

Abstract
Ketamine is the prototype for a new generation of glutamate-based antidepressants that rapidly alleviate depression within hours of treatment. Over the past decade, there has been replicated evidence demonstrating the rapid and potent antidepressant effects of ketamine in treatment-resistant depression. Moreover, preclinical and biomarker studies have begun to elucidate the mechanism underlying the rapid antidepressant effects of ketamine, offering a new window into the biology of depression and identifying a plethora of potential treatment targets. This article discusses the efficacy, safety, and tolerability of ketamine, summarizes the neurobiology of depression, reviews the mechanisms underlying the rapid antidepressant effects of ketamine, and discusses the prospects for next-generation rapid-acting antidepressants.
Keywords: depression, synaptic plasticity, BDNF, mTOR, biomarker
INTRODUCTION
Major depressive disorder (MDD) is a leading cause of disability throughout the world (1). In the United States, the estimated lifetime prevalence is ~17% (2). More than 20 different antidepressant medications, all of which target monoaminergic systems, are currently available. However, the efficacy of these medications is limited, with a substantial proportion of patients failing to achieve a sustained remission (3). Moreover, the full clinical benefit of these traditional antidepressants is only achieved following weeks to months of treatment (4). Therefore, there is a clear and urgent need for rapid-acting antidepressants with robust efficacy in patients who are refractory to traditional antidepressants.
Ketamine is the prototype for a new generation of antidepressants that rapidly alleviate MDD symptoms and display efficacy in patients who are refractory to currently available treatments. Ketamine is a noncompetitive N-methyl-D-aspartate receptor (NMDA) glutamate receptor antagonist used for induction and maintenance of anesthesia. Approximately 15 years ago, we found that low (subanesthetic) doses of this drug administered intravenously began to reduce depression symptoms within 4 h of administration in severely treatment-resistant depressed patients (5). This finding has since been replicated in multiple controlled studies by several research groups (6, 7). These rapid and potent antidepressant effects were also demonstrated in patient groups known to respond poorly to current antidepressants, such as patients diagnosed with bipolar disorder and patients with depressive symptoms that did not respond to electroconvulsive therapy (8, 9). In this review, we briefly discuss the efficacy, safety, and tolerability of ketamine in depressed patients. We then review the neurobiology of depression and describe the mechanisms believed to underlie the rapid antidepressant effects of ketamine. Recently discovered effects of ketamine on molecular pathways involved in synaptogenesis and on brain circuitry critical to affective regulation are summarized. Clinical biomarkers related to the rapid antidepressant effects of ketamine are presented. We conclude by considering the potential implications of ketamine and other rapid-acting antidepressants for the treatment of mood disorders.
THE RAPID ANTIDEPRESSANT EFFECTS OF KETAMINE
In the late 1980s, we and other groups revisited the psychopharmacology of ketamine to link NMDA receptor dysfunction to schizophrenia symptomatology (10, 11) and enhanced NMDA receptor function to alcoholism (12). By the mid-1990s, we extended this conceptual approach to depression (5). Although we were aware of prior evidence implicating NMDA receptors in the pathophysiology and treatment of depression (13), we were surprised to observe that antidepressant effects emerged so rapidly following the administration of a single ketamine dose and persisted for so long (5). The antidepressant effects tend to emerge 1–2 h after the acute perceptual disturbances of ketamine have abated and can persist for two weeks or longer in some patients even though the plasma redistribution half-life is approximately 4 min and overall terminal plasma half-life is 1–3 h (14).
To date, five placebo-controlled studies have replicated the rapid antidepressant effects of ketamine in MDD and in bipolar depression (5, 7, 8, 15, 16). These studies infused 0.5 mg/kg of ketamine intravenously over 40 min. The antidepressant effects were evident within 4 h of treatment and sustained for 3–7 days, with a response rate of approximately 40–60% at 24 h post treatment. Although ketamine was generally well tolerated, mild to moderate transient adverse effects were observed. In particular, ketamine induced transient perceptual disturbances, dissociation, euphoria, dysphoria, and/or anxiety during infusion. Physical adverse effects included nausea, dizziness, and minimal increases in blood pressure and heart rate. These adverse effects abate within an hour of stopping ketamine infusion and are gone completely within two hours. Numerous open-label and case series have reported comparable efficacy, safety, and tolerability following a single infusion of ketamine (17). A limitation of early placebo-controlled ketamine trials is the functional unblinding of treatment status due to the acute side effects of ketamine. This limitation has been partially mitigated in a recent controlled trial that demonstrated robust rapid antidepressant effects of ketamine compared to midazolam (an anesthetic benzodiazepine) as an active comparator, which optimized blinding to treatment status (6).
Unfortunately, relatively little is known about the long-term safety and efficacy of repeated ketamine dosing, which appears to extend the benefits of single ketamine infusions. Published open-label case reports suggest that repeated ketamine infusions may safely extend the benefits of ketamine for several months (18), and unpublished clinical observations suggest that these benefits may be extended for a year or more (19). The limited pilot data currently available are consistent with this view in suggesting that up to six infusions of low-dose ketamine administered once, twice, or three times per week are efficacious in maintaining and prolonging treatment response (20–24). A larger controlled study of repeated ketamine administration was recently completed (NCT01627782), and the results will be available in the coming months. Other studies reported rapid antidepressant effects following a single administration of various routes and doses of ketamine, including 0.2 mg/kg intravenous bolus (25), 50 mg intranasal (26), and 0.5 mg/kg or 0.25 mg/kg intramuscular injection (27). In depressed patients, ketamine has antisuicidal properties with rapid reduction in suicidal ideation within hours of a single infusion (25, 28–31). However, the durability and generalizability of this observation to nondepressed populations remain to be determined. To date, there are few clinical predictors of the antidepressant effects of ketamine. Increased response to ketamine has been described in patients with a family history of alcoholism (32, 33), a finding that could be related to the enhanced NMDA receptor function in this population (34).
SYNAPTIC HOMEOSTASIS AND THE NEUROBIOLOGY OF DEPRESSION
This review focuses on mechanisms of synaptic plasticity that link the neurobiology of depression to the therapeutic effects of ketamine. The term synaptic plasticity applies to the mechanisms through which neural circuits regulate their excitability and connectivity, particularly in the context of adaptation, e.g., processes of development, learning, coping with stress, and aging (35). These phenomena are accomplished by regulating synaptic strength [e.g., changing the number of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors] and synaptic number (e.g., altering dendritic spine density and shape). Of note, synaptic and extrasynaptic NMDA receptors [see sidebar, N-Methyl-D-Aspartate (NMDA) Receptors] have opposing effects on synaptic plasticity, promoting or reducing synaptic strength, respectively (36). Synaptic plasticity can be “local,” such as long-term potentiation, or “global,” such as homeostatic plasticity (37). The latter is of particular relevance to clinical depression, which is associated with reduced prefrontal synaptic connectivity (see below). A major form of homeostatic plasticity is “synaptic scaling,” which regulates the overall strength of neuronal synaptic connectivity. For example, a prolonged increase in neuronal activities produces a downscaling in overall synaptic strength (38). Among many other mechanisms (37), synaptic scaling is regulated by inflammatory cytokines (e.g., tumor necrosis factor) (39) and by neurotrophins [e.g., brain-derived neurotrophic factor (BDNF)] (40); alteration in both factors has been associated with depression (41, 42).
