

Abstract
Optimum clinical protocols require systemic delivery of oncolytic viruses in the presence of an intact immune system. We show that preconditioning with immune modulators, or loading virus onto carrier cells ex vivo, enhances virus-mediated antitumor activity. Our early trials of systemic reovirus delivery showed that after infusion reovirus could be recovered from blood cells—but not from plasma—suggesting that rapid association with blood cells may protect virus from neutralizing antibody. We therefore postulated that stimulation of potential carrier cells directly in vivo before intravenous viral delivery would enhance delivery of cell-associated virus to tumor. We show that mobilization of the CD11b+ cell compartment by granulocyte macrophage-colony stimulating factor immediately before intravenous reovirus, eliminated detectable tumor in mice with small B16 melanomas, and achieved highly significant therapy in mice bearing well-established tumors. Unexpectedly, cytokine conditioning therapy was most effective in the presence of preexisting neutralizing antibody. Consistent with this, reovirus bound by neutralizing antibody effectively accessed monocytes/macrophages and was handed off to tumor cells. Thus, preconditioning with cytokine stimulated recipient cells in vivo for enhanced viral delivery to tumors. Moreover, preexisting neutralizing antibody to an oncolytic virus may, therefore, even be exploited for systemic delivery to tumors in the clinic.
Introduction
Oncolytic virotherapy is based on the concept that a replicating virus introduced into a tumor will rapidly spread through and lyse that tumor, with targeted replication being possible through natural, or engineered, selectivity.1 Encouragingly, several viruses are currently entering later-stage clinical trials, and a randomized phase III study (OPTiM) using herpes simples virus therapy for melanoma has achieved its primary endpoint, with a durable response rate of 16% seen in patients receiving herpes simples virus compared with 3% in the control arm.2 Trials of this sort have also highlighted the multicomponent role of the immune system on the efficiency of virotherapy. Thus, antiviral immune responses clearly impair virus delivery to tumors after systemic administration and can restrict replication/oncolysis.3,4,5 On the other hand, virus replication does not always correlate with therapy,6,7 and tumor clearance often requires immune effectors against tumor8,9 and/or virus.4,6,7,8,9,10,11 However, the development of protocols for systemic delivery, in the presence of an intact immune system, to metastatic tumors remains to be a major clinical challenge.1,12,13,14 In this respect, many barriers to efficient systemic delivery exist, including the tumor vasculature,15,16,17 virus inactivation (including by neutralizing antibody (NAb)), mislocalization, sequestration, and inadequate extravasation.13,18,19,20
In our own studies, we have developed the use of reovirus as a systemically delivered oncolytic agent in both preclinical models9,13,21,22,23,24,25,26 and in early-phase clinical trials.14,27,28,29,30 Reovirus has direct oncolytic activity against many human/murine tumor cells,29,31 partly because of disruption of the RNA-dependent protein kinase-mediated antiviral response in malignant cells.32,33 In addition, we have shown that antitumor therapy is directly associated with immune activation by virus replication in tumors.24,25 To mimic the clinical challenges of systemic delivery of oncolytic viruses, we developed a murine model in which injection of reovirus into subcutaneous (s.c.) B16 melanomas generates therapy, but intravenous (i.v.) reovirus does not.13 However, we demonstrated that i.v. virus could achieve significant activity by conditioning the host with immune modulators (IL-2/Treg depletion or cyclophosphamide),13,21,22 or by conditioning the tumor vasculature for increased reovirus localization/replication after i.v. delivery.9,23 In addition, we,12,26,34,35 and others,36,37 have successfully used carrier cells of different types, loaded ex vivo, to protect viruses from neutralization and chaperone them into tumors.19 However, translation of strategies that require in vitro expansion of carrier cells, which are subsequently loaded with a replicating oncolytic virus, before i.v. delivery, is currently expensive and complex from a regulatory perspective.
From our ongoing clinical program, we have shown in a phase Ib, biological endpoint clinical study (REO13) that, after i.v. injection of reovirus before planned resection of colorectal cancer liver metastases, reovirus could be specifically detected in patient tumors at the time of surgery despite the presence of NAb in the circulation at baseline in all patients.38 Moreover, the REO13 study also demonstrated that, after systemic reovirus administration, replication-competent virus could be retrieved from mononuclear cells, granulocytes, and platelets within patient blood, but not from the plasma. These data suggested that although free reovirus is rapidly neutralized by NAb after i.v. injection, it may be successfully transported to tumors via protective carriage by blood cells. Therefore, on the basis of these clinical observations, we hypothesized that i.v. injection of reovirus results in rapid adhesion to, or infection of, blood cells, which can protect the virus from neutralization, including by NAb; moreover, it may be possible to stimulate specific cell compartments before i.v. virus injection such that virus adhesion occurs selectively to a population of cell carriers which can traffic, and deliver virus, to tumors. Consistent with this hypothesis, we show here that, after i.v. administration into mice, reovirus associated predominantly with CD11b+ cells and that stimulation of this compartment with granulocyte macrophage colony-stimulating factor (GM-CSF) before reovirus delivery significantly enhanced antitumor therapy by an immune-mediated mechanism dependent on both natural killer (NK) cells and monocytes/macrophages. Interestingly, however, GM-CSF conditioning therapy was most effective in the presence of preexisting NAb. Our data are significant in that they show that preexisting NAb to an oncolytic virus may actually be exploited for systemic delivery to tumors and extend the previous use of ex vivo–loaded cell carriers to a new concept of directed in vivo cell loading, thereby representing a readily testable, and translatable, method to enhance systemic delivery of oncolytic viruses in patients.
Results
Systemic delivery of reovirus in the presence of neutralizing antibodies
Similar to the findings of our biological endpoint clinical study,38 2 minutes after i.v. injection of reovirus, infectious virus was recovered from both plasma and cells of non–reovirus-immune mice, but could only be recovered from the cellular fraction in virus-immune mice (Figure 1a). By 30 minutes after infusion, although very low levels (nonimmune), or no (virus-immune), virus was recovered from plasma, infectious virus was still associated with the cell fraction, with significantly increased levels recoverable from blood cells from virus-immune mice compared with mice with no NAb (P < 0.01) (Figure 1a). Moreover, substantial levels of infectious virus were still recovered from blood cells up to 1 hour after injection of virus in both groups, although the difference between virus-immune and nonimmune mice was reduced by this time point (P = 0.05) (Figure 1a). At 30 minutes postvirus injection, −75% of the cell-associated reovirus recovered from virus-immune mice was in the CD11b+ cell (monocyte/macrophage) fraction (Figure 1b). These data suggest that although free virus is rapidly nullified in the circulation, blood cells, and predominantly CD11b+ cells, might protect virus for potential delivery to tumors.
Figure 1.

Reovirus administered i.v. is rapidly neutralized by Nab, but functional virus can be found associated with CD11b+ cells. (a) C57Bl/6 mice were injected with 107 TCID50 reovirus i.v. 2, 30, or 60 minutes later mice (three mice per time point); blood was collected by cardiac puncture and reovirus titer was determined by plaque assay. (b) Thirty minutes after i.v. injection of 107 TCID50 reovirus, the intact packed cell fraction was harvested and reovirus titer was determined by plaque assay. In separate mice, the CD11b+ and CD11b− fractions were recovered by microbead separation and titered for reovirus.
