

Abstract
Na+/Ca2+ exchanger (NCX) is a plasma membrane transporter that, by regulating Ca2+ and Na+ homeostasis, contributes to brain stroke damage. The objectives of this study were to investigate whether there might be miRNAs in the brain able to regulate NCX1 expression and, thereafter, to set up a valid therapeutic strategy able to reduce stroke-induced brain damage by regulating NCX1 expression. Thus, we tested whether miR-103-1, a microRNA belonging to the miR-103/107 family that on the basis of sequence analysis might be a potential NCX1 regulator, could control NCX1 expression. The role of miR-103-1 was assessed in a rat model of transient cerebral ischemia by evaluating the effect of the correspondent antimiRNA on both brain infarct volume and neurological deficits. NCX1 expression was dramatically reduced when cortical neurons were exposed to miR-103-1. This alleged tight regulation of NCX1 by miR-103-1 was further corroborated by luciferase assay. Notably, antimiR-103-1 prevented NCX1 protein downregulation induced by the increase in miR-103-1 after brain ischemia, thereby reducing brain damage and neurological deficits. Overall, the identification of a microRNA able to selectively regulate NCX1 in the brain clarifies a new important molecular mechanism of NCX1 regulation in the brain and offers the opportunity to develop a new therapeutic strategy for stroke.
Introduction
MicroRNAs (miRNAs) are small, 17 to 23 nucleotides, noncoding RNAs able to control protein translation by binding to the complementary seed sequences in the 3′-untraslated regions (3′-UTRs) of mRNAs.1 Owing to their small size, relative easiness of delivery, sequence specificity in recognizing their targets, and multitarget properties, miRNAs represent promising therapeutic options for several CNS disorders such as Alzheimer's disease, multiple sclerosis, Parkinson's disease, ALS, and cerebral ischemia.2 In particular, cerebral ischemia triggers a multifaced cascade of biochemical events mediated by alterations in molecular processes such as translation and transcription.3 To date, no exhaustive reports are available on the miRNA microarray profiling of the ischemic brain. Nevertheless, several reports have demonstrated the role of specific miRNAs in neuronal differentiation, neurogenesis, neural cell specification, and neurodevelopmental function.4,5,6,7,8
Interestingly, among those genes whose expression is influenced by cerebral ischemia is the sodium–calcium exchanger-1 (NCX1), a ubiquitous plasma membrane protein regulating cellular calcium and sodium homeostasis in the brain.3,9,10,11
In general, all three isoforms of this exchanger, NCX1, NCX2, and NCX3, are involved in some serious diseases characterized by a dysregulation of ionic homeostasis; including stroke, Alzheimer's disease, multiple sclerosis, and epilepsy.12,13,14,15,16 Recently, studies have shown that brain damage worsens in ischemic rats and mice in which NCX is either downregulated, knocked out, or pharmacologically blocked.3,17 By contrast, when NCX is selectively stimulated, the increase in NCX activity ameliorates the consequences of ischemic brain damage.10,11,15,17,18 Therefore, the identification of new compounds capable of increasing NCX activity may be a suitable strategy to limit the extension of ischemic brain damage. In addition, and, more specifically, an alternative and rational strategy capable of counteracting the reduction in NCX1 expression and activity induced by stroke could be the identification of miRNAs or antimiRNAs able to increase NCX1 expression and thus mitigate the consequences of ischemic stroke.
To this aim, we tested the hypothesis that miR-103-1, a microRNA belonging to the family known as miR-103/107 (ref. 19), that on the basis of sequence analysis might be a potential NCX1 regulator, mediates NCX1 expression. NCX protein expression was evaluated by western blotting in PC12, BHK cell lines, and primary neuronal cultures transfected with miR-103-1. The role of miR-103-1 was assessed in an in vivo rat model of transient cerebral ischemia (tMCAo) by evaluating both the expression of miR-103-1 in the cortex and striatum of ischemic rats and the effect of intracerebroventricular (icv) infusion of the correspondent antimiRNA on brain infarct volume and on neurological deficits.
Results
miR-103-1 interacts with the 3′UTRs of NCX1 RNA messenger
By using, in the HOCTAR database, the query “Slc8a1” for isoform 1 of sodium/calcium exchanger, we obtained a list of all the intragenic miRNAs predicted to bind to the selected target gene. Intragenic miRNAs, which inversely correlated with the target NCX1 transcripts, were ranked as described later in Methods Section. By analyzing miRNA affinity for the 3′UTRs of the NCX1 messenger of the first 50 percentile of the ranking list, we were able to identify miR-103-1 as a potential candidate. This miRNA paired perfectly with 8 nucleotides of its sequence (seed nucleus) to the 3′ UTR of rat NCX1 (Figure 1a), with a release of free interaction energy equal to −28.5 kcal/mol by heteroduplex formation. Moreover, other two sites of miRNA-mRNA interactions of 5 and 6 nucleotides in length, respectively, of perfect base pairing were observed; we thus hypothesized that these sites may cooperate in the miRNA modulation of NCX1 gene transcript because of their close proximity to each other. Thanks to these interaction data we also hypothesized that miR-103-1 most likely interacts with NCX1.
Figure 1.

miR-103-1 regulates NCX1 by targeting 3′-UTR of Ncx1 mRNA. (a) Predicted miR-103-1 binding sites in the 3′-UTRs of NCX1 mRNA. NCX1 contains 3 sites of annealing with candidate miRNA. Site position relative to the beginning of the 3′-UTRs is indicated. Target sequences are highlighted by a box, and base pairing between miR-103-1 and the target site is marked by vertical lines. The most relevant sites of miRNA-mRNA interaction (8 bp of interaction) relatively to the beginning of the 3′-UTRs region of NCX1 (rattus norvegicus) is shown. (b) Luciferase activity assay has been evaluated in PC12 cells by coexpressing the vector cointaining 3′ UTRs of NCX1 and candidate miRNA (miR-103-1). The activity of luciferase enzyme was normalized to β-galactosidase activity and compared with empty vector measurements (pmiR empty). This experiment was performed in triplicate and is representative of three independent experiments. Results show that a strong decrease in luciferase activity is observed when the pmirGLO vector, containing sites 2 and 3 out of three total interaction sites, was cotransfected with mirR-103-1 at 150 nmol/l in PC12 cells. The panel shows that site 1 is ineffective in NCX1 mRNA modulation by miRNA. Data are expressed as mean ± SEM. *P < 0.01.
