

Abstract
Patients with recessive dystrophic epidermolysis bullosa (RDEB) have severe, incurable skin fragility, blistering, and multiple skin wounds due to mutations in the gene encoding type VII collagen (C7), the major component of anchoring fibrils mediating epidermal-dermal adherence. Nearly 10–25% of RDEB patients carry nonsense mutations leading to premature stop codons (PTCs) that result in truncated C7. In this study, we evaluated the feasibility of using aminoglycosides to suppress PTCs and induce C7 expression in two RDEB keratinocyte cell lines (Q251X/Q251X and R578X/R906) and two primary RDEB fibroblasts (R578X/R578X and R163X/R1683X). Incubation of these cells with aminoglycosides (geneticin, gentamicin, and paromomycin) resulted in the synthesis and secretion of a full-length C7 in a dose-dependent and sustained manner. Importantly, aminoglycoside-induced C7 reversed the abnormal RDEB cell phenotype and incorporated into the dermal-epidermal junction of skin equivalents. We further demonstrated the general utility of aminoglycoside-mediated readthrough in 293 cells transiently transfected with expression vectors encoding 22 different RDEB nonsense mutations. This is the first study demonstrating that aminoglycosides can induce PTC readthrough and restore functional C7 in RDEB caused by nonsense mutations. Therefore, aminoglycosides may have therapeutic potential for RDEB patients and other inherited skin diseases caused by nonsense mutations.
Introduction
Dystrophic epidermolysis bullosa (DEB) is an inherited skin disease. Affected individuals demonstrate severe skin fragility at birth, with subsequent blisters, erosions, and scarring from insignificant trauma.1 The disease is caused by mutations in the COL7A1 gene that encodes type VII collagen (C7), the major component of anchoring fibrils (AFs).2,3,4,5,6 AFs are structures in the dermal-epidermal junction (DEJ) of human skin that are responsible for dermal-epidermal adherence. In the absence of sufficient numbers of functional AFs, minor trauma to the skin results in dermal-epidermal disadherence and blistering of the skin. Repetitive cycles of blistering, erosions, milia formation, fibrosis, and scarring may ultimately lead to aggressive squamous cell carcinomas, the main cause of fatality in RDEB patients. Unfortunately, RDEB is currently incurable.
Over 700 distinct mutations have been identified in DEB patients.7,8 These mutations include missense, frameshift, insertion, deletion, and nonsense changes. The devastating Hallopeau-Siemens recessive subtype is caused by premature termination codon (PTC) mutations on both COL7A1 alleles, while the less severe non-Hallopeau-Siemens recessive DEB (RDEB-nonHS) genotype is due to one PTC mutation on one allele and one missense mutation on the other allele.2,3,7 PTCs may be caused by nonsense, frameshift, or splice site mutations that result in the formation of an unstable mRNA transcript that subsequently undergoes nonsense-mediated mRNA decay, or the formation of truncated polypeptides that cannot assemble into functional AFs.
More than 1,800 human-inherited diseases are due to nonsense mutations leading to shortened proteins. According to the Human Gene Mutation Database, 12% of all mutations reported are nonsense mutations that result in primary PTCs.9 Recent in vitro and in vivo studies have revealed that aminoglycoside antibiotics can suppress primary PTCs and produce full length functional protein in several genetic disorders such as cystic fibrosis (CF), Duchenne's muscular dystrophy (DMD), hemophilia, β-thalassemia, spinal muscular atrophy, Hurler syndrome, and retinitis pigmentosa.9,10,11,12,13,14
It is estimated that between 10 and 25% of disease-causing COL7A1 mutations represent nonsense mutations that create a PTC in the reading frame and result in a truncated, nonfunctional protein.7,8 Therefore, a therapeutic approach to overcome primary PTC mutations could be of considerable benefit to these patients. In this study, we tested the hypothesis that aminoglycoside antibiotics might be potentially useful in the treatment of RDEB caused by nonsense mutations. Using two RDEB keratinocyte cell lines harboring nonsense mutations and primary fibroblast cultures from two RDEB patients with nonsense mutations, we showed that aminoglycosides (G418, gentamicin, and paramomycin) were able to induce PTC readthrough and restore a functional full-length C7 by increasing the stability of C7 mRNA. Aminoglycoside restored C7 in RDEB skin cells reversed the abnormal cell motility associated with RDEB. Using cultured human skin equivalents composed of RDEB cells, normal cells, or RDEB cells treated with aminoglycosides, we found that the C7 produced by aminoglycoside-treated RDEB cells incorporated into the DEJ of the skin equivalents. Lastly, we used site-directed mutagenesis to generate 22 known RDEB nonsense mutations and transfected these constructs into human 293 cells. Aminoglycoside treatment of these cells induced PTC readthrough and induction of full-length C7 of varying degrees in all 22 nonsense construct transfected cells. We believe that this is the first study, to our knowledge, demonstrating that aminoglycosides can induce PTC readthrough and restore functional C7 in RDEB caused by nonsense mutations. Our results suggest that aminoglycoside-mediated nonsense-suppression therapy may provide a novel and noninvasive option for the treatment of RDEB patients carrying nonsense mutations.
