

Abstract
Modeling the structure and dynamics of large macromolecules remains a critical challenge. Molecular dynamics (MD) simulations are expensive because they model every atom independently, and are difficult to combine with experimentally derived knowledge. Assembly of molecules using fragments from libraries relies on the database of known structures and thus may not work for novel motifs. Coarse-grained modeling methods have yielded good results on large molecules but can suffer from difficulties in creating more detailed full atomic realizations. There is therefore a need for molecular modeling algorithms that remain chemically accurate and economical for large molecules, do not rely on fragment libraries, and can incorporate experimental information. RNABuilder works in the internal coordinate space of dihedral angles and thus has time requirements proportional to the number of moving parts rather than the number of atoms. It provides accurate physics-based response to applied forces, but also allows user-specified forces for incorporating experimental information. A particular strength of RNABuilder is that all Leontis-Westhof basepairs can be specified as primitives by the user to be satisfied during model construction. We apply RNABuilder to predict the structure of an RNA molecule with 160 bases from its secondary structure, as well as experimental information. Our model matches the known structure to 10.2 Angstroms RMSD and has low computational expense.
Index Terms: Internal coordinate mechanics, molecular, structure, dynamics, RNA, modeling, prediction, linear, scaling
1 INTRODUCTION
RNA plays a pervasive role in gene regulation and expression. Messenger RNA provides the template for protein synthesis, but also contains structures which regulate translation [1]. The ribosome is responsible for reading the genetic code and for synthesizing proteins. The spliceosome, a complex of RNA and protein, removes noncoding introns but can also select specific exons in the context of alternative splicing. In recent years, there has been an explosion of knowledge in RNA epigenetics [2], [3], with other functions of RNA emerging [4], [5]. RNA is thus central to life.
However, our understanding of RNA structure and function is limited because the molecules are difficult to crystallize [6]. Compounding the problem, RNAs are prone to misfolding and long-lived kinetic traps [7], [8]. Theoretical approaches for folding and dynamics are challenged by the role of counterions, the size of the molecules, the delicate energetic balance between alternative conformations, and the long times required to equilibrate during folding.
Several existing methods can predict the structures of smaller RNAs using knowledge-based methods, which depend on the statistical analysis of available experimental structural data. Fragment Assembly of RNA (FARNA) samples trinucleotide fragments from a database and screens these structures using a coarse-grained potential [9]. MC-Sym samples four-nucleotide cycles with a given combination of base pairs and assembles them into a final molecule [10]. These fragment assembly methods can have difficulty forming closed structures because of the difficulty in finding a combination of fragments consistent with them. Discrete Molecular Dynamics (DMD) uses a reduced representation of RNAs with three pseudoatoms per nucleotide and has been reported to predict the structure of tRNA with good accuracy; results for P4/P6 are expected to appear soon. The Nucleic Acid Simulation Tool (NAST) represents RNA with one pseudoatom per residue and can fold the (~160 nt) P4/P6 domain of the Tetrahymena group I intron to 16:3 Å RMSD, representing the best computational prediction prior to that of the current work [11]. NAST required 300 cpu-hours, whereas the present method converges in a few hours on a single processor, while also obtaining much greater accuracy. The C2A program allows NAST structures to be refined to atomic detail, as a starting point for further refinement [44]. In this paper, we present results with RNABuilder, an internal coordinate (IC) dynamics code. Because of its internal coordinate framework, its computational requirements for the most part grow linearly with system size, and thus, it can handle significantly larger molecules than those described here. RNABuilder does not use fragment libraries and therefore is not limited by their lack of structural diversity. A key design feature of RNABuilder is to allow the user to make all decisions about modeling, and so it is not an entirely automated method (in contrast to some of the methods reviewed above), but instead provides a powerful toolkit for making and testing structural hypotheses. The user determines which secondary structural and tertiary base-pairing forces will be applied, which regions will be rigid rather than flexible, and how sterics will be treated. This flexible framework supports a wide variety of applications beyond those described here.
In this work, we first describe the open source Simbody IC mechanics library that forms the base of RNABuilder. We then describe the Molmodel API that provides a molecular interface to Simbody. RNABuilder is built on Molmodel and incorporates specific knowledge of RNA bases and their geometry and physics; we describe its force field, handling of steric exclusion, various supported polymers, operating parameters, and performance. In a prior preliminary report, RNABuilder was used to generate accurate all-atoms structures of tRNA, the P4/P6 domain of the Tetrahymena ribozyme [12], and the entire Azoarcus ribozyme [13]. In this paper, we demonstrate the use of a reduced-atom representation of the RNA polymer to model the P4/P6 domain, the resulting structure has accuracy comparable to that of our previously published all-atoms structure [13] and is fully converged within a few hours on a laptop computer.
The Simbody library underlying RNABuilder is an internal (or generalized) coordinate code. Most popular dynamical methods (MD, DMD), in contrast, use Cartesian coordinates, each atom or particle is an independent point mass with three x,y,z coordinates specifying its location with respect to a fixed origin. However, dynamics can be computed in any coordinate set q in which Newton’s second law can be satisfied, with the Cartesian coordinates obtained as needed via known functions x(q), y(q), z(q). For example, we can use a normal mode basis in which q is a set of modal frequencies [14].
Under an IC scheme [15], atoms are partitioned onto rigid bodies, and the bodies are interconnected via joints into an open tree structure, with the coordinates q representing nonlinear joint coordinates relating each body to its “parent” body within the tree. (Algebraic constraints g (q) = 0 are adjoined if there are interbody loops.) The bond lengths and bond angles are typically fixed, leaving the bodies free to rotate only about rotatable bond axes. However, a major strength of this scheme is the freedom to choose these mobilities explicitly. If the internal motion of an entire molecular domain is unimportant or stable enough to be considered immobile, all of the rotatable bonds therein can be rigidified, turning the entire domain into a single unit whose rigid body motion can be computed very economically. On the other hand, where accuracy of details matters and the molecule is more dynamic, local bonds can be freed, as would be done in Cartesian coordinate mechanics.