Over the past two decades, convergent evidence highlighted the role of synaptic homeostasis in the pathophysiology and treatment of depression (43, 44). In particular, prolonged stress and depression have been associated with neuronal atrophy and overall synaptic depression in the prefrontal cortex (PFC) and the hippocampus (45–47), while other brain regions such as the amygdala and nucleus accumbens show changes consistent with neuronal hypertrophy and synaptic potentiation (48, 49). These synaptic changes are believed to result from stress-induced altered glutamate release and astroglial loss leading to neurotrophic factor deficits and to sustained increase in extracellular glutamate. The excess gluatmate precipitates excitotoxicity, altered synaptic strength, reduced dendritic spine density, dendritic retraction, and reduced dendritic branching in the PFC (44, 50) (Figure 1). It has been proposed that downregulation of PFC activity leads to gain of function in other brain regions negatively controlled by the PFC, such as the amygdala, a brain region associated with increased anxiety and hypothalamic-pituitary-adrenal axis reactivity (51).
Figure 1.
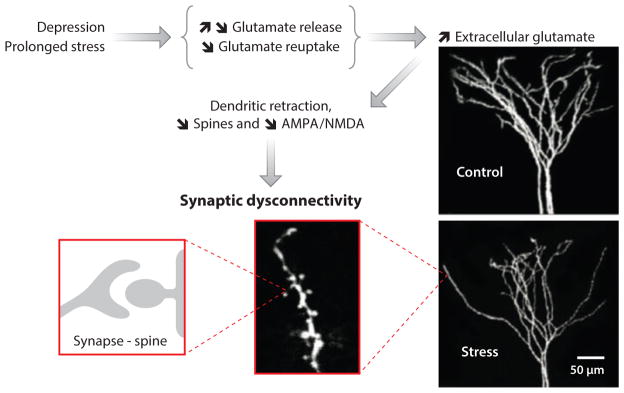
A schematic representation of the proposed neurobiological model of depression. In this model, prolonged stress and depression alter prefrontal glutamate release and reduce glutamate uptake, leading to increased extracellular glutamate and excitotoxicity. High levels of extracellular glutamate precipitate neuronal atrophy through dendritic retraction, reduced dendritic arborization, decreased spine density, and reduced synaptic strength. An example of the effect of prolonged stress on dendritic arborization and length in rats is shown on the right.
N-METHYL-D-ASPARTATE (NMDA) RECEPTORS.
NMDA receptors are glutamate ion channel receptors that allow the passage of Ca2+ and Na+ into the cell and K+ out of the cell. NMDA receptor activation requires coactivation of the glutamate and glycine sites of the receptor, as well as membrane depolarization to remove Mg2+ from the receptor’s channel. Each NMDA receptor is composed of four subunits. To date, seven subunit types have been identified (GluN1, GluN2A, GluN2B, GluN2C, GluN2D, GluN3A, GluN3B). NMDA receptor functions vary according to their subunit composition and subcellular location. They are typically located in the synapse or in the peri- or extrasynaptic space.
Convergent evidence shows opposing effects of synaptic and extrasynaptic NMDA receptors. Synaptic NMDA receptors promote synaptic formation and neuronal survival. In contrast, extrasynaptic NMDA receptor activation promotes synaptic atrophy and neuronal death by altering nuclear calcium. This deregulates target gene expression, leading to mitochondrial dysfunction, reduced dendritic length and arborization, and synaptic loss. Distinct features of synaptic and extrasynaptic NMDA receptors may facilitate the development of extrasynaptic NMDA receptor modulators. For example, extrasynaptic NMDA receptors are enriched with GluN2B subunits and are predominantly coactivated by glycine, whereas synaptic NMDA receptors predominantly contain GluN2A and are coactivated primarily by D-serine.
In this model, prefrontal synaptic deficits and the subsequent neuronal dysconnectivity are critical to the progress and treatment of depression. Molecular studies have started to identify signaling pathways implicated in the observed stress-related synaptic dysfunction. It has been found that synaptic deficits are precipitated by reduction in neurotrophic factors such as BDNF (41) and by inhibition of the mammalian target of rapamycin complex 1 (mTORC1) signaling pathway (52). Inhibition of mTORC1 signaling or reduction of BDNF leads to depressive-like behavior and blocks the effect of antidepressants in animal models of depression (41, 52). Enhancing mTORC1 signaling or increasing BDNF produces antidepressant effects in preclinical studies (41, 52). In humans, reduced central and peripheral BDNF levels were found in depressed patients (41, 53), and a functional variant of BDNF polymorphism (Val66Met) has been related to depression, especially in males (54).
Together these data posit that enhancement of BDNF and mTORC1 signaling leading to prefrontal synaptic formation (synaptogenesis), and reversal of stress- and depression-induced neuronal atrophy and synaptic dysconnectivity, are required for efficacious antidepressant treatment. Traditional antidepressants, targeting monoaminergic systems, were found to increase BDNF and synaptogenesis (41, 44). However, these effects were only evident following chronic treatment, which is in line with the delayed antidepressant response to these drugs in humans. Therefore, it is proposed that rapid-acting antidepressants would need to directly target the induction of mTORC1 signaling, the increase of BDNF levels, and the ultimate enhancement of prefrontal synaptogenesis.
Synaptogenesis and the Rapid Antidepressant Effects of Ketamine
The actions of ketamine are unique in the sense that the antidepressant response emerges after the acute symptoms produced by ketamine abate and after the drug has been metabolized, i.e., the antidepressant effects emerge as a reaction to the acute effects. Recent animal studies have begun to elucidate downstream effects of ketamine that may underlie the beneficial effects in depressed patients. In brief, ketamine’s antagonism of the glutamatergic NMDA receptor is the first step in a cascade of events that includes rapid increases in presynaptic glutamate release, enhanced regional activity in excitatory networks, and ultimately marked changes in synaptic plasticity and connectivity. More specifically, a series of recent studies in rodents has demonstrated that low-dose ketamine administration rapidly triggers three consecutive events: first, a presynaptic disinhibition of glutamatergic neurons, which leads to a glutamate surge; second, an increased activation of the AMPA glutamate receptor, combined with the blockade of extrasynaptic NMDA –receptors; and third, a postsynaptic activation of neuroplasticity-related signaling pathways involving BDNF and mTORC1, which results in overall synaptogenesis and synaptic potentiation (55–57). Among other postsynaptic signaling pathways (43), the ketamine-induced synaptogenesis involves the inhibition of the eukaryotic elongation factor 2 (eEF2) kinase, leading to reduced eEF2 phosphorylation and subsequently increased BDNF translation (55). Together, these acute effects of ketamine treatment rapidly oppose the stress-induced prefrontal neuronal atrophy and synaptic dysconnectivity (58) (Figure 2).
Figure 2.

Prefrontal synaptic connectivity during normal mood, depression, and after remission of depression. In euthymic individuals, stimulus and circuit activities maintain and regulate synaptic strength. Following prolonged stress and depression, an overall synaptic dysconnectivity is observed along with significant reduction in glutamate neurotransmission, excitatory amino acids transporters (EAATs), BDNF expression and release, and mTORC1 signaling. Ketamine’s rapid restoration of prefrontal synaptic connectivity is believed to result from the following consecutive events. (1) Blockade of NMDA receptors located on inhibitory GABAergic interneurons, leading to a stimulus-independent widespread prefrontal glutamate surge. (2) Activation of AMPA receptors combined with blockade of extrasynaptic NMDA receptors. (3) Increased BDNF release and activation of mTORC1 signaling, which in turn increases protein synthesis and AMPA cycling. Abbreviations: ⦿, activate; ∅, block;
 , decrease; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BDNF, brain-derived neurotrophic factor; GABA, γ-aminobutyric acid; mTORC1, mammalian target of rapamycin complex 1.
