and D) IPTG. Bar equals 2 μm.
Discussion
To investigate whether the periplasmic murein amidases in E. coli are specific components of the septal ring complex, we studied their subcellular localization in live cells using fusions to GFP. This was possible because, as shown here, both AmiA and AmiC are routed to the periplasm via the Tat system. That this might be the case was suggested by the similar cell-chaining phenotype observed for both Ami− and Tat− mutants (Heidrich et al., 2001; 2002; Höltje and Heidrich, 2001; Stanley et al., 2001), and by the presence of Tat-targeting motifs in the predicted signal peptides of preAmiA and preAmiC (Fig. 1A). Accordingly, we showed that GFP fusions to these amidases (i) are functional, fluorescent and concentrated at the cell periphery in wild-type cells; and (ii) fail to mature and remain throughout the cytoplasm in Tat−cells. The failure of Tat− cells to export AmiA and AmiC to the periplasm is very likely to explain their chaining phenotype.
Unlike the predicted signal sequences of preAmiA and preAmiC, that of preAmiB does not contain a recognizable Tat-targeting motif (Fig. 1A). This suggests that AmiB is exported in a Tat-independent fashion, which is supported by the ability of pTB33 [Plac∷amiB–gfp] to suppress the chaining phenotype of Tat− cells. Moreover, the fact that chaining of Tat− cells could be suppressed by pTB33, but not by pTB32 [Plac∷amiA–gfp] or pTB28 [Plac∷amiC–gfp], supports our contention that the chaining phenotype of Tat− cells results primarily from a failure to export AmiA and AmiC. In an accompanying paper, Ize et al. (2003) provide additional independent evidence that also leads to the conclusion that AmiA and AmiC are Tat substrates whereas AmiB is not.
Interestingly, the periplasmic distribution patterns of AmiA–GFP and AmiC–GFP differed significantly. AmiA–GFP appeared to be present throughout the periplasm in all cells. In contrast, AmiC–GFP appeared to be present throughout the periplasm only in non-constricting cells. In cells with a visible constriction, the fusion was found almost exclusively in a ring at the site of constriction, suggesting that AmiC becomes a specific component of the septal ring organelle. Accordingly, AmiC rings failed to form in filaments depleted for FtsN, indicating that recruitment of AmiC requires the prior association of FtsN with the ring, and rendering AmiC the latest known recruit to the division apparatus to date. Compared with other E.coli septal ring components, AmiC is unique in several other respects. Our finding that virtually all cells showing an AmiC ring also show signs of constriction indicates that its recruitment may not occur until after the initiation of constriction. In addition, AmiC is so far the only known septal ring component that is entirely periplasmic, and also the only one that is known to be non-essential, although it is clearly important for the timely completion of division.
Together with murein glycosylases and peptidases, the murein amidases belong to a group of diverse enzymes, collectively referred to as autolysins, that cleave covalent linkages in the cell wall of Gram-negative and Gram-positive organisms. In addition to cell separation, autolysins have been proposed to play important roles in growth, maturation (re)modelling and recycling of the murein layer during vegetative growth (Höltje, 1998; Höltje and Heidrich, 2001), but firm evidence for the involvement of autolysins in these additional processes is still lacking (Heidrich et al., 2002). Many autolysins have a modular structure and possess accessory domains in addition to a catalytic one. In Gram-positive bacteria, a number of these accessory domains are known to be important for cell wall binding, and some of these target their cognate autolysin to polar or septal regions of the wall (Navarre and Schneewind, 1999). These include the choline-binding domains of the Streptococcus pneumoniae LytA amidase and LytB glucosaminidase (Diaz et al., 1989; De Las Rivas et al., 2002), the GW modules of Listeria monocytogenes Ami (Braun et al., 1997) and of Staphylococcus aureus Atl, a bifunctional autolysin with amidase and glucosaminidase domains (Yamada et al., 1996; Baba and Schneewind, 1998), and the CBD domain of the L. monocytogenes bacteriophage A118 endolysin Ply118 (Loessner et al., 2002). The exact targets recognized by most of these cell wall-binding domains are not known, although some component of teichoic or lipoteichoic acids is involved in some cases (Höltje and Tomasz, 1975; Jonquieres et al., 1999).