Results
The ability of aminoglycosides to produce full-length C7 in RDEB cells is dose dependent
In order to test the feasibility of aminoglycosides as a treatment for RDEB caused by nonsense mutations, we first tested a number of aminoglycosides including G418, gentamicin and paromomycin on two established RDEB keratinocyte cell lines, RDEB1 and RDEB2. These cells exclusively harbor C7 nonsense mutations. The RDEB1 is heterozygous for R578X and Q906X mutations whereas RDEB2 is homozygous for Q251X mutations. RDEB keratinocytes were incubated with increasing concentrations of aminoglycosides for 48 hours and cell lysates were prepared and subjected to immunoblot analysis using an antibody to the non-collagenous domain 1 (NC1) of C7. As shown in Figure 1, each drug had an effective concentration range that induced the production of a full-length, 290 kDa C7 α chain in a dose-dependent manner. As expected, untreated parent cells entirely lacked C7 expression. Optimal concentrations leading to the maximum readthrough and full-length C7 production for G418, gentamicin, and paromomycin were 8, 400, and 1,000 μg/ml, respectively, in both cell lines. Under these optimal concentrations, quantification showed that G418 restored full-length C7 at the levels of 24.8 and 17.2% of that observed in normal keratinocytes for RDEB1 and RDEB 2 respectively. For gentamicin, the readthrough levels were 22.3 and 14.4% for RDEB1 and RDEB2 respectively. For paromomycin, the readthrough levels were 22.6 and 23.5% for RDEB1 and RDEB2 respectively. Cellular cytotoxicity was not observed under any of the aminoglycoside concentrations tested above (Supplementary Figure S1). In addition, we also tested three additional aminoglycosides, amikacin, neomycin, and tobramycin, and found that they failed to induce readthrough and restore C7 production in either cell line (data not shown).
Figure 1.
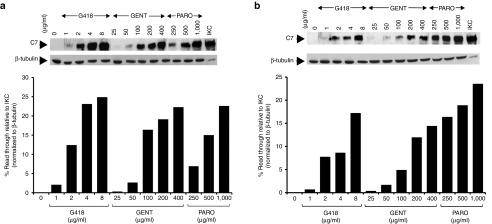
Aminoglycosides mediate dose-dependent induction of full-length C7 production in recessive dystrophic epidermolysis bullosa (RDEB) keratinocytes. RDEB keratinocytes, RDEB1 (a) and RDEB2 (b), were treated with increasing concentrations of G418, gentamicin (GENT), and paromomycin (PARO), as indicated, for 48 hours. Cell lysates were prepared and then subjected to 4–12% SDS-PAGE, followed by immunoblot analysis with a rabbit polyclonal antibody to the NC1 domain of C7 or anti-β-tubulin (loading control) antibody. Last lane represents 25% level of C7 produced from immortalized normal human keratinocytes (IKC). Please note that all three agents induced full-length C7 production in a dose-dependent manner in both cell lines. We have performed three independent experiments and similar results were obtained.
Intracellular and extracellular accumulation of full-length C7 is sustained after one treatment
We next determined how long the effects of a single administration could be maintained within the RDEB keratinocytes and whether this protein would be properly excreted into the extracellular space. RDEB keratinocytes were incubated with growth medium containing the optimal concentration of gentamicin (400 μg/ml) or G418 (8 μg/ml) for 48 hours, and then switched to drug-free growth medium. Media and cell lysates were collected on subsequent days following this initial treatment and subjected to immunoblot analyses using an antibody to NC1. As shown in Figure 2a, a single dose of either aminoglycoside induced RDEB1 to produce full-length, 290 kDa C7 for up to 2 weeks. This sustained production was also observed in the conditioned media (Figure 2b). The maximum levels of C7 induction were observed at day 5 within the cells and day 6 in the media. This indicates that C7 production due to aminoglycoside-induced readthrough is sustained.
Figure 2.
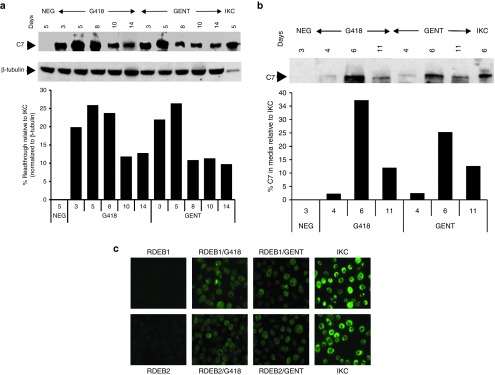
Aminoglycosides induce sustained full-length C7 production. (a) RDEB1 keratinocytes were incubated with growth media in the absence (NEG) or presence of G418 (8 μg/ml) or gentamicin (400 µg/ml) for 48 hours and then changed to drug-free growth media. Cell lysates were prepared at various times after treatment, as indicated, and then subjected to 4–12% SDS-PAGE followed by immunoblot analysis with a rabbit polyclonal antibody to the NC1 domain of C7 or an anti-β-tubulin antibody (internal loading control). Cell lysates from day 5 untreated cells were run as negative control (NEG). Note that both gentamicin and G418 induced sustained C7 production with readily detectable levels observed even 2 weeks after a single treatment. We have performed three independent experiments and similar results were obtained. (b) RDEB1 keratinocytes were incubated with growth media containing either G418 (8 μg/ml) or gentamicin (400 μg/ml) for 48 hours and then changed to media without drugs. The conditioned medium was harvested at various time points indicated after drug treatment and concentrated. Conditioned medium from untreated cells harvest 72 hours was used as negative control (NEG). Equal amounts of each sample were separated on a 4–12% SDS-PAGE and analyzed by immunoblotting with a rabbit polyclonal antibody against the NC1 domain of C7. Last lane represents 20% level of C7 secreted from immortalized normal human keratinocytes (IKC) harvested at day 6. We have performed three independent experiments and similar results were obtained. (c) RDEB1 and RDEB2 keratinocytes were either untreated or treated with G418 (8 µg/ml) or gentamicin (400 µg/ml) for 48 hours and then subjected to immunofluorescence staining with an affinity purified polyclonal antibody to the NC1 domain of C7. Note that both G418 and gentamicin induced C7 expression at levels 15–30% of that observed in immortalized normal human keratinocytes (IKC). In contrast, untreated RDEB keratinocytes lacked C7 staining. We have performed three independent experiments and similar results were obtained.