, decrease; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; BDNF, brain-derived neurotrophic factor; GABA, γ-aminobutyric acid; mTORC1, mammalian target of rapamycin complex 1.
In rodents, microdialysis and electrophysiological studies consistently indicate that low doses of ketamine and other NMDA receptor antagonists induce a “glutamate surge” in the PFC (59–64). This glutamate surge has been confirmed by 13C magnetic resonance spectroscopy (MRS) studies suggesting an increase in glutamate/glutamine cycling, as reflected by the 13C incorporation into glutamate following injection of a subanesthetic dose of ketamine (65). At anesthetic doses of ketamine, there are no increases and may even be decreases in extracellular glutamate and in glutamate cycling (60, 65). Of interest, synaptogenesis and the antidepressant effects of ketamine are also limited to subanesthetic doses (56). Hence, there is a dose-response parallel between the glutamate surge, synaptogenesis, and the antidepressant effects of ketamine.
Further evidence supporting the crucial role of the surge of glutamate neurotransmission in ketamine’s effect comes from a well-replicated finding in rodents demonstrating that AMPA receptor activation is required for the antidepressant effects of ketamine (55, 56, 66, 67). Moreover, we have found that blocking group II metabotropic receptors exerts mTORC1-dependent rapid antidepressant-like effects in a fashion similar to ketamine (68), presumably by precipitating a glutamate surge. Finally, scopolamine, an anticholinergic drug that was recently found to have rapid antidepressant effects in depressed patients (69), also precipitates a prefrontal glutamate surge, increases mTORC1 signaling, and promotes neurogenesis (70). Similar to those of ketamine, these rapid antidepressant effects were mTORC1 and synaptogenesis dependent and were blocked by an AMPA receptor antagonist (70). Taken together, these preclinical data identified novel therpeutic targets for rapid induction of antidepressant-like effects. However, additional studies are needed to investigate the safety and optimal dosing of repeated ketamine administration, as well as other maintenance strategies, to extend the antidepressant effects following single ketamine administration.
CLINICAL BIOMARKERS AND THE RAPID ANTIDEPRESSANT EFFECTS OF KETAMINE
Several biological measures have been utilized in clinical studies to characterize treatment response and to gain insight into neural substrates underlying ketamine’s rapid antidepressant effects. These biomarkers can be generally clustered in three categories: (a) biomarkers of synaptic strength and prefrontal excitability, (b) biomarkers of restoration of neurotrophic function, and (c) biomarkers of ketamine-induced glutamate surge.
Synaptic Strength and Prefrontal Excitability
Magnetoencephalography studies found higher antidepressant responses to ketamine in depressed patients with pretreatment high rostral anterior cingulate activity in response to exposure to fearful faces (71). Conversely, high pregenual anterior cingulate activity during a working memory task predicted poor response to ketamine (72). Interestingly, patients with poor pretreatment connectivity between the pregenual anterior cingulate and the left amygdala showed better response to ketamine treatment (72). Moreover, responders, but not nonresponders, to ketamine treatment showed increased stimulus-evoked somatosensory cortical excitability ~6 h post treatment. This finding was interpreted as evidence of a positive relationship between ketamine-induced synaptic potentiation and response to treatment (73). Additional evidence associating synaptic potentiation with ketamine antidepressant effects comes from an electroencephalography (EEG) study, which examined sleep slow waves as putative markers of synaptic plasticity. The study provided evidence of increased synaptic strength the night after ketamine treatment of depressed patients (74). Additionally, synaptic plasticity changes were related to BDNF and treatment response (see below). Using proton magnetic resonance spectroscopy (1H-MRS), the same research group found an enhanced antidepressant effect of ketamine in depressed patients with low pretreatment frontal glx/glutamate ratio, a presumable surrogate of glutamine (75). However, there was no correlation between pretreatment frontal glutamate and treatment response (75). Similarly, 1H-MRS acquired in depressed patients at baseline, 3 h, and 48 h after ketamine infusion found no relationship between ketamine’s effect and occipital glutamate, glutamine, and GABA (15).
Restoration of Neurotrophic Function
Given the known role of BDNF in the pathophysiology and treatment of depression, several studies have investigated the relationship between ketamine effects and peripheral BDNF levels or a polymorphism of the BDNF gene. An early pilot study found no relationship between serum BDNF and the effects of ketamine in patients with MDD (76). Another small pilot study in bipolar depression found no differences between pretreatment BDNF levels in responders and nonresponders to ketamine. However, following treatment, serum levels of BDNF were significantly reduced in nonresponders (77). A more recent, relatively large trial did find a positive relationship between peripheral BDNF levels and the antidepressant effect of ketamine in depressed patients. Four hours post ketamine infusion, plasma BDNF increased in responders compared to nonresponders (78). Using EEG sleep slow waves as a marker of plasticity, another clinical study found a positive relationship between enhanced synaptic strength and increased plasma BDNF 4 h post ketamine administration. This correlation was found in patients who responded to ketamine treatment but not in nonresponders (74). In addition, a significant increase in plasma BDNF was observed in the total group (74). A recent genetic study examined the relationship between a functional variant of BDNF (Val66Met; rs6265) and response to ketamine treatment. The Met allele blocks the processing and activity-dependent release of BDNF and is therefore a loss-of-function allele. The study reported enhanced antidepressant effect in patients with the Val/Val BDNF variant compared to Met carriers. Together, these findings corroborate preclinical evidence implicating BDNF in the rapid antidepressant effects of ketamine (55, 57). Other studies investigated plasma ketamine and its active metabolite norketamine; however, no associations with treatment response were detected (33, 79, 80). Pretreatment homocysteine, folic acid, and vitamin B12 were examined in patients with bipolar depression, but only B12 was associated with treatment response. Patients with high levels of B12 responded better to ketamine (81).
Ketamine-induced Glutamate Surge
The above data provided clinical evidence for a direct relationship between prefrontal activities, synaptic plasticity, BNDF, and the rapid antidepressant effects of ketamine. However, to our knowledge, there are no clinical data directly relating the ketamine-induced glutamate surge to the antidepressant effects of the drug. In humans, the demonstration of a glutamate surge has been largely examined in healthy volunteers and was driven by interest in the relationship between ketamine-induced glutamate transmission changes and the acute perceptual side effects of ketamine, which mimic core symptoms of schizophrenia. The presence of a glutamate surge and its association with the psychotomimetic effects of ketamine in humans have been suggested by studies showing that glutamate release inhibitors, such as lamotrigine or group II metabotropic agonists, attenuate these perceptual effects (82–85). Interestingly, some depression studies, but not all, have found a positive correlation between the acute perceptual adverse effects and the antidepressant effects of ketamine post treatment (80, 86). Thanks to the strong relationship between glutamate cycling and energy consumption (87), positron emission tomography (PET) studies provide indirect evidence of altered glutamate neurotransmission showing increased neural metabolism, particularly in the PFC, following the administration of low-dose ketamine (88–90). 1H-MRS studies in healthy subjects reported a transient increase in glutamate or glutamine levels in the PFC (91, 92), although not a consistent one (93). This inconsistency could be due to the fact that 1H-MRS measures the total (intra- and extracellular) glutamate level, which may reflect processes other than glutamatergic neurotransmission. In addition, at low- and mid-field strengths (1.5–4.7 Tesla), the 1H spectroscopy signal from glutamate is difficult to distinguish from glutamine (94). Future studies in depressed patients using higher-magnetic-field strength (e.g., 7 Tesla) 1H-MRS and/or using 13C MRS to measure glutamate/glutamine cycling would be required to determine the relationship between acute glutamate transmission changes and the antidepressant effects of ketamine.