Here, we show that the N-terminal nonamidase domain of E. coli AmiC (TAmiC) is both necessary and sufficient for targeting the enzyme to the septal ring. As such, TAmiC is the first autolysin-targeting domain identified in Gram-negative bacteria. Database searches indicate that TAmiC is unrelated to any of the known Gram-positive targeting domains, but that TAmiC-like domains are present in (predicted) murein amidases from several other Gram-negative organisms (not shown). The septal target recognized by TAmiC is not yet known. Purified AmiC was found preferentially to destroy ring-like accumulations of septal murein present in sacculi purified from Ami− cell chains (Heidrich et al., 2001). Although it is not known whether this apparent preference for septal murein was dictated by the targeting domain, it is possible that TAmiC directly recognizes some murein structure that is present transiently during septation. Alternatively, TAmiC might bind one or more of the other known septal ring proteins, or other molecules that associate with the organelle and still remain to be identified. Future experiments are required to distinguish between these and other possibilities.
An interesting question is why AmiA and AmiC are exported by the Tat system instead of using the more general Sec pathway, which is presumably used by AmiB. Many Tat substrates contain bulky cofactors such as iron–sulphur clusters that must be assembled in the cytoplasm (Berks et al., 2000). The periplasmic amidases are not predicted to contain such cofactors, but some evidence indicates that they may require a divalent transition metal such as Zn2+ for activity (Shida et al., 2001). It is possible, therefore, that AmiA and AmiC must coordinate a metal ion before export and that this requires at least partial folding of the amidase polypeptide. Another possibility is that AmiA and AmiC simply fold too rapidly to be compatible with Sec-mediated export. Finally, it is possible that the particular mode of export is rather irrelevant to amidase function per se, in which case the use of Tat by AmiA and AmiC may merely be the result of evolutionary happenstance. The use of a different transport mechanism by AmiB would seem to favour this possibility. Further investigation will be required to determine which, if any, of the above possibilities is correct.
This work presents the first sublocalization of periplasmic proteins in live cells. In this regard, it is interesting to note that the AmiC–GFP rings in dividing cells were only very poorly preserved by chemical fixation protocols (Harry et al., 1995; Addinall et al., 1996) that are routinely used to sublocalize cytoplasmic and inner membrane proteins by immunological techniques (data not shown). Whether the location of other periplasmic proteins will be equally difficult to fix remains to be seen. In either case, it can be expected that Tat-targeted GFP fusions will be valuable tools for studying the distribution of other periplasmic proteins involved in cell division and other important cell envelope-associated processes.
Experimental procedures
Plasmids and phages
Plasmids pMLB1113 (de Boer et al., 1989), pCH50 (Hale and de Boer, 1997), pCH148 (Hale et al., 2000), pKD13 and pKD46 (Datsenko and Wanner, 2000) and pBAD18 (Guzman et al., 1995) have been described before.
Plasmids presented in Fig. 1 are derivatives of the medium-copy vector pMLB1113 [bla lacIq Plac∷lacZ] and encode AmiA, AmiB or (portions of) AmiC fused to GFPmut2 (Cormack et al., 1996) via linker peptide LEDPPAEF. Inserts were generated by polymerase chain reaction (PCR). Unless indicated otherwise, MG1655 chromosomal DNA was used as template.