The use of IC models for biopolymers has a long history; including the construction of physical ball and stick models that only allow dihedral rotations, as used by early crystallographers [45]. Levinthal [16] also used IC models in early computer models. Levitt et al. [17], Noguti and Go [18], and Li and Scheraga [19] used them to compute physical properties, but did not compute forward dynamics. Mazur et al. [20] derived equations of motion in internal coordinates but claimed that they required O(n3) time to solve—time proportional to the cube of the number of atoms. For that reason, these methods were largely abandoned in favor of atomistic molecular dynamics. The discovery of recursive methods to solve IC equations of motion in O(n) time (linear in the number of independent bodies) made these methods practical for dynamics. An O(n) multibody method developed for spacecraft was applied to proteins in [21], and related methods are useful for investigating polymers in all-torsion coordinates [22], [23], and large protein domain motions in reduced-torsion coordinates [24]. Methods for x-ray [25] and NMR [15], [26] structure refinement also use IC methods with specialized force fields. Although these results are promising, the methods are technically difficult to develop and progress has been hampered by the lack of available open source codes. Recently, we developed the Simbody multibody code to address this need [27], [28].
2 METHODS
In this section, we describe the software architecture of RNABuilder. The bottom layer is provided by the Simbody internal coordinate mechanics library. The Molmodel layer facilitates connection to Simbody by providing molecular objects. RNABuilder provides an interface with which the user can easily control simulation conditions, specify flexibility, and add sterics and base pairing forces.
2.1 Simbody Internal Coordinate Mechanics Code
We implemented RNABuilder using the SimTK simulation toolkit. SimTK provides a variety of tools for physical simulation including its internal coordinate rigid body dynamics toolkit, Simbody, and a molecular modeling API, Molmodel. These are summarized in Fig. 2. Working in internal coordinates allows one to embed joint constraints directly within the coordinate basis. Thus, the flexibility of the molecule is controlled by choice of coordinates. Our method allows the user to change which parts are rigid at different stages of the algorithm. We have used this in prior work to rigidify threading templates, onto which a flexible molecule of unknown structure is aligned [12]. Similarly, conformational change in a molecule of known structure could be modeled by rigidifying domains but leaving hinge regions flexible, and subsequently aligning the domains to a template or enforcing specific contacts. Lastly, if part of a molecule is of known or converged structure, that part can be made rigid leaving only the region of unknown structure flexible for economy.
Fig. 2. Software architecture of RNABuilder.

Simbody is a general-purpose multibody mechanics library that implements efficient O(n) mechanics which can be used at any length scale and is not specific or limited to biological systems. MolModel extends Simbody and provides functionality for building molecular models, and for applying atomic forces and chemical constraints. RNABuilder has an interface which allows the user to easily apply forces, set flexibility, and control a molecular simulation. Simbody, Molmodel, and RNABuilder are open source packages distributed by the Stanford Simbios Center.
Conventional (Cartesian coordinate) molecular dynamics treats each atom as a separate body with three degrees of freedom. To make the system more rigid, one must use a constraint algorithm, such as SHAKE [37] or LINCS [39], to remove unwanted degrees of freedom. In this conventional view, everything is free to move unless restricted by additional constraint equations. For highly constrained systems, these algorithms can be very inefficient.
In contrast, Simbody describes a system as a set of rigid bodies interconnected by mobilizers, as depicted in Fig. 1. In this view, a rigid body has no inherent degrees of freedom and cannot move. The only degrees of freedom present in the system are those explicitly granted by a mobilizer. For example, a pin mobilizer (used to represent a torsional bond) defines only a single degree of freedom, the rotation angle around its axis. The more rigid the system is, the fewer degrees of freedom there are, and the more efficient the simulation becomes. More precisely, the ith mobilizer defines a small number (1–6) of generalized coordinates qi and generalized speeds ui, depending on the number of degrees of freedom introduced by that mobilizer. These are aggregated into q = {qi} and u = {ui} which are the complete set of nq internal coordinates and n internal velocities. In this set of coordinates, the multibody tree system’s equations of motion are as follows:
| (1) |
where M is the n × n composite system mass matrix, f is n generalized forces including the contributions of applied and Coriolis forces, N is an nq × n block diagonal kinematic coupling matrix, and t is time. If the system is also subject to constraints, its equations of motion become
| (2) |
where g(t,q) is a set of m constraint equations, G = ∂g/∂q and the Lagrange multipliers λ represent the unknown constraint forces. In the constrained case, the generalized coordinates span a larger motion space than is allowed for the bodies, so they must be actively restricted to move only in the manifold represented by the constraint equations g. Note that (1) is just a set of ordinary differential equations (ODEs) while (2) is a numerically challenging set of mixed differential and algebraic equations (DAEs) of index 3 [29], [30]. Simbody provides numerical integrators that can efficiently integrate this system through time.
Fig. 1. An internal coordinate multibody system.
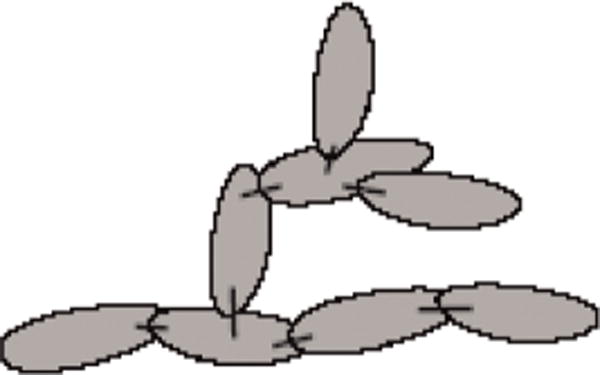
Mobilizers (represented by sticks) define relative motion between bodies (represented by filled ellipses).
In conventional molecular dynamics, the coordinates all control components (atoms) that are of a similar scale and there are a very large number of them, so stability requirements do not vary much from step to step. Consequently, choosing a numerical integration method that runs at a fixed step size limited by the worst-case step’s stability requirement is reasonable. In internal coordinate multibody systems, on the other hand, the coordinates vary dramatically in their effects, there are far fewer of them, and coordinate coupling and gyroscopic effects introduce strong state dependence. Thus, activity from step to step can vary widely. Consequently, variable step, error-controlled numerical integrators are standard for use with multibody systems. This permits effective step size to be determined by the average requirements of the model over the whole simulation, typically permitting much larger steps than required by the worst individual steps, without loss of accuracy or stability. Simbody provides a variety of such integrators; we use the fourth order Runge-Kutta-Merson integrator for this work. This is a variable step size integrator that uses five force evaluations in each time step to produce a fourth order accurate trajectory and a third order accurate error estimate. It adaptively chooses the size of each time step to maintain a desired level of accuracy while still taking the smallest number of time steps possible. Runge-Kutta-Merson integration is known to perform well on rough potentials, such as result from using the collision-detecting contact spheres described later in this work. RNABuilder also offers the Velocity Verlet integrator. This explicitly conserves energy when running in the optional fixed time step mode, but in knowledge-based simulation using a thermostat, energy conservation is not relevant. The choice of integrator makes a difference in computational expense but has little effect on the final result. Constraints are stabilized using the method of coordinate projection [30].