Immunoblot results were further confirmed by immunofluorescence analysis. As shown in Figure 2c, without any treatment, there is virtually no detectable C7 in either RDEB cell line. Nevertheless, C7-positive cells were detected in G418- or gentamicin-treated cells with a level of C7 expression at 16–30% or 15–22%, respectively, of that seen in normal human keratinocytes, similar to the results obtained from immunoblot analysis. These results indicate that both G418 and gentamicin were capable of mediating PTC readthrough in both RDEB keratinocyte lines.
Aminoglycoside treatment increases the C7 message level in RDEB keratinocytes
It has been suggested that nonsense mutations give rise to unstable mRNA transcripts, which subsequently undergo nonsense-mediated mRNA decay, leading to the production of little or no functional protein.15 One mechanism by which aminoglycosides facilitate PTC read through may be by suppressing nonsense-mediated mRNA decay. We next determined if aminoglycosides affect C7 mRNA levels using quantitative real-time polymerase chain reaction (RT-PCR). As shown in Figure 3, the amounts of C7 mRNA in the two RDEB keratinocyte lines were decreased to 40–46% of the levels seen in normal human keratinocytes. Incubating the RDEB keratinocytes with either 8 μg/ml G418 or 400 μg/ml gentamicin for 24 hours increased C7 mRNA levels to 93–105% of that in normal human keratinocytes. For paromomycin-treated cells, the mRNA levels were increased to 74 and 68% of the normal levels for RDEB1 and RDEB2 respectively. Interestingly, aminoglycosides did not affect the normal levels of C7 mRNA on normal human keratinocytes. These results indicate that aminogycoside-induced PTC readthrough and C7 production in RDEB keratinocytes are mediated by increasing mRNA stability.
Figure 3.

Aminoglycosides increase COL7A1 mRNA levels in recessive dystrophic epidermolysis bullosa (RDEB) keratinocytes. mRNAs were isolated from normal immortalized human keratinocytes (IKC) or RDEB1 and RDEB2 keratinocytes either untreated or treated with G418 (8 µg/ml) or gentamicin (400 µg/ml) for 24 hours and then subjected to real-time RT-PCR. The relative amounts of C7 mRNA levels compared with β-actin mRNA levels are shown as means + SD from two independent experiments. In each experiment, samples were run in triplicate.
Aminoglycosides are able to suppress PTC and induce full-length C7 production in RDEB fibroblasts
Two principal cell types in skin, epidermal keratinocytes, and dermal fibroblasts, produce C7. In order to test whether aminoglycosides can also induce PTC readthrough and restore C7 production in dermal fibroblasts, we cultured primary dermal fibroblasts from two RDEB patients, RDEB3 and RDEB4. The RDEB3 patient is homozygous for the R578X nonsense mutations whereas the RDEB4 patient is heterozygous for R163X and R1683X mutations. RDEB fibroblasts were incubated with increasing concentrations of G418, gentamicin, and paromomycin for 48 hours and cell lysates were prepared and subjected to immunoblot analysis. As shown in Figure 4, all three aminoglycosides mediated a dose-dependent PTC readthrough and full-length C7 production in both RDEB3 and RDEB4 fibroblasts. The optimal doses for G418, gentamicin, and paromomycin were 100, 800, and 2,000 μg/ml, respectively. These concentrations are significantly higher than those used for RDEB keratinocytes. Nevertheless, under all of these aminoglycoside concentrations, we did not observe any cytotoxicity in the treated cells (data not shown). Under these optimal concentrations, quantification showed that G418 restored the full-length C7 at the levels of 43.6 and 19% of that observed in normal fibroblasts for RDEB3 and RDEB4 respectively. For gentamicin, the readthrough levels were 45.6 and 14% for RDEB3 and RDEB4 respectively. For paromomycin, the readthrough levels were 15.2 and 12% for RDEB3 and RDEB4 respectively. RDEB3 harboring homozygous R578X mutations exhibited stronger responses to all three aminoglycosides than RDEB4 with heterozygous R163X and R1683X mutations. These results indicate that aminoglycosides were also capable of inducing readthrough and restoration of C7 production in RDEB fibroblasts.
Figure 4.
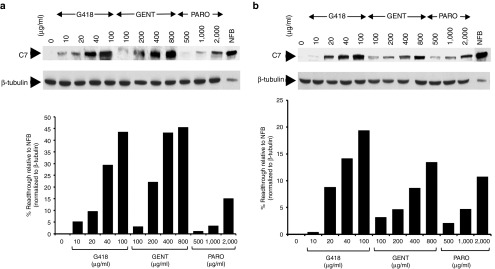
Aminoglycosides mediate dose-dependent induction of full-length C7 production in recessive dystrophic epidermolysis bullosa (RDEB) fibroblasts. RDEB fibroblasts, RDEB3 (a) and RDEB4 (b), established from two RDEB patients harboring nonsense mutations were treated with increasing concentrations of G418, gentamicin, and paromomycin as indicated for 48 hours. Cell lysates were prepared and then subjected to 4–12% SDS-PAGE, followed by immunoblot analysis with a rabbit polyclonal antibody to the NC1 domain of C7 or an anti-β-tubulin (loading control) antibody. Last lane represents 25% level of C7 produced from normal human fibroblasts (NFB). Note that all three agents induced full-length C7 production in a dose-dependent manner in both patients' fibroblasts. We have performed three independent experiments and similar results were obtained.