For pTB27 [Plac∷PamiC∷amiC–gfp], pTB28 [Plac∷amiC–gfp], pTB32 [Plac∷amiA–gfp], pTB33 [Plac∷amiB–gfp], pTB34 [Plac∷TamiC–gfp] and pTB37 [Plac∷SSamiC–gfp], the appropriate PCR product was treated with XbaI and XhoI and used to replace the XbaI–XhoI zipA fragment of pCH151 [Plac∷zipA–gfp]. Plasmid pCH151 had been obtained by replacing the 948 bp XbaI–XhoI fragment of pCH148 with the 1026 bp XbaI–XhoI fragment of pCH50. Primers used were (XbaI and XhoI sites underlined): 5′-CCTGTCTAGAGCGACTTTTTTATCAGTAATGCCGTG-3′ and 5′-CCTGCTCGAGTCCCCTTCTCGCCAGCGTCGC-3′ (pTB27); 5′-CCTGTCTAGAGCGTCTTTCGCTAAAGTTTCCGGTC-3′ and 5′-CCTGCTCGAGTCCCCTTCTCGCCAGCGTCGC-3′ (pTB28); 5′-CCTGTCTAGAGGAACTTTATTATTACAACTCAGGC-3′ and 5′-CCTGCTCGAGTCGCTTTTTCGAATGTGCTTTCTGG-3′ (pTB32); 5′-CCTGTCTAGAGGCGCGTTTAGCCGGTTAACCT-3′ and 5′-CCTGCTCGAGGTTTGGCAGCGTGCGATCTGG-3′ (pTB33); 5′-CCTGTCTAGAGCGTCTTTCGCTAAAGTTTCCGGTC-3′ and 5′-CCTGCTCGAGACGATCGCGCCCTGCTTTACCC-3′ (pTB34); and 5′-CCTGTCTAGAAACTCAGGAGATATACCATGTCAGGATCCAACACTGCAATC-3′ and 5′-TCTCCCTCGAGACCAGATCTCGCAgctagcGAGACCTGACTTACGCTCAATAGCC-3′ (pTB37). Plasmid pTB28 was used as template to generate the pTB37 insert. This 143 nt fragment contains the strong T7 gene10 ribosome binding site (RBS; indicated by * in Fig. 1) upstream of the amiC signal sequence (codons 1–32). In addition, codons 29 and 30 contain silent mutations to generate an NheI site (lower case in the primer sequence above). The NheI site was incorporated so that pTB37 could be used to fuse heterologous open reading frames to SSamiC and to generate pTB39 and pTB41.
For pTB39 [Plac∷amiC–gfp] and pTB41 [Plac∷CamiC–gfp], the appropriate PCR product was inserted into pTB37 using NheI and XhoI. Primers used were (NheI and XhoI sites underlined): 5′-GTCTCTGCTAGCTGCGGTCAGCCAGGTCGTGGCG-3′ and 5′-CCTGCTCGAGTCCCCTTCTCGCCAGCGTCGC-3′ (pTB39); and 5′-GTCTCTGCTAGCTGCCCTTGCGCCGGTCGCC-3′ and 5′-CCTGCTCGAGTCCCCTTCTCGCCAGCGTCGC-3′ (pTB41).
Plasmid pTB29 [aph araC PBAD] is a derivative of pKD13 containing the araBAD promoter and araC adjacent to aph (KanR). Both aph and araC are transcribed in the direction opposite to that of PBAD-directed transcription. The majority of pKD13 was amplified with the primers 5′-CCGGGTACCATTCCGGGGATCCGTCGACC-3′ and 5′-CCGGAATTCGGCGATGACGCATCCTCACG-3′, yielding a 3389 nt fragment flanked by SalI (present in the amplified portion of the fragment) and EcoRI (underlined). A 1286 bp fragment flanked by the same sites and containing the araBAD promoter and araC was amplified from pBAD18 with primers 5′-CTCTGTCGACCCCTATGCTACTCCGTCAAGC-3′ and 5′-CGGGTACCGAGCTCGAATTCGC-3′. After digestion with EcoRI and SalI, the fragments were ligated to generate pTB29.
Phages λTB27 and λTB28 were obtained by crossing pTB27 and pTB28, respectively, with λNT5 (de Boer et al., 1989).
Strains
Strains MG1655 [rph1] (Guyer et al., 1981), MC4100 [araD139 Δ(argF-lac)U169 rpsL150 relA1 flbB5301 deoC1 ptsF25 rbsR] (Casadaban, 1976), B1LKO [MC4100 ΔtatC] (Bogsch et al., 1998), DY329 [rph1 IN(rrnD-rrnE) Δ(argF-lac)U169 nadA∷Tn10 gal490 λcI857 Δ(cro-bioA)] (Yu et al., 2000) and MHD52 [ΔamiA∷cat ΔamiB ΔamiC∷aph araD139 Δ(ara-leu)7696 galE15 galK16 Δ(lac)X74 rpsL hsdR2 urA mcrB1] (Heidrich et al., 2001) have been described previously.
Strains TB28 [MG1655 lacIZYA < > frt] and TB54 [DY329 PftsN < > (aph araC PBAD)] were constructed by λ Red-mediated recombineering (Datsenko and Wanner, 2000; Yu et al., 2000). The symbol < > denotes DNA replacement (Yu et al., 2000), and frt a scar sequence remaining after eviction of the aph cassette by FLP recombinase (Datsenko and Wanner, 2000).