In addition to time integration, it is also possible to do Monte Carlo (MC) simulations in RNABuilder. However, in internal coordinate mechanics a small torsion angle change close to the root of the biopolymer can be amplified into a large displacement of the other end of the chain, thus MC has a high rejection rate in this context. RNABuilder is nonetheless a useful testbed for exploring the viability of such approaches.
RNABuilder uses steric interactions that are represented by elastic spheres implemented with Simbody’s contact modeling features. Spheres are placed at points on the molecule, and apply a repulsive force when they come into contact with each other. Unlike the nonbonded interactions in conventional molecular force fields, this is a very short-range interaction that goes to zero as soon as the spheres are no longer in contact. This allows it to be calculated very quickly. The force on overlapping spheres is calculated with Simbody’s Hunt-Crossley contact model [31], [32]. The radii of the spheres is chosen to optimize the quality of ideal A-form helices, but can be altered by the user in the supplied RNABuilder parameter file.
Force calculations in Simbody are implemented in a modular way allowing different forces to be added together in arbitrary combinations. We compute the total forces f(t,q,u) in the system by combining the modular component forces. RNABuilder uses three main force subsystems: 1) contact forces for the collision detecting spheres used to prevent steric clashes, 2) a base pairing module which uses a force-torque pair to bring bases into the desired interaction geometry [12], and 3) a Tinker-style force field with Amber99 [33] parameters. The user can separately control the strength of all terms of the latter; for economy only the bond stretching term is active by default. Temperature is maintained with one of two thermostats: 1) a simple velocity rescaling thermostat, which periodically rescales the values of all generalized speeds (and hence, the Cartesian velocities of the rigid bodies in the system) to enforce the correct total kinetic energy, or 2) a Nosé-Hoover thermostat [34], [35], which is a deterministic (i.e, force-based) thermostat that maintains a Boltzmann distribution of energy.
2.2 Molmodel Extension to Simbody
As mentioned earlier, RNABuilder is a program for modeling and simulating coarse-grained RNA molecules. RNABuilder exploits the Molmodel and Simbody libraries of the Simbios tool kit (SimTK, Fig. 2). Simbody is a software library and API for simulation of articulated rigid body systems using multibody mechanics (see earlier section). Molmodel is a molecular modeling API layered on Simbody for modeling and simulating molecules, with customizable flexibility, including all-atom Cartesian models, internal coordinate models, fully rigid molecules, and hybrid models.
Rigidification of molecular structures can be done at many scales and results in enhanced performance and simplified simulation. At the fine scale, rigidification of bond lengths is customary in many contemporary molecular simulations [36], [37], [38], [39]. At the coarser scale, domains or entire molecules may be rigidified for a variety of modeling reasons including but not limited to computational economy [12].
An important challenge in coarse-grained molecular modeling is choosing which groups of atoms to combine into rigid clusters. This choice is comparatively simple for small functional groups such as aromatic rings, where chemical constraints suggest groups of atoms with minimal freedom to move relative to one another. At larger scales, secondary structure elements such as DNA and RNA duplexes and protein alpha helices may be modeled as rigid units. At even larger scales, individual domains and the boundaries between them may be modeled as rigid [40] or sets of rigid components connected with flexible hinges (determined experimentally or inferred computationally [41]). RNABuilder generally assumes that bases are fully rigid. There is a single ring closing bond in the ribose ring which is free to change length, angle, and dihedral to accommodate puckering motions; the force field’s bond stretching term keeps this bond length within the normal range. All remaining bonds have fixed lengths and angles but are free to rotate (Fig. 3).
Fig. 3. Default Bond Mobility for Molmodel and RNABuilder RNA residues.
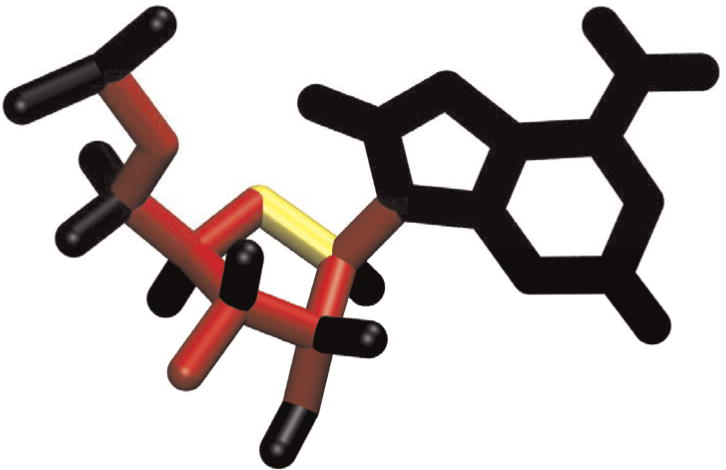
The backbone bonds, most ribose ring bonds, the C2′-O2′ bond, and the glycosidic nitrogen bond are set to Torsion (fixed bond lengths and angles, red), while the O4′-C1′ bond is set to Free (no restriction, yellow). The base bonds and bonds of all single-coordinated atoms (hydrogens and some phosphate oxygens) are set to Rigid (no freedom, black).
2.3 Enforcing Base-Base Interactions
RNABuilder can apply interactions between bases which at equilibrium reproduce any of the base pair types classified in the Leontis-Stombaugh-Westhof catalog [42]. These consist of a force and torque which tend to align an attachment frame on the first residue’s base, with a body frame on the second residue’s base (centered on the glycosidic nitrogen, see Fig. 4). Once these frames are aligned, the desired base pairing geometry is recapitulated. The task of parameterizing the force field is thus primarily that of choosing the position and orientation of the attachment frame with respect to a frame of reference fixed on the first residue’s glycosidic nitrogen, to reproduce a desired base-pairing geometry. Accordingly, RNABuilder’s parameter file contains the X,Y,Z distances and rotation angles of attachment frames A1 needed to generate any of the Leontis and Westhof base pairs [42], as well as stacking and other interactions. We also provide a program to determine these parameters for any additional interactions the user may wish to model, given the 3D atomic coordinates of two residues engaged in the interaction.
