

Abstract
Enzyme-free signal amplification has enabled sensitive in vitro detection of biomolecules such as proteins and nucleic acids. However, monitoring targets of interest in live cells via enzyme-free amplification is still challenging, especially for analytes with low concentrations. To the best of our knowledge, this paper reports the first attempt to perform mRNA imaging inside live cells, using a nonenzymatic hairpin DNA cascade reaction for high signal gain, termed a hairpin DNA cascade amplifier (HDCA). In conventional nucleic acid probes, such as linear hybridization probes, mRNA target signaling occurs in an equivalent reaction ratio (1:1), whereas, in HDCA, one mRNA target is able to yield multiple signal outputs (1:m), thus achieving the goal of signal amplification for low-expression mRNA targets. Moreover, the recycled mRNA target in the HDCA serves as a catalyst for the assembly of multiple DNA duplexes, generating the fluorescent signal of reduced MnSOD mRNA expression, thus indicating amplified intracellular imaging. This programmable cascade reaction presents a simple and modular amplification mechanism for intracellular biomarkers of interest, providing a significant boost to the search for clues leading to the accurate identification and effective treatment of cancers.
Differences in relative biomarker expression levels of cancer-related mRNAs reveal vital information about tumor progression for studies in the fields of molecular biology, medical diagnosis, and drug delivery.1,2 However, visualization of cellular mRNAs, especially mRNAs with low expression levels, remains a challenge owing to their inadequate signal output which complicates quantification. Biological techniques, such as reverse transcriptase-polymerase chain reaction (RT-PCR) and in situ hybridization (ISH), need to extract mRNAs from cells or fix cells prior to analysis. Therefore, developing signal amplification strategies inside live cells is the key to effective monitoring of cellular mRNAs with low copy numbers. Current enzymatic signal amplification methods, e.g., loop-mediated isothermal amplification (LAMP),3 strand displacement amplification (SDA),4 and rolling circle amplification (RCA),5,6 offer extraordinary signal enhancement for trace amounts of the target in vitro. For example, Li et al. developed a toehold-initiated RCA approach for visualization of individual microRNAs in situ.7 However, the exotic enzymes that are necessary for rolling circle amplification impede its further utility inside live cells. Thus, the goal becomes molecular engineering of nonenzymatic signal amplification methods, especially nucleic acids based systems, for intracellular mRNA imaging.8−11
Pierce et al. designed a programmable hybridization chain reaction (HCR) for amplified imaging of multiplex mRNA expression, both inside fixed cells and in zebrafish embryos.12 However, fluorescence “always-on” hairpin nucleic acid probes are typically used in the hybridization chain reaction. Consequently, the strong fluorescence background leakage resulting from metastable hairpin probes must be removed by a washing step before signal acquisition. Unfortunately, the wash procedure can only be carried out in fixed cells or tissues, but not live cells. To visualize low-expression mRNA with high signal gain in live cells, two requirements must be satisfied in the molecular engineering of nucleic acid probes: signal switch from “OFF” to “ON” and enzyme-free amplification upon target mRNA binding.
Owing to recent advances in molecular engineering and programming of DNA nanotechnology, nucleic acid hybridization-based nanocircuits have allowed the detection of small-molecule or nucleic acid targets with tens- or even hundreds-fold catalytic amplification.13−17 In this work, we propose to utilize a hairpin DNA cascade amplifier (HDCA) for the imaging of mRNAs with low expression levels in live cells. In particular, this HDCA has two major components: a catalytic element consisting of two hairpin-shaped metastable DNA substrates, whose conformational transformations can be catalytically triggered upon target mRNA binding, and the reporting moiety containing a hybridized DNA duplex with a fluorophore (Rep-F) and quencher (Rep-Q). As shown in Figure 1, in the absence of target mRNA, two metastable hairpin DNA substrates, H1 and H2, remain intact, since their cross-reactivity is effectively blocked by intramolecular hybridization. However, formation of a stable hybridized duplex between H1 and H2 can be triggered by target mRNA, which, instead of being consumed, is released to catalyze the formation of other H1–H2 duplexes. The H1–H2 duplex driven by enthalpy decrease further dehybridizes the reporting moiety, resulting in a restoration of the initially quenched fluorescent signal. Similar to the catalyst in a chemical reaction, the target mRNA sequence plays a catalytic role in this hairpin DNA cascade reaction. In conventional nucleic acid probes, such as linear hybridization probes or molecular beacons, mRNA target signaling occurs in an equivalent reaction ratio (1:1), whereas, in the HDCA, one mRNA target is able to yield multiple signal outputs (1:m), thus achieving the goal of signal amplification for low-expression mRNA targets.