For TB28, the aph cassette from pKD13 was amplified with the primers 5′-CACCATCGAATGGCGCAAAACCTTTCGCGGTATGGCATGAATTCCGGGGATCCGTCGACC-3′ and 5′-CGTATCAGGCAATTTTTATAATTTAAACTGACGATTCAACGTGTAGGCTGGAGCTGCTTCG-3′. The resulting 1345 bp fragment was flanked on either side by 40 bp of sequence homologous to the lac locus (underlined in the primer sequences). This fragment was electroporated into MG1655/pKD46, and recombinants, in which the lac locus (from 36 bp upstream of lacI to 15 bp upstream of the lacA stop codon) is replaced by aph, were selected by plating on LB-Kan as described previously (Datsenko and Wanner, 2000). The resulting lacIZYA < > aph allele was transduced to MG1655 with P1, and the aph cassette was evicted as described previously (Datsenko and Wanner, 2000).
For TB54, the (aph araC PBAD) cassette from pTB29 was amplified with the primers 5′-TCCGGGGATCAACACGCGCGTTACCTTAAAGTAAACCTTTGTGTAGGCTGGAGCTGCTTC-3′ and 5′-ACATAATCTCGTTGTGCCACTATCGTTTCGCTGTATTTATATCGCCGAATTCGCTAGCCC-3′. The resulting 2601 bp fragment was flanked on either side by 40 bp of sequence homologous to the region upstream of ftsN (underlined in the primer sequences). This fragment was electroporated into DY329 as described previously (Yu et al., 2000). Recombinants, in which the ftsN promoter region (from 20 to 80 bp upstream of the ftsN start codon) is replaced by the (aph araC PBAD) cassette, were selected on LB-Kan plates containing 0.2% arabinose.
Strains TB36 [TB28 ΔamiC∷aph] and TB53 [TB36 ΔamiA∷cat] were constructed by P1-mediated transduction of ΔamiC∷aph and ΔamiA∷cat from MHD52 to TB28 and TB36 respectively.
Growth conditions and microscopy
Cells were grown at 30°C. Division phenotypes of TB53 and B1LKO were determined on saturated overnight cultures in LB supplemented with various amounts of IPTG. For immunoblotting studies on fractionated cell extracts, cells were grown in LB supplemented with 0.2% maltose and various amounts of IPTG to an OD600 between 0.5 and 0.6. For the sublocalization studies and the immunoblotting studies on whole-cell extracts, cells were grown to an OD600 between 0.4 and 0.7 in M9 minimal medium supplemented with 0.2% maltose, 0.2% casamino acids and 50 μM thiamine. For TB54/pTB28, 40 μM biotin, 50 μM nicotinic acid and arabinose (as indicated) were also included. Where appropriate, antibiotics were used at 10 (Cam), 25 (Kan) or 50 μg ml−1 (Amp).
Microscopy was performed essentially as described previously (Johnson et al., 2002).
Cell fractionation, preparation of extracts and immunoblotting
Before blotting, proteins in whole-cell and fractionated cell extracts were resolved on 10% T, 2.7% C SDS-PAGE gels, except for the upper part of Fig. 4A, for which an 8% T, 2.7% C gel was used.
Whole-cell extracts were prepared as described before (Hale and de Boer, 1999). Extracts were adjusted to be equivalent to a culture of 20 OD600 units, and 7.5 μl was loaded per lane.
To fractionate cells of strains TB28/pTB32 and TB28/pTB34 into periplasmic and non-periplasmic components, they were grown as described above. Each culture was split into two samples (20 ml each), and cells were pelleted by centrifugation at 2000 g for 5 min. The cells in each pellet were resuspended in 250 μl of SP buffer (25% sucrose, 30 mM Tris-HCl, pH 8.0) and supplemented with 250 μl of dH2O. The cells in one of the suspensions remained unfractionated, whereas those in the other were induced to form spheroplasts by the addition of 10 μl each of EDTA (0.25 M) and lysozyme (10 mg ml−1). The conversion to spheroplasts was monitored by microscopy and was judged complete after about 3 min at room temperature. TB28/pTB34 spheroplasts were pelleted immediately by centrifugation at 16 000 g for 20 s. The TB28/pTB32 spheroplast suspension was brought to 250 mM NaCl and incubated on ice for 5 min before centrifugation. This latter step significantly enhanced the release of periplasmic AmiA–GFP (data not shown). The supernatant was reserved as the periplasmic fraction, and the pellet was resuspended in 500 μl of 0.5× SP buffer. An aliquot of 500 μl of 2× sample buffer (Johnson et al., 2002) was added to each sample (unfractionated control, periplasmic fraction and spheroplast pellet). Samples were placed in a boiling water bath for 3 min and were sonicated briefly to reduce viscosity. 10 μl aliquots were loaded per lane.