Fig. 4. Enforcing base pairs.
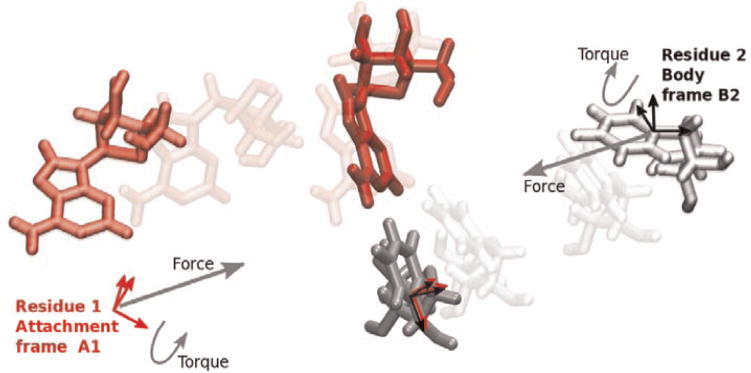
The base pairing interaction includes a force and a torque which act to align the attachment frame A1 of residue 1 with the body frame B2 of residue 2. Equilibrated configuration is shown in bold colors in the center.
The translational force used to bring A1 and B1 together was introduced in [12] and will be adjusted here. But it is not enough to align A1 and B1 translationally, they must also align rotationally. The rotation that must be applied to align it with A1 is computed as:
| (3) |
| (4) |
Here, we are given GRB1 and GRB2, the body frame orientations with respect to ground, which are known as functions of the generalized coordinates q, and B1RA1 the constant orientation of attachment frame A1 in residue 1’s body frame, which is known from our model of the particular base pairing interaction being enforced.
From Euler’s rotation theorem, A1RB2 can be expressed as a rotation of scalar angle θ about an axis (vector of unit length) . The following potential is intended to minimize θ as well as r, the translational distance between A1 and B2 (Fig. 5)
| (5) |
where
| (6) |
| (7) |
Fig. 5. The RNABuilder base-pairing potential.
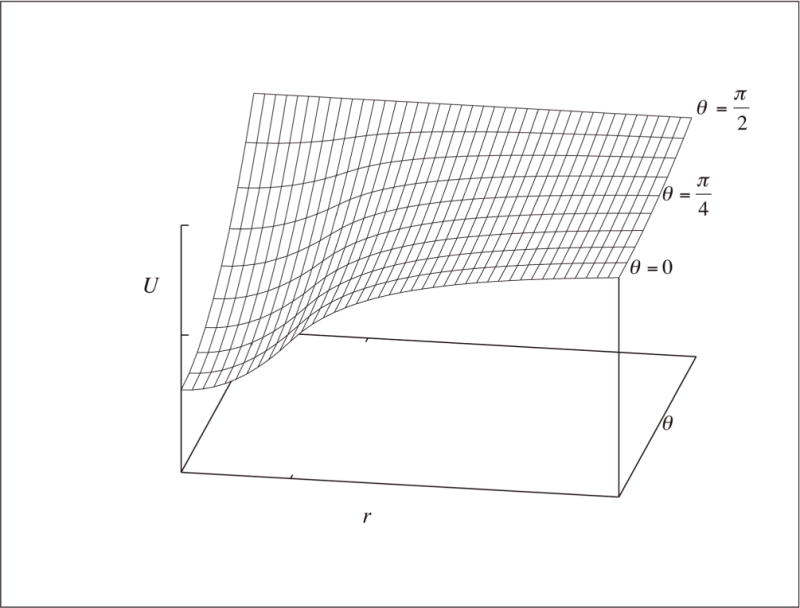
At short range (r < c) the potential is quadratic with r, while at long range it is goes like the inverse, approaching zero at infinite r for all θ Note the inflection point at r = c and θ = 0, where U = k (tick marks). The dependence on θ is quadratic everywhere.
The constants κ and k are set separately for each interaction type in the parameter file, where κ is typically positive and k is typically negative for this potential type. The radial range c is set globally by the user in the input file, as is the scaling factor m. The function g is harmonic at short range (as are most potentials), decays with inverse radius at long range (like the electrostatic forces that may drive folding), and has a maximum derivative (hence maximum force) at its inflection point at c (reminiscent of the Lennard-Jones potential, which also has an inflection point). The force is then
| (8) |
where
| (9) |
The translational force is therefore
| (10) |
Where we dropped the negative sign because of the sense of . The angular dependence of this expression did not appear in [13].
As a technical matter, we do not apply the force to A1 directly but instead on to the body origin of the first base, O1. Moving the point of application in this way results in a torque, which must then be removed. Similarly, forces are not applied to B2 but to O2. The location of the body origin depends on bond mobilities and is generally unknown to the user. The adjusted torques are thus
| (11) |
Where the first term on the right-hand side in each expression is recognizable from (8) and the second constitutes the described adjustment.
The user should also be aware of units and the meaning of physical quantities. RNABuilder inherits its system of units (picoseconds, nanometers, kJ/mol, and Daltons) from Molmodel. The depth k and range c of the potential are motivated by physicochemical experiments and statistical studies [12]. However, time, energy, and temperature are not physically meaningful since the interactions are imposed by the user and the dimensionality of the kinematics is reduced; further the treatment of sterics is very approximate as we will discuss next.
2.4 Steric Exclusion
RNABuilder models steric exclusion using two collision-detecting contact sphere schemes designed to prevent atomic nuclei from approaching each other too closely. The two schemes are as follows:
In the reduced (SelectedAtoms) sterics scheme, we apply Contact spheres to the phosphorus, C4*, and glycosidic nitrogen atoms. The user can modify the identities of the atoms (up to four atoms), as well as the radius and effective stiffness of the sphere to be applied to each. We determined the default radius of each of these three spheres by iteratively forming 10 base pair helices and minimizing the root-mean-square deviation (RMSD) with respect to idealized helices generated using the make_na server [43]. The reduced scheme is designed for economy at some cost in accuracy (note the lack of spheres in the interior of the helix) but often reproduces structures comparable to those obtained with a fuller scheme.