CONCLUSION AND FUTURE DIRECTIONS
Five decades of antidepressant research focused on the monoaminergic system and wrestled with the fact that monoaminergic antidepressants require weeks to months to produce their full therapeutic effects. In addition, these antidepressants were effective in only a fraction of depressed patients. The well-replicated finding of rapid antidepressant effects following a single infusion of the glutamate NMDA receptor antagonist ketamine demonstrated that rapid (within hours) antidepressant effects are possible and that targeting the glutamatergic system may offer a truly novel class of antidepressants. To date, a wealth of controlled and open-label studies have demonstrated the efficacy and tolerability of a single infusion of ketamine in rapidly treating severely refractory depressed patients. However, considering the abuse liability of ketamine and the known toxicity following daily intake of high doses (95), ketamine administration remains a research procedure with potential risks (see Future Issues, below).
Although the large majority of research to date has focused on ketamine’s antidepressant effects, emerging evidence also highlights the utility of the drug in rapidly reducing suicidality (28) and in alleviating post-traumatic stress disorder symptoms (96). Ketamine is also being studied as a treatment for obsessive-compulsive disorder and cocaine dependence (97–99). An intriguing subject for future research is the investigation of the procognitive correlates of the ketamine-induced enhanced prefrontal plasticity demonstrated in rodents and supported by clinical studies (56, 73, 74). Ketamine research offers a new window into the biology of depression and provides new therapeutic targets to achieve rapid antidepressant effects. The research outlined in this review may lead to new valuable treatments that are safe, rapid in relieving depression and suicidality, and effective for those who are suffering treatment-refractory depression.
SUMMARY POINTS.
An emerging body of well-replicated evidence has demonstrated the rapid antidepressant effects of ketamine in treatment-refractory patients.
Although a single infusion of ketamine appears to be safe, the long-term safety of repeated ketamine dosing is not fully known.
Prolonged stress and depression are associated with neuronal atrophy and overall synaptic depression in the PFC.
Enhancing BDNF and mTORC1 signaling leads to prefrontal synaptic formation and reversal of stress- and depression-induced neuronal atrophy and synaptic dysconnectivity. This appears to be a required step for efficacious antidepressant treatment.
The rapid antidepressant effects of ketamine are triggered by three consecutive –events: first, a presynaptic disinhibition of glutamatergic neurons leading to a glutamate –surge; second, an increased activation of AMPA receptors, combined with blockade of extrasynaptic NMDA –receptors; and third, a postsynaptic activation of neuroplasticity-related signaling pathways involving BDNF and mTORC1, resulting in restoration of prefrontal synaptic connectivity.
As a prototype for rapid-acting antidepressants, ketamine has provided an exciting new direction that may offer hope of rapid therapeutics for patients who are suffering from depression.
FUTURE ISSUES.
What are the optimal dose and preferable route of administration of ketamine?
At what frequency and dose does repeated ketamine administration stop being beneficial and become harmful?
What is the best strategy to maintain treatment response following ketamine infusion?
Will ketamine-induced synaptic plasticity lead to enhanced cognitive functions following a single infusion?
Will the anti-suicidal properties of ketamine be of clinical value in the emergency setting?
Will the ketamine-induced rapid antidepressant effects and enhanced synaptic plasticity facilitate and augment response to cognitive behavioral therapy?
Acknowledgments
The authors gratefully acknowledge the State of Connecticut Department of Mental Health and Addiction Services for its support of the Abraham Ribicoff Research Facilities of the Connecticut Mental Health Center; the Department of Veterans Affairs, via its funding of the VA National Center for PTSD; the National Institute of Mental Health (MH17871, MH093897, MH101498, MH081211); and the National Center for Advancing Translational Science (CTSA Grant Number UL1 RR024139).
Glossary
- MDD
major depressive disorder
- NMDA
N-methyl-D-aspartate
- AMPA
α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid
- Dendritic spines
small dendritic protrusions on which the majority of synapses are formed
- Neurotrophic factor
a protein that regulates the growth, differentiation, and survival of neurons
- BDNF
brain-derived neurotrophic factor
- mTORC1
mammalian target of rapamycin complex 1
- Group II metabotropic receptor (mGluR2/3)
a presynaptic receptor known to inhibit glutamate release
- glx
the combined unresolved signal of glutamate and glutamine
- Lamotrigine
an anticonvulsant drug known to inhibit glutamate release
Footnotes
DISCLOSURE STATEMENT
Dr. Abdallah has received research funding or consultation fees from the Brain and Behavior Research Foundation (NARSAD), the American Psychiatric Foundation, and Genentech.
Dr. Krystal has served as a scientific consultant to the following companies (the Individual Consultant Agreements listed are <$10,000 per year): Aisling Capital, Astellas Pharma Global Development, AstraZeneca Pharmaceuticals, Biocortech, Brintnall & Nicolini, Easton Associates, Gilead Sciences, GlaxoSmithKline, Janssen Pharmaceuticals, Lundbeck Research USA, Medivation, Merz Pharmaceuticals, MK Medical Communications, Hoffmann–La Roche, SK Holdings, Sunovion Pharmaceuticals, Takeda Industries, and Teva Pharmaceutical Industries. He is on the Scientific Advisory Board for the following companies: Abbott Laboratories, Bristol-Myers-Squibb, Eisai, Eli Lilly, Forest Laboratories, Lohocla Research Corporation, Mnemosyne Pharmaceuticals, Naurex, Pfizer Pharmaceuticals, and Shire Pharmaceuticals. He holds <$150 in exercisable warrant options with Tetragenex Pharmaceuticals. He is on the Board of Directors of the Coalition for Translational Research in Alcohol and Substance Use Disorders. He was the principal investigator of a multicenter study in which Janssen Research Foundation provided drug and some support to the Department of Veterans Affairs. He is Editor of Biological Psychiatry. He has a patent on dopamine and noradrenergic reuptake inhibitors in treatment of schizophrenia (patent number 5447948) and is a coinventor with Dr. Gerard Sanacora on a filed patent application by Yale University related to targeting the glutamatergic system for the treatment of neuropsychiatric disorders (PCTWO06108055A1). He has a patent pending on intranasal administration of ketamine to treat depression.
Dr. Sanacora has received consulting fees from AstraZeneca, Avanier Pharmaceuticals, Bristol-Myers-Squibb, Eli Lilly, Hoffman–La Roche, Naurex, Noven Pharmaceuticals, and Takeda Industries over the last 24 months. He has also received additional research contracts from AstraZeneca, Bristol-Myers-Squibb, Eli Lilly, Johnson & Johnson, Hoffman–La Roche, Merck, Naurex, and Servier over the last 24 months. Free medication was provided to Dr. Sanacora for an NIH-sponsored study by Sanofi-Aventis. He holds shares in BioHaven Pharmaceuticals Holding Company and is a coinventor on a filed patent application by Yale University (PCTWO06108055A1).