To confirm that miR-103-1 directly binds to and represses ncx1 expression, we used a luciferase gene reporter assay. We made three different pmiR reporter constructs: construct 1 contained the full length 3-UTR of ncx1 (ncx1 3′-UTR FL), construct 2 contained site 1 (ncx1 3′-UTR site 1), and construct 3 contained sites 2 and 3 (ncx1 3′-UTR site 2&3). No difference in luciferase activity was found between PC12 cells transfected with the control vector (pmirGLO) and PC12 cells transfected with the vector containing the 3-UTRs of ncx1 (Figure 1b). In contrast, the cotransfection of miR-103-1 at 100 and 150 nmol/l resulted in 50% reduction in luciferase activity in cells transfected either with ncx1 3′-UTR FL or with ncx1 3′-UTR site 2&3. Cells trasfected with ncx1 3′-UTR site 1 containing only site 1 sequence were not sensitive to miRNA exposure either at 100 or at 150 nmol/l (Figure 1b).
miR-103-1 transfection causes a reduction in NCX1 expression in BHK-NCX1 cells, in PC12 cells and in cortical neurons
To verify whether NCX1 was actually a target for miR-103-1, we treated BHK cells transfected with NCX1 with increasing concentrations of miR-103-1 in the presence of lipofectamine. The transient transfection with miR-103-1 remarkably downregulated NCX1 in BHK-NCX1 cells. In particular, this reduction was concentration and time-dependent (Figure 2a).
Figure 2.

Representative western blot of NCX1 protein levels and densitometric quantification of BHK-NCX1, PC12 cell, and cortical neurons transfected with miR-103-1 at different time intervals and concentrations. (a) NCX1 expression in BHK-NCX1 cells transfected with miR-103-1. Values are normalized with respect to α-tubulin. MiR-103-1 mimic transfection in BHK-NCX1 cell clones determined a concentration dependent modulation in NCX1 protein levels at 24 hours. A parallel trend of downregulation of NCX1 protein at 48 hours after transfection in BHK-NCX1 cells is evidenced. (b) MiRNA mimic was tested in PC12 cells at several time intervals from transient transfection (24, 48, and 72 hours) and at different miRNA concentration (10, 100, and 150 nmol/l). Results show that miR-103-1 is able to act as a potent repressor of NCX1 protein expression in particular when used at 100 nmol/l concentration after 48 hours of transfection; this effect is more impressive at 72 hours. (c) Finally, experiment of mir-103-1 overexpression was made 72 hours after miRNA transfection in cortical neurons from rat embryos. All values are expressed as mean ± SEM of three independent experimental sessions. *P < 0.05 versus their respective controls.
To prove the species specificity, we transfeced miR-103-1 in rat pheocromocytoma PC12 cells. Twenty-four hours after miRNA exposure, 10 and 100 nmol/l miR-103-1 did not reduce NCX1 expression. However, 48 and 72 hours later, the same miRNA concentrations induced a significant concentration-dependent reduction of 49 and 93.8%, respectively, in NCX1 expression (Figure 2b). Since miR-103 belongs to the miRNA family known as miR-103/107 (ref. 20) and differs from miR-107 by 1 nucleotide, we tested the hypothesis that (i) miR-107 was able to control NCX1 expression in a manner similar to miR-103, (ii) anti-miR-103 was able to act on miR-103 and miR-107, and (iii) anti-miR-107 was able to act on miR-103 and miR-107. The results obtained in PC12 confirmed the interchangability of miR-103 and miR-107 (Supplementary Figure S1).
Finally, we investigated NCX1 regulation by miR-103-1 in primary cultures of cortical neurons. In these cells, a 72-hour incubation with 100 nmol/l of miR-103-1 reduced NCX1 expression by 50% (Figure 2c).
Anti-miR-103-1 significantly and selectively upregulates NCX1 protein expression after ischemia in the brain cortex and striatum of ischemic rats
To test whether anti-miR-103-1 affected only NCX1 expression or also the other two NCX proteins expressed in the brain, the effects of anti-miR-103-1 treatment were also evaluated on NCX2 and NCX3 expression.
As expected, anti-miR-103-1 upregulated NCX1 protein expression in the ipsilateral cortex and striatum of rats subjected to ischemic stroke, compared to rats treated with the negative control and to sham-operated animals (Figure 3a). Interestingly, icv administration of anti-miR-103-1 in ischemic rats influenced neither NCX2 protein expression (Figure 3b), nor NCX3 protein expression (Figure 3c) in the ipsilateral brain cortex and striatum of ischemic rats.
Figure 3.

NCX1 isoform-specificity of anti-miR-103-1 treatment evaluated by western blotting 24 hours after ischemia induction. (a–c) Expression levels of NCX1 (a), NCX2 (b), and NCX3 (c) in brain cortex (left side) and striatum (right side) of ischemic rats treated with negative control (Neg CTL) or anti-miR-103-1 9 μg/kg (AntimiRNA) evaluated 24 hours after ischemia. *P < 0.05 versus sham-operated group; **P < 0.05 versus sham-operated group and Neg CTL group. Each column represents the mean ± SEM. n = 5 animals per group. Data were normalized on the basis of α-tubulin levels and expressed as percentage of sham-operated controls (Sham).
A time–course analysis of miR-103-1 levels and NCX1 expression after stroke revealed that miR-103-1 levels inversely correlated with NCX1 expression. Indeed, miR-103-1 expression levels were dramatically increased by 300% 24 hours after stroke induction (Figure 4a). Conversely, NCX1 expression was reduced by 30% (Figure 4b).
Figure 4.
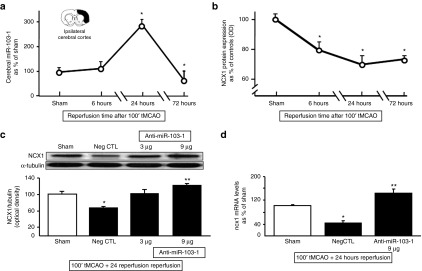
Time-course of miR-103-1, NCX1 protein, and NCX1 mRNA in rat ischemic brain cortex; identification of effective anti-miR-103-1 dose. (a) Time-course of miR-103-1 expression levels in cerebral cortex samples harvested from rats subjected to 100' tMCAO and sacrificed at 6, 24, and 72 hours. Results are expressed as fold changes of expression of miR-103-1 compared to sham. (b) Time-course NCX1 expression levels in cerebral cortex samples harvested from rats subjected to 100' tMCAO and sacrificed at 6, 24, and 72 hours. Results are expressed as fold changes of expression of NCX1 compared to sham and normalized for α-tubulin. n = 3–4 animals per group. *P < 0.05 versus sham-operated group in both expression profiles. Each point of the graphic line represents the mean ± SEM. (c) A quantitative analysis of NCX1 protein expression levels in ipsilateral damaged brain area of rats subjected to tMCAO and treated with negative control (Neg CTL), anti-miR-103-1 2 µg/kg, and anti-miR-103-1 9 µg/kg. A representative western blotting is included on the top of panel (c). (d) Changes in expression levels of ncx1 mRNA from ischemic brain cortex samples of rats subjected to tMCAO and treated with negative control (NegCTL), and anti-miR-103-1 9 µg/kg (anti-miR-103-1 9) compared to ncx1 mRNA expression in sham-operated animals. n = 5 per group. Data were normalized on the basis of α-tubulin levels and expressed as percentage of sham-operated controls (Sham). *P < 0.05 versus sham-operated group; **P < 0.05 versus negative control group and versus sham-operated group. mRNA levels are expressed as percentage of sham-operated controls (Sham). Each column represents the mean ± SEM.