Immunoblotting was performed essentially as described previously (Johnson et al., 2002), with the following modifications. For the detection of GFP fusions, the primary antibodies were goat anti-GFP (Rockland) diluted 1:10 000, and the secondary antibodies were rabbit anti-goat IgG conjugated with horseradish peroxidase (Rockland) diluted 1:50 000. For the detection of FtsZ and MalE, the primary antibodies were rabbit anti-FtsZ (Hale and de Boer, 1999) and anti-MalE (New England Biolabs), respectively, both of which were used at a dilution of 1:10 000. The secondary antibodies were goat anti-rabbit IgG conjugated with horse-radish peroxidase (Sigma) used at a dilution of 1:50 000. Blots were developed with the Super Signal West Pico system (Pierce).
Acknowledgments
We thank Don Court, Cynthia Hale, Christoph Heidrich, Joachim-Volker Höltje, Tracy Palmer, Phil Rather and Barry Wanner for strains and plasmids, Tracy Palmer for communicating results before publication, and members of our laboratory for support and helpful comments. This work was supported by NIH grant GM57059. Thomas G. Bernhardt is a Damon Runyon Fellow supported by the Damon Runyon Cancer Research Foundation (DRG-1698-02).
References
- Adam M, Fraipont C, Rhazi N, Nguyen-Disteche M, Lakaye B, Frere JM, et al. The bimodular G57–V577 polypeptide chain of the class B penicillin-binding protein 3 of Escherichia coli catalyzes peptide bond formation from thiolesters and does not catalyze glycan chain polymerization from the lipid II intermediate. J Bacteriol. 1997;179:6005–6009. doi: 10.1128/jb.179.19.6005-6009.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addinall SG, Bi E, Lutkenhaus J. FtsZ ring formation in fts mutants. J Bacteriol. 1996;178:3877–3884. doi: 10.1128/jb.178.13.3877-3884.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Addinall SG, Cao C, Lutkenhaus J. FtsN, a late recruit to the septum in Escherichia coli. Mol Microbiol. 1997;25:303–309. doi: 10.1046/j.1365-2958.1997.4641833.x. [DOI] [PubMed] [Google Scholar]
- Baba T, Schneewind O. Targeting of muralytic enzymes to the cell division site of Gram-positive bacteria: repeat domains direct autolysin to the equatorial surface ring of Staphylococcus aureus. EMBO J. 1998;17:4639–4646. doi: 10.1093/emboj/17.16.4639. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bateman A, Birney E, Cerruti L, Durbin R, Etwiller L, Eddy SR, et al. The Pfam protein families database. Nucleic Acids Res. 2002;30:276–280. doi: 10.1093/nar/30.1.276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berks BC, Sargent F, Palmer T. The Tat protein export pathway. Mol Microbiol. 2000;35:260–274. doi: 10.1046/j.1365-2958.2000.01719.x. [DOI] [PubMed] [Google Scholar]
- Bi E, Lutkenhaus J. FtsZ ring structure associated with division in Escherichia coli. Nature. 1991;354:161–164. doi: 10.1038/354161a0. [DOI] [PubMed] [Google Scholar]
- de Boer PAJ, Crossley RE, Rothfield LI. A division inhibitor and a topological specificity factor coded for by the minicell locus determine proper placement of the division septum in E. coli. Cell. 1989;56:641–649. doi: 10.1016/0092-8674(89)90586-2. [DOI] [PubMed] [Google Scholar]
- Bogsch EG, Sargent F, Stanley NR, Berks BC, Robinson C, Palmer T. An essential component of a novel bacterial protein export system with homologues in plastids and mitochondria. J Biol Chem. 1998;273:18003–18006. doi: 10.1074/jbc.273.29.18003. [DOI] [PubMed] [Google Scholar]
- Braun L, Dramsi S, Dehoux P, Bierne H, Lindahl G, Cossart P. InlB: an invasion protein of Listeria monocytogenes with a novel type of surface association. Mol Microbiol. 1997;25:285–294. doi: 10.1046/j.1365-2958.1997.4621825.x. [DOI] [PubMed] [Google Scholar]