Reovirus/neutralizing antibody complexes remain available for transfer to tumor cells
Consistent with the hypothesis that CD11b+ cells may act as in vivo carriers for reovirus delivery to tumors, anti-reovirus NAb abrogated productive reovirus infection of B16 tumor cells, PDCA+ dendritic cells (DCs) and T cells in vitro. In contrast, productive infection of CD11b+ cells was maintained in the presence of NAb (Figure 2a,b). CD11b+ cells, DC, and T cells, preloaded with reovirus, were all able to hand virus off to B16 cells upon coculture (Figure 2c), with no significant inhibition by NAb (Figure 2d), consistent with our previous observation that murine T cells and DC carrier cells can protect virus from NAb for hand-off and subsequent replication in target tumor cells.12,26,34,35 However, reovirus preincubated with NAb before loading onto carriers cells was only inefficiently delivered to B16 cells by DC, whereas T cells were unable to hand-off virus at all under these conditions. In contrast, CD11b+ cells loaded with NAb/reovirus complexes still efficiently passed the virus on to tumor cells (Figure 2e). Taken together, these data show that NAb does not prevent direct infection of CD11b+ cells and that reovirus taken up in the form of NAb/reovirus complexes by this same population remains available for hand-off to tumor cells.
Figure 2.
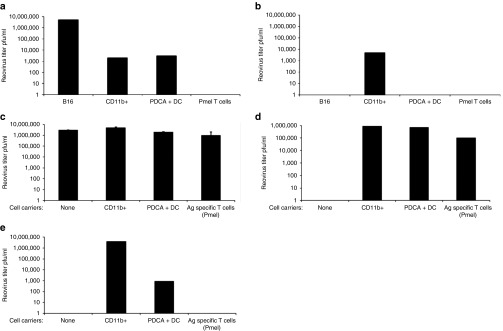
Reovirus–NAb complexes can be transferred by CD11b+ cells for productive tumor cell infection. (a,b) Target cells (as indicated) were infected with reovirus at MOI 1 in the (a) absence or (b) presence (100 µl immune serum) of reovirus NAb (in the same experiment). After 72 hours, reovirus titer was determined by plaque assay from the cells and supernatant. Representative of three separate experiments. (c,d) Carrier cells were loaded with reovirus at MOI 1 for 2 hours at 4 °C, washed twice in PBS, and cocultured with B16 tumor targets in (c) the absence or (d) presence of reovirus NAb (in the same experiment). After 72 hours, reovirus titer was determined by plaque assay from the cells and supernatant. Representative of two separate experiments. (e) Carrier cells were loaded with reovirus–NAb complexes at MOI 1 and cocultured with B16 targets. After 72 hours, reovirus titer was determined by plaque assay from the cells and supernatant. Representative of three separate experiments.
GM-CSF mobilization of CD11b+ cells into tumors in vivo
CD11b+ cells seemed to form the major potential depot for sequestration of reovirus and NAb/reovirus complexes after i.v. injection (Figures 1 and 2). Therefore, as a prelude to loading these cells in vivo with i.v. injected virus, we first tested whether it would be possible to use cytokine preconditioning to enhance their numbers and subsequent trafficking into tumors. Reovirus-immune mice bearing 10-day established s.c. B16 tumors were conditioned with three daily injections of GM-CSF, a cytokine known both to mobilize CD11b+ cells in vivo and to have potent antitumor vaccinating effects.39,40 This treatment significantly increased infiltration of CD11b+ cells into tumors compared with phosphate-buffered saline (PBS) treatment (P < 0.01) (Figure 3a). Furthermore, conditioning with GM-CSF, followed by two daily injections of reovirus, increased viral delivery to the tumors (Figure 3b), a finding consistent with GM-CSF–enhancing CD11b+ cells as in vivo carriers of i.v. injected reovirus into tumors. Importantly, no toxicity was seen in any mice with any of the treatments in this or other in vivo experiments in this study.
Figure 3.

GM-CSF mobilizes CD11b+ cells into tumorsin vivoleading to enhanced reovirus delivery to tumors. (a) C57Bl/6 mice were vaccinated i.p. with reovirus (2 × 107 TCID50). After 14 days, mice were seeded with s.c. B16 tumors. Mice bearing 10-day established s.c. B16 tumors were given three daily injections of PBS or GM-CSF; after 48 hours, tumors were harvested, dissociated, and analyzed for CD45+/CD11b+ infiltrating cells by FACS. Reovirus-vaccinated, tumor-bearing mice as in a were given three daily injections of PBS or GM-CSF followed by two daily injections of PBS or reovirus i.v. After 72 hours, tumors (b) were harvested and reovirus titer determined. The third PBS/Reo-treated mouse had no detectable virus in the explanted tumor, which may have been due to technical reasons with virus injection and/or the low levels of virus which reached the tumor in this animal in the absence of GM-CSF conditioning. FACS, fluorescence-activated cell sorting.
GM-CSF conditioning with systemic reovirus eliminates small tumors
We next investigated whether the combination of in vivo viral sequestration of NAb/reovirus complexes by CD11b+ cells, along with GM-CSF-mediated enhancement of virus delivery to s.c. tumors, translated into better therapy upon systemic virus delivery. A single cycle of GM-CSF/REO (three daily injections of GM-CSF followed by two daily injections of reovirus) generated no significant therapy of s.c. B16 tumors compared with PBS, GM-CSF alone or reovirus alone, in mice with no prior immunity to reovirus (Figure 4a). By contrast, where mice with 3-day established s.c. B16 tumors were immunized with reovirus 2 weeks before tumor implantation and, therefore, had high anti-reovirus NAb, between 80 and 100% were free of detectable tumor long term after a single cycle of GM-CSF/REO (Figure 4a). However, virus-naive, and preimmune mice, bearing 5-day established tumors were treated effectively (P < 0.0001 compared with reovirus-immune, or naive, controls alone) by giving three cycles, rather than just one, of GM-CSF/REO (Figure 4b). In separate studies, we found that the maximal NAb response to reovirus is achieved by day 7–9 after the first reovirus injection (Figure 4c), which is consistent with the timing of peak NAb levels in patients.38 Therefore, we believe that the ability of three cycles of GM-CSF/REO to treat virus-naive mice was probably due to the generation of high levels of NAb as a result of the first cycle of reovirus treatment, which effectively means that the second and third cycles were being delivered to reovirus-immune animals. Consistent with this hypothesis, a single cycle of GM-CSF/REO was ineffective as a treatment in mice previously vaccinated with the completely separate vesicular stomatitis virus (VSV) (Figure 4d); however, three cycles of GM-CSF/REO were still able to treat 5-day established s.c. B16 tumors in mice previously vaccinated with VSV, although significantly less well than if the mice had no competing antiviral antibodies before treatment (P = 0.01 compared with treatment of virus-naive or only reovirus-preimmune mice) (Figure 4b). Three cycles of GM-CSF/REO were also effective at treating large, 10-day established s.c. B16 tumors (P < 0.0001 compared with GM-CSF or reovirus alone) in virus-immune mice, although fewer mice were free of detectable tumor long term compared with when treatment was initiated at a smaller tumor size (Figure 4e). Importantly, therapy in all of these models was only effective when GM-CSF preceded reovirus (not shown).
Figure 4.