Dr. Duman has received consulting fees or investigator-initiated research support from Taisho, Eli Lilly, Lundbeck Research USA, Forest, Sunovion, Johnson & Johnson, and Naurex.
Contributor Information
Chadi G. Abdallah, Email: chadi.abdallah@yale.edu.
Gerard Sanacora, Email: gerard.sanacora@yale.edu.
Ronald S. Duman, Email: ronald.duman@yale.edu.
John H. Krystal, Email: john.krystal@yale.edu.
LITERATURE CITED
- 1.Collins PY, Patel V, Joestl SS, et al. Grand challenges in global mental health. Nature. 2011;475:27–30. doi: 10.1038/475027a. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kessler RC, Berglund P, Demler O, et al. Lifetime prevalence and age-of-onset distributions of DSM-IV disorders in the National Comorbidity Survey Replication. Arch Gen Psychiatry. 2005;62:593–602. doi: 10.1001/archpsyc.62.6.593. [DOI] [PubMed] [Google Scholar]
- 3.Rush AJ, Trivedi MH, Wisniewski SR, et al. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: a STAR*D report. Am J Psychiatry. 2006;163:1905–17. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- 4.Katz MM, Tekell JL, Bowden CL, et al. Onset and early behavioral effects of pharmacologically different antidepressants and placebo in depression. Neuropsychopharmacology. 2004;29:566–79. doi: 10.1038/sj.npp.1300341. [DOI] [PubMed] [Google Scholar]
- 5.Berman RM, Cappiello A, Anand A, et al. Antidepressant effects of ketamine in depressed patients. Biol Psychiatry. 2000;47:351–54. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- 6.Murrough JW, Iosifescu DV, Chang LC, et al. Antidepressant efficacy of ketamine in treatment-resistant major depression: a two-site randomized controlled trial. Am J Psychiatry. 2013;170:1134–42. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zarate CA, Jr, Singh JB, Carlson PJ, et al. A randomized trial of an N-methyl-D-aspartate antagonist in treatment-resistant major depression. Arch Gen Psychiatry. 2006;63:856–64. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- 8.Diazgranados N, Ibrahim L, Brutsche NE, et al. A randomized add-on trial of an N-methyl-D-aspartate antagonist in treatment-resistant bipolar depression. Arch Gen Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Ibrahim L, Diazgranados N, Luckenbaugh DA, et al. Rapid decrease in depressive symptoms with an N-methyl-D-aspartate antagonist in ECT-resistant major depression. Prog Neuropsychopharmacol Biol Psychiatry. 2011;35:1155–59. doi: 10.1016/j.pnpbp.2011.03.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Krystal JH, Karper LP, Seibyl JP, et al. Subanesthetic effects of the noncompetitive NMDA antagonist, ketamine, in humans. Psychotomimetic, perceptual, cognitive, and neuroendocrine responses. Arch Gen Psychiatry. 1994;51:199–214. doi: 10.1001/archpsyc.1994.03950030035004. [DOI] [PubMed] [Google Scholar]
- 11.Domino EF, Chodoff P, Corssen G. Pharmacologic effects of Ci-581, a new dissociative anesthetic, in man. Clin Pharmacol Ther. 1965;6:279–91. doi: 10.1002/cpt196563279. [DOI] [PubMed] [Google Scholar]
- 12.Krystal JH, Petrakis IL, Limoncelli D, et al. Altered NMDA glutamate receptor antagonist response in recovering ethanol-dependent patients. Neuropsychopharmacology. 2003;28:2020–28. doi: 10.1038/sj.npp.1300252. [DOI] [PubMed] [Google Scholar]
- 13.Skolnick P, Layer RT, Popik P, et al. Adaptation of N-methyl-D-aspartate (NMDA) receptors following antidepressant treatment: implications for the pharmacotherapy of depression. Pharmacopsychiatry. 1996;29:23–26. doi: 10.1055/s-2007-979537. [DOI] [PubMed] [Google Scholar]
- 14.Mion G, Villevieille T. Ketamine pharmacology: an update (pharmacodynamics and molecular aspects, recent findings) CNS Neurosci Ther. 2013;19:370–80. doi: 10.1111/cns.12099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Valentine GW, Mason GF, Gomez R, et al. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [1H]-MRS. Psychiatry Res. 2011;191:122–27. doi: 10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zarate CA, Jr, Brutsche NE, Ibrahim L, et al. Replication of ketamine’s antidepressant efficacy in bipolar depression: a randomized controlled add-on trial. Biol Psychiatry. 2012;71:939–46. doi: 10.1016/j.biopsych.2011.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Aan Het Rot M, Zarate CA, Jr, Charney DS, Mathew SJ. Ketamine for depression: Where do we go from here? Biol Psychiatry. 2012;72:537–47. doi: 10.1016/j.biopsych.2012.05.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Blier P, Zigman D, Blier J. On the safety and benefits of repeated intravenous injections of ketamine for depression. Biol Psychiatry. 2012;72:e11–12. doi: 10.1016/j.biopsych.2012.02.039. [DOI] [PubMed] [Google Scholar]
- 19.Szymkowicz SM, Finnegan N, Dale RM. A 12-month naturalistic observation of three patients receiving repeat intravenous ketamine infusions for their treatment-resistant depression. J Affect Disord. 2013;147:416–20. doi: 10.1016/j.jad.2012.10.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Murrough JW, Perez AM, Pillemer S, et al. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biol Psychiatry. 2013;74:250–56. doi: 10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Diamond PR, Farmery AD, Atkinson S, et al. Ketamine infusions for treatment resistant depression: a series of 28 patients treated weekly or twice weekly in an ECT clinic. J Psychopharmacol. 2014;28:536–44. doi: 10.1177/0269881114527361. [DOI] [PubMed] [Google Scholar]
- 22.Rasmussen KG, Lineberry TW, Galardy CW, et al. Serial infusions of low-dose ketamine for major depression. J Psychopharmacol. 2013;27:444–50. doi: 10.1177/0269881113478283. [DOI] [PubMed] [Google Scholar]
- 23.Shiroma PR, Johns B, Kuskowski M, et al. Augmentation of response and remission to serial intravenous subanesthetic ketamine in treatment resistant depression. J Affect Disord. 2014;155:123–29. doi: 10.1016/j.jad.2013.10.036. [DOI] [PubMed] [Google Scholar]
- 24.Segmiller F, Ruther T, Linhardt A, et al. Repeated S-ketamine infusions in therapy resistant depression: a case series. J Clin Pharmacol. 2013;53:996–98. doi: 10.1002/jcph.122. [DOI] [PubMed] [Google Scholar]
- 25.Larkin GL, Beautrais AL. A preliminary naturalistic study of low-dose ketamine for depression and suicide ideation in the emergency department. Int J Neuropsychopharmacol. 2011;14:1127–31. doi: 10.1017/S1461145711000629. [DOI] [PubMed] [Google Scholar]