- Buddelmeijer N, Judson N, Boyd D, Mekalanos JJ, Beckwith J. YgbQ, a cell division protein in Escheichia coli and Vibrio cholerae, localizes in codependent fashion with FtsL to the division site. Proc Natl Acad Sci USA. 2002;99:6316–6321. doi: 10.1073/pnas.092128499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casadaban MJ. Transposition and fusion of the lac genes to selected promoters in Escherichia coli using bacteriophage lambda and Mu. J Mol Biol. 1976;104:541–555. doi: 10.1016/0022-2836(76)90119-4. [DOI] [PubMed] [Google Scholar]
- Chen JC, Beckwith J. FtsQ, FtsL and FtsI require FtsK, but not FtsN, for co-localization with FtsZ during Escherichia coli cell division. Mol Microbiol. 2001;42:395–413. doi: 10.1046/j.1365-2958.2001.02640.x. [DOI] [PubMed] [Google Scholar]
- Cormack BP, Valdivia RH, Falkow S. FACS-optimized mutants of the green fluorescent protein (GFP) Gene. 1996;173:33–38. doi: 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Datsenko KA, Wanner BL. One-step inactivation of chromosomal genes in Escherichia coli K-12 using PCR products. Proc Natl Acad Sci USA. 2000;97:6640–6645. doi: 10.1073/pnas.120163297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Las Rivas B, Garcia JL, Lopez R, Garcia P. Purification and polar localization of pneumococcal LytB, a putative endo-beta-N-acetylglucosaminidase: the chain-dispersing murein hydrolase. J Bacteriol. 2002;184:4988–5000. doi: 10.1128/JB.184.18.4988-5000.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeLisa MP, Samuelson P, Palmer T, Georgiou G. Genetic analysis of the twin arginine translocator secretion pathway in bacteria. J Biol Chem. 2002;277:29825–29831. doi: 10.1074/jbc.M201956200. [DOI] [PubMed] [Google Scholar]
- Diaz E, Garcia E, Ascaso C, Mendez E, Lopez R, Garcia JL. Subcellular localization of the major pneumococcal autolysin: a peculiar mechanism of secretion in Escherichia coli. J Biol Chem. 1989;264:1238–1244. [PubMed] [Google Scholar]
- Feilmeier BJ, Iseminger G, Schroeder D, Webber H, Phillips GJ. Green fluorescent protein functions as a reporter for protein localization in Escherichia coli. J Bacteriol. 2000;182:4068–4076. doi: 10.1128/jb.182.14.4068-4076.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gueiros-Filho FJ, Losick R. A widely conserved bacterial cell division protein that promotes assembly of the tubulin-like protein FtsZ. Genes Dev. 2002;16:2544–2556. doi: 10.1101/gad.1014102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guyer MS, Reed RR, Steitz JA, Low KB. Identification of a sex-factor-affinity site in E. coli as gamma delta. Cold Spring Harb Symp Quant Biol. 1981;45:135–140. doi: 10.1101/sqb.1981.045.01.022. [DOI] [PubMed] [Google Scholar]
- Guzman LM, Belin D, Carson MJ, Beckwith J. Tight regulation, modulation, and high-level expression by vectors containing the arabinose PBAD promoter. J Bacteriol. 1995;177:4121–4130. doi: 10.1128/jb.177.14.4121-4130.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. Direct binding of FtsZ to ZipA, an essential component of the septal ring structure that mediates cell division in E. coli. Cell. 1997;88:175–185. doi: 10.1016/s0092-8674(00)81838-3. [DOI] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. Recruitment of ZipA to the septal ring of Escherichia coli is dependent on FtsZ, and independent of FtsA. J Bacteriol. 1999;181:167–176. doi: 10.1128/jb.181.1.167-176.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, de Boer PAJ. ZipA is required for recruitment of FtsK, FtsQ, FtsL, and FtsN to the septal ring in Escherichia coli. J Bacteriol. 2002;184:2552–2556. doi: 10.1128/JB.184.9.2552-2556.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hale CA, Rhee AC, de Boer PAJ. ZipA-induced bundling of FtsZ polymers mediated by an interaction between C-terminal domains. J Bacteriol. 2000;182:5153–5166. doi: 10.1128/jb.182.18.5153-5166.2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harry EJ, Pogliano K, Losick R. Use of immunofluorescence to visualize cell-specific gene expression during sporulation in Bacillus subtilis. J Bacteriol. 1995;177:3386–3393. doi: 10.1128/jb.177.12.3386-3393.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heidrich C, Templin MF, Ursinus A, Merdanovic M, Berger J, Schwarz H, et al. Involvement of N-acetylmuramyl-l-alanine amidases in cell separation and antibiotic-induced autolysis of Escherichia coli. Mol Microbiol. 2001;41:167–178. doi: 10.1046/j.1365-2958.2001.02499.x. [DOI] [PubMed] [Google Scholar]