Anti-miR-103-1, icv infused, prevented NCX1 reduction induced by stroke in the ipsilateral cortex and strongly increased NCX1 protein levels compared both to ischemic animals treated with negative control miRNA and to sham-operated animals (Figure 4c).
Notably, the effect on NCX1 protein expression was mirrored by the levels of NCX1 mRNA. Indeed, anti-miR-103-1, icv infused, increased NCX1 mRNA levels (Figure 4d).
To further demonstrate the similarities between miR-103 and miR-107, the same experiments were repeated with miR-107 and the effects observed were replicated also with this miRNA (Supplementary Figure S2).
Anti-miR-103-1 exerts a strong neuroprotective effect on ischemic damage
To investigate the effect of miR-103-1 on brain damage induced by tMCAO, anti-miR-103-1 was icv infused in rats subjected to ischemia. Anti-miR-103-1 icv infused, 24 hours before stroke onset reduced the extent of brain ischemia by ~60% (19.1 ± 3.4) compared to rats subjected to 100 minutes of tMCAO and treated with vehicle (51.0 ± 6.3) or with negative control (59.5 ± 7.9) (Figure 5a). Interestingly, the reduction in the ischemic volume induced by anti-miR-103-1 was mirrored by amelioration in general and focal scores (Figure 5b). Notably, a similar neuroprotective effect was observed when anti-miR-103-1 infusion started after stroke onset and rats were euthanized 48 hours later. Indeed, anti-miR-103-1, icv infused for 48 hours (9 µg/kg), starting 20' after transient ischemia induction, reduced the extent of brain ischemia by ~60% (24.6 ± 6.4) compared to rats subjected to 100 minutes of tMCAO and treated with negative control (59.3 ± 2.1) (Figure 6a). Also, in this case, the reduction in the ischemic volume induced by anti-miR-103-1 was mirrored by amelioration in general and focal scores (Figure 6b).
Figure 5.

Quantification of infarct volume after 100 minutes of tMCAO and administration of Anti-miR-103-1 coupled with performance of general and focal neurological deficits after anti-miR drug treatment in ischemic rats. (a) Effect of anti-miR-103-1 9 µg/kg on infarct volume of rats subjected to 100' tMCAO compared to ischemic rats treated with vehicle or with negative control (Neg CTL). On the top side are reported representative coronal brain slices indicating the extension of the infarct area (circled area). (b) Effect of anti-miR-103-1 9 µg/kg (Anti-miRNA 103-1) on general and focal scores evaluated when rats were euthanized 24 hours after tMCAO (horizontal line indicates the mean neurological scores). *P < 0.05 versus 100 minutes of MCAO (vehicle) and versus ischemic rats treated with negative control. n = 5–6 animals per group. Each column represents the mean ± SEM.
Figure 6.
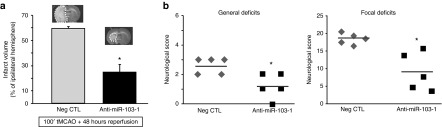
Quantification of infarct volume after 100 minutes of tMCAO and administration of Anti-miR-103-1, started 20' after ischemia induction coupled with performance of general and focal neurological deficits after Anti-miR drug treatment in ischemic rats. (a) Effect of anti-miR-103-1 9 µg/kg on infarct volume of rats subjected to 100' tMCAO compared to ischemic rats treated with vehicle or with negative control (Neg CTL). The administration of anti-miR-103-1 has been started 20' after tMCAO induction. Rats were sacrified 48 hours after ischemia to ensure the administration of 9 µg (1 µl/hour) of anti-miR. (b) Effect of anti-miR-103-1 9 µg/kg (AntimiRNA 103-1) delayed administered on general and focal scores evaluated when rats were euthanized 48 hours after tMCAO. *P < 0.05 versus 100 minutes of MCAO (vehicle) and versus ischemic rats treated with negative control. n = 5–6 animals per group. Each column represents the mean ± SEM.
Among the behavioral tests that were improved by anti-miR-103-1 treatment, it was observed an amelioration in body symmetry maintenance and in movement coordination, a reduction in the tendency of making compulsive circles, and an improvement of spontaneous activity.
Discussion
In the present paper, we identified a family of microRNA known as miRNA-103/107 as a brain miRNA able to selectively modulate NCX1 expression. In particular, we found that these microRNAs, by reducing NCX1 expression, worsened ischemic damage induced by tMCAO. More interestingly, anti-miR-103-1, by sequestering miR-103-1, induced an upregulation of NCX1 protein expression, thus exerting a remarkable neuroprotective effect in stroke even when administered after stroke onset. Interestingly, this action was accompanied by a marked improvement in general and focal neurological scores of animals subjected to stroke.
Notable, anti-miR-103-1 exerted a neuroprotective effect through NCX1 activation without affecting the expression of the other two NCX brain isoforms, NCX2 and NCX3, both of which are also involved in brain ischemia.21,22
The explanation of the neuroprotective role exerted by NCX1 and observed in the present study lies on our previous in vivo and in vitro studies. Indeed, in in vivo studies we have shown that brain damage worsens in ischemic rats in which NCX1 is either downregulated by antisense strategy17 or pharmacologically blocked.3 The neuroprotective action of NCX1 is further highlighted by our ischemic preconditioning experiments, in which an endogenous intervention causes neuroprotection by eliciting an increase in NCX1 expression and activity under in vivo and in vitro conditions.18,24 In addition, the protection elicited by ischemic preconditioning is remarkably reduced by NCX1 silencing. Several other findings obtained in in vivo models of stroke with drugs modulating NCX activity, further corroborate NCX1 neuroprotective role.15 However, these data have often been questioned by the lack of selectivity toward the different NCX isoforms, thereby hampering the conclusions drawn by these pharmacological tools.3,15,17,18 Furthermore, several papers suggested a neurodetrimental role for NCX1. In fact, it has been shown that pharmacological inhibition of NCX activity protects the brain against stroke30,31 and a smaller infarct volume was observed in ischemic C57BL/6J mice heterozygous for ncx131. However, it has also been demonstrated that these drugs are not selective for NCX,32 and that the heterozygous genetic ablation of ncx1 does not influence infarct volume in 129/SV background mice subjected to 90 minutes of tMCAo and 24 hours of reperfusion.33
At the cellular level, we demonstrated that in cortical neurons exposed to oxygen and glucose deprivation,23,24 the antiporter NCX1, working in the reverse mode, promotes Ca2+-influx and favors Ca2+-refilling into the endoplasmic reticulum, which is depleted under these anoxic conditions, thus allowing neurons to delay the apoptotic process.23 Moreover, under these conditions, NCX1 also contributes to counteracting [Na+]i overload, which causes cell swelling and neuronal death.25
Another molecular event that explains the neuroprotective effect exerted by anti-miR-103-1 on NCX1 is that this isoform is the target of the prosurvival pathways PI3K/AKT and MAPK26,27 and that the NCX1 promoter is activated by transcriptional factors, which are activated during stroke, such as HIF-1 and REST.28,29
In this scenario, anti-miR-103-1 emerged in our study as the first compound capable of selectively increasing NCX1 expression in the brain.