Figure 1.
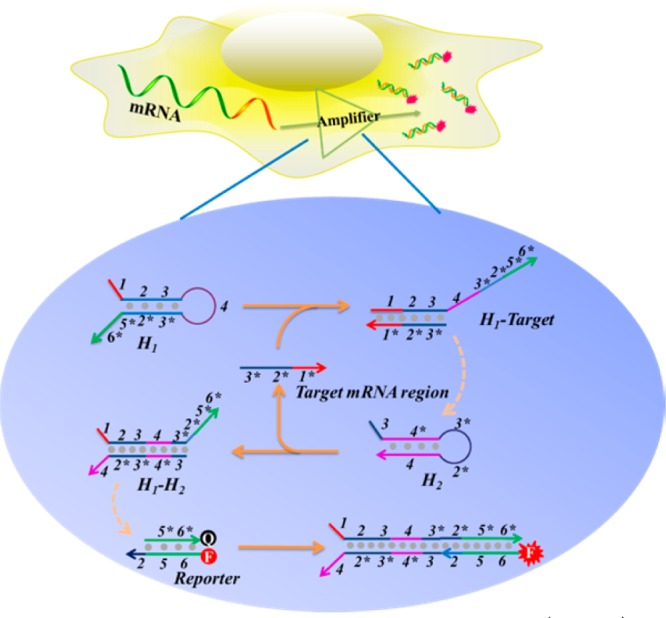
Illustration of hairpin DNA cascade amplifier (HDCA) for catalytic signal enhancement of specific mRNA expression in living cells. After transfection into live cells, the catalytic HDCAs are initiated by specific target mRNA to repeatedly yield many H1–H2 duplexes, which further destabilize the Reporter moiety and fluoresce inside the cells.
The basic concept of catalyzed hairpin substrates was introduced by Pierce et al. and Chen et al.9,14 However, to the best of our knowledge, this principle has not been applied to catalytic amplification of specific mRNA targets inside live cells. To improve the HDCA in live cell systems, two molecular design concerns had to be resolved: intracellular delivery of HDCA components and inhibition of false positive fluorescence signals resulting from degradation by cellular nucleases. The question of cytosolic delivery of multiple DNA sequences, e.g., DNA-based logic gates and unnatural nucleotide-modified molecular beacons, has been answered by using the highly efficient transfection approach.18,19 To improve the thermostability and nuclease resistance of the HDCA system for long-term imaging of mRNAs at physiological temperature (37 °C), four locked nucleic acid (LNA) nucleotides are incorporated into the fluorophore-labeled reporting moiety.20 In this way, the HDCA system targeting a specific mRNA can be efficiently transfected into live cells for accurate catalytic amplification without interference from cellular nucleases and proteins.
To demonstrate our hairpin DNA cascade amplifier, manganese superoxide dismutase (MnSOD) mRNA, which is involved in malignant phenotype and tumor proliferation, was chosen as the specific target model.21,22 According to the selected region of human MnSOD mRNA (GenBank Accession No. NM-000636), a pair of metastable DNA hairpin structures (H1 and H2) was rationally designed. As shown in Figure 1, MnSOD mRNA hybridization to the exposed toehold domain 1 of H1 gradually initiates a strand displacement, generating a H1–MnSOD mRNA intermediate through domain hybridization (1-2-3 and 3*-2*-1*). Released toehold domain 3* in the H1–MnSOD mRNA intermediate further triggers branch migration on domain 3-4*-3*-2* of H2, followed by displacement of MnSOD mRNA for the next catalytic round. Domain 2*-5*-6* on the H1–H2 duplex is fully complementary to Rep-F that lights up inside live cells. Thus, the intracellular MnSOD mRNA can trigger hybridization between hairpin H1 and H2 for multiple cycles, providing enzyme-free signal amplification for monitoring specific mRNA of interest in live cells.