- Heidrich C, Ursinus A, Berger J, Schwarz H, Holtje JV. Effects of multiple deletions of murein hydrolases on viability, septum cleavage, and sensitivity to large toxic molecules in Escherichia coli. J Bacteriol. 2002;184:6093–6099. doi: 10.1128/JB.184.22.6093-6099.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltje JV. Growth of the stress-bearing and shape-maintaining murein sacculus of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:181–203. doi: 10.1128/mmbr.62.1.181-203.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Höltje JV, Heidrich C. Enzymology of elongation and constriction of the murein sacculus of Escherichia coli. Biochimie. 2001;83:103–108. doi: 10.1016/s0300-9084(00)01226-8. [DOI] [PubMed] [Google Scholar]
- Höltje JV, Tomasz A. Specific recognition of choline residues in the cell wall teichoic acid by the N-acetylmuramyl-l-alanine amidase of Pneumococcus. J Biol Chem. 1975;250:6072–6076. [PubMed] [Google Scholar]
- Ishino F, Matsuhashi M. Peptidoglycan synthetic activities of highly purified penicillin-binding protein 3 in Escherichia coli: a septum-forming reaction sequence. Biochem Biophys Res Commun. 1981;101:905–911. doi: 10.1016/0006-291x(81)91835-0. [DOI] [PubMed] [Google Scholar]
- Ize B, Stanley NR, Buchanan G, Palmer T. Role of the Escherichia coli Tat pathway in outer membrane integrity. Mol Microbiol. 2003 doi: 10.1046/j.1365-2958.2003.03504.x. [DOI] [PubMed] [Google Scholar]
- Johnson JE, Lackner LL, de Boer PAJ. Targeting of DMinC/MinD and DMinC/DicB complexes to septal rings in Escherichia coli suggests a multistep mechanism for MinC-mediated destruction of nascent FtsZ-rings. J Bacteriol. 2002;184:2951–2962. doi: 10.1128/JB.184.11.2951-2962.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jonquieres R, Bierne H, Fiedler F, Gounon P, Cossart P. Interaction between the protein InlB of Listeria monocytogenes and lipoteichoic acid: a novel mechanism of protein association at the surface of Gram-positive bacteria. Mol Microbiol. 1999;34:902–914. doi: 10.1046/j.1365-2958.1999.01652.x. [DOI] [PubMed] [Google Scholar]
- Liu Z, Mukherjee A, Lutkenhaus J. Recruitment of ZipA to the division site by interaction with FtsZ. Mol Microbiol. 1999;31:1853–1861. doi: 10.1046/j.1365-2958.1999.01322.x. [DOI] [PubMed] [Google Scholar]
- Loessner MJ, Kramer K, Ebel F, Scherer S. C-terminal domains of Listeria monocytogenes bacteriophage murein hydrolases determine specific recognition and high-affinity binding to bacterial cell wall carbohydrates. Mol Microbiol. 2002;44:335–349. doi: 10.1046/j.1365-2958.2002.02889.x. [DOI] [PubMed] [Google Scholar]
- Lutkenhaus J, Addinall SG. Bacterial cel division and the Z ring. Annu Rev Biochem. 1997;66:93–116. doi: 10.1146/annurev.biochem.66.1.93. [DOI] [PubMed] [Google Scholar]
- Margolin W. Themes and variations in prokaryotic cell division. FEMS Microbiol Rev. 2000;24:531–548. doi: 10.1111/j.1574-6976.2000.tb00554.x. [DOI] [PubMed] [Google Scholar]