Aminoglycosides correct the hypermotility characteristic of RDEB fibroblasts
The suppression of PTC is mediated by a mispairing between the stop codon and near-cognate aminoacyl tRNA.16 Since the amino acid inserted at the PTC may differ from the residue encoded in the wild-type protein and the readthrough protein may have less function compared to normal if another amino acid has been inserted at the PTC, we next asked whether the full-length C7 protein produced after aminoglycoside treatment of RDEB fibroblasts is functional. We showed previously that RDEB cells demonstrated hypermotility when compared with normal human fibroblasts.17 To determine if aminoglycoside-induced C7 can reverse the hypermotility, characteristic of RDEB cells, we subjected normal and RDEB fibroblasts, either untreated or treated with G418 or gentamicin, to a well-established fibroblast migration assay. Figure 5a (top panel) shows representative microscopic fields of normal human fibroblasts and the two RDEB primary fibroblasts (RDEB3 and RDEB4), before and after aminoglycoside exposure. Consistent with previous studies, both RDEB3 and RDEB4 fibroblasts exhibited enhanced motility in comparison with normal human fibroblasts. In contrast, aminoglycoside-treated RDEB fibroblasts exhibited normalized cellular motility similar to normal human fibroblasts.
Figure 5.

Aminoglycoside-induced C7 reverses fibroblast hypermotility. RDEB3 and RDEB4 fibroblasts were either untreated or treated with G418 (40 µg/ml), or gentamicin (400 µg/ml) for 48 hours and then subjected to a colloidal gold salt migration assay using collagen I as a matrix. (a) The top panels are representative fields photographed at ×40 under dark field optics. (b) The bottom panels are computer-generated migration indices (MIs). The migration index is the percentage of the total field area occupied by migration tracks. Error bars, SE of three different experiments. Note that both RDEB3 and RDEB4 fibroblasts showed hypermotility in comparison with normal human dermal fibroblasts (NFB). In contrast, aminoglycoside-treated RDEB3 and RDEB4 cells demonstrated reduced motility to levels similar to normal human fibroblasts.
Cellular motility can be quantitated by a migration index (MI), which is calculated as the percentage of the microscopic field consumed by motility tracks. As shown in Figure 5b (bottom panel), the MIs were 39.87 and 37.2 for RDEB3 and RDEB4 compared with 21.9 for normal human fibroblasts. G418-treated RDEB3 and RDEB4 produced MIs of 21.32 and 24.37, similar to that of normal cells. Similarly, gentamicin-treated fibroblasts generated MIs of 22.29 and 27.68, respectively for RDEB3 and RDEB4. Therefore, both G418 and gentamicin effectively promoted PTC readthrough and produced functional C7 that corrected the hypermotility phenotype that is characteristic of RDEB cells.
Aminoglycoside-induced C7 localizes to the DEJ of RDEB skin equivalents
After restoring C7 in two dimensional cultures of RDEB cells and demonstrating the correction of the RDEB abnormal cellular phenotype, we sought to determine if aminoglycoside-induced C7 could incorporate into the DEJ of in vitro three dimensional, organotypic skin equivalents (SEs). To test this, we used an in vitro three-dimensional SE model, where acellular swine dermis was infused with fibroblasts and then seeded with keratinocytes as described previously.18 In this series of experiments, the SEs were constructed with parental RDEB cells, RDEB cells treated with aminoglycosides, or normal human keratinocytes and fibroblasts. Three weeks after the SEs were established in culture, they were subjected to immunofluorescence staining using a monoclonal antibody specific to human C7 that does not cross react with porcine C7. As shown in Figure 6, after 3 weeks of culture at an air interface, immunolabeling of SEs composed of RDEB cells treated with gentamicin revealed tight linear staining at the DEJ between the keratinocytes and the dermal equivalent. This pattern was identical to SEs composed of normal human keratinocytes and fibroblasts. As expected, there was no C7 expression in the SEs composed of parental RDEB keratinocytes and fibroblasts. Quantification showed that the amount of C7 deposited at the DEJ in SEs composed of aminoglycoside treated RDEB1 and RDEB2 keratinocytes combined with RDEB4 fibroblasts was ~60 and 40%, respectively, of the level of C7 produced from SEs composed of normal keratinocytes and fibroblasts. We conclude that gentamicin-induced C7 is capable of incorporating into the DEJ in vitro.
Figure 6.

Aminoglycoside-induced C7 incorporates into the dermal-epidermal junction (DEJ) of in vitro skin equivalents. The cyrosections were subjected to immunofluorescent labeling using a monoclonal antibody (NP185) specific to human C7. Left panels are skin equivalents composed of untreated recessive dystrophic epidermolysis bullosa (RDEB) keratinocytes (RDEB1 or RDEB2) combined with untreated RDEB fibroblasts (RDEB4). Middle panels are skin equivalents composed of RDEB keratinocytes (RDEB1 or RDEB2) treated with gentamicin (400 μg/ml) combined with RDEB fibroblasts (RDEB4). Right panels are skin equivalents composed of normal human fibroblasts combined with normal human keratinocytes. d, dermis; e, epidermis. Arrows indicate DEJ. We have performed three independent experiments and similar results were obtained.