In the full (AllHeavyAtomSterics) scheme, every atom except for the hydrogens gets a contact sphere, all with the same radius and stiffness, which are user adjustable parameters. The default radius was similarly optimized by minimizing RMSD with respect to an idealized helix (Fig. 6).
Fig. 6. Simple helices generated using the reduced (left) and full (right) schemes.

Parameters used are: temperature 1 K, simulation time 10 ns, forceMultiplier 10. The reduced scheme applies 1:75 Å spheres to P and C4*, and 1:35 Å spheres to the glycosidic nitrogen. The choice of atom identities (up to four atoms) and radii and stiffnesses of contact spheres is user adjustable for each residue type. Resulting helix is within 1:88 Å RMSD of an idealized helix generated using NAB. The full scheme applies 1:34 Å spheres to all atoms except hydrogen. The resulting helix is within 1:05 Å RMSD of the NAB helix. With no sterics at all, we obtained 1:96 Å RMSD.
2.5 Protein Modeling
RNABuilder allows the user to create protein as well as RNA chains. The base pairing force field and reduced sterics scheme discussed were devised specifically for RNA. However, all other RNABuilder features are applicable to proteins as well. The user may create a chain by specifying its sequence in single letter code using the alphabet of 20 canonical amino acid types, read in its structure from a file, apply the full (AllHeavyAtomSterics) sterics, control the flexibility for any stretch of residues, restrain any residue to ground or to any other residue, etc. The objective is not to provide full modeling functionality for proteins at this time, but rather to provide a basic treatment of the protein component of protein-RNA complexes such as the ribosome. As an example, in ongoing work we flexibilized only key hinge points in a ribosome (the “neck” region, and base of the beak and L1 stalk) from a species A, while leaving the remaining RNA domains rigid. The protein components were rigid and fixed to the corresponding RNA domains. We then aligned the semiflexible ribosome with a fully rigid ribosome from a species B in a different conformational state. The result was a ribosome from species A in a conformation previously observed in species B, obtained at low computational cost.
2.6 Modeling in Stages
RNABuilder allows user control of modeling parameters, including temperature, size, and number of reporting intervals, the weight (often zero) to be applied to any of the Amber99 [33] force field terms, and others. These can be grouped into “stages” to apply a multistep strategy in a single modeling run. For example, the program can change the temperature in successive stages. It can apply forces, bond mobilities, and sterics in a sequence specified by the user. These features allow the user to rigidify regions after they converge (saving computer time), apply forces at different times to prescribe a folding sequence, and apply time-ordered temperature profiles to create a simulated annealing profile.
2.7 Computational Complexity and Memory Requirements
As mentioned, the computer time requirement for integrating the equations of motion is O(n) for n mobilities (degrees of freedom) in Simbody’s formulation of multibody mechanics. We measured the amount of time required in RNABuilder to compute 1 ps of dynamics for extended chains of RNA of varying lengths. The calculation was done on a single core of an Intel Nehalem processor of an 8-core Mac Pro. We plotted this for rigid chains as well as flexible chains with and without AllHeavyAtomSterics applied; the time requirements were approximately O(n) in each case. Considerable savings can be had by rigidifying bonds in the molecule (Fig. 7).
Fig. 7. Relative scaling of per-step computer time versus number of residues.
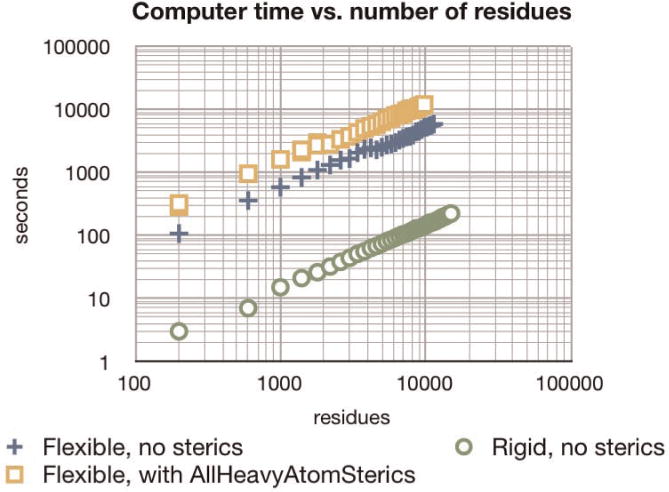
In all cases, we generated two complementary RNA strands of varying lengths and ran for 1 ps at each length. To isolate just per-step costs, we used a Velocity Verlet integrator with 1 fs fixed time steps, no thermostat, and no Molecular Dynamics force field. The rigid (green circles) strands had no internal DOFs, only the 12 rigid-body DOFs; also no forces were applied. The flexible strands (blue plusses and orange squares) had Watson-Crick RNABuilder forces pulling complementary bases on opposite strands together. All three runs showed approximately O(n) scaling. Considerable savings resulted from rigidifying the strands. The user is advised that absolute performance is dependent on simulation conditions; thus relative times are most meaningful.
Memory requirements are also an important aspect because they determine the maximum size of molecules which can be treated. In order to find the size limit, we created extended chains as before, with base pairing forces applied, on a MacBook Pro with a 2.93 GHz Intel processor. RNABuilder was compiled in 32 bit mode enabling the use of 4 GB of RAM; if available, more memory can be addressed by compiling in 64 bit mode. We found that with the current implementation, the RNA Biopolymer required about 226 KBytes of real memory per residue; we were able to instantiate chains of at least 13,000 residues before running out of free memory (Fig. 8).
Fig. 8. Memory requirement of RNA Biopolymer.
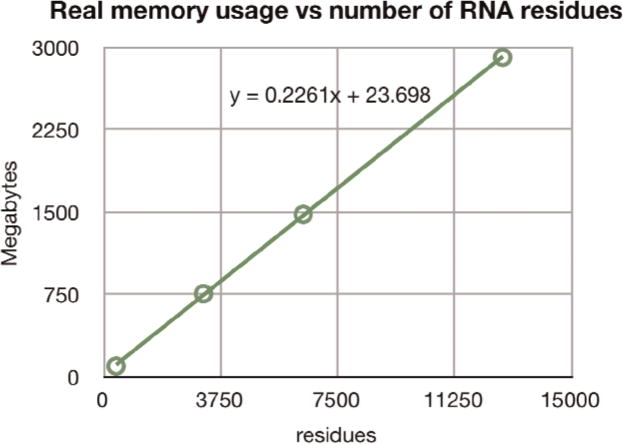
Running in 32 bit mode on a laptop, we were able to generate polymers of at least 13,000 residues, with each residue requiring some 226 KB. More memory or optimization of data structures would enable treatment of even larger molecules.