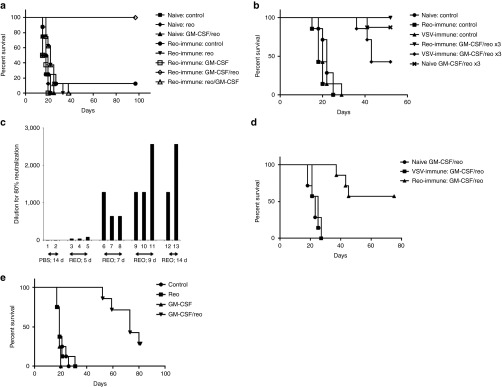
GM-CSF conditioning followed by systemic reovirus is an effective therapy in mice preimmunized with reovirus. (a) C57Bl/6 mice were vaccinated i.p. with either PBS (virus naive) or reovirus (2 × 107 TCID50) (preimmune). After 14 days, mice were seeded with s.c. B16 tumors and 3 days later were treated with once cycle of PBS/PBS, GM-CSF/PBS, PBS/reovirus, or GM-CSF/reovirus treatment (three daily injections of GM-CSF i.p. followed by two daily injections of reovirus i.v.) (eight total groups, three treatments virus naive or five treatments preimmune). Survival (tumor <1.0 cm diameter) with time is shown. (b) C57Bl/6 mice, vaccinated 14 days earlier with PBS, reovirus (2 × 107 TCID50) (preimmune) or VSV (106 pfu i.v.) (VSV immune), were seeded with s.c B16 tumors. After 5 days, these tumor bearing mice were treated with three cycles of GM-CSF/reovirus. Additional groups of mice were vaccinated with reovirus and then treated with three cycles of PBS/reovirus, GM-CSF/PBS, or PBS/PBS. Survival (tumor <1.0 cm diameter) with time is shown. (c) C57Bl/6 mice were vaccinated i.p. with either PBS (two mice) or reovirus (2 × 107 TCID50) (2–3 per group). After 5, 7, 9, or 14 days, levels of anti-reovirus NAb were measured as described in Materials and Methods (preincubation of serum from PBS-treated mice with reovirus was not able to reduce the toxicity to L929 cells to any greater extent than DMEM). (d) C57Bl/6 mice were vaccinated i.p. with either PBS (PBS(VAC)), VSV (2 × 107 pfu VSV-GFP) (VSV(VAC)) or reovirus (2 × 107 TCID50) (REO(VAC)). After 14 days, mice were seeded with s.c. B16 tumors and 5 days later (compared with treatment of 3-day established tumors in a) were treated with one cycle of GM-CSF/reovirus treatment (three daily injections of GM-CSF i.p. followed by two daily injections of reovirus i.v.). Survival (tumor <1.0 cm diameter) with time is shown. (e) C57Bl/6 mice bearing 10-day established s.c. B16 tumors were treated with three cycles of PBS/PBS, GM-CSF/PBS, PBS/reovirus, or GM-CSF/reovirus. Survival (tumor <1.0 cm diameter) with time is shown.
GM-CSF-activated immune cells are cytotoxic to reovirus-resistant tumor cells in the presence of NAb–reovirus complexes
We used the B16ova variant of B16, which we have previously shown to be largely resistant to oncolysis by reovirus due to low expression of the viral receptor JAM-A,25 to separate direct viral cytotoxicity from immune-mediated cytotoxicity against tumor cells. When immune effector cells from pooled lymph node cells and splenocytes (LN/splenocytes) from C57Bl/6 mice were cocultured with B16ova cells expressing green fluorescent protein (GFP; B16ova-GFP) alone, minimal cytotoxicity was seen relative to controls (not shown) (designated 100% survival of GFP+ve B16ova cells) (Figure 5a, (i)). The addition of reovirus led to immune-mediated killing of −42% of the B16ova-GFP cells, leaving −58% alive (Figure 5a, (ii)), consistent with the innate cytotoxicity of reovirus-activated human mononuclear cells against human melanoma cell lines in vitro.41 Treatment of the LN/splenocyte cells with GM-CSF alone did not stimulate immune cytotoxicity against B16ova-GFP in the absence of reovirus (Figure 5a, (iii)), although −57% of the tumor cells were killed by immune-mediated cytotoxicity in the presence of reovirus together with GM-CSF (Figure 5a, (iv)), i.e., similar levels to that seen with reovirus alone in Figure 5a, (ii). Preincubation of reovirus with NAb completely abrogated the reovirus alone-induced immune killing of B16ova-GFP cells in culture (Figure 5a, (ii) versus Figure 5a, (v)); however, treatment of LN/splenocytes with GM-CSF, followed by incubation with NAb–reovirus immune complexes, led to killing of >98% of the target tumor cells (Figure 5a, (vi)). GM-CSF-activated LN/splenocytes also seemed to be cytotoxic in the presence of reovirus preincubated with NAb against VSV (Figure 5a, (vii)) (albeit less so than with anti-reovirus NAb), although this effect was not reproducibly significant (Figure 7b). The NAb–reovirus immune complex-induced cytotoxicity was not affected when GM-CSF-activated LN/splenocyte cultures were depleted of either CD4 or CD8 T cells, but was completely inhibited by depletion of either NK cells or CD11b+ cells (Figure 5b). In addition, the cytotoxicity could be reconstituted only by the addition of both NK cells and CD11b+ cells together, but not separately (Figure 5c). Separation of the GM-CSF-activated LN/splenocyte cultures and the B16ova-GFP targets in transwells prevented any killing of B16 tumor cells in the presence of NAb–reovirus complexes (data not shown). Finally, cytotoxicity of GM-CSF-activated LN/splenocyte cultures against B16ova-GFP targets in the presence of NAb–reovirus complexes correlated with the production of tumor necrosis factor-alpha (TNF-α) in the cocultures, which was again dependent on the presence of both NK cells and CD11b+ cells (Figure 5d).
Figure 5.

GM-CSF is required for activation of LN/splenocytes for tumor cell killing in vitro by NAb–reovirus complexes and is dependent on NK cells and CD11b+ cells. (a) B16ova cells expressing GFP, which are resistant to direct reovirus killing, were cultured for 24 hours at a 1:10 ratio with LN/splenocyte cells ± 10 ng/ml GM-CSF. After 24 hours, reovirus, reovirus/anti-reovirus-NAb (100 µl immune serum) complexes or reovirus/anti-VSV-NAb complexes were added at MOI 1 and, after a further 6 hours, cells and supernatants were harvested. (a) (i)–(vii): The number of viable GFP-expressing cells was determined by FACS as a measure of immune-mediated cytotoxicity against the B16ova cells, and (d) TNF-α in the supernatants was determined by ELISA. (b) LN/splenocyte effector cells were first depleted of CD4+ T cells, CD8+ T cells, NK cells, or CD11b+ cells, as indicated, before culture as described in a. (c) Intact LN/splenocyte cells, or isolated NK cells, CD11b+, or combined NK + CD11b+ cells were cocultured with target tumor cells as in a, and the number of viable GFP-expressing cells was determined by FACS as a measure of cytotoxicity against the B16ova cells. FACS, fluorescence-activated cell sorting.
Figure 7.
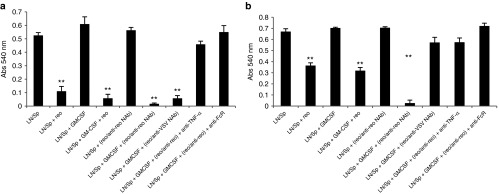
Immune mediated tumor cell killing is dependent on Fc receptors and TNF-α. A total of 103 (a) B16tk (reo-sensitive) or (b) B16ova (relatively reo-insensitive) cells were cocultured in triplicates with 104 splenocytes/LN cells ± GM-CSF (10 ng/ml). After 24 hours, reovirus, reovirus/anti-reovirus-Nab or reovirus/anti-VSV-NAb at MOI 1, ± anti-TNF-α Ab or anti-FcR CD16/CD32 antibody (1 µg/million cells) as indicated, was added to the wells. After a further 24 hours, cultures were washed three times with PBS to remove nonadherent cells, and cell killing was assayed using an MTT assay (absorbance at 540 nm). Graph shows mean ± SD. **P < 0.01. Representative of two separate experiments.
GM-CSF conditioning enhances Fc receptor expression and NAb–reovirus immune complex transfer from CD11b+ cells
To further explore the mechanism(s) by which GM-CSF may enhance reovirus therapy in the context of NAb, we next addressed the effects of GM-CSF on Fc receptor expression by immune cell populations in vitro. GM-CSF treatment of bulk LN/splenocyte cultures upregulated expression of a representative Fc receptor (FcγR1) across the whole cell population, as measured by RTPCR. Statistically significant increased FcR expression was observed between cultures treated with GM-CSF compared with untreated controls (mean +1.0, ±0, lanes 5–7; compared with mean −2.3, ±0.58, lanes 1–3) (P < 0.01) (Figure 6a). Depletion of specific cell populations from these cultures suggested that CD11b+ cells (rather than lymphocytes or neutrophils) were responsible for the majority of the GM-CSF-induced Fc receptor overexpression in the whole cell population (CD11b+ depleted cultures (mean −3, ±0, lanes 21–23) (P < 0.01)) (Figure 6a). Consistent with this, GM-CSF treatment of isolated CD11b+ cells upregulated expression of FcγR1; significantly increased expression was observed upon GM-CSF treatment (mean 3.33 ± 0.58) compared with non–GM-CSF-treated CD11b+ cells (mean 0.667 ± 0.58) (P = 0.01) (Figure 6b) and, consistent with Figure 1e, also further enhanced their ability to pass on infectious virus from NAb–reovirus complexes, for replication in target B16 tumor cells in culture (Figure 6c). This is consistent with a central role for CD11b+ cells in the binding and delivery of reovirus to tumor cells in vivo in the presence of NAb. Preincubating CD11b+ cells with serum from control C57Bl/6 mice before loading with NAb–reovirus immune complexes did not block the ability of GM-CSF activated CD11b+ cells to transfer infectious virus to target cells (Figure 6c). However, preincubating with serum from VSV-vaccinated C57Bl/6 mice before loading with NAb–reovirus immune complexes significantly inhibited transfer of infectious virus from both nonconditioned and GM-CSF-conditioned CD11b+ cells to target B16 cells (Figure 6c). These data suggest that GM-CSF-enhanced Fc receptor expression mediates the association of NAb–reovirus immune complexes with CD11b+ cells, which is then responsible for the ability of infectious reovirus to be transferred to target tumor cells both in vitro (Figure 6) and in vivo (Figures 3 and 4). Furthermore, this reovirus hand-off to tumor cells can be inhibited by NAb against an alternative oncolytic virus, most likely via competition for Fc receptors on carrier CD11b+ cells.
Figure 6.

GM-CSF conditioning enhances FcγR1 receptor expression and NAb–reovirus immune complex transfer from CD11b+ cells. (a) Intact LN/splenocyte cultures (each lane representing cultures from an individual mouse, three per treatment), or cultures depleted of specific cell populations as indicated, were cultured ± GM-CSF 10 ng/ml), and FcγR1 expression was determined by qRTPCR. Representative of four separate experiments. (b) Microbead-purified CD11b+ cells were treated ± GM-CSF and FcγR1 expression was determined as in a. Representative of two separate experiments. (c) Purified CD11b+ cells, ± GM-CSF, were treated as indicated, washed in PBS, and cocultured with B16 cells. After 72 hours, reovirus titer was determined by plaque assay from the cells and supernatant. (d) B16 tumor cells, or magnetic bead-purified CD11b+ cells, were infected with reovirus at an MOI 1 ± reovirus NAb. After 2 hours, cells were washed three times in PBS, cultured, and reovirus titers were determined by plaque assay from the cells and supernatant at the time points indicated. Graph shows mean ± SD and is representative of two separate experiments.
Reovirus titers increased with time after infection of B16 cultures, an effect which was completely blocked by the presence of anti-reo NAb (Figure 6d). Initial infection of CD11b+ cells also led to increasing release of virus with time, although at a slower rate than from B16 tumor targets, indicating that the CD11b+ cells were truly infected with virus (Figure 6d). Interestingly, this level of virus production with time was not inhibited by anti-reo NAb (Figure 6d), consistent with the hypothesis that virus bound to NAb may provide an alternative route of infection of these cells.
Finally, we confirmed the role of TNF-α and Fc receptors in the immune-mediated killing of target tumor cells. Thus, free reovirus, or reovirus with NAb against a different virus (VSV), led to direct viral-induced cell killing of the highly reovirus-sensitive B16tk cell line, but much less so against the relatively reovirus-insensitive B16ova (Figure 7a,b). However, reovirus complexed with NAb against itself was still highly potent in activating GM-CSF-activated splenocyte/LN cultures to kill both B16ova (as in Figure 5a, (vi)) and B16tk targets (Figure 7a,b). Consistent with the data of Figures 5d and 6a, these effects were largely blocked by either an anti-FcR antibody or by a neutralizing anti-TNF-α antibody (Figure 7a,b).
Cytokine conditioning is dependent on monocytes/macrophages and NK cells in vivo and is widely applicable
Antitumor activity of GM-CSF/reovirus therapy in reovirus-immune mice was dependent on both NK cells and Ly-6C+ve cells (expressed on neutrophils and monocytes), but not on Ly-6G+ve (expressed on neutrophils), and on NK cells (Figure 8a), confirming that key effector cells for GM-CSF conditioning were monocytes/macrophages rather than granulocytes. Moreover, the efficacy of GM-CSF/reovirus therapy was not restricted to the B16 melanoma model, because three cycles of GM-CSF/reovirus eliminated detectable tumor in seven of eight reovirus-immune mice bearing 5-day established s.c. TC2 prostate tumors; no elimination of detectable tumors was seen in mice treated with either GM-CSF or reovirus alone (Figure 8b). Finally, on basis of the success of GM-CSF preconditioning, we also screened other clinically applicable cytokines for possible use as an adjunct to systemic delivery of reovirus. In this respect, although not as effective as GM-CSF, preconditioning of preimmune mice bearing s.c. B16 tumors with IL-2 before i.v. reovirus generated survival advantage (P = 0.02) over cytokine alone (Figure 8c). Of interest, G-CSF preconditioning, which expands granulocyte/neutrophil populations but not monocytes,42,43 did not improve reovirus therapy, providing further confirmation of the involvement of monocytes/macrophages as key effector cells in this strategy.
Figure 8.

GM-CSF conditioning requires NK cells and monocytes/macrophages in vivo and is applicable to other tumor models and cytokines. (a) Reovirus-immune C57Bl/6 mice bearing 5-day established s.c. B16 tumors were depleted of CD8, CD4, NK, Ly-6G+, or Ly-6C+ve immune cell subsets (days 6 and 7; 13 and 14, 20 and 21) and received three cycles of GM-CSF/reovirus treatment (days 6–10, 13–17, and 20–24). (b) C57Bl/6 mice were vaccinated i.p. with reovirus (2 × 107 TCID50). After 14 days, mice were seeded with s.c. TC2 tumors and, 6 days later, treated with three cycles of PBS/PBS, PBS/reovirus, GM-CSF/PBS, or GM-CSF/reovirus. (c) Reovirus-immune C57Bl/6 mice bearing 5-day established s.c. B16 tumors were treated with one cycle of cytokine conditioning, ± reovirus, as indicated. In all cases, tumor size was monitored and animals were killed when tumors reached 1.0 cm diameter.
Discussion
We have previously shown in a phase Ib, biological endpoint trial (REO13), that reovirus administered i.v. to patients is rapidly neutralized in the plasma, but can be recovered associated with blood cells.38 This clinical result led us to hypothesize that blood cells loaded in situ after systemic administration would protect virus from antiviral NAb and that, if a population of tumor homing cells could be selectively activated before i.v. virus, this may lead to effective delivery to tumor in patients. Because systemically administered reovirus was found associated with peripheral blood mononuclear cell, granulocytes, and platelets in patients,38 we used preconditioning with GM-CSF, which expands both monocyte/macrophage and granulocyte populations,39,40 to increase a pool of potential virus carriers within the blood which may also traffic to tumors and, therefore, improve therapy of i.v. delivered reovirus. In addition, the known antitumor vaccinating effects of GM-CSF make it an attractive candidate for use in patients with cancer as an immune adjuvant to oncolytic virus delivery, the precedent for which has already been established with the genetic modification of the two most clinically advanced oncolytic viruses currently in clinical trials—vaccinia (JX-594) and herpes (T-Vec)—to coexpress this cytokine.44,45
We confirmed that, after i.v. administration in reovirus-immune animals, reovirus was recovered from the cellular, but not the plasma, fractions of blood, consistent with our findings in our clinical REO13 study (Figure 1). The majority of viral retention was by CD11b+ cells (which in mice is expressed not only on monocytes/macrophages but also by other cell types, including myeloid-derived suppressor cells), again consistent with our clinical findings, in which the highest titer of virus was retrieved from mononuclear cells. Interestingly, CD11b+ cells, unlike T cells or DC, could be infected by reovirus even in the presence of NAb (Figure 2b). Although all three populations protected preloaded virus from NAb for productive infection of target tumor cells (Figure 2d), CD11b+ cells were most effective at handing off preformed NAb–reovirus complexes to target tumor cells (Figure 2e). Taken together, these data showed that NAb does not prevent direct infection of CD11b+ cells, and reovirus taken up in the form of NAb–reovirus complexes, presumably the major form of virus present in the blood almost immediately after i.v. injection, remains available for hand-off to tumor cells.
Therefore, we tested whether GM-CSF, a widely used clinical agent that boosts both monocyte/macrophage and granulocyte populations, would first enhance in vivo reovirus-loaded CD11b+ cell infiltration into tumors and, hence, virus delivery. Pre-, but not post-, conditioning with GM-CSF increased both intratumoral CD11b+ cell numbers and reovirus titers (Figure 3a,b) in reovirus preimmune mice, consistent with GM-CSF-mediated modulation of the tumor microenvironment.45
A single cycle of preconditioning with GM-CSF, followed by reovirus, eliminated detectable tumor in most mice bearing 3- to 5-day established melanomas, but only in mice that were already preimmune to the virus (Figure 4a). These data were initially surprising, given the paradigm that antiviral NAb acts to block viral delivery in vivo and will, therefore, compromise, rather than enhance, oncolytic viral therapy. We have seen that this is clearly the case in the absence of preconditioning with GM-CSF, where the presence of NAb decreased the low levels of i.v. reovirus that reach s.c. B16 tumors.13 Nonetheless, the presence of preexisting NAb—even without GM-CSF conditioning—did not completely ablate systemically administered reovirus from reaching tumors either in mice (Figure 3b) or in patients.14,27,28,29,30 However, the combination of GM-CSF pretreatment, with preexisting NAb before virus administration, enhanced virus delivery to tumors by up to 2 logs (Figure 3b), indicating that GM-CSF was enhancing the levels of potential virus carrier cells, the phenotype of the virus carriers, or both. These results may also be consistent with those in which an autologous, cell-based, rhabdovirus-infected anti-leukaemia vaccine was more effective if animals were already virus-immune, although the mechanism in that model remains undefined.46 In direct support of the critical role of preexisting NAb in these therapeutic effects, the significant difference of efficacy between preimmunized and virus-naive mice (or mice initially vaccinated against a different virus) was diminished after multiple cycles of GM-CSF/reovirus (Figure 4b). This correlated with the appearance of anti-reovirus NAb after the first treatment cycle, which provided the necessary antiviral vaccination to improve subsequent rounds of therapy (Figure 4c).
To investigate any adjuvant immune-activating contributions of GM-CSF treatment to overall antitumor efficacy, we used a variant of B16 that is relatively resistant to direct reovirus cytolysis25 in vitro to address bystander innate immune-mediated antitumor effects of GM-CSF/reovirus. A mixed immune cell population from lymph nodes and spleens of mice killed B16ova-GFP cells in vitro most effectively, when they were first activated with GM-CSF and then incubated with NAb–reovirus complexes (Figure 5a, (vi)). A similar, although less potent, effect was seen when reovirus was mixed with anti-VSV NAb (Figure 5a, (vii)), suggesting that even the presence of nonspecific antibodies can enhance the cytotoxicity of immune cells in the presence of both GM-CSF and virus, perhaps via NAb engagement of activating FcR receptors on CD11b+ cells. Both NK cells and CD11b cells (but neither CD4 nor CD8 cells) were required for killing activated by GM-CSF/NAb–reovirus, and cytotoxicity correlated with the production of TNF-α (Figure 5b–d); moreover, GM-CSF enhanced FcγR1 expression on CD11b+ cells and increased the ability of isolated CD11b+ cells to pass infectious reovirus from NAb–reovirus complexes to tumor cells (Figure 6). In contrast to the positive effect of anti-VSV NAb mixed with reovirus enhancing the cytotoxicity of immune effectors (Figure 5a, (vii)), anti-VSV NAb inhibited transfer of NAb–reovirus complexes from CD11b+ cells to target tumor cells (Figure 6c); hence, the effects of irrelevant NAb are likely to be various in vivo and may differ with regard to hand-off of replicating virus (inhibitory) and immune-mediated therapy (activating). Overall, these in vitro findings were consistent with differential in vivo depletion studies, which showed that NK cells and monocytes/macrophages, but not granulocytes or CD4/CD8 cells, were required for effective therapy using GM-CSF conditioning followed by systemic reovirus administration (Figure 8a).
Finally, we showed that prostate tumors, and melanoma, could be treated effectively in preimmune mice with GM-CSF followed by reovirus (Figure 8b) and that IL-2 (but significantly not G-CSF) enhanced therapy, although not as effectively as GM-CSF (Figure 8c). This is consistent with a predominant role for monocytes/macrophages rather than granulocytes/T cells as innate effectors in reovirus immunovirotherapy. Experiments are now underway to test whether cytokine conditioning, with GM-CSF or other cytokines, is effective with oncolytic viruses other than reovirus, and the range of tumor types and anatomical locations against which such a strategy is effective.
Our results here are significant in several respects. First, they extend the previous use of ex vivo–loaded cell carriers12,26,34,35,47 to generate a new concept of directed in vivo cell loading. Clinically, the ex vivo expansion and loading of cell carrier populations is time consuming, expensive, and challenging from a regulatory perspective; however, directed in vivo cell loading offers a potentially simpler, cheaper, and effective way to enhance systemic delivery of oncolytic viruses with, at least in the models reported here, considerable antitumor efficacy. Second, our data also raise the intriguing possibility that manipulation of preexisting NAb to an oncolytic virus may actually be exploited for systemic delivery to tumors. For example and paradoxically, our results predict that prevaccination of patients (along with subsequent cytokine conditioning), before i.v. treatment with oncolytic viruses, may lead to significantly more effective delivery of oncolytic viruses to patient tumors and therapy, than without prior induction of NAb. Interestingly, such a prevaccination step was mandated in the highly encouraging OPTiM trial of T-Vec (whose primary clinical endpoint has been met), in which a herpes simples virus oncolytic virus was engineered to express GM-CSF, although the rationale there was to limit viral toxicity rather than to improve therapy. Finally, because both GM-CSF and reovirus have been used extensively in patients, clinical testing of the GM-CSF/reovirus regimen represents a readily testable and translatable protocol.
In summary, our data are consistent with a model in which cytokine preconditioning expands and activates a population of immune cells, which can then be loaded directly in vivo with systemically delivered reovirus (Figure 9). After i.v. injection of reovirus, viral sequestration by blood cells, and in particular CD11b+ monocytes/macrophages, protects reovirus from neutralization. Contrary to the paradigm that preexisting NAb will universally inhibit in vivo delivery of virus to tumors, CD11b+ cells preserve the infectious properties of i.v. injected reovirus for hand-off to target tumor cells. In addition to the enhanced delivery of reovirus by GM-CSF-conditioned, CD11b+ immune cells carrying NAb–reovirus immune complexes into tumors, GM-CSF also activates a potent immune cytotoxicity against tumor cells, which is dependent on NK, and CD11b+ cells, associated with TNF-α secretion. Thus, a combination strategy of pretreatment with GM-CSF, followed by systemic administration of reovirus, offers a promising alternative to ex vivo cell loading, or intratumoral injection of oncolytic viruses for the treatment of cancer, which, paradoxically, may be enhanced by the presence of antiviral NAb. We are currently taking this readily testable approach forward into the clinic, by building on our preexisting platform of clinical trials using systemic delivery of reovirus.14,27,28,29,30
Figure 9.

Proposed mechanism for GM-CSF conditioning to enhance systemic reovirus therapy. (a) Monocyte/macrophages are activated by GM-CSF to upregulate expression of Fc receptors. After systemic delivery of reovirus in an immunized animal, reovirus is bound by anti-reovirus NAb, forming complexes that are loaded onto monocytes/macrophages via binding to their Fc receptors. Increased trafficking of macrophages, induced by GM-CSF, facilitates their delivery to tumors where the virus is handed off for infection of tumor cells. (b) Tumor cell killing is effected by two mechanisms. Once handed off from macrophages, the virus infects and replicates within the tumor, leading to oncolysis and release of new viral particles. In addition, GM-CSF-activated macrophages traffic to tumors, where they bind newly released, tumor-associated reovirus via upregulated Fc receptors. Macrophage–NK interactions at the tumor site promote increased tumor cell killing.
Materials and Methods
Cell lines. Murine B16 melanoma cells (H2-Kb)23 were grown in Dulbecco's modified Eagle medium (Life Technologies, Grand Island, NY) supplemented with 10% (v/v) fetal calf serum (Life Technologies) and l-glutamine (Life Technologies). TRAMP-C2 (TC2) cells are derived from a prostate tumor that arose in a TRAMP mouse and were characterized by Dr Esteban Celis. TRAMP-C2 cells grow in an androgen-independent manner and are routinely grown as tumors in C57Bl/6 male mice.48,49 All cell lines were monitored routinely and found to be free of Mycoplasma infection.
Reovirus. Wild-type reovirus type 3 (Dearing strain) stock titers were measured by plaque assays on L929 cells.14,38 For in vivo studies, reovirus was administered i.v. at 2 × 107 TCID50 per injection. For virus titration from tumors, tumors were harvested from mice, weighed, and lysed (three freeze-thaw cycles within 2 hours of removal). Virus in lysates was titered on L929 cells and expressed as TCID50/mg tissue.13
In vivo experiments. Six- to eight-week-old female C57Bl/6 mice were purchased from Jackson Laboratories (Bar Harbor, Maine). All in vivo studies were approved by the Mayo IACUC. Mice were challenged subcutaneously with 5 × 105 B16 melanoma cells or with 2 × 105 TC2 prostate tumor cells in 100 μl PBS (HyClone). Tumors were measured three times per week, and mice were euthanized when tumors reached 1.0 cm diameter.
In vivo immune depletions. Immune cell depletions were performed by intraperitoneal injections (0.1 mg per mouse) of antibody (Monoclonal Antibody Core Facility, Mayo Clinic unless otherwise stated) to CD8 (Lyt 2.43), CD4 (GK1.5), NK cells (antibody to asialo-GM-1, Cedarlane), Ly-6G+ neutrophils (IA8), Ly6C+ cells (neutrophils, monocytes) (RB6-8C5), or IgG control (ChromPure Rat IgG, Jackson ImmunoResearch). Fluorescence-activated cell sorting analysis of spleens and lymph nodes confirmed subset-specific depletions (data not shown).
In vivo cytokine conditioning. A single cycle of cytokine conditioning consisted of GM-CSF (300 ng per injection,) or rhIL-2 (25,000 U) (R&D Systems, Minneapolis, MN) i.p. for three consecutive days, followed by i.v. reovirus at 2 × 107 TCID50 per injection for 2 days. After 2 days rest, this cycle was repeated either once or twice.
Antibody titration from mouse serum. Preheated mouse antiserum was mixed with an equal volume of reovirus (predetermined as killing 80% of target L929 cells) and incubated at 37 °C for 2 hours to allow antibody to bind to virus. The virus/antibody mix was transferred to L929 monolayers, and cell survival was assayed at 48 hours by 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide assay. An anti-reovirus polyclonal antiserum was used as a positive control. The neutralizing titer is the highest dilution of serum that blocks the killing of L929 cells.14
In vitro cell fractionation. NK cells, CD11b+ monocytes, or PDCA-1+ plasmacytoid DCs were purified/depleted from spleens and lymph nodes of C57Bl/6 mice using magnetic sorting with the NK Cell Isolation Kit II, anti-CD11b or anti-mPDCA-1 microbeads (Miltenyi Biotec, Auburn, CA) as directed by the manufacturer. Pmel-1 transgenic mice (C57Bl/6 background) express the Vα1/Vβ13 T cell receptor that recognizes amino acids 25–33 of gp100 of pmel-17 presented by H2-Db MHC class I molecules.50 Pmel-1 breeding colonies were purchased from The Jackson Laboratory at 6–8 weeks of age. Naive Pmel-1 T cells were isolated from the spleens and lymph nodes of Pmel-1 transgenic mice. Single cell suspensions were prepared by crushing tissues through a 100-μm filter, and red blood cells were removed by incubation in Ammonium-Chloride-Potassium lysing buffer (sterile distilled H2O containing 0.15 mol/l NH4Cl, 1.0 mmol/l KHCO3, and 0.1 mmol/l EDTA adjusted to pH 7.2–7.4) for 2 minutes. CD8+ T cells were isolated using the MACS CD8a (Ly-2) microbead magnetic cell sorting system (Miltenyi Biotec, Auburn, CA).
Quantitative RTPCR. RNA was extracted from LN/splenocyte cultures, intact or depleted of specific cell populations, using the Qiagen RNeasy kit (Qiagen, Valencia, CA). cDNA was made from 1 μg total cellular RNA using the First Strand cDNA Synthesis Kit (Roche, Indianapolis, IN). qRTPCR was performed using a LightCycler480 SYBR Green I Master kit and a LightCycler480 instrument (Roche) according to the manufacturer's instructions. Typically, RNA was prepared from equal numbers of cells from each sample (usually 5,000 cells) and reverse transcribed as described above. PCR (primers at 0.5 µM) was run with diluted cDNA samples (neat, 1:10, 1:100, 1:1000). GAPDH amplification was used as a control for equal loading of target cDNAs. The threshold cycle (Ct) at which amplification of the target sequence was detected was used to compare the relative levels of mRNA between samples. Relative quantities of the target gene mRNA were normalized with Ct of GAPDH amplification. mGAPDH sense: TCATGACCACAGTCCATGCC, mGAPDH antisense: TCAGCTCTGGGATGACCTTG.
MTT assay. A total of 103 B16tk or B16ova target cells were seeded into wells in triplicate in a 96-well plate. A total of 104 mixed splenocytes/lymph node cells from C57Bl/6 mice were added per well and treated with or without GM-CSF at 10 ng/ml. After 24 hours reovirus, reovirus/anti-reovirus NAb or reovirus/anti-VSV NAb were added at MOI 1. In addition, some cocultures were incubated with the anti-TNF-α Ab (AF-410-NA, R&D systems, at 0.5 µg/ml) or with the anti-FcR CD16/CD32 antibody (2.4G2 (BD Pharmingen) at 1 µg/million cells). After 24 hours, cultures were washed three times with PBS to remove nonadherent cells, and cell killing was assayed using an MTT assay (Cell Proliferation Kit I, Roche) according to the manufacturer's instructions; absorbance was read at 540 nm.
Reovirus infection of CD11b and B16 cells. A total of 104 magnetic bead purified CD11b or B16 tumor cells were seeded in triplicate wells of a 96-well plate, treated for 24 hours with 10 ng/ml GM-CSF, and then infected with reovirus at MOI 1 in the absence or presence of reovirus NAb. After 2 hours, cells were washed three times in PBS to remove free virus, and the cells were cultured in fresh medium. Cells and supernatants were harvested at 0, 24, 48, 72, 96, and 120 hours postinfection, and reovirus titers were determined by plaque assay on L929 cells.
Statistics. Survival curves were analyzed by the Log-Rank test. All other data were analyzed by the two-tailed t-test. Statistical significance was set at P < 0.05 for all experiments.
Acknowledgments
We thank Toni Higgins for expert secretarial assistance. This work was supported by the Richard M Schulze Family Foundation, the Mayo Foundation, Cancer Research, UK, the National Institutes of Health grants R01 CA107082, R01CA130878, and R01 CA132734, and a grant from Terry and Judith Paul.
References
- Cattaneo R, Miest T, Shashkova EV, Barry MA. Reprogrammed viruses as cancer therapeutics: targeted, armed and shielded. Nat Rev Microbiol. 2008;6:529–540. doi: 10.1038/nrmicro1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hans Ingemar Andtbacka R, Collichio FA, Amatruda T, Senzer NN, Chesney J, Delman KA, et al. 2013OPTiM: A randomized phase III trial of talimogene laherparepvec (T-VEC) versus subcutaneous (SC) granulocyte-macrophage colony-stimulating factor (GM-CSF) for the treatment (tx) of unresected state IIIB/C and IV melanoma J Clin Oncol 31 (June 20 supplement). [Google Scholar]
- Altomonte J, Ebert O. Replicating viral vectors for cancer therapy: strategies to synergize with host immune responses. Microb Biotechnol. 2012;5:251–259. doi: 10.1111/j.1751-7915.2011.00296.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Melcher A, Parato K, Rooney CM, Bell JC. Thunder and lightning: immunotherapy and oncolytic viruses collide. Mol Ther. 2011;19:1008–1016. doi: 10.1038/mt.2011.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stanford MM, Breitbach CJ, Bell JC, McFadden G. Innate immunity, tumor microenvironment and oncolytic virus therapy: friends or foes. Curr Opin Mol Ther. 2008;10:32–37. [PubMed] [Google Scholar]
- Galivo F, Diaz RM, Thanarajasingam U, Jevremovic D, Wongthida P, Thompson J, et al. Interference of CD40L-mediated tumor immunotherapy by oncolytic vesicular stomatitis virus. Hum Gene Ther. 2010;21:439–450. doi: 10.1089/hum.2009.143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Galivo F, Diaz RM, Wongthida P, Thompson J, Kottke T, Barber G, et al. Single-cycle viral gene expression, rather than progressive replication and oncolysis, is required for VSV therapy of B16 melanoma. Gene Ther. 2010;17:158–170. doi: 10.1038/gt.2009.161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Breitbach CJ, Paterson JM, Lemay CG, Falls TJ, McGuire A, Parato KA, et al. Targeted inflammation during oncolytic virus therapy severely compromises tumor blood flow. Mol Ther. 2007;15:1686–1693. doi: 10.1038/sj.mt.6300215. [DOI] [PubMed] [Google Scholar]
- Kottke T, Hall G, Pulido J, Diaz RM, Thompson J, Chong H, et al. Antiangiogenic cancer therapy combined with oncolytic virotherapy leads to regression of established tumors in mice. J Clin Invest. 2010;120:1551–1560. doi: 10.1172/JCI41431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wongthida P, Diaz RM, Galivo F, Kottke T, Thompson J, Pulido J, et al. Type III IFN interleukin-28 mediates the antitumor efficacy of oncolytic virus VSV in immune-competent mouse models of cancer. Cancer Res. 2010;70:4539–4549. doi: 10.1158/0008-5472.CAN-09-4658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz RM, Galivo F, Kottke T, Wongthida P, Qiao J, Thompson J, et al. Oncolytic immunovirotherapy for melanoma using vesicular stomatitis virus. Cancer Res. 2007;67:2840–2848. doi: 10.1158/0008-5472.CAN-06-3974. [DOI] [PubMed] [Google Scholar]
- Qiao J, Kottke T, Willmon C, Galivo F, Wongthida P, Diaz RM, et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat Med. 2008;14:37–44. doi: 10.1038/nm1681. [DOI] [PubMed] [Google Scholar]
- Qiao J, Wang H, Kottke T, White C, Twigger K, Diaz RM, et al. Cyclophosphamide facilitates antitumor efficacy against subcutaneous tumors following intravenous delivery of reovirus. Clin Cancer Res. 2008;14:259–269. doi: 10.1158/1078-0432.CCR-07-1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- White CL, Twigger KR, Vidal L, De Bono JS, Coffey M, Heinemann L, et al. Characterization of the adaptive and innate immune response to intravenous oncolytic reovirus (Dearing type 3) during a phase I clinical trial. Gene Ther. 2008;15:911–920. doi: 10.1038/gt.2008.21. [DOI] [PubMed] [Google Scholar]
- Dvorak HF. Vascular permeability factor/vascular endothelial growth factor: a critical cytokine in tumor angiogenesis and a potential target for diagnosis and therapy. J Clin Oncol. 2002;20:4368–4380. doi: 10.1200/JCO.2002.10.088. [DOI] [PubMed] [Google Scholar]
- Ellis LM, Hicklin DJ. VEGF-targeted therapy: mechanisms of anti-tumour activity. Nat Rev Cancer. 2008;8:579–591. doi: 10.1038/nrc2403. [DOI] [PubMed] [Google Scholar]
- Thorpe PE. Vascular targeting agents as cancer therapeutics. Clin Cancer Res. 2004;10:415–427. doi: 10.1158/1078-0432.ccr-0642-03. [DOI] [PubMed] [Google Scholar]
- Fisher K. Striking out at disseminated metastases: the systemic delivery of oncolytic viruses. Curr Opin Mol Ther. 2006;8:301–313. [PubMed] [Google Scholar]
- Harrington K, Vile R. Virus smuggling, tax evasion and tumor assassination. Nat Med. 2006;12:507–509. doi: 10.1038/nm0506-507. [DOI] [PubMed] [Google Scholar]
- Fulci G, Breymann L, Gianni D, Kurozomi K, Rhee SS, Yu J, et al. Cyclophosphamide enhances glioma virotherapy by inhibiting innate immune responses. Proc Natl Acad Sci U S A. 2006;103:12873–12878. doi: 10.1073/pnas.0605496103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Galivo F, Wongthida P, Diaz RM, Thompson J, Jevremovic D, et al. Treg depletion-enhanced IL-2 treatment facilitates therapy of established tumors using systemically delivered oncolytic virus. Mol Ther. 2008;16:1217–1226. doi: 10.1038/mt.2008.83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Thompson J, Diaz RM, Pulido J, Willmon C, Coffey M, et al. Improved systemic delivery of oncolytic reovirus to established tumors using preconditioning with cyclophosphamide-mediated Treg modulation and interleukin-2. Clin Cancer Res. 2009;15:561–569. doi: 10.1158/1078-0432.CCR-08-1688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Chester J, Ilett E, Thompson J, Diaz R, Coffey M, et al. Precise scheduling of chemotherapy primes VEGF-producing tumors for successful systemic oncolytic virotherapy. Mol Ther. 2011;19:1802–1812. doi: 10.1038/mt.2011.147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Errington F, Ilett EJ, Morgan RS, Scott KJ, Kottke T, et al. Tumor infection by oncolytic reovirus primes adaptive antitumor immunity. Clin Cancer Res. 2008;14:7358–7366. doi: 10.1158/1078-0432.CCR-08-0831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Prestwich RJ, Ilett EJ, Errington F, Diaz RM, Steele LP, Kottke T, et al. Immune-mediated antitumor activity of reovirus is required for therapy and is independent of direct viral oncolysis and replication. Clin Cancer Res. 2009;15:4374–4381. doi: 10.1158/1078-0432.CCR-09-0334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ilett EJ, Bárcena M, Errington-Mais F, Griffin S, Harrington KJ, Pandha HS, et al. Internalization of oncolytic reovirus by human dendritic cell carriers protects the virus from neutralization. Clin Cancer Res. 2011;17:2767–2776. doi: 10.1158/1078-0432.CCR-10-3266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Comins C, Spicer J, Protheroe A, Roulstone V, Twigger K, White CM, et al. REO-10: a phase I study of intravenous reovirus and docetaxel in patients with advanced cancer. Clin Cancer Res. 2010;16:5564–5572. doi: 10.1158/1078-0432.CCR-10-1233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KJ, Karapanagiotou EM, Roulstone V, Twigger KR, White CL, Vidal L, et al. Two-stage phase I dose-escalation study of intratumoral reovirus type 3 dearing and palliative radiotherapy in patients with advanced cancers. Clin Cancer Res. 2010;16:3067–3077. doi: 10.1158/1078-0432.CCR-10-0054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harrington KJ, Vile RG, Melcher A, Chester J, Pandha HS. Clinical trials with oncolytic reovirus: moving beyond phase I into combinations with standard therapeutics. Cytokine Growth Factor Rev. 2010;21:91–98. doi: 10.1016/j.cytogfr.2010.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vidal L, Pandha HS, Yap TA, White CL, Twigger K, Vile RG, et al. A phase I study of intravenous oncolytic reovirus type 3 Dearing in patients with advanced cancer. Clin Cancer Res. 2008;14:7127–7137. doi: 10.1158/1078-0432.CCR-08-0524. [DOI] [PubMed] [Google Scholar]
- Yap TA, Brunetto A, Pandha H, Harrington K, Debono JS. Reovirus therapy in cancer: has the orphan virus found a home. Expert Opin Investig Drugs. 2008;17:1925–1935. doi: 10.1517/13543780802533401. [DOI] [PubMed] [Google Scholar]
- Coffey MC, Strong JE, Forsyth PA, Lee PW. Reovirus therapy of tumors with activated Ras pathway. Science. 1998;282:1332–1334. doi: 10.1126/science.282.5392.1332. [DOI] [PubMed] [Google Scholar]
- Hirasawa K, Nishikawa SG, Norman KL, Coffey MC, Thompson BG, Yoon CS, et al. Systemic reovirus therapy of metastatic cancer in immune-competent mice. Cancer Res. 2003;63:348–353. [PubMed] [Google Scholar]
- Harrington K, Alvarez-Vallina L, Crittenden M, Gough M, Chong H, Diaz RM, et al. Cells as vehicles for cancer gene therapy: the missing link between targeted vectors and systemic delivery. Hum Gene Ther. 2002;13:1263–1280. doi: 10.1089/104303402760128504. [DOI] [PubMed] [Google Scholar]
- Qiao J, Wang H, Kottke T, Diaz RM, Willmon C, Hudacek A, et al. Loading of oncolytic vesicular stomatitis virus onto antigen-specific T cells enhances the efficacy of adoptive T-cell therapy of tumors. Gene Ther. 2008;15:604–616. doi: 10.1038/sj.gt.3303098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Power AT, Bell JC. Cell-based delivery of oncolytic viruses: a new strategic alliance for a biological strike against cancer. Mol Ther. 2007;15:660–665. doi: 10.1038/sj.mt.6300098. [DOI] [PubMed] [Google Scholar]
- Thorne SH, Negrin RS, Contag CH. Synergistic antitumor effects of immune cell-viral biotherapy. Science. 2006;311:1780–1784. doi: 10.1126/science.1121411. [DOI] [PubMed] [Google Scholar]
- Adair RA, Roulstone V, Scott KJ, Morgan R, Nuovo GJ, Fuller M, et al. Cell carriage, delivery, and selective replication of an oncolytic virus in tumor in patients. Sci Transl Med. 2012;4:138ra77. doi: 10.1126/scitranslmed.3003578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dranoff G. GM-CSF-based cancer vaccines. Immunol Rev. 2002;188:147–154. doi: 10.1034/j.1600-065x.2002.18813.x. [DOI] [PubMed] [Google Scholar]
- Dranoff G, Jaffee E, Lazenby A, Golumbek P, Levitsky H, Brose K, et al. Vaccination with irradiated tumor cells engineered to secrete murine granulocyte-macrophage colony-stimulating factor stimulates potent, specific, and long-lasting anti-tumor immunity. Proc Natl Acad Sci U S A. 1993;90:3539–3543. doi: 10.1073/pnas.90.8.3539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adair RA, Scott KJ, Fraser S, Errington-Mais F, Pandha H, Coffey M, et al. Cytotoxic and immune-mediated killing of human colorectal cancer by reovirus-loaded blood and liver mononuclear cells. Int J Cancer. 2013;132:2327–2338. doi: 10.1002/ijc.27918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babalola CP, Nightingale CH, Nicolau DP. Adjunctive efficacy of granulocyte colony-stimulating factor on treatment of Pseudomonas aeruginosa pneumonia in neutropenic and non-neutropenic hosts. J Antimicrob Chemother. 2004;53:1098–1100. doi: 10.1093/jac/dkh237. [DOI] [PubMed] [Google Scholar]
- Han B, Unsinger J, Liu F, Link DC, Bessler M. G-CSF induced progenitor mobilization in mice with PIGA- blood cells. Hematol J. 2004;5:347–352. doi: 10.1038/sj.thj.6200383. [DOI] [PubMed] [Google Scholar]
- Heo J, Reid T, Ruo L, Breitbach CJ, Rose S, Bloomston M, et al. Randomized dose-finding clinical trial of oncolytic immunotherapeutic vaccinia JX-594 in liver cancer. Nat Med. 2013;19:329–336. doi: 10.1038/nm.3089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaufman HL, Bines SD. OPTIM trial: a Phase III trial of an oncolytic herpes virus encoding GM-CSF for unresectable stage III or IV melanoma. Future Oncol. 2010;6:941–949. doi: 10.2217/fon.10.66. [DOI] [PubMed] [Google Scholar]
- Conrad DP, Tsang J, Maclean M, Diallo JS, Le Boeuf F, Lemay CG, et al. Leukemia cell-rhabdovirus vaccine: personalized immunotherapy for acute lymphoblastic leukemia. Clin Cancer Res. 2013;19:3832–3843. doi: 10.1158/1078-0432.CCR-12-3199. [DOI] [PubMed] [Google Scholar]
- Kottke T, Diaz RM, Kaluza K, Pulido J, Galivo F, Wongthida P, et al. Use of biological therapy to enhance both virotherapy and adoptive T-cell therapy for cancer. Mol Ther. 2008;16:1910–1918. doi: 10.1038/mt.2008.212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boisgerault N, Kottke T, Pulido J, Thompson J, Diaz RM, Rommelfanger-Konkol D, et al. Functional cloning of recurrence-specific antigens identifies molecular targets to treat tumor relapse. Mol Ther. 2013;21:1507–1516. doi: 10.1038/mt.2013.116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kottke T, Errington F, Pulido J, Galivo F, Thompson J, Wongthida P, et al. Broad antigenic coverage induced by vaccination with virus-based cDNA libraries cures established tumors. Nat Med. 2011;17:854–859. doi: 10.1038/nm.2390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaluza K, Kottke T, Diaz RM, Rommelfanger D, Thompson J, Vile RG. Adoptive transfer of cytotoxic T lymphocytes targeting two different antigens limits antigen loss and tumor escape. Hum Gene Ther. 2012;131:844–854. doi: 10.1089/hum.2012.030. [DOI] [PMC free article] [PubMed] [Google Scholar]