- 26.Lapidus KA, Levitch CF, Perez AM, et al. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biol Psychiatry. 2014 doi: 10.1016/j.biopsych.2014.03.026. In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Chilukuri H, Reddy NP, Pathapati RM, et al. Acute antidepressant effects of intramuscular versus intravenous ketamine. Indian J Psychol Med. 2014;36:71–76. doi: 10.4103/0253-7176.127258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Price RB, Iosifescu DV, Murrough JW, et al. Effects of ketamine on explicit and implicit suicidal cognition: a randomized controlled trial in treatment-resistant depression. Depress Anxiety. 2014;31:335–43. doi: 10.1002/da.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biol Psychiatry. 2009;66:522–26. doi: 10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.DiazGranados N, Ibrahim LA, Brutsche NE, et al. Rapid resolution of suicidal ideation after a single infusion of an N-methyl-D-aspartate antagonist in patients with treatment-resistant major depressive disorder. J Clin Psychiatry. 2010;71:1605–11. doi: 10.4088/JCP.09m05327blu. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Thakurta RG, Das R, Bhattacharya AK, et al. Rapid response with ketamine on suicidal cognition in resistant depression. Indian J Psychol Med. 2012;34:170–75. doi: 10.4103/0253-7176.101793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Luckenbaugh DA, Ibrahim L, Brutsche N, et al. Family history of alcohol dependence and antidepressant response to an N-methyl-D-aspartate antagonist in bipolar depression. Bipolar Disord. 2012;14:880–87. doi: 10.1111/bdi.12003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Phelps LE, Brutsche N, Moral JR, et al. Family history of alcohol dependence and initial antidepressant response to an N-methyl-D-aspartate antagonist. Biol Psychiatry. 2009;65:181–84. doi: 10.1016/j.biopsych.2008.09.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Petrakis IL, Limoncelli D, Gueorguieva R, et al. Altered NMDA glutamate receptor antagonist response in individuals with a family vulnerability to alcoholism. Am J Psychiatry. 2004;161:1776–82. doi: 10.1176/ajp.161.10.1776. [DOI] [PubMed] [Google Scholar]
- 35.Citri A, Malenka RC. Synaptic plasticity: multiple forms, functions, and mechanisms. Neuropsychopharmacology. 2008;33:18–41. doi: 10.1038/sj.npp.1301559. [DOI] [PubMed] [Google Scholar]
- 36.Hardingham GE, Bading H. Synaptic versus extrasynaptic NMDA receptor signalling: implications for neurodegenerative disorders. Nat Rev Neurosci. 2010;11:682–96. doi: 10.1038/nrn2911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Turrigiano G. Homeostatic synaptic plasticity: local and global mechanisms for stabilizing neuronal function. Cold Spring Harb Perspect Biol. 2012;4:a005736. doi: 10.1101/cshperspect.a005736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Turrigiano G. Too many cooks? Intrinsic and synaptic homeostatic mechanisms in cortical circuit refinement. Annu Rev Neurosci. 2011;34:89–103. doi: 10.1146/annurev-neuro-060909-153238. [DOI] [PubMed] [Google Scholar]
- 39.Beattie EC, Stellwagen D, Morishita W, et al. Control of synaptic strength by glial TNFalpha. Science. 2002;295:2282–85. doi: 10.1126/science.1067859. [DOI] [PubMed] [Google Scholar]
- 40.Rutherford LC, Nelson SB, Turrigiano GG. BDNF has opposite effects on the quantal amplitude of pyramidal neuron and interneuron excitatory synapses. Neuron. 1998;21:521–30. doi: 10.1016/s0896-6273(00)80563-2. [DOI] [PubMed] [Google Scholar]
- 41.Duman RS, Monteggia LM. A neurotrophic model for stress-related mood disorders. Biol Psychiatry. 2006;59:1116–27. doi: 10.1016/j.biopsych.2006.02.013. [DOI] [PubMed] [Google Scholar]
- 42.Dantzer R, O’Connor JC, Freund GG, et al. From inflammation to sickness and depression: when the immune system subjugates the brain. Nat Rev Neurosci. 2008;9:46–56. doi: 10.1038/nrn2297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Duman RS, Aghajanian GK. Synaptic dysfunction in depression: potential therapeutic targets. Science. 2012;338:68–72. doi: 10.1126/science.1222939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Popoli M, Yan Z, McEwen BS, Sanacora G. The stressed synapse: the impact of stress and gluco-corticoids on glutamate transmission. Nat Rev Neurosci. 2012;13:22–37. doi: 10.1038/nrn3138. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kang HJ, Voleti B, Hajszan T, et al. Decreased expression of synapse-related genes and loss of synapses in major depressive disorder. Nat Med. 2012;18:1413–17. doi: 10.1038/nm.2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Bessa JM, Ferreira D, Melo I, et al. The mood-improving actions of antidepressants do not depend on neurogenesis but are associated with neuronal remodeling. Mol Psychiatry. 2009;14:764–73. doi: 10.1038/mp.2008.119. [DOI] [PubMed] [Google Scholar]
- 47.Yuen EY, Wei J, Liu W, et al. Repeated stress causes cognitive impairment by suppressing glutamate receptor expression and function in prefrontal cortex. Neuron. 2012;73:962–77. doi: 10.1016/j.neuron.2011.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bessa JM, Morais M, Marques F, et al. Stress-induced anhedonia is associated with hypertrophy of medium spiny neurons of the nucleus accumbens. Transl Psychiatry. 2013;3:e266. doi: 10.1038/tp.2013.39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Vyas A, Pillai AG, Chattarji S. Recovery after chronic stress fails to reverse amygdaloid neuronal hypertrophy and enhanced anxiety-like behavior. Neuroscience. 2004;128:667–73. doi: 10.1016/j.neuroscience.2004.07.013. [DOI] [PubMed] [Google Scholar]
- 50.Krystal JH, Sanacora G, Duman RS. Rapid-acting glutamatergic antidepressants: the path to ketamine and beyond. Biol Psychiatry. 2013;73:1133–41. doi: 10.1016/j.biopsych.2013.03.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Savitz J, Drevets WC. Bipolar and major depressive disorder: neuroimaging the developmental-degenerative divide. Neurosci Biobehav Rev. 2009;33:699–771. doi: 10.1016/j.neubiorev.2009.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Ota KT, Liu RJ, Voleti B, et al. REDD1 is essential for stress-induced synaptic loss and depressive behavior. Nat Med. 2014;20:531–35. doi: 10.1038/nm.3513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sen S, Duman R, Sanacora G. Serum brain-derived neurotrophic factor, depression, and antidepressant medications: meta-analyses and implications. Biol Psychiatry. 2008;64:527–32. doi: 10.1016/j.biopsych.2008.05.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Verhagen M, van der Meij A, van Deurzen PA, et al. Meta-analysis of the BDNF Val66Met polymorphism in major depressive disorder: effects of gender and ethnicity. Mol Psychiatry. 2010;15:260–71. doi: 10.1038/mp.2008.109. [DOI] [PubMed] [Google Scholar]
- 55.Autry AE, Adachi M, Nosyreva E, et al. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Li N, Lee B, Liu RJ, et al. mTOR-dependent synapse formation underlies the rapid antidepressant effects of NMDA antagonists. Science. 2010;329:959–64. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Liu RJ, Lee FS, Li XY, et al. Brain-derived neurotrophic factor Val66Met allele impairs basal and ketamine-stimulated synaptogenesis in prefrontal cortex. Biol Psychiatry. 2012;71:996–1005. doi: 10.1016/j.biopsych.2011.09.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Li N, Liu RJ, Dwyer JM, et al. Glutamate N-methyl-D-aspartate receptor antagonists rapidly reverse behavioral and synaptic deficits caused by chronic stress exposure. Biol Psychiatry. 2011;69:754–61. doi: 10.1016/j.biopsych.2010.12.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Abekawa T, Honda M, Ito K, Koyama T. Effects of NRA0045, a novel potent antagonist at dopamine D4, 5-HT2A, and alpha1 adrenaline receptors, and NRA0160, a selective D4 receptor antagonist, on phencyclidine-induced behavior and glutamate release in rats. Psychopharmacology. 2003;169:247–56. doi: 10.1007/s00213-003-1517-8. [DOI] [PubMed] [Google Scholar]