Another aspect of the present study is that the correlation between miR-103-1 and NCX1 levels of expression was manifest only at one time point. In particular, we observed a clear inverse correlation between miR-103-1 and NCX1 levels of expression in the brain 24 hours after the ischemic insult, whereas no such correlation was detected at earlier time intervals and actually disappeared at later time intervals. A possible explanation for this discrepancy is that cerebral ischemia is a complex pathological phenomenon that implies the intervention of other molecular pathways such as caspase cascade and calpain activation. Therefore a verisimilar hypothesis is that although miR-103-1 represents an important NCX1 regulator, the expression of this antiporter is also influenced by other transductional and transcriptional factors, including REST,29 HIF,28 and calpains.34 On the other hand, although a single miRNA may effectively repress the expression of a single messenger RNA, such as the ncx1 gene, it may also repress the production of several other proteins. In particular, miR-103-1, identified in the present study as an NCX1 modulator, is an intronic miRNA contained in three PANK (pantothenate kinase) loci of the human genome. Recently, Martello and his colleagues demonstrated that the miR-103/107 family is able to target Dicer, a key component of the miRNA processing machinery, thus causing a global reduction in miRNA abundance in the cytoplasm.19,35,36,37 Accordingly, our working hypothesis is that the increasing levels of endogenous miR-103-1 occurring 24 hours after ischemia onset trigger a general reduction in miRNA global abundance. This effect might dramatically influence stroke progression by increasing the expression of proteins normally downregulated by those miRNAs that are no longer synthesized. In the present study, the increased expression of miR-103-1 reduced NCX1, thus worsening ischemic outcome. On the contrary, anti-miR was able to counteract ischemic brain damage by preventing stroke-induced downregulation of NCX1 expression.
Besides miR-103-1, two other miRNAs, miR-145 and miR-214, have been reported to regulate NCX1 expression.38,39 However, their activity appears to be cardiac-tissue specific, being restricted within the heart. Indeed, we did not find a direct correlation between the expression levels of these two miRNAs and NCX1 in the brain (data not shown).
Besides the obvious potential therapeutic perspectives for the use of anti-miR-103-1 in the development of future ischemic stroke treatments, the identification of a specific cerebral miRNA, related to stroke progression, can also represent a useful strategy to identify peripheral biomarkers for stroke patients. Indeed, the availability of a biomarker easily detectable in the blood and temporarily related to the pathophysiological process of stroke might be a useful probe to follow the evolution of stroke lesions as well as to evaluate the clinical efficacy of drugs which ameliorate ischemic brain damage through NCX1 activation.
Overall, the identification of a microRNA able to selectively regulate cerebral NCX1 clarifies a new important molecular mechanism involved in NCX1 regulation in the brain. Indeed, our results may pave the way for the development of new diagnostic tools and more promising and effective therapeutic strategies for stroke.
Materials and Methods
Specific miRNA identification. HOCTAR database was used to identify a list of miRNAs potentially able to interact with NCX1 sequence. In brief, this database allows predictions of miRNA-RNA interactions on the basis of an inverse relationship between a miRNA and its mRNA target. This database has been developed for sequence-based human miRNA target recognition, but the high degree of sequence homology between mRNA of NCX1 in homo sapiens and in rattus norvegicus allowed us to use it for the recognition of putative microRNA interactors. This resource is based on the assumption that intronic microRNAs (60–70% of total miRNAs in a cell) are part of a single transcriptional unit within confining genes; therefore, monitoring the expression of a neighboring gene is possible to obtain an indication of the expression of the embedded miRNA.40 Inverse correlation information between all host genes and target genes of embedded miRNAs derives from experimental evidence on miRNA-mRNA interactions previously validated by microarray experiments. This procedure suggests that the miRNAs contained in their host gene directly modulate the inversely correlated target genes.
Thus, in the present study, using HOCTAR database, we were able to predict the existence of a strict complementarity between NCX1 and miR-103-1. Such prediction was based on the Watson-Crick annealing between the “seed sequence” of the miRNA of interest and the target sequence on NCX1 mRNA. This was tested by pairing software IBM RNA 22 (http://cbcsrv.watson.ibm.com/rna22.html).41
Drugs and chemicals. miRIDIAN microRNA hsa-miR-103a-3p mimics (C-300522-03-0005 5 nmol) corresponding to the mirbase accession (MIMAT0000101 and miRIDIAN Mimic Transfection Control with Dy547 (Cp-004500-01-05 5nmol)) were purchased from Thermo Fisher Scientific (Waltham, MA). Locked nucleic acids (LNA) anti-miR-103-1 (414336-00 predesigned miRCURY LNA microRNA Inhibitor, 5 nmol) and negative control anti-miR (199004-00, microRNA miRCURY LNA Power Antisense Control A, 5 nmol) were purchased from Exiqon (Vedbaek, Denmark).
Cell cultures. Baby hamster kidney (BHK) cells stably transfected with canine cardiac NCX1, containing two miRNA 103-1 binding sites (site 1 756–761 and site 2 2812–2818), rat pheochromocytoma cells (PC-12 cells), and cortical neurons from brains of 14-day-old rat embryos were prepared as previously described.14,24,42,43
Transfection of BHK cells, PC12, and cortical neurons. PC12 cells and BHK were transfected with 10, 50, 100, and 150 nmol/l of hsa-mir-103-1 Mimic and with 10, 50, 100, and 150 nmol/l of microRNA Mimic Trasfection Control according to the manufacturer's protocol (Applied Biosystems, Carlsbad, CA). After 5 hours of incubation, the medium was replaced, and 24, 48, and 72 hours after transfection, cells were harvested and used for western blot or polymerase chain reaction (PCR) real-time analysis. Conversely, rat cortical neurons were transfected with 150 nmol/l of hsa-mir-103-1 for 72 hours.27,44
Western blot analysis. PC12 cells, BHK cells, rat cortical neurons, and rat brain samples were homogenized in a lysis buffer (50 mmol/l Tris–HCl, pH 7.5, 100 mmol/l NaCl, 1% Triton X-100) containing protease and phosphatase inhibitors. After centrifugation at 12,000 g at 4 °C for 5 minutes, the supernatants were collected. Protein concentration was estimated using the Bradford reagent. Then, 50 μg of protein was mixed with a Laemmli sample buffer and boiled at 95 °C for 5 minutes. The samples were resolved by sodium dodecyl sulfate–polyacrylamide gel electrophoresis and transferred to polyvinylidene difluoride membranes. Blots were probed with antibodies to NCX1 (1:1,000, Swant, Marly, Switzerland), NCX2 (1:1,000, Alpha Diagnostic, San Antonio, TX), and NCX3 (1:2,000, a kind gift from Prof Philipson and Prof Nicoll, UCLA, Los Angeles, CA), and α-tubulin (1:2,000; Abcam, MA) diluted in Tris-buffered saline (TBS-T) 1% bovine serum albumin overnight (4 °C).