Aminoglycosides induce PTC readthrough and full-length C7 expression in multiple RDEB nonsense mutations
The efficacy of readthrough is thought to be dependent on the type of aminoglycoside used, the sequence of the PTC, and the nucleotide base immediately downstream of the stop codon.12 Mutational analysis has revealed at least 70 distinct RDEB nonsense mutations.7,8 To evaluate the general potential utility of aminoglycoside therapy for other reported RDEB nonsense mutations, we used site-directed mutagenesis to introduce 22 nonsense RDEB mutations into a C7 expression vector and transfected these constructs into human 293 cells. Figure 7a is a schematic of the C7 molecule showing the location of each of the newly introduced nonsense mutations.
Figure 7.

Aminoglycosides induced full-length C7 expression in 293 cells transfected with multiple cDNA constructs coding for recessive dystrophic epidermolysis bullosa (RDEB) nonsense mutants. (a) The 2944 amino acid sequence of C7 consists of a central triple-helical domain (TH), flanked by a large amino-terminal noncollagenous domain, NC1, and a smaller carboxyl-terminal noncollagenous domain, NC2. Twenty-two C7 nonsense mutants, generated by site directed mutagenesis, are shown in the schematic with the approximate positions of the mutations indicated by arrows. (b,c) 293 cells were transfected with cDNA expression vectors coding for wild type C7 and the C7 nonsense mutants as indicated for 24 hours. Transfected cells were then incubated with (b) G418 (200 μg/ml) or (c) gentamicin (1 mg/ml) for an additional 48 hours. Cell lysates were prepared and then subjected to 4–12% SDS-PAGE followed by immunoblot analysis using an anti-NC1 antibody or an anti-β-tubulin antibody (loading control). Last lane represents 50% level of C7 produced from 293 cells transfected with expression vector for wild type C7. Note that both G418 and gentamicin induced various level of premature stop codons readthrough and full-length C7 production in 293 cells transfected with all C7 nonsense mutant expression vectors. We have performed three independent experiments and similar results were obtained.
To assess the efficiency of aminoglycoside-induced readthrough in these various nonsense mutations, we incubated 293 cells that were transiently transfected with a single nonsense mutant construct with G418 or gentamicin for 48 hours, and then prepared cell lysates for immunoblot analysis using an anti-NC1 antibody. We selected concentrations of 200 μg/ml for G418 and 1,000 μg/ml for gentamicin, which gave the highest readthrough activity in 293 cells without affecting the viability of the cells (data not shown). As shown in Figure 7b,c, treatment with G418 or gentamicin promoted PTC readthrough and induced full-length C7 production in 100% of the 293 cells transfected with the indicated nonsense mutant C7 constructs. Densitometry analysis revealed that different RDEB mutants exhibited different degrees of PTC readthrough and that C7 levels ranged from 5% - 80% of that obtained from 293 cells transfected with the wild type C7 construct. In addition, in the absence of aminoglycosides, there is no detectable basal readthrough of C7 in 293 cells transfected with any of the C7 nonsense mutant constructs (Supplementary Figure S2). Thus, both G418 and gentamicin were able to induce PTC readthrough and restore C7 production in 293 cells transfected with C7 expression constructs harboring all 22 different RDEB associated nonsense mutations. Our data demonstrate that all 22 naturally occurring RDEB PTC mutations tested here are amenable to suppression by aminoglycosides, but with different levels of efficacy.
Aminoglycoside-induced full-length C7 has the same structural properties of wild type C7
Next, we sought to determine whether C7 produced as a result of aminoglycoside-mediated readthrough of PTCs has the same structural properties as wild-type C7. In order to obtain C7 from the cells for structural analysis, we generated stable cell lines from four nonsense constructs, R137X, Q251X, R578X, and Q906X, all of which demonstrated high levels of PTC readthrough in the 293 cell transiently transfected system. C7 was then purified from conditioned media as described.19 Purified recombinant C7 protein was run on 4–12% SDS-PAGE under nonreducing conditions and subsequently subjected to immunoblot analysis using an antibody to the non-collagenous domain 2 (NC2) of C7. As shown in Figure 8a, under nonreducing conditions, both wild type and aminoglycoside-induced full-length C7 products migrated at 900 kDa, the expected size of a C7 trimer.
Figure 8.

Aminoglycoside-induced C7 forms stable triple helices. (a) Purified wild-type C7 (C7) and aminoglycoside-induced C7 from four nonsense constructs as indicated were subjected to 4–12% SDS-PAGE, followed by immunoblot analysis with a polyclonal antibody to the NC2 domain. Proteins were nonreduced before loading onto gels. The positions of molecular weight markers, and trimer (T) of C7 are indicated. (b) A schematic diagram of the C7 α-chain. The triple helical domain (TH) of C7 contains a 39 amino acid of nonhelical interruption (hinge region). This site of C7 is sensitive to protease digestion, and the helix can be cleaved into carboxyterminal P1 and aminoterminal P2 fragments. Each fragment represents approximately one-half of the TH domain. The protease cleavage sites (P) are indicated by arrows. The recognition region for a polyclonal antibody to the TH domain (α-TH) is also indicated. (c) Purified wild-type C7 (C7) and aminoglycoside-induced C7 from four nonsense constructs, as indicated, were treated with chymotrypsin and analyzed by 6% SDS-PAGE followed by immunoblot analysis with a polyclonal antibody to the TH domain. The positions of molecular weight markers, the 200-kDa intact TH domain and the 120-kDa carboxyl-terminal half of the TH fragment (P1) are indicated.