- Nanninga N. Morphogenesis of Escherichia coli. Microbiol Mol Biol Rev. 1998;62:110–129. doi: 10.1128/mmbr.62.1.110-129.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navarre WW, Schneewind O. Surface proteins of Gram-positive bacteria and mechanisms of their targeting to the cell wall envelope. Microbiol Mol Biol Rev. 1999;63:174–229. doi: 10.1128/mmbr.63.1.174-229.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nielsen H, Engelbrecht J, Brunak S, von Heijne G. Identification of prokaryotic and eukaryotic signal peptides and prediction of their cleavage sites. Protein Eng. 1997;10:1–6. doi: 10.1093/protein/10.1.1. [DOI] [PubMed] [Google Scholar]
- Pedersen LB, Angert ER, Setlow P. Septal localization of penicillin-binding protein 1 in Bacillus subtilis. J Bacteriol. 1999;181:3201–3211. doi: 10.1128/jb.181.10.3201-3211.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pedro MA, Quintela JC, Höltje J-V, Schwarz H. Murein segregation in Escherichia coli. J Bacteriol. 1997;179:2823–2834. doi: 10.1128/jb.179.9.2823-2834.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Pedro MA, Donachie WD, Holtje JV, Schwarz H. Constitutive septal murein synthesis in Escherichia coli with impaired activity of the morphogenetic proteins RodA and penicillin-binding protein 2. J Bacteriol. 2001;183:4115–4126. doi: 10.1128/JB.183.14.4115-4126.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pichoff S, Lutkenhaus J. Unique and overlapping roles for ZipA and FtsA in septal ring assembly in Escherichia coli. EMBO J. 2002;21:685–693. doi: 10.1093/emboj/21.4.685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothfield L, Justice S, García-Lara J. Bacterial cell division. Annu Rev Genet. 1999;33:423–448. doi: 10.1146/annurev.genet.33.1.423. [DOI] [PubMed] [Google Scholar]
- Santini CL, Bernadac A, Zhang M, Chanal A, Ize B, Blanco C, Wu LF. Translocation of jellyfish green fluorescent protein via the Tat system of Escherichia coli and change of its periplasmic localization in response to osmotic up-shock. J Biol Chem. 2001;276:8159–8164. doi: 10.1074/jbc.C000833200. [DOI] [PubMed] [Google Scholar]
- Shida T, Hattori H, Ise F, Sekiguchi J. Mutational analysis of catalytic sites of the cell wall lytic N-acetylmuramoyl-l-alanine amidases CwlC and CwlV. J Biol Chem. 2001;276:28140–28146. doi: 10.1074/jbc.M103903200. [DOI] [PubMed] [Google Scholar]
- Stanley NR, Findlay K, Berks BC, Palmer T. Escherichia coli strains blocked in Tat-dependent protein export exhibit pleiotropic defects in the cell envelope. J Bacteriol. 2001;183:139–144. doi: 10.1128/JB.183.1.139-144.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas JD, Daniel RA, Errington J, Robinson C. Export of active green fluorescent protein to the periplasm by the twin-arginine translocase (Tat) pathway in Escherichia coli. Mol Microbiol. 2001;39:47–53. doi: 10.1046/j.1365-2958.2001.02253.x. [DOI] [PubMed] [Google Scholar]
- Vollmer W, Höltje JV. Morphogenesis of Escherichia coli. Curr Opin Microbiol. 2001;4:625–633. doi: 10.1016/s1369-5274(01)00261-2. [DOI] [PubMed] [Google Scholar]
- Wang X, Huang J, Mukherjee A, Cao C, Lutkenhaus J. Analysis of the interaction of FtsZ with itself, GTP, and FtsA. J Bacteriol. 1997;179:5551–5559. doi: 10.1128/jb.179.17.5551-5559.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wientjes FB, Nanninga N. Rate and topography of peptidoglycan synthesis during cell division in Escherichia coli: concept of a leading edge. J Bacteriol. 1989;171:3412–3419. doi: 10.1128/jb.171.6.3412-3419.1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yamada S, Sugai M, Komatsuzawa H, Nakashima S, Oshida T, Matsumoto A, Suginaka H. An autolysin ring associated with cell separation of Staphylococcus aureus. J Bacteriol. 1996;178:1565–1571. doi: 10.1128/jb.178.6.1565-1571.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu D, Ellis HM, Lee EC, Jenkins NA, Copeland NG, Court DL. An efficient recombination system for chromosome engineering in Escherichia coli. Proc Natl Acad Sci USA. 2000;97:5978–5983. doi: 10.1073/pnas.100127597. [DOI] [PMC free article] [PubMed] [Google Scholar]