Analysis by real-time PCR. Total RNA was extracted from cells with TRIZOL according to the manufacturer's protocol (Invitrogen, Monza Italy). The cDNA was synthesized from 5 µg of total RNA extracted from cells using reverse transcriptase MultiScribe for the retrotranscription polymerase reaction. The semiquantitative polymerase reaction was performed as previously described.28 The pairs of oligonucleotides used were 5′-ACCACCAAGACTACAGTGCG-3′ and 5′-TTGGAAGCTGGTCTGTCTCC-3′ for NCX1; 5′-CCTGCTGGATTACATTAAAGCACTG-3′ and 5′-CCTGAAGTACTCATTATAGTCAAG-3′ for the HPRT gene. The differences in mRNA content between groups were calculated as previously described.45 The microRNA extraction from brain samples of cerebral cortex and striatum was achieved using Mirvana miRNA Isolation Kit according to manufacturer's protocol (Applied Biosystems). Rodents were euthanized up to 6, 24, and 72 hours after ischemia induction, and samples were collected. The amplification and the normalization of the microRNA of interest was performed by real-time PCR (qRT-PCR). TaqMan probes were used (Life Technologies, Monza, Italy). miRNA assay for rno-miR-103-1 (batch ID 4427975 miRNA mature sequence detection), miRNA assay for rno-miR-107 (batch ID 4427975), and miRNA assay for rno-miR 4.5S (H) as endogenous control (batch ID 001716) were carried out. For both miRNAs of interest, a primer for reverse transcription of cDNA from RNA extraction (TaqMan MicroRNA Reverse Transcription Kit, Applied Biosystems) and a pair of PCR primers (forward and reverse), optimized for the detection and sensitive amplification of specific miRNAs by RT-PCR, were provided.
Luciferase assay for miR-103-1. To construct the luciferase reporter plasmids ncx1 3′-UTR FL (genomic coordinates chr6: 4420046-4421221), ncx1 3′-UTR site 1, and ncx1 3′-UTR site 2&3, each 3′-UTR sequence of NCX1 was amplified from rat genomic DNA by PCR and ligated into pCR 2.01 vector (Invitrogen) according to the directional TOPO cloning kit (Invitrogen) protocol, and subsequently cloned into Nhe1 and XhoI site of the pmirGlo plasmid (Promega, Madison,WI). The sequences of primers used in the aforementioned construction were ncx1 3′-UTR FL FW 5′-GCTGATGGAATCCAGCTTCAAG-3′, RV 5′-GCTTCTTAGAAGTGAGCGTGCAG-3′, ncx1 3′-UTR site 1 FW 5′-CCATCATCTCCCATCATCGAG-3′, RV 5′-CGATCCCGAATCCACTCACC-3′, ncx1 3′-UTR site 2&3 FW 5′-GCAGCAGGATGATTAACC-3′, RV 5′-CTCACTTATGTCTTCAATGTCC-3′ FW. The correct sequences and orientation of all constructs were verified by sequencing (Primm, Milan, Italy). Transient transfections of PC12 cells were performed using Lipofectamine 2000 (Invitrogen) according to the manufacturer's instructions. For the luciferase assay, cells were plated at a density of 2 × 105 cells/well in 24-well plates and cotransfected after 24 hours with 0.2 μg pGL3 reporter plasmid and 0.002 μg renilla plasmid (Promega) or 0.1 μg pmirGLO reporter plasmid (Promega), as well as 100 and 150 nmol/l miRNA mimics per well. Luciferase activities were measured 24 hours after transfection using the Dual luciferase reporter assay system (Promega) and a GloMax Luminometer (Promega). All experiments were performed in triplicate with data averaged from at least three independent experiments.
In vivo experimental groups. One hundred and three Sprague–Dawley male rats (Charles River Laboratories, Calco, Varese, Italy) weighing 250 to 300 g were housed under diurnal lighting conditions (12-hour darkness/light). Experiments were performed according to the international guidelines for animal research. The experimental protocol was approved by the Animal Care Committee of the “Federico II” University of Naples.
Transient focal ischemia and anti-miR administration. Transient focal ischemia was induced, as previously described46 by individuals who did not implant osmotic pumps in animals. In brief, suture occlusion of the middle cerebral artery (MCA) was performed in male rats anesthetized with 1.5% sevofluorane, 70% N2O, and 28.5% O2. Ischemia was induced surgically as follows. A 5-O surgical monofilament nylon suture (Doccol, Sharon, MA) was inserted from the external carotid artery into the internal carotid artery and advanced into the circle of Willis up to the branching point of the MCA, thereby occluding the MCA.47 Achievement of ischemia was confirmed by monitoring regional cerebral blood flow in the area of the right MCA. Cerebral blood flow was monitored through a disposable microtip fiber optic probe (diameter 0.5 mm) connected through a Master Probe to a laser Doppler computerized main unit (PF5001; Perimed, Järfälla, Sweden) and analyzed using PSW Perisoft 2.5.46 Animals not showing a cerebral blood flow reduction of at least 70% were excluded from the experimental group, as well as animals that died after ischemia induction (Supplementary Table S1). Rectal temperature was maintained at 37 ± 0.5 °C with a thermostatically controlled heating pad and lamp. All surgical procedures were performed under an operating stereomicroscope.
Rats were divided into three experimental groups: (i) sham-operated rats receiving icv infusion of vehicle, (ii) ischemic rats receiving icv infusion of anti-miR-103-1 at different dosages, and (iii) ischemic rats receiving icv infusion of negative control anti-miR. All animals were killed 24 hours or 48 hours after the 100 minutes tMCAO by an overdose of sevoflurane, to evaluate either the infarct volume or protein expression.
The continuous release of anti-miR-103-1 into brain lateral ventricle was achieved by using osmotic pumps (Alzet, Palo Alto, CA). The osmotic pumps were prefilled with anti-miR-103-1, anti-miR-107 or negative control anti-miR in a blinded manner by individuals who did not performe tMCAO surgery on animals. Implantation of the osmotic pump frame was carried out in rats positioned on a stereotaxic apparatus 24 hours before the induction of transient ischemia.17 The osmotic pump was connected to a brain infusion kit (Alzet, n° 0004760) made of a stainless steel cannula that was implanted into the right lateral ventricle using the stereotaxic coordinates from the bregma: 0.4 mm caudal, 2 mm lateral, and 2 mm below the dura and secured to the skull with dental cement.46,48,49 The pump was placed in the skin fold on the neck of the rat. The 48-hour anti-miR infusion allowed us to overcome problems related to the short half-life of miRNA (ca. 1–3,5 hours). Anti-miR-103-1 and the negative control anti-miR were diluted to the final concentration in saline solution (0.9% NaCl g/l) previously filtered (Microglass filters). The anti-miR was icv administered at the concentrations of 3 µmol/l (2 µg/kg body weight) and 8 µmol/l (9 µg/kg body weight) starting either 24 hours before ischemia induction or 20 minutes after ischemia onset. In both cases, the release of anti-miR by the osmotic pump within the rat cerebral ventricle was set up at a speed of 1 µl/hour. Rectal temperature was maintained at 37 ± 0.5 °C with a thermostatically controlled heating pad during the whole surgical procedure. In the animals, a catheter was inserted into the femoral artery to measure arterial blood gases before and after ischemia (Rapid Laboratory 860; Chiron Diagnostic, Emeryville, CA). All surgical procedures were performed under an operating stereomicroscope.
Evaluation of the infarct volume and of neurological deficit scores. Animals were killed with sevoflurane overdose 24 or 48 hours after ischemia. Brains were quickly removed, sectioned coronally at 1 mm intervals, and stained by immersion in the vital dye (2%) 2,3,5-triphenyltetrazolium hydrochloride. The infarct volume was calculated by summing the infarction areas of all sections and by multiplying the total by slice thickness. To avoid that edema could affect the infarct volume value, we expressed infarct volume as percentage of the infarct by dividing the infarct volume by the total ipsilateral hemispheric volume.10,17 Neurological scores were evaluated 24 or 48 hours after reperfusion according to the following two scales: a general neurological scale and a focal neurological scale. In the general score, the following six general neurological functions were evaluated: (i) hair conditions (0–2), (ii) position of ears (0–2), (iii) eyes conditions (0–4), (iv) posture (0–4), (v) spontaneous activity (0–4), and (vi) epileptic behavior (0–12). For each of the six general functions measured, animals received a score depending on the severity of the symptoms, higher is the score worse is the rat condition. The scores of investigated items were then summed to provide a total general score ranging from 0 to 28. In the focal score, the following seven areas were assessed: (i) body symmetry, (ii) gait, (iii) climbing, (iv) circling behavior, (v) front limb symmetry, (vi) compulsory circling, and (vii) whisker response. For each of these items, animals were rated between 0 and 4 depending on the severity. The seven items were then summed to give a total focal score ranging between 0 and 28.50 Infarct volumes and neurological scores were evaluated in a blinded manner by individuals who did not perform the surgical procedures.
Statistical analysis. The data were evaluated as means ± SEM. Statistically significant differences among means were determined by analysis of variance followed by Student–Newman–Keuls test. The threshold for statistical significance data was set at P < 0.05.
The data related to focal and general neurological deficits, being nonparametric data, were analyzed using the nonparametric Kruskal–Wallis test, followed by the Nemenyi test for the nonparametric multiple comparison. Statistical significance was accepted at the 95% confidence level (P < 0.05).
SUPPLEMENTARY MATERIAL Figure S1. Effect of miRNA-107 on NCX1 expression in PC12 cells. Figure S2. Effect of anti-miRNA-107 on NCX1 expression in brain samples harvested from ischemic rats. Table S1. Number of animals included in the different experimental groups or excluded because died or because cerebral blood flow (CBF) did not reach values less than 70% compared to pre-stroke condition.
Acknowledgments
The authors thank Paola Merolla, for language editing of the paper, and Mr Vincenzo Grillo and Mr Carmine Capitale for technical support. This work was supported by the following grant: PON 01_01602 by MIUR to A.L.
Supplementary Material
Effect of miRNA-107 on NCX3 expression in PC12 cells.
Effect of anti-miRNA-107 on NCX1 expression in brain samples harvested from ischemic rats.
Number of animals included in the different experimental groups or excluded because died or because cerebral blood flow (CBF) did not reach values less than 70% compared to pre-stroke condition.
References
- Esteller M. Non-coding RNAs in human disease. Nat Rev Genet. 2011;12:861–874. doi: 10.1038/nrg3074. [DOI] [PubMed] [Google Scholar]
- Gambari R, Fabbri E, Borgatti M, Lampronti I, Finotti A, Brognara E, et al. Targeting microRNAs involved in human diseases: a novel approach for modification of gene expression and drug development. Biochem Pharmacol. 2011;82:1416–1429. doi: 10.1016/j.bcp.2011.08.007. [DOI] [PubMed] [Google Scholar]
- Annunziato L, Pignataro G, Di Renzo GF. Pharmacology of brain Na+/Ca2+ exchanger: from molecular biology to therapeutic perspectives. Pharmacol Rev. 2004;56:633–654. doi: 10.1124/pr.56.4.5. [DOI] [PubMed] [Google Scholar]
- Osada H, Takahashi T. MicroRNAs in biological processes and carcinogenesis. Carcinogenesis. 2007;28:2–12. doi: 10.1093/carcin/bgl185. [DOI] [PubMed] [Google Scholar]
- Lim LP, Lau NC, Garrett-Engele P, Grimson A, Schelter JM, Castle J, et al. Microarray analysis shows that some microRNAs downregulate large numbers of target mRNAs. Nature. 2005;433:769–773. doi: 10.1038/nature03315. [DOI] [PubMed] [Google Scholar]
- Kim J, Krichevsky A, Grad Y, Hayes GD, Kosik KS, Church GM, et al. Identification of many microRNAs that copurify with polyribosomes in mammalian neurons. Proc Natl Acad Sci USA. 2004;101:360–365. doi: 10.1073/pnas.2333854100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kosik KS, Krichevsky AM. The Elegance of the MicroRNAs: A Neuronal Perspective. Neuron. 2005;47:779–782. doi: 10.1016/j.neuron.2005.08.019. [DOI] [PubMed] [Google Scholar]
- Schratt GM, Tuebing F, Nigh EA, Kane CG, Sabatini ME, Kiebler M, et al. A brain-specific microRNA regulates dendritic spine development. Nature. 2006;439:283–289. doi: 10.1038/nature04367. [DOI] [PubMed] [Google Scholar]
- Boscia F, Gala R, Pignataro G, de Bartolomeis A, Cicale M, Ambesi-Impiombato A, et al. Permanent focal brain ischemia induces isoform-dependent changes in the pattern of Na+/Ca2+ exchanger gene expression in the ischemic core, periinfarct area, and intact brain regions. J Cereb Blood Flow Metab. 2006;26:502–517. doi: 10.1038/sj.jcbfm.9600207. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Tortiglione A, Scorziello A, Giaccio L, Secondo A, Severino B, et al. Evidence for a protective role played by the Na+/Ca2+ exchanger in cerebral ischemia induced by middle cerebral artery occlusion in male rats. Neuropharmacology. 2004;46:439–448. doi: 10.1016/j.neuropharm.2003.09.015. [DOI] [PubMed] [Google Scholar]
- Tortiglione A, Pignataro G, Minale M, Secondo A, Scorziello A, Di Renzo GF, et al. Na+/Ca2+ exchanger in Na+ efflux-Ca2+ influx mode of operation exerts a neuroprotective role in cellular models of in vitro anoxia and in vivo cerebral ischemia. Ann N Y Acad Sci. 2002;976:408–412. doi: 10.1111/j.1749-6632.2002.tb04768.x. [DOI] [PubMed] [Google Scholar]
- Craner MJ, Newcombe J, Black JA, Hartle C, Cuzner ML, Waxman SG. Molecular changes in neurons in multiple sclerosis: altered axonal expression of Nav1.2 and Nav1.6 sodium channels and Na+/Ca2+ exchanger. Proc Natl Acad Sci USA. 2004;101:8168–8173. doi: 10.1073/pnas.0402765101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ketelaars SO, Gorter JA, Aronica E, Wadman WJ. Calcium extrusion protein expression in the hippocampal formation of chronic epileptic rats after kainate-induced status epilepticus. Epilepsia. 2004;45:1189–1201. doi: 10.1111/j.0013-9580.2004.03304.x. [DOI] [PubMed] [Google Scholar]
- Pannaccione A, Secondo A, Molinaro P, D'Avanzo C, Cantile M, Esposito A, et al. A new concept: Aβ1-42 generates a hyperfunctional proteolytic NCX3 fragment that delays caspase-12 activation and neuronal death. J Neurosci. 2012;32:10609–10617. doi: 10.1523/JNEUROSCI.6429-11.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Molinaro P, Cantile M, Cuomo O, Secondo A, Pannaccione A, Ambrosino P, et al. Neurounina-1, a novel compound that increases Na+/Ca2+ exchanger activity, effectively protects against stroke damage. Mol Pharmacol. 2013;83:142–156. doi: 10.1124/mol.112.080986. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Sirabella R, Anzilotti S, Di Renzo G, Annunziato L. Does Na+/Ca2+ exchanger, NCX, represent a new druggable target in stroke intervention. Transl Stroke Res. 2014;5:145–155. doi: 10.1007/s12975-013-0308-8. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Gala R, Cuomo O, Tortiglione A, Giaccio L, Castaldo P, et al. Two sodium/calcium exchanger gene products, NCX1 and NCX3, play a major role in the development of permanent focal cerebral ischemia. Stroke. 2004;35:2566–2570. doi: 10.1161/01.STR.0000143730.29964.93. [DOI] [PubMed] [Google Scholar]
- Pignataro G, Boscia F, Esposito E, Sirabella R, Cuomo O, Vinciguerra A, et al. NCX1 and NCX3: two new effectors of delayed preconditioning in brain ischemia. Neurobiol Dis. 2012;45:616–623. doi: 10.1016/j.nbd.2011.10.007. [DOI] [PubMed] [Google Scholar]
- Martello G, Rosato A, Ferrari F, Manfrin A, Cordenonsi M, Dupont S, et al. A MicroRNA targeting dicer for metastasis control. Cell. 2010;141:1195–1207. doi: 10.1016/j.cell.2010.05.017. [DOI] [PubMed] [Google Scholar]
- Trajkovski M, Hausser J, Soutschek J, Bhat B, Akin A, Zavolan M, et al. MicroRNAs 103 and 107 regulate insulin sensitivity. Nature. 2011;474:649–653. doi: 10.1038/nature10112. [DOI] [PubMed] [Google Scholar]
- Jeon D, Yang YM, Jeong MJ, Philipson KD, Rhim H, Shin HS. Enhanced learning and memory in mice lacking Na+/Ca2+ exchanger 2. Neuron. 2003;38:965–976. doi: 10.1016/s0896-6273(03)00334-9. [DOI] [PubMed] [Google Scholar]
- Molinaro P, Cuomo O, Pignataro G, Boscia F, Sirabella R, Pannaccione A, et al. Targeted disruption of Na+/Ca2+ exchanger 3 (NCX3) gene leads to a worsening of ischemic brain damage. J Neurosci. 2008;28:1179–1184. doi: 10.1523/JNEUROSCI.4671-07.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sirabella R, Secondo A, Pannaccione A, Scorziello A, Valsecchi V, Adornetto A, et al. Anoxia-induced NF-kappaB-dependent upregulation of NCX1 contributes to Ca2+ refilling into endoplasmic reticulum in cortical neurons. Stroke. 2009;40:922–929. doi: 10.1161/STROKEAHA.108.531962. [DOI] [PubMed] [Google Scholar]
- Scorziello A, Santillo M, Adornetto A, Dell'aversano C, Sirabella R, Damiano S, et al. NO-induced neuroprotection in ischemic preconditioning stimulates mitochondrial Mn-SOD activity and expression via Ras/ERK1/2 pathway. J Neurochem. 2007;103:1472–1480. doi: 10.1111/j.1471-4159.2007.04845.x. [DOI] [PubMed] [Google Scholar]
- Annunziato L, Pignataro G, Boscia F, Sirabella R, Formisano L, Saggese M, et al. ncx1, ncx2, and ncx3 gene product expression and function in neuronal anoxia and brain ischemia. Ann N Y Acad Sci. 2007;1099:413–426. doi: 10.1196/annals.1387.050. [DOI] [PubMed] [Google Scholar]
- Formisano L, Saggese M, Secondo A, Sirabella R, Vito P, Valsecchi V, et al. The two isoforms of the Na+/Ca2+ exchanger, NCX1 and NCX3, constitute novel additional targets for the prosurvival action of Akt/protein kinase B pathway. Mol Pharmacol. 2008;73:727–737. doi: 10.1124/mol.107.042549. [DOI] [PubMed] [Google Scholar]
- Sirabella R, Secondo A, Pannaccione A, Molinaro P, Formisano L, Guida N, et al. ERK1/2, p38, and JNK regulate the expression and the activity of the three isoforms of the Na+ /Ca2+ exchanger, NCX1, NCX2, and NCX3, in neuronal PC12 cells. J Neurochem. 2012;122:911–922. doi: 10.1111/j.1471-4159.2012.07838.x. [DOI] [PubMed] [Google Scholar]
- Valsecchi V, Pignataro G, Del Prete A, Sirabella R, Matrone C, Boscia F, et al. NCX1 is a novel target gene for hypoxia-inducible factor-1 in ischemic brain preconditioning. Stroke. 2011;42:754–763. doi: 10.1161/STROKEAHA.110.597583. [DOI] [PubMed] [Google Scholar]
- Formisano L, Guida N, Valsecchi V, Pignataro G, Vinciguerra A, Pannaccione A, et al. NCX1 is a new rest target gene: role in cerebral ischemia. Neurobiol Dis. 2013;50:76–85. doi: 10.1016/j.nbd.2012.10.010. [DOI] [PubMed] [Google Scholar]
- Jeffs GJ, Meloni BP, Bakker AJ, Knuckey NW. The role of the Na(+)/Ca(2+) exchanger (NCX) in neurons following ischaemia. J Clin Neurosci. 2007;14:507–514. doi: 10.1016/j.jocn.2006.07.013. [DOI] [PubMed] [Google Scholar]
- Morimoto N, Kita S, Shimazawa M, Namimatsu H, Tsuruma K, Hayakawa K, et al. Preferential involvement of Na+/Ca2+ exchanger type-1 in the brain damage caused by transient focal cerebral ischemia in mice. Biochem Biophys Res Commun. 2012;429:186–190. doi: 10.1016/j.bbrc.2012.10.114. [DOI] [PubMed] [Google Scholar]
- Reuter H, Henderson SA, Han T, Matsuda T, Baba A, Ross RS, et al. Knockout mice for pharmacological screening: testing the specificity of Na+-Ca2+ exchange inhibitors. Circ Res. 2002;91:90–92. doi: 10.1161/01.res.0000027529.37429.38. [DOI] [PubMed] [Google Scholar]
- Luo J, Wang Y, Chen H, Kintner DB, Cramer SW, Gerdts JK, et al. A concerted role of Na+ -K+ -Cl- cotransporter and Na+/Ca2+ exchanger in ischemic damage. J Cereb Blood Flow Metab. 2008;28:737–746. doi: 10.1038/sj.jcbfm.9600561. [DOI] [PubMed] [Google Scholar]
- Bano D, Young KW, Guerin CJ, Lefeuvre R, Rothwell NJ, Naldini L, et al. Cleavage of the plasma membrane Na+/Ca2+ exchanger in excitotoxicity. Cell. 2005;120:275–285. doi: 10.1016/j.cell.2004.11.049. [DOI] [PubMed] [Google Scholar]
- Kumar MS, Lu J, Mercer KL, Golub TR, Jacks T. Impaired microRNA processing enhances cellular transformation and tumorigenesis. Nat Genet. 2007;39:673–677. doi: 10.1038/ng2003. [DOI] [PubMed] [Google Scholar]
- Lu J, Getz G, Miska EA, Alvarez-Saavedra E, Lamb J, Peck D, et al. MicroRNA expression profiles classify human cancers. Nature. 2005;435:834–838. doi: 10.1038/nature03702. [DOI] [PubMed] [Google Scholar]
- Ozen M, Creighton CJ, Ozdemir M, Ittmann M. Widespread deregulation of microRNA expression in human prostate cancer. Oncogene. 2008;27:1788–1793. doi: 10.1038/sj.onc.1210809. [DOI] [PubMed] [Google Scholar]
- Aurora AB, Mahmoud AI, Luo X, Johnson BA, van Rooij E, Matsuzaki S, et al. MicroRNA-214 protects the mouse heart from ischemic injury by controlling Ca2+ overload and cell death. J Clin Invest. 2012;122:1222–1232. doi: 10.1172/JCI59327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tritsch E, Mallat Y, Lefebvre F, Diguet N, Escoubet B, Blanc J, et al. An SRF/miR-1 axis regulates NCX1 and annexin A5 protein levels in the normal and failing heart. Cardiovasc Res. 2013;98:372–380. doi: 10.1093/cvr/cvt042. [DOI] [PubMed] [Google Scholar]
- Gennarino VA, Sardiello M, Avellino R, Meola N, Maselli V, Anand S, et al. MicroRNA target prediction by expression analysis of host genes. Genome Res. 2009;19:481–490. doi: 10.1101/gr.084129.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennarino VA, Sardiello M, Mutarelli M, Dharmalingam G, Maselli V, Lago G, et al. HOCTAR database: a unique resource for microRNA target prediction. Gene. 2011;480:51–58. doi: 10.1016/j.gene.2011.03.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secondo A, Staiano RI, Scorziello A, Sirabella R, Boscia F, Adornetto A, et al. BHK cells transfected with NCX3 are more resistant to hypoxia followed by reoxygenation than those transfected with NCX1 and NCX2: Possible relationship with mitochondrial membrane potential. Cell Calcium. 2007;42:521–535. doi: 10.1016/j.ceca.2007.01.006. [DOI] [PubMed] [Google Scholar]
- Greene LA, Tischler AS. Establishment of a noradrenergic clonal line of rat adrenal pheochromocytoma cells which respond to nerve growth factor. Proc Natl Acad Sci USA. 1976;73:2424–2428. doi: 10.1073/pnas.73.7.2424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Secondo A, Pannaccione A, Cataldi M, Sirabella R, Formisano L, Di Renzo G, et al. Nitric oxide induces [Ca2+]i oscillations in pituitary GH3 cells: involvement of IDR and ERG K+ currents. Am J Physiol Cell Physiol. 2006;290:C233–C243. doi: 10.1152/ajpcell.00231.2005. [DOI] [PubMed] [Google Scholar]
- Formisano L, Noh KM, Miyawaki T, Mashiko T, Bennett MV, Zukin RS. Ischemic insults promote epigenetic reprogramming of mu opioid receptor expression in hippocampal neurons. Proc Natl Acad Sci USA. 2007;104:4170–4175. doi: 10.1073/pnas.0611704104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignataro G, Meller R, Inoue K, Ordonez AN, Ashley MD, Xiong Z, et al. In vivo and in vitro characterization of a novel neuroprotective strategy for stroke: ischemic postconditioning. J Cereb Blood Flow Metab. 2008;28:232–241. doi: 10.1038/sj.jcbfm.9600559. [DOI] [PubMed] [Google Scholar]
- Longa EZ, Weinstein PR, Carlson S, Cummins R. Reversible middle cerebral artery occlusion without craniectomy in rats. Stroke. 1989;20:84–91. doi: 10.1161/01.str.20.1.84. [DOI] [PubMed] [Google Scholar]
- Vemuganti R. The MicroRNAs and Stroke: No Need to be Coded to be Counted. Transl Stroke Res. 2010;1:158–160. doi: 10.1007/s12975-010-0030-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dharap A, Bowen K, Place R, Li LC, Vemuganti R. Transient focal ischemia induces extensive temporal changes in rat cerebral microRNAome. J Cereb Blood Flow Metab. 2009;29:675–687. doi: 10.1038/jcbfm.2008.157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark WM, Lessov NS, Dixon MP, Eckenstein F. Monofilament intraluminal middle cerebral artery occlusion in the mouse. Neurol Res. 1997;19:641–648. doi: 10.1080/01616412.1997.11740874. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Effect of miRNA-107 on NCX3 expression in PC12 cells.
Effect of anti-miRNA-107 on NCX1 expression in brain samples harvested from ischemic rats.
Number of animals included in the different experimental groups or excluded because died or because cerebral blood flow (CBF) did not reach values less than 70% compared to pre-stroke condition.