We then examined if these trimers are stable and have a proper triple helical conformation in their collagenous domain. Both wild-type and aminoglycoside-induced C7 products were treated with chymotrypsin and then subjected to immunoblot analysis using a polyclonal antibody against the triple helical (TH) domain of C7. Figure 8b shows the schematic protease digestion patterns of C7. It is expected that the triple helical (TH) domain of the C7 mostly resists digestion, except for a partial cleavage into the P1 and P2 fragments at the site of the hinge region in the center of the TH domain, as described by Burgeson.5 As shown in Figure 8c, digestion of the 290-kDa wild-type C7 with chymotrypsin yielded two characteristic bands, one corresponding to the 200-kDa intact TH domain, and one corresponding to the 120-kDa carboxy-terminal half of the triple helical fragment, the P1 band. Similarly, chymotrypsin digestion of C7s purified from the 293 cells transfected with the four nonsense C7 constructs and exposed to aminoglycosides produced the same two digestion products with identical size and quantity as wild type C7. These data indicated that the aminoglycoside-induced C7 is configured into a triple helix like wild-type C7.
Discussion
Various therapeutic strategies have been suggested for RDEB based on preclinical observations in animal models, including intradermal injection of allogeneic dermal fibroblasts or gene-corrected RDEB fibroblasts,20,21 intradermal injection of lentiviral vectors expressing C7,22 intradermal injection of human recombinant C7,19,23 and transplantation of gene-corrected keratinocyte autografts.17,24 In addition, the feasibility of administering cells or protein systemically via intravenous injection has been evaluated25,26 A number of proof-of-principle clinical trials have been initiated in patients with RDEB, including bone marrow/stem cell transplantation and intradermal injection of allogeneic fibroblasts into RDEB lesional skin.21,27 To date, however, none of these therapies have been shown to be consistently effective, and some have associated morbidity and mortality. Using aminoglycoside therapy in RDEB patients with nonsense mutations has never been attempted. Nevertheless, aminoglycoside therapy, if effective, would be attractive because it is relatively noninvasive and direct. In this study, we evaluated the feasibility of aminoglycosides to suppress PTCs and restore C7 expression and function for RDEB. Using RDEB keratinocytes and fibroblasts containing nonsense mutations, we demonstrated that aminoglycosides were able to bypass PTCs and produce full-length C7 protein in a dose-dependent and sustained fashion. The aminoglycoside-induced C7 protein in RDEB fibroblasts corrected the abnormal RDEB cellular phenotype, hypermotility, that is characteristic of RDEB cells. In addition, we showed that aminoglycosides were capable of promoting various degrees of PTC readthrough and inducing C7 expression in human 293 cells transfected with C7 expression constructs harboring 22 different known RDEB nonsense mutations. Our results suggest that aminoglycoside mediated nonsense-suppression therapy may provide a novel option for the treatment of RDEB patients carrying nonsense mutations.
The goal of any RDEB therapy is generating the expression of functional, full-length C7 and having it properly located in the DEJ of skin such that it provides good epidermal-dermal adherence. Several lines of evidence indicate that about 35% of the normal level of C7 is necessary for good epidermal-dermal adherence. First, a morphometric analysis of C7 in RDEB patients' skin revealed that their anchoring fibrils were decreased at least 77% below normal skin.28 Second, studies from Kern et al. in RDEB-like C7 knockout mice showed that restoration of C7 to 35% of normal was sufficient for reasonable epidermal-dermal adherence against shear forces, while adherence was compromised below this.29 Third, family members of RDEB patients who are heterozygous carriers of a COL7A1 null mutations and have 50% of the normal complement of C7 and AFs are phenotypically normal with no skin fragility or mechanobullous disease. Therefore, restoration of C7 above 35% of the level seen in normal skin will be our therapeutic goal. In this paper, we showed that both G418 and gentamicin could induce PTC read through and restore full-length C7 production in RDEB keratinocyte cell lines and primary RDEB fibroblast cultures at levels as high as 45% of normal skin cells with a single treatment. In addition, the amounts of C7 restored in RDEB fibroblasts were sufficient to correct the abnormal hypermotility cellular phenotype of RDEB cells. More importantly, using an in vitro SE model, we found that aminoglycoside-treated RDEB1 and RDEB2 keratinocytes combined into SEs with RDEB fibroblasts produced C7 that incorporated into the DEJ at 60 and 40% of levels seen in SEs derived from normal cells (Figure 6).
Systemic administration of aminoglycosides frequently causes ototoxicity and/or renal impairment.30,31 Nevertheless, RDEB is primarily a skin and mucosa disease, which lends itself to the possibility of using intralesionally injected or topically administered medications. The transdermal, intralesional, or topical administration of aminoglycosides might be effective in RDEB patients with C7 nonsense mutations and would likely significantly reduce the systemic exposure of the drug and subsequent side effects. In this regard, it is important to point out that the therapeutic concentration of topical gentamicin is 100-fold higher than that recommended for therapeutic plasma concentrations.32 Interestingly, the topical application of 0.1% gentamicin cream to patients with Hailey-Hailey disease, a blistering disease of the skin inherited in an autosomal dominant fashion, has produced promising results.32 Transdermal delivery of gentamicin combined with depilatory agent-treatment of skin has been shown to be able to penetrate skin and deliver gentamicin to target areas equivalent to systemic administration.33 Further, because C7 and AF structures are extremely stable once formed and have a slow turnover,5 the administration of aminoglycosides may not need to be constant. That is, perhaps aminoglycosides could be administered to the RDEB patient until new blistering ceases and new AFs are formed and then discontinued for weeks or months.