- 60.Moghaddam B, Adams B, Verma A, Daly D. Activation of glutamatergic neurotransmission by ketamine: a novel step in the pathway from NMDA receptor blockade to dopaminergic and cognitive disruptions associated with the prefrontal cortex. J Neurosci. 1997;17:2921–27. doi: 10.1523/JNEUROSCI.17-08-02921.1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Moghaddam B, Adams BW. Reversal of phencyclidine effects by a group II metabotropic glutamate receptor agonist in rats. Science. 1998;281:1349–52. doi: 10.1126/science.281.5381.1349. [DOI] [PubMed] [Google Scholar]
- 62.Lorrain DS, Baccei CS, Bristow LJ, et al. Effects of ketamine and N-methyl-D-aspartate on glutamate and dopamine release in the rat prefrontal cortex: modulation by a group II selective metabotropic glutamate receptor agonist LY379268. Neuroscience. 2003;117:697–706. doi: 10.1016/s0306-4522(02)00652-8. [DOI] [PubMed] [Google Scholar]
- 63.Bonaventure P, Aluisio L, Shoblock J, et al. Pharmacological blockade of serotonin 5-HT(7) receptor reverses working memory deficits in rats by normalizing cortical glutamate neurotransmission. PLOS ONE. 2011;6:e20210. doi: 10.1371/journal.pone.0020210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Lopez-Gil X, Babot Z, Amargos-Bosch M, et al. Clozapine and haloperidol differently suppress the MK-801-increased glutamatergic and serotonergic transmission in the medial prefrontal cortex of the rat. Neuropsychopharmacology. 2007;32:2087–97. doi: 10.1038/sj.npp.1301356. [DOI] [PubMed] [Google Scholar]
- 65.Chowdhury GM, Behar KL, Cho W, et al. 1H-[13C]-nuclear magnetic resonance spectroscopy measures of ketamine’s effect on amino acid neurotransmitter metabolism. Biol Psychiatry. 2012;71:1022–25. doi: 10.1016/j.biopsych.2011.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Koike H, Iijima M, Chaki S. Involvement of AMPA receptor in both the rapid and sustained antidepressant-like effects of ketamine in animal models of depression. Behav Brain Res. 2011;224:107–11. doi: 10.1016/j.bbr.2011.05.035. [DOI] [PubMed] [Google Scholar]
- 67.Maeng S, Zarate CA, Jr, Du J, et al. Cellular mechanisms underlying the antidepressant effects of ketamine: role of alpha-amino-3-hydroxy-5-methylisoxazole-4-propionic acid receptors. Biol Psychiatry. 2008;63:349–52. doi: 10.1016/j.biopsych.2007.05.028. [DOI] [PubMed] [Google Scholar]
- 68.Dwyer JM, Lepack AE, Duman RS. mTOR activation is required for the antidepressant effects of mGluR2/3 blockade. Int J Neuropsychopharmacology. 2011;15:429–34. doi: 10.1017/S1461145711001702. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Furey ML, Drevets WC. Antidepressant efficacy of the antimuscarinic drug scopolamine: a randomized, placebo-controlled clinical trial. Arch Gen Psychiatry. 2006;63:1121–29. doi: 10.1001/archpsyc.63.10.1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Voleti B, Navarria A, Liu RJ, et al. Scopolamine rapidly increases mammalian target of rapamycin complex 1 signaling, synaptogenesis, and antidepressant behavioral responses. Biol Psychiatry. 2013;74:742–49. doi: 10.1016/j.biopsych.2013.04.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Salvadore G, Cornwell BR, Colon-Rosario V, et al. Increased anterior cingulate cortical activity in response to fearful faces: a neurophysiological biomarker that predicts rapid antidepressant response to ketamine. Biol Psychiatry. 2009;65:289–95. doi: 10.1016/j.biopsych.2008.08.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Salvadore G, Cornwell BR, Sambataro F, et al. Anterior cingulate desynchronization and functional connectivity with the amygdala during a working memory task predict rapid antidepressant response to ketamine. Neuropsychopharmacology. 2010;35:1415–22. doi: 10.1038/npp.2010.24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Cornwell BR, Salvadore G, Furey M, et al. Synaptic potentiation is critical for rapid antidepressant response to ketamine in treatment-resistant major depression. Biol Psychiatry. 2012;72:555–61. doi: 10.1016/j.biopsych.2012.03.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Duncan WC, Sarasso S, Ferrarelli F, et al. Concomitant BDNF and sleep slow wave changes indicate ketamine-induced plasticity in major depressive disorder. Int J Neuropsychopharmacology. 2013;16:301–11. doi: 10.1017/S1461145712000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Salvadore G, van der Veen JW, Zhang Y, et al. An investigation of amino-acid neurotransmitters as potential predictors of clinical improvement to ketamine in depression. Int J Neuropsychopharmacology. 2011;15:1063–72. doi: 10.1017/S1461145711001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Machado-Vieira R, Yuan P, Brutsche N, et al. Brain-derived neurotrophic factor and initial antidepressant response to an N-methyl-D-aspartate antagonist. J Clin Psychiatry. 2009;70:1662–66. doi: 10.4088/JCP.08m04659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rybakowski JK, Permoda-Osip A, Skibinska M, et al. Single ketamine infusion in bipolar depression resistant to antidepressants: Are neurotrophins involved? Hum Psychopharmacol. 2013;28:87–90. doi: 10.1002/hup.2271. [DOI] [PubMed] [Google Scholar]
- 78.Haile CN, Murrough JW, Iosifescu DV, et al. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. Int J Neuropsychopharmacology. 2014;17:331–36. doi: 10.1017/S1461145713001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Zarate CA, Jr, Brutsche N, Laje G, et al. Relationship of ketamine’s plasma metabolites with response, diagnosis, and side effects in major depression. Biol Psychiatry. 2012;72:331–38. doi: 10.1016/j.biopsych.2012.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Sos P, Klirova M, Novak T, et al. Relationship of ketamine’s antidepressant and psychotomimetic effects in unipolar depression. Neuro Endocrinol Lett. 2013;34:287–93. [PubMed] [Google Scholar]
- 81.Permoda-Osip A, Dorszewska J, Bartkowska-Sniatkowska A, et al. Vitamin B12 level may be related to the efficacy of single ketamine infusion in bipolar depression. Pharmacopsychiatry. 2013;46:227–28. doi: 10.1055/s-0033-1349861. [DOI] [PubMed] [Google Scholar]
- 82.Anand A, Charney DS, Oren DA, et al. Attenuation of the neuropsychiatric effects of ketamine with lamotrigine: support for hyperglutamatergic effects of N-methyl-D-aspartate receptor antagonists. Arch Gen Psychiatry. 2000;57:270–76. doi: 10.1001/archpsyc.57.3.270. [DOI] [PubMed] [Google Scholar]
- 83.Deakin JF, Lees J, McKie S, et al. Glutamate and the neural basis of the subjective effects of ketamine: a pharmaco-magnetic resonance imaging study. Arch Gen Psychiatry. 2008;65:154–64. doi: 10.1001/archgenpsychiatry.2007.37. [DOI] [PubMed] [Google Scholar]
- 84.Krystal JH, Mathew SJ, D’Souza DC, et al. Potential psychiatric applications of metabotropic glutamate receptor agonists and antagonists. CNS Drugs. 2010;24:669–93. doi: 10.2165/11533230-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 85.Krystal JH, Abi-Saab W, Perry E, et al. Preliminary evidence of attenuation of the disruptive effects of the NMDA glutamate receptor antagonist, ketamine, on working memory by pretreatment with the group II metabotropic glutamate receptor agonist, LY354740, in healthy human subjects. Psychopharmacology. 2005;179:303–9. doi: 10.1007/s00213-004-1982-8. [DOI] [PubMed] [Google Scholar]
- 86.Luckenbaugh DA, Niciu MJ, Ionescu DF, et al. Do the dissociative side effects of ketamine mediate its antidepressant effects? J Affect Disord. 2014;159:56–61. doi: 10.1016/j.jad.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hyder F, Rothman DL, Bennett MR. Cortical energy demands of signaling and nonsignaling components in brain are conserved across mammalian species and activity levels. Proc Natl Acad Sci USA. 2013;110:3549–54. doi: 10.1073/pnas.1214912110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Breier A, Malhotra AK, Pinals DA, et al. Association of ketamine-induced psychosis with focal activation of the prefrontal cortex in healthy volunteers. Am J Psychiatry. 1997;154:805–11. doi: 10.1176/ajp.154.6.805. [DOI] [PubMed] [Google Scholar]
- 89.Vollenweider FX, Leenders KL, Oye I, et al. Differential psychopathology and patterns of cerebral glucose utilisation produced by (S)- and (R)-ketamine in healthy volunteers using positron emission tomography (PET) Eur Neuropsychopharmacology. 1997;7:25–38. doi: 10.1016/s0924-977x(96)00042-9. [DOI] [PubMed] [Google Scholar]
- 90.Vollenweider FX, Leenders KL, Scharfetter C, et al. Metabolic hyperfrontality and psy-chopathology in the ketamine model of psychosis using positron emission tomography (PET) and [18F]fluorodeoxyglucose (FDG) Eur Neuropsychopharmacology. 1997;7:9–24. doi: 10.1016/s0924-977x(96)00039-9. [DOI] [PubMed] [Google Scholar]
- 91.Rowland LM, Bustillo JR, Mullins PG, et al. Effects of ketamine on anterior cingulate glutamate metabolism in healthy humans: a 4-T proton MRS study. Am J Psychiatry. 2005;162:394–96. doi: 10.1176/appi.ajp.162.2.394. [DOI] [PubMed] [Google Scholar]
- 92.Stone JM, Dietrich C, Edden R, et al. Ketamine effects on brain GABA and glutamate levels with 1H-MRS: relationship to ketamine-induced psychopathology. Mol Psychiatry. 2012;17:664–65. doi: 10.1038/mp.2011.171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Taylor MJ, Tiangga ER, Ni Mhuircheartaigh R, Cowen P. Lack of effect of ketamine on cortical glutamate and glutamine in healthy volunteers: a proton magnetic resonance spectroscopy study. J Psychopharmacol. 2011;26:733–37. doi: 10.1177/0269881111405359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yang S, Hu J, Kou Z, Yang Y. Spectral simplification for resolved glutamate and glutamine measurement using a standard STEAM sequence with optimized timing parameters at 3, 4, 4.7, 7, and 9.4T. Magn Reson Med. 2008;59:236–44. doi: 10.1002/mrm.21463. [DOI] [PubMed] [Google Scholar]
- 95.Morgan CJ, Curran HV Independent Scientific Committee on Drugs. Ketamine use: a review. Addiction. 2012;107:27–38. doi: 10.1111/j.1360-0443.2011.03576.x. [DOI] [PubMed] [Google Scholar]
- 96.Feder A, Parides MK, Murrough JW, et al. Efficacy of intravenous ketamine for treatment of chronic posttraumatic stress disorder: a randomized clinical trial. JAMA Psychiatry. 2014;71:681–88. doi: 10.1001/jamapsychiatry.2014.62. [DOI] [PubMed] [Google Scholar]
- 97.Rodriguez CI, Kegeles LS, Levinson A, et al. Randomized controlled crossover trial of ketamine in obsessive-compulsive disorder: proof-of-concept. Neuropsychopharmacology. 2013;38:2475–83. doi: 10.1038/npp.2013.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Bloch MH, Wasylink S, Landeros-Weisenberger A, et al. Effects of ketamine in treatment-refractory obsessive-compulsive disorder. Biol Psychiatry. 2012;72:964–70. doi: 10.1016/j.biopsych.2012.05.028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Dakwar E, Levin F, Foltin RW, et al. The effects of subanesthetic ketamine infusions on motivation to quit and cue-induced craving in cocaine-dependent research volunteers. Biol Psychiatry. 2014;76:40–46. doi: 10.1016/j.biopsych.2013.08.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
RELATED RESOURCES
- 1.Bading H. Nuclear calcium signalling in the regulation of brain function. Nat Rev Neurosci. 2013;14:593–608. doi: 10.1038/nrn3531. [DOI] [PubMed] [Google Scholar]
- 2.Gladding CM, Raymond LA. Mechanisms underlying NMDA receptor synaptic/extrasynaptic distribution and function. Mol Cell Neurosci. 2011;48:308–20. doi: 10.1016/j.mcn.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 3.Hardingham GE, Bading H. The yin and yang of NMDA receptor signalling. Trends Neurosci. 2003;26:81–89. doi: 10.1016/S0166-2236(02)00040-1. [DOI] [PubMed] [Google Scholar]
- 4.Kavalali ET, Monteggia LM. Synaptic mechanisms underlying rapid antidepressant action of ketamine. Am J Psychiatry. 2012;169:1150–56. doi: 10.1176/appi.ajp.2012.12040531. [DOI] [PubMed] [Google Scholar]
- 5.Lin YC, Koleske AJ. Mechanisms of synapse and dendrite maintenance and their disruption in psychiatric and neurodegenerative disorders. Annu Rev Neurosci. 2010;33:349–78. doi: 10.1146/annurev-neuro-060909-153204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Liu RJ, Fuchikami M, Dwyer JM, et al. GSK-3 inhibition potentiates the synaptogenic and antidepressant-like effects of subthreshold doses of ketamine. Neuropsychopharmacology. 2013;38:2268–77. doi: 10.1038/npp.2013.128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Mabb AM, Ehlers MD. Ubiquitination in postsynaptic function and plasticity. Annu Rev Cell Dev Biol. 2010;26:179–210. doi: 10.1146/annurev-cellbio-100109-104129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Niciu MJ, Henter ID, Luckenbaugh DA, et al. Glutamate receptor antagonists as fast-acting therapeutic alternatives for the treatment of depression: ketamine and other compounds. Annu Rev Pharmacol Toxicol. 2014;54:119–39. doi: 10.1146/annurev-pharmtox-011613-135950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Niswender CM, Conn PJ. Metabotropic glutamate receptors: physiology, pharmacology, and disease. Annu Rev Pharmacol Toxicol. 2010;50:295–322. doi: 10.1146/annurev.pharmtox.011008.145533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Paoletti P, Bellone C, Zhou Q. NMDA receptor subunit diversity: impact on receptor properties, synaptic plasticity and disease. Nat Rev Neurosci. 2013;14:383–400. doi: 10.1038/nrn3504. [DOI] [PubMed] [Google Scholar]