The catalytic feasibility of the hairpin DNA cascade amplifier was first verified by using the DNA analog of the mRNA target through native polyacrylamide gel electrophoresis (PAGE). As shown in Figure 2, in the proof-of-concept assay, the cross-interaction between H1 and H2 is effectively blocked without addition of target DNA. However, after introducing target DNA, hybridization of H1 and H2 is initiated, resulting in a stable H1–H2 duplex. More importantly, the presence of target DNA can facilitate the hybridization yield of the H1–H2 duplex in contrast to that formed from the annealing of hairpin H1 and H2.
Figure 2.
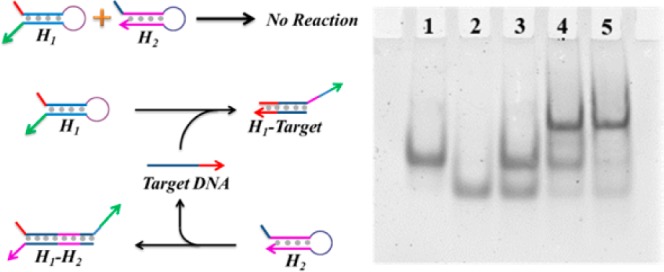
Native PAGE gel of catalytic hairpin DNA cascade reaction without and with addition of target DNA. Lane 1: H1; Lane 2: H2; Lane 3: mixture of H1 and H2; Lane 4: annealing of H1 and H2; Lane 5: H1 + H2 + target DNA.
To drive the equilibrium of the hairpin DNA cascade reaction to yield more H1–H2 duplexes, the concentration ratio between H2 and H1 was first optimized in vitro. With a constant H1 probe quantity, various concentrations of H2 probe were titrated, followed by measurement of the restored reporter signal. The result revealed elevated fluorescence intensity with increasing concentration of the H2 probe, especially ∼15% more enhancement when the H2 quantity was in 3-fold excess over the H1 probe in contrast to the equivalent quantity between H1 and H2 (Figure S1). To avoid an unnecessary leakage reaction without target DNA, the reaction ratio between H2 and H1 (4:1) was maintained throughout the subsequent buffer and cellular experiments. In addition, conventional nucleic acid probes can yield false positive signals in a cellular environment, mostly from endogenous nuclease degradation. As shown in Figure S2, the regular DNA reporter showed a rapid fluorescence increase monitored in cell lysate solution within 2 h. However, the locked nucleic acid modified reporter displayed improved resistance against enzymatic digestion. Therefore, the metastable hairpin DNA structures with the LNA-modified reporter were applied to subsequent assays in vitro and measurement of intracellular MnSOD mRNA expression in breast cancer cell lines (MDA-MB-231).
The catalytic amplification of our HDCA system triggered by target DNA was compared to two conventional nucleic acid probes: a linear hybridization probe (LHP) and a molecular beacon (MB). The catalytic amplification turnover of HDCA was over 40-fold at 0.5 nM target DNA, which was better than the molecular beacon and linear hybridization probe at low target concentration (Figure S4). As shown in Figure 3, the fluorescence enhancement between HDCA and LHP increased with elevated concentrations of target DNA, reaching 17-fold at 10 nM target DNA. A similar tendency was also observed between HDCA and MB. Even at the target concentration as low as 500 pM, HDCA still showed a 4-fold fluorescence enhancement over LHP and MB, indicating enhanced catalytic amplification efficacy of the hairpin DNA cascade amplifier (1:m) over these commonly used nucleic acid molecular probes (1:1). Although the overall reaction rate of HDCA was slower than that of LHP or MB, the fluorescence background could be well resolved by incorporation of the LNA-modified reporter. Next, the target DNA specificity of HDCA was demonstrated in relation to its capacity to differentiate single-nucleotide polymorphisms (SNP). To accomplish this, fully complementary, single- and double-mismatched target DNA sequences were assessed with HDCA against the fully matched DNA target, as shown in Figure S5, revealing a significant differentiation between the fully matched DNA target and either the single- (7.4-fold) or double-mismatched DNA target (11.2-fold).
Figure 3.
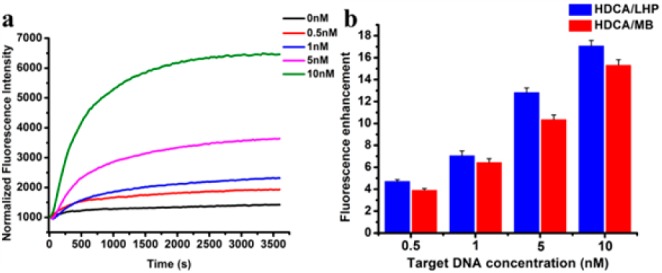
(a) Fluorescence kinetics of hairpin DNA cascade reaction with various concentrations of target DNA (0, 0.5, 1, 5, 10 nM). (b) Fluorescence intensity amplification capability of hairpin DNA cascade reaction relative to a linear hybridization probe (HDCA/LHP) and molecular beacon (HDCA/MB).
Having demonstrated the amplification effectiveness of the HDCA in buffer solution, we next studied its imaging capability in live cells. First, a control hairpin DNA cascade amplifier (cHDCA) and control linear hybridization probe (cLHP) were designed with scrambled sequences, but similar conformations, as references. After transfection into MDA-MB-231 cells via lipofectamine 3000, the HDCA and cHDCA were incubated with cells at 37 °C for 2 h (Figures 4a and S6a). An intense fluorescence signal of Rep-F from HDCA was observed, in contrast to the negligible signal from the control probe, indicating excellent specificity of the rationally designed HDCA for target MnSOD mRNA in live cells. The catalytic amplification efficacy of HDCA was further tested against both the linear hybridization probe (LHP) and corresponding control probe (cLHP). When using LHP, the result showed much lower fluorescence intensity, approximately equal to the background signals for HDCA and cHDCA, respectively (Figures 4b and S6b). To quantify the intracellular signal amplification of HDCA in a large cell population, MDA-MB-231 cells transfected with the HDCA or LHP were treated with trypsin, followed by characterization by analytical flow cytometry (Figure S7).23 Samples of 10 000 cells were counted and compared based on cell-associated fluorescence intensity. As shown in Figure 4c, HDCA showed an average 7.4-fold fluorescence enhancement over that of cells transfected with the linear hybridization probe. Due to the careful design and gel purification of HDCA sequences, the leaking reaction of the HDCA system that was tested in the non-MnSOD mRNA expressed IMR-90 cells can be greatly suppressed (Figure S8). The flow cytometry data are consistent with the confocal imaging results, both demonstrating the catalytic amplification ability of HDCA for intracellular mRNA imaging.
Figure 4.
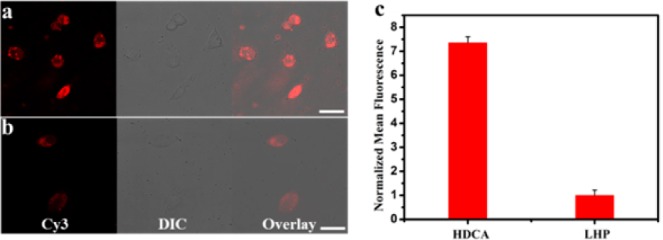
Intracellular imaging of MnSOD mRNA in MDA-MB-231 cell line with HDCA and LHP. Confocal laser microscopy images of MDA-MB-231 cells transfected with (a) HDCA and (b) LHP, followed by incubation at 37 °C for 2 h. (c) Cell-associated fluorescence of MDA-MB-231 cells treated with HDCA and LHP, as measured by flow cytometry. Scale bar: 25 μm.
To further verify that the HDCA could be used to monitor low expression levels of the mRNA target, live MDA-MB-231 cells were treated with 100 μM cordycepin to downregulate the expression level of MnSOD mRNA.22 Since Western blot analysis can provide accurate information about steady-state MnSOD mRNA expression, the reduced expression of MnSOD mRNA by cordycepin was verified by Western blot analysis before performing the intracellular measurement. In contrast to constant β-actin mRNA expression, a significantly reduced MnSOD mRNA expression at ∼30% of normal was confirmed in MDA-MB-231 cells postincubation with 100 μM cordycepin, as shown in Figure 5a. Furthermore, the hairpin DNA cascade amplifier was challenged to image such significantly reduced MnSOD mRNA expression. The confocal results indicate that the HDCA still produced an observable fluorescence signal in MDA-MB-231 cells treated with 100 μM cordycepin inhibitor. However, only a negligible fluorescence signal was observed from cells transfected with the linear hybridization probe under the same conditions (Figures 5b–c and S9). Further statistical analysis of the imaging data from HDCA-transfected cells showed a 3.1-fold increase in fluorescence intensity over that shown by LHP, demonstrating that HDCA is capable of monitoring a low amount of mRNA expression with the aid of enzyme-free nucleic acid signal amplification inside live cells. With further molecular engineering and introduction of multiplexed fluorescent labeling, this hairpin DNA cascade amplifier can be used to realize simultaneous and quantitative imaging of multiple mRNA targets in live cells.24 More importantly, aptamers, due to their programmability and unique molecular recognition to specific targets, including small metabolites and cellular proteins, can be adapted to design DNA cascade reactions for intracellular targets other than mRNAs.
Figure 5.
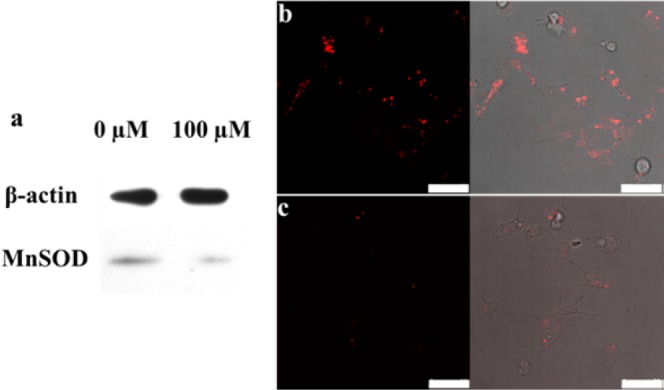
Live cell imaging of MnSOD mRNA with a low-expression level. (a) Western blot analysis of cordycepin-inhibited MnSOD mRNA expression in MDA-MB-231 cells. Confocal microscopy fluorescence images of MDA-MB-231 cells treated with 100 μM cordycepin, followed by transfection and incubation with (b) HDCA and (c) LHP at 37 °C for 2 h. Scale bar: 50 μm.
In summary, we have molecularly engineered a programmable catalytic hairpin DNA cascade reaction for amplified imaging of specific mRNA targets with low expression levels in living cells. After transfection into live cells, the endogenous mRNA triggered assembly of the hairpin H1 and H2 duplex, resulting in a fluorescence signal. More significantly, the specific mRNA target could catalyze the regeneration of multiple H1–H2 duplexes in repeated cycles, achieving enzyme-free signal amplification in situ. By using this strategy, a more intense response of the hairpin DNA cascade amplifier to a specific mRNA target was demonstrated and further compared with conventional nucleic acid probes, such as the linear hybridization probe. Moreover, reduced specific gene expression after exposure to a small-molecule inhibitor was visualized by the HDCA in contrast to the linear hybridization probe which showed only a negligible response, indicating that the HDCA is capable of recognizing low amounts of mRNA target. The metastable hairpin DNA cascade amplifier can be easily reconfigured and engineered for imaging of other endogenous targets of interest, including intracellular proteins and small molecules. Finally, real-time monitoring of very low levels of mRNA molecules in living cells will improve the accuracy of early cancer diagnosis and, hence, the specificity of therapeutic intervention.
Acknowledgments
This work is supported by grants awarded by the National Key Scientific Program of China (2011CB911000 and 2013CB932702), NSFC grants (NSFC 21221003 and NSFC 21327009), and China National Instrumentation Program 2011YQ03012412, and by the National Institutes of Health (GM079359 and CA133086). C.W. acknowledges support from the American Chemical Society, Division of Analytical Chemistry Fellowship, sponsored by the Society for Analytical Chemists of Pittsburgh. The authors thank Dr. K. R. Williams for the manuscript review.
Supporting Information Available
Detailed experimental procedures, DNA sequences, and supplementary data. This material is available free of charge via the Internet at http://pubs.acs.org.
The authors declare no competing financial interest.
Funding Statement
National Institutes of Health, United States
Supplementary Material
References
- Tyagi S. Nat. Methods 2009, 6, 331. [DOI] [PubMed] [Google Scholar]
- Spiller D. G.; Wood C. D.; Rand D. A.; White M. R. H. Nature 2010, 465, 736. [DOI] [PubMed] [Google Scholar]
- Notomi T.; Okayama H.; Masubuchi H.; Yonekawa T.; Watanabe K.; Amino N.; Hase T. Nucleic Acids Res. 2000, 28, e63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker G. T.; Fraiser M. S.; Schram J. L.; Little M. C.; Nadeau J. G.; Malinowski D. P. Nucleic Acids Res. 1992, 20, 1691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao W.; Ali M. M.; Brook M. A.; Li Y. Angew. Chem., Int. Ed. 2008, 47, 6330. [DOI] [PubMed] [Google Scholar]
- Ali M. M.; Li F.; Zhang Z.; Zhang K.; Kang D.-K.; Ankrum J. A.; Le X. C.; Zhao W. Chem. Soc. Rev. 2014, 43, 3324. [DOI] [PubMed] [Google Scholar]
- Deng R.; Tang L.; Tian Q.; Wang Y.; Lin L.; Li J. Angew. Chem., Int. Ed. 2014, 53, 2389. [DOI] [PubMed] [Google Scholar]
- Dirks R. M.; Pierce N. A. Proc. Natl. Acad. Sci. U.S.A. 2004, 101, 15275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B.; Ellington A. D.; Chen X. Nucleic Acids Res. 2011, 39, e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.; Briggs N.; McLain J. R.; Ellington A. D. Proc. Natl. Acad. Sci. U.S.A. 2013, 110, 5386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.; Zhu G.; Wu C.; Zhu Z.; Chen T.; Zhang X.; Tan W. ACS Nano 2013, 7, 2312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi H. M. T.; Chang J. Y.; Trinh L. A.; Padilla J. E.; Fraser S. E.; Pierce N. A. Nat. Biotechnol. 2010, 28, 1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D. Y.; Turberfield A. J.; Yurke B.; Winfree E. Science 2007, 318, 1121. [DOI] [PubMed] [Google Scholar]
- Yin P.; Choi H. M. T.; Calvert C. R.; Pierce N. A. Nature 2008, 451, 318. [DOI] [PubMed] [Google Scholar]
- Jiang Y.; Li B.; Milligan J. N.; Bhadra S.; Ellington A. D. J. Am. Chem. Soc. 2013, 135, 7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han D.; Kang H.; Zhang T.; Wu C.; Zhou C.; You M.; Chen Z.; Zhang X.; Tan W. Chem.—Eur. J. 2014, 20, 5866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jung C.; Ellington A. D. Acc. Chem. Res. 2014, 47, 1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ke G.; Wang C.; Ge Y.; Zheng N.; Zhu Z.; Yang C. J. J. Am. Chem. Soc. 2012, 134, 18908. [DOI] [PubMed] [Google Scholar]
- Hemphill J.; Deiters A. J. Am. Chem. Soc. 2013, 135, 10512. [DOI] [PubMed] [Google Scholar]
- Wang L.; Yang C. J.; Medley C. D.; Benner S. A.; Tan W. J. Am. Chem. Soc. 2005, 127, 15664. [DOI] [PubMed] [Google Scholar]
- Church S. L.; Grant J. W.; Ridnour L. A.; Oberley L. W.; Swanson P. E.; Meltzer P. S.; Trent J. M. Proc. Natl. Acad. Sci. U.S.A. 1993, 90, 3113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Qiu L.; Wu C.; You M.; Han D.; Chen T.; Zhu G.; Jiang J.; Yu R.; Tan W. J. Am. Chem. Soc. 2013, 135, 12952. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.; Chen T.; Han D.; You M.; Peng L.; Cansiz S.; Zhu G.; Li C.; Xiong X.; Jimenez E.; Yang C. J.; Tan W. ACS Nano 2013, 7, 5724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santangelo P. J.; Nix B.; Tsourkas A.; Bao G. Nucleic Acids Res. 2004, 32, e57. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.