As mentioned above, systemic exposure of aminoglycosides in humans frequently generates untoward side effects. Nevertheless, recently, there has been the discovery of nonaminoglycoside derivatives with much more potent PTC readthrough potential than gentamicin and without the side effects.34,35 One of the leading compounds that have been extensively evaluated is PTC124, which has minimal toxicity and can be orally administered. PTC124 is more efficient than gentamicin in stable cell lines and promotes dystrophin expression in primary muscle cells from DMD patients.36 It also restored 24–29% of the wild-type protein function in a CF mouse model.37,38 In the study herein, we evaluated PTC124 in our RDEB keratinocytes and fibroblasts harboring five different nonsense mutations but failed to observe any readthrough despite using a broad range of concentrations (data not shown), which is consistent with a recent study showing that PTC124 was not able to induce PTC readthrough in two C7 cDNA constructs harboring nonsense mutations.39
Several studies have shown that the type of the stop codon (UGA>UAG>UAA), as well as the immediate downstream nucleotides (C>U>G>A), determines the relative readthrough ability of any particular PTC mutation. Specifically, the stop codon UGA followed by a C is most susceptible to aminoglycoside-mediated readthrough.9,40 In our analysis of 22 RDEB PTC mutations (16 containing UGA and 6 containing UAG) using our transient transfection system, we did not observe an apparent correlation between the readthrough capacity and the type, context of each stop codon, or proximity to exon–intron boundaries (Supplementary Table S1). There was a range of 5–80% of C7 produced in 293 cells transfected with various nonsense constructs in response to aminoglycoside treatment. Since 16 out of 22 of our generated nonsense mutations were comprised of the UGA-G stop codon, it was surprising to see such readthrough variance. It was also interesting to note that a number of “less optimal” PTC mutations bearing the UAG stop codon such as Q189X (UAGC), Q906X (UAGG), Q641X (UAGA), and Q251X (UAGU) displayed greater readthrough activity than several UGA-G mutations. In keeping with our observations, a recent study with an in-depth statistical analysis of gentamicin-induced readthrough efficiency with 66 nonsense mutations showed that the type of the stop codon did not affect the response to gentamicin. They found, however, that the presence of a cytosine at the +4 position and a uracil residue immediately upstream from the PTC promoted higher levels of readthrough activity than other nucleotides.41 In our study herein, aminoglycoside-mediated readthrough efficacy of the 22 RDEB nonsense mutations did not follow this rule. Unfortunately, the set of mutations analyzed thus far does not give us enough statistical power to analyze the possible correlation between readthrough efficiency and the nature of the PTC and surrounding nucleotides. We did, however, observe a correlation between the location of the PTC along the COL7A1 gene encoding for C7 and readthrough efficacy. More specifically, of the 16 UGA-G mutations studied here, those mutations occurring early in the reading frame (amino acid less than 1,000) are more readily bypassed than those occurring later. For example, a patient with a R20X mutation and 51% response to gentamicin might be expected to respond better than a person with a R1933X mutation with only an 11% response. This may be relevant since an adequate therapeutic response in RDEB is estimated to be a restoration of C7 at the DEJ to ~35% of the normal level. A systematic examination of all 70 RDEB nonsense mutations, including more mutations from suboptimal PTCs, or the creation of synthetic mutations encompassing all varieties of PTCs at specific locations along C7, would be necessary for a more definitive answer about how the type of stop codon and its surrounding nucleotide context affect the readthrough efficiency. Our data presented here, however, may provide useful information for guiding “personalized therapy” of RDEB patients harboring the specific nonsense mutations reported in this study.
In summary, our data provide evidence that the treatment of RDEB cells carrying various PTCs in the COL7A1 gene with aminoglycosides results in the re-expression of sufficient functional C7 protein that can reverse the abnormal RDEB cellular phenotype and incorporate into the DEJ of cultured SEs. Additionally, we further demonstrated the general utility of aminoglycoside-mediated readthrough in 293 cells transiently transfected with 22 different RDEB nonsense mutations. Therefore, our study provides the proof of concept for using aminoglycosides to suppress PTCs and induce C7 expression in RDEB patients with nonsense mutations. The utility of aminoglycosides for treating PTC mutations associated with RDEB needs to be further evaluated in clinical trials. In addition, the systematic screening for aminoglycoside derivatives that maintain their capacity to suppress disease-causing PTCs in RDEB but also have reduced toxicity, should be of considerable clinical interest since about 10–25% of RDEB patients harbor nonsense mutations. Lastly, therapy based on suppressing PTCs may also be applied to other inherited skin diseases caused by nonsense mutations.
Materials and Methods
Cell cultures. The human embryonic kidney cell line 293 T was cultured in Dulbecco's modified essential medium (DMEM) supplemented with 10% fetal bovine serum. Two immortalized RDEB keratinocyte lines, RDEB1 heterozygous for R578X and Q906X mutations and RDEB2 homozygous for the Q251X mutation were cultured in low calcium, serum-free keratinocyte growth medium supplemented with bovine pituitary extract, and epidermal growth factor (SFM; GIBCO BRL, Gaithersburg, MD) as described by Boyce and Ham42 and modified by O'Keefe and Chiu.43 RDEB keratinocytes were immortalized using the E6 and E7 genes of human papillomavirus type 6 as described.17 Primary dermal fibroblasts from two RDEB patients, RDEB3 homozygous for R578X mutations and RDEB4 heterozygous for R163X and R1683X mutations were established from the patients' skin biopsies as described44 and cultured in DMEM/Ham's F12 (1:1) supplemented with 10% fetal bovine serum. Normal human fibroblasts from neonatal foreskin were initiated into culture as described previously.17 Primary fibroblasts were passaged as they reached confluence and all experiments were performed on cells between passages 4 and 6.