3 APPLICATION: FOLDING P4/P6
In prior work [13], we folded the P4/P6 domain of the Tetrahymena group I intron by applying base pairing contacts obtained from experiments and calculations that did not make explicit reference to the crystallographic structure. These included UV-crosslinking, dimethyl sulfate and other protection assays, NMR, structural bioinformatics, and phylogenetics. These results provide information not only of the residues involved but often also of the specific type of interaction. For example, the tetraloop—11-nucleotide receptor motif can be detected from sequence and strictly follows a known 3D pattern across molecules and organisms. Also, NMR can provide the structure of small fragments of a molecule even when the larger molecule has not been solved crystallographically. RNABuilder provides a means to turn most of this information into putative 3D molecular structures. In this work, we describe engineering aspects of the P4P6 simulation.
The calculation was run on a single core of an Intel Nehalem processor of an 8-core Mac Pro. We repeated the run four times with randomized initial velocities and obtained 9.3, 10.1, 11.2, and 10:3 Å RMSD, averaged over the last nanosecond of the simulation (Fig. 9). This is some 6 Å lower than the best previously published computational prediction. Each run required about 10.5 hours of computer time. The correct overall geometry was recovered with the main discrepancy being in the geometry and topology around P5c. L5c in the reference structure is contacting the neighboring molecule in the crystal, making the position of helix P5c incorrect [11]. This issue makes it unlikely that the accuracy of any method will approach 0 Å. All regions of the molecule converged on roughly the same time scale and so no particular rigidification was applied; we found that in such cases rigidification beyond that of the default configuration (Fig. 3) diminished accuracy. See Fig. 10, for a representative structure and additional simulation parameters.
Fig. 9. RMSD and number of satisfied contacts versus simulation time.

We folded P4P6 four times, randomizing velocities at the beginning of each stage. We initially folded the P5abc domain separately from the rest of the molecule, then pulled the two domains together after 4.8 ns. From the 2.9 to the 6.7 ns mark, base pairing interactions were turned off for 6 ps periods and back on for 114 ps periods, repeatedly, to escape kinetic traps. This led to the periodic broken contacts in the graph. The contacts were mostly converged after 6.7 ns. Total simulation time was 9.6 ns. In three runs, all 166 base pairing contacts were successfully enforced; in one only 162 were enforced.
Fig. 10.

Predicted structure of P4/P6 (right), compared to the crystallographically obtained structure (left). To generate the predicted structure, sterics were treated with the SelectedAtoms scheme. The temperature was 10 K. All Amber99 force field terms were turned off except for bond stretching, which was scaled to 0.1 of its default value. The RNABuilder base-wise forces were scaled by a factor of 20.
4 CONCLUSIONS
Previously, we showed that RNABuilder can generate accurate structures of transfer RNA, P4/P6 [12], and the entire Azoarcus group I intron [13]. In this work, we modified the potential slightly and described engineering aspects of folding P4/P6. The results highlight the advantages of using internal coordinate multibody mechanics. By reducing the degrees of freedom and using collision-detecting spheres rather than physical pairwise interactions, we achieved convergence in a few hours. The RMSD was 6 Å lower than that of previously published methods, and the computational expense was an order of magnitude lower. RNABuilder enables user control of modeling choices, including base-interaction forces, steric schemes, and runtime parameters. Our empirical results confirm that the computational complexity of RNABuilder is O(n) and that rigidification greatly reduces cost. Our results suggest that RNABuilder will be useful for modeling much larger RNA structures, up to around 13,000 residues on a laptop in 32 bit mode.
5 AVAILABILITY
Binary distributions (for Linux, Mac, and Windows) of RNABuilder are available for download from the RNA-Toolbox project at https://simtk.org/home/rnatoolbox, which also provides source code. A tutorial is available in the “Downloads” section. The Simbios National Center provides software support and workshops for using RNABuilder. This work was done using RNABuilder revision 284, Molmodel revision 650, and Simbody revision 1030.
Acknowledgments
The work was supported by the National Institutes of Health through the NIH Roadmap for Medical Research Grant U54 GM072970. We thank Jeanette Schmidt for comments on the manuscript, and Rick Russell for access to computational resources.
Biographies

Samuel Coulbourn Flores received the undergraduate degree in mechanical and electrical engineering from the Technological Institute of Monterrey, Mexico, and the PhD degree in physics from the Mark Gerstein’s lab, Yale University. He developed new servers to predict protein flexibility and motion, hosted on http://molmovdb.org. He recently completed an appointment as a Distinguished Simbios Fellow in Russ Altman’s lab at Stanford, where he wrote the RNABuilder code described in this work. He is now an assistant professor at Uppsala University, Sweden.

Michael A. Sherman received the BS degree in electrical engineering and computer science from the University of California, Berkeley, in 1978. He began his long professional software development career with Hewlett-Packard in Silicon Valley. He then joined supercomputer startup Elxsi in 1981, where he developed and then managed development of operating system software. Subsequently, he cofounded Symbolic Dynamics, serving as its president and chief software architect for more than a decade, where he wrote and marketed the still widely used SD/FAST multibody simulation tool for mechanical, aerospace, robotics, and biomechanics applications. This software was acquired by Parametric Technology Corp. (PTC) in 1989, where he was a consultant and chief software architect for several PTC mechanical engineering simulation products. In 2000, he cofounded and was CEO of Protein Mechanics, a venture capital funded biotechnology startup, to apply multibody technology to the simulation of medically relevant biomolecules. Protein Mechanics was sold to a pharmaceutical firm, and he joined the Simbios Center at its inception in 2004 as its chief software architect, where he has been responsible for the design and development of the open-source SimTK Core biosimulation toolkit.

Christopher M. Bruns received the PhD degree in biochemistry and computer science from Cornell University. He is a computational biologist at the Stanford University. He was an accomplished macromolecular crystallographer for 10 years before moving into software engineering, bioinformatics, and scientific visualization.

Peter Eastman received the PhD degree in applied physics from Stanford, in 2000. He is a scientific software engineer in Bioengineering Department at the Stanford University. He spent seven years writing commercial bioinformatics software at Agilent Technologies and Silicon Genetics. He works on a variety of physics-based simulation software packages, including OpenMM and SimTK.

Russ Biagio Altman received the AB degree from the Harvard College, the MD degree from the Stanford Medical School, and the PhD degree in medical information sciences from Stanford. He is a professor of bioengineering, genetics, and medicine (and of computer science, by courtesy) and chairman in the Bioengineering Department at the Stanford University. His primary research interests are in the application of computing technology to basic molecular biological problems of relevance to medicine. He particularly interested in informatics methods for advancing pharmacogenomics, the study of how human genetic variation impacts drug response (e.g., http://www.pharmgkb.org/). Other work focuses on the analysis of functional sites within macromolecules and the application of algorithms for determining the structure, dynamics, and function of biological macromolecules (http://features.stanford.edu/). He has been the recipient of the US Presidential Early Career Award for Scientists and Engineers and a National Science Foundation CAREER Award. He is a fellow of the American College of Physicians, the American College of Medical Informatics, and the American Institute of Medical and Biological Engineering. He is a past-president, founding board member, and fellow of the International Society for Computational Biology, and an organizer of the Annual Pacific Symposium on Biocomputing. He leads one of seven NIH-supported National Centers for Biomedical Computation, focusing on physics-based simulation of biological structures (http://simbios.stanford.edu/). He won the Stanford Medical School graduate teaching Award in 2000. He is a member of the Institute of Medicine of the National Academies.
Footnotes
For information on obtaining reprints of this article, please send e-mail to: tcbb@computer.org, and reference IEEECS Log Number TCBB-2010-03-0074.
Contributor Information
Samuel Coulbourn Flores, Email: samuelfloresc@gmail.com, Department of Cell and Molecular Biology, Uppsala University, Box 596, 751 24 Uppsala, Sweden.
Michael A. Sherman, Email: msherman@stanford.edu, Department of Bioengineering, Stanford University, Stanford, CA 94305-5448
Christopher M. Bruns, Email: cmbruns@rotatingpenguin.com, Department of Bioengineering, Stanford University, Stanford, CA 94305-5448
Peter Eastman, Email: peastman@stanford.edu, Department of Bioengineering, Stanford University, Stanford, CA 94305-5448.
Russ Biagio Altman, Email: russ.altman@stanford.edu, Department of Bioengineering, Stanford University, Stanford, CA 94305-5448.
References
- 1.Roth A, Breaker RR. The Structural and Functional Diversity of Metabolite-Binding Riboswitches. Ann Rev Biochemistry. 2009;78:305–34. doi: 10.1146/annurev.biochem.78.070507.135656. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Bartel DP. MicroRNAs: Genomics, Biogenesis, Mechanism, and Function. Cell. 2004;116(2):281–297. doi: 10.1016/s0092-8674(04)00045-5. [DOI] [PubMed] [Google Scholar]
- 3.Carthew RW, Sontheimer EJ. Origins and Mechanisms of miRNAs and siRNAs. Cell. 2009;136(4):642–655. doi: 10.1016/j.cell.2009.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Katayama S, et al. Antisense Transcription in the Mammalian Transcriptome. Science. 2005;309(5740):1564–1566. doi: 10.1126/science.1112009. [DOI] [PubMed] [Google Scholar]
- 5.Carninci P, et al. The Transcriptional Landscape of the Mammalian Genome. Science. 2005;309(5740):1559–1563. doi: 10.1126/science.1112014. [DOI] [PubMed] [Google Scholar]
- 6.Ferre-D’Amare AR, Zhou K, Doudna JA. A General Module for RNA Crystallization. J Molecular Biology. 1998;279(3):621–631. doi: 10.1006/jmbi.1998.1789. [DOI] [PubMed] [Google Scholar]
- 7.Treiber DK, Williamson JR. Exposing the Kinetic Traps in RNA Folding. Current Opinion in Structural Biology. 1999;9(3):339–345. doi: 10.1016/S0959-440X(99)80045-1. [DOI] [PubMed] [Google Scholar]
- 8.Russell R. RNA Misfolding and the Action of Chaperones. Frontiers in Bioscience. 2008;13:1–20. doi: 10.2741/2557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Das R, Baker D. Automated De Novo Prediction of Native-Like RNA Tertiary Structures. Proc Nat’l Academy of Sciences USA. 2007;104(37):14664–14669. doi: 10.1073/pnas.0703836104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Parisien M, Major F. The MC-Fold and MC-Sym Pipeline Infers RNA Structure from Sequence Data. Nature. 2008;452(7183):51–55. doi: 10.1038/nature06684. [DOI] [PubMed] [Google Scholar]
- 11.Jonikas MA, et al. Coarse-Grained Modeling of Large RNA Molecules with Knowledge-Based Potentials and Structural Filters. RNA. 2009;15(2):189–199. doi: 10.1261/rna.1270809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Flores S, Wan Y, Russell R, Altman RB. Predicting RNA Structure by Multiple Template Homology Modeling. Proc Pacific Symp Biocomputing. 2010 doi: 10.1142/9789814295291_0024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Flores S, Altman RB. Turning Limited Experimental Information Into 3D Models of RNA. RNA. 2010;16:1769–1778. doi: 10.1261/rna.2112110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Sweet CR, et al. Normal Mode Partitioning of Langevin Dynamics for Biomolecules. J Chemical Physics. 2008;128(14):145101–145113. doi: 10.1063/1.2883966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Schwieters CD, Clore GM. Internal Coordinates for Molecular Dynamics and Minimization in Structure Determination and Refinement. J Magnetic Resonance. 2001;152(2):288–302. doi: 10.1006/jmre.2001.2413. [DOI] [PubMed] [Google Scholar]
- 16.Levinthal C. Molecular Model-Building by Computer. Scientific Am. 1966;214(6):42–52. doi: 10.1038/scientificamerican0666-42. [DOI] [PubMed] [Google Scholar]
- 17.Levitt M, Sander C, Stern PS. Protein Normal-Mode Dynamics: Trypsin Inhibitor, Crambin, Ribonuclease and Lysozyme. J Molecular Biology. 1985;181(3):423–447. doi: 10.1016/0022-2836(85)90230-x. [DOI] [PubMed] [Google Scholar]
- 18.Noguti T, Go N. Efficient Monte Carlo Method for Simulation of Fluctuating Conformations of Native Proteins. Biopolymers. 1985;24(3):527–546. doi: 10.1002/bip.360240308. [DOI] [PubMed] [Google Scholar]
- 19.Li Z, Scheraga HA. Monte Carlo-Minimization Approach to the Multiple-Minima Problem in Protein Folding. Proc Nat’l Academy of Sciences USA. 1987;84(19):6611–6615. doi: 10.1073/pnas.84.19.6611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mazur AK, Dorofeev VE, Abagyan RA. Derivation and Testing of Explicit Equations of Motion for Polymers Described by Internal Coordinates. J Computational Physics. 1991;92(2):261–272. [Google Scholar]
- 21.Jain A, Vaidehi N, Rodriguez G. A Fast Recursive Algorithm for Molecular Dynamics Simulation. J Computational Physics. 1993;106(2):258–268. [Google Scholar]
- 22.Vaidehi N, Jain A, Goddard WAI. Constant Temperature Constrained Molecular Dynamics: The Newton-Euler Inverse Mass Operator Method. J Physical Chemistry. 1996 Jun;100(25):10508–10517. [Google Scholar]
- 23.Mathiowetz AM, et al. Protein Simulations Using Techniques Suitable for Very Large Systems: The Cell Multipole Method for Nonbond Interactions and the Newton-Euler Inverse Mass Operator Method for Internal Coordinate Dynamics. Proteins: Structure, Function, and Genetics. 1994;20(3):227–247. doi: 10.1002/prot.340200304. [DOI] [PubMed] [Google Scholar]
- 24.Vaidehi N, Goddard WA. Domain Motions in Phosphoglycerate Kinase Using Hierarchical NEIMO Molecular Dynamics Simulations. J Physical Chemistry A. 2000;104(11):2375–2383. [Google Scholar]
- 25.Rice LM, Brunger AT. Torsion Angle Dynamics: Reduced Variable Conformational Sampling Enhances Crystallographic Structure Refinement. Proteins. 1994;19(4):277–90. doi: 10.1002/prot.340190403. [DOI] [PubMed] [Google Scholar]
- 26.Guntert P, Mumenthaler C, Wuthrich K. Torsion Angle Dynamics for NMR Structure Calculation with the New Program DYANA. J Molecular Biology. 1997;273(1):283–298. doi: 10.1006/jmbi.1997.1284. [DOI] [PubMed] [Google Scholar]
- 27.Schmidt JP, et al. The Simbios National Center: Systems Biology in Motion. Proc IEEE. 2008 Aug;96(8):1266–1280. doi: 10.1109/JPROC.2008.925454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Sherman MA. Simbody Home Page. 2009 https://simtk.org/home/simbody.
- 29.Brenan KE, Campbell SL, Petzold LR. Numerical Solution of Initial-Value Problems in Differential-Algebraic Equations. Vol. 8. North Holland; 1989. p. 210. [Google Scholar]
- 30.Eich E. Results for a Coordinate Projection Method Applied to Mechanical Systems with Algebraic Constraints. SIAM J Numerical Analysis. 1993;30(5):1467–1482. [Google Scholar]
- 31.Johnson KL. Contact Mechanics. Cambridge Univ Press; 1985. ch 4 (Section 4.2) [Google Scholar]
- 32.Hunt KH, Crossley FRE. Coefficient of Restitution Interpreted as Damping in Vibroimpact. ASME J Applied Mechanics, Series E. 1975;42:440–445. [Google Scholar]
- 33.Wang JM, Cieplak P, Kollman PA. How Well Does a Restrained Electrostatic Potential (RESP) Model Perform in Calculating Conformational Energies of Organic and Biological Molecules? J Computational Chemistry. 2000;21:1049–1074. [Google Scholar]
- 34.Nosé S. A Unified Formulation of the Constant Temperature Molecular Dynamics Methods. J Chemical Physics. 1984;81:511–519. [Google Scholar]
- 35.Hoover W. Canonical Dynamics: Equilibrium Phase-Space Distributions. Physical Rev A. 1985;31:1695–1697. doi: 10.1103/physreva.31.1695. [DOI] [PubMed] [Google Scholar]
- 36.Ryckaert JP, Ciccotti G, Berendsen HJC. Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of N-Alkanes. J Computational Physics. 1977;23(3):327–341. [Google Scholar]
- 37.Andersen HC. Rattle: A “Velocity” Version of the Shake Algorithm for Molecular Dynamics Calculations. J Computational Physics. 1983;52(1):24–34. [Google Scholar]
- 38.Miyamoto S, Kollman PA. Settle: An Analytical Version of the SHAKE and RATTLE Algorithm for Rigid Water Models. J Computational Chemistry. 1992;13(8):952–962. [Google Scholar]
- 39.Hess B, Bekker H, Berendsen HJC, Fraaije JGEM. LINCS: A Linear Constraint Solver for Molecular Simulations. J Computational Chemistry. 1997;18(12):1463–1472. [Google Scholar]
- 40.Flores S, et al. The Database of Macromolecular Motions: New Features Added at the Decade Mark. Nucleic Acids Res. 2006;34:D296–D301. doi: 10.1093/nar/gkj046. Database Issue. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Flores SC, et al. HingeMaster: Normal Mode Hinge Prediction Approach and Integration of Complementary Predictors. Proteins. 2008;73(2):299–319. doi: 10.1002/prot.22060. [DOI] [PubMed] [Google Scholar]
- 42.Leontis NB, Stombaugh J, Westhof E. The Non-Watson-Crick Base Pairs and Their Associated Isostericity Matrices. Nucleic Acids Res. 2002;30(16):3497–3531. doi: 10.1093/nar/gkf481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Stroud J. The Make-Na Server. 2010 http://structure.usc.edu/make-na/server.html.
- 44.Jonikas MA, Radmer RJ, Altman RB. Knowledge-Based Instantiation of Full Atomic Detail Into Coarse-Grain RNA 3D Structural Models. Bioinformatics. 2009;25(24):3259–3266. doi: 10.1093/bioinformatics/btp576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Astbury WT. The Structure of Biological Tissues as Revealed by X-Ray Diffraction Analysis and Electron Microscopy. British J Radiology. 1949;22:355–365. doi: 10.1259/0007-1285-22-259-355. [DOI] [PubMed] [Google Scholar]