Aminoglycoside treatment and immunoblot analysis. In experiments where an aminoglycoside was used to induce PTC readthrough, keratinocytes or fibroblasts from normal or RDEB patients at 60–70% confluency were exposed to G418 (1–200 μg/ml, Life Technologies, Carlsbad, CA), gentamicin (25–800 μg/ml, Sigma, St Louis, MO) or paromomycin (500–2,000 μg/ml, Sigma) for 48 hours. For 293 cells transiently transfected with cDNA expression constructs containing nonsense mutations, aminoglycosides were added to cells 24 hours after transfection.
To determine the cellular expression of readthrough C7 protein, cellular extracts were prepared 48 hours after incubating with the above drugs as described and subjected to 4–12% SDS-PAGE (Bio-Rad, Hercules, CA). Proteins were then electrotransferred onto a nitrocellulose membrane. The presence of C7 was detected with polyclonal antibodies to the NC1 domain of C7, followed by a horseradish peroxidase-conjugated goat anti-rabbit IgG and enhanced chemiluminescence detection reagent (GE Healthcare, Buckinghamshire, UK).
For the sustainability of aminoglycoside treatment on C7 expression, the media containing G418 or gentamicin were removed at 48 hours after incubation, replaced with drug-free, fresh growth medium, and the cellular extracts were prepared and evaluated at various time points for C7 expression.
To determine the aminoglycoside-induced C7 secretion into the medium, the cells were treated with G418 or gentamicin for 48 hours and the medium was changed to serum-free medium containing 150 μm ascorbic acid. The cultures were maintained for various time points. The media were collected, equilibrated to 5 mmol/l ethylenediaminetetraacetic acid, 50 µmol/l N-ethylmaleimide and 50 µmol/l phenylmethylsulfonyl fluoride, concentrated 10- to 15-fold (Centricon-100, Amicon, Beverly, MA) and subjected to 4–12% SDS-PAGE followed by immunoblot analysis as described above.
Cell proliferation assay. For the aminoglycoside cytotoxicity assay, RDEB keratinocytes or fibroblasts were seeded in 12-well plates in triplicate at a density of 90,000 per well. At 24 hours after seeding, medium was changed to one containing G418 (1–100 μg/ml), gentamicin (0–800 μg/ml), or paromomycin (0–4,000 μg/ml). At 48 hours, cells were collected by trypsinzation, incubated with 0.2% trypan blue, and manually counted live cells.
Immunofluorescence staining of cell cultures. Keratinocytes were plated in TissueTek chamber slides (Nunc, Naperville, IL) on polylysine at 37 °C for 18 hours. Cells were immersed in periodate-lysine-paraformaldehyde fixative for 10 minutes at room temperature, washed several times with phosphate-buffered saline (PBS) to remove fixative, and then permeabilized and blocked by incubating in PBS with 3% bovine serum albumin, 1% saponin, and 10% normal goat serum for 15 minutes at room temperature. The cells were incubated with an affinity-purified polyclonal antibody to the NC1 domain of C7 at a dilution of 1:200 in a humidified chamber for 2 hours, then washed three times with PBS, 1% saponin, counterstained with a fluorescein isothiocyanate-conjugated goat antibody to rabbit IgG (1:200 dilution) for 1 hour (Organon Teknika-Capel) and washed. The cells were then examined and photographed with a Zeiss epiluminating immunofluorescence microscopy. All images were photographed using the same camera and at identical exposure times. Mean fluorescence intensity was calculated for each sample using Image J (Rasband WS, NIH, Bethesda, MD; http://rsb.info.nih.gov/ij/) as described.21
Quantitative RT-PCR. Keratinocytes were grown until confluent and treated with gentamicin (400 μg/ml) or G418 (8 μg/ml) for 24 hours. Total RNAs were extracted by Aurum total RNA kit as recommended by the manufacturer (Bio-Rad). RNA was quantified with a Nanodrop apparatus and its quality was analyzed on an agarose gel. To amplify the C7 mRNA, RT-PCR was carried out using the following primers: forward (GGTCCTAGCTGACGGCTTTT) and reverse (GGATGAGGAGCCATCCAGT) (product length of 174bp). β-actin was also amplified using the following primers: forward (CCACACTGTGCCCATCTACG) and reverse (AAGATCTTCATGAGGTAGTCA). RT-PCR assays were then performed with LightCycler technology (Roche Manheim, Manheim, Germany). Experiments were carried out in triplicate with the total mRNA from duplicate cell cultures. Student's t-test was used for the determination of P values.
Cell migration assay. Fibroblast migration was assessed as described by Woodley et al.45 Briefly, colloidal gold salts were immoblized on coverslips and coated with type I collagen (15 µg/ml). Normal human dermal fibroblasts, untreated parental RDEB fibroblasts or aminoglycoside-treated RDEB fibroblasts were suspended, plated on the coverslips and allowed to migrate for 16–20 hours. The cells were fixed in 0.1% formaladehyde in PBS and examined under dark field optics with a video camera attached to a computer equipped with image capture capability. The computer analyzes 15 nonoverlapping fields in each experimental condition with NIH Image 1.6 and determines the percentage area of each field consumed by cell migration tracks, a so-called Migration Index. Confirmation of a difference in migration as statistically significant requires rejection of the null hypothesis of no difference between mean migration indices obtained from replicate sets at the P = 0.05 level with a Student's t-test.
Establishment of in vitro organotypic SEs and immunofluorescence microscopy analysis. Establishment of an in vitro skin coculture model was performed as previously described.18 Briefly, 2 × 106 normal human fibroblasts or fibroblasts from RDEB4 patient carrying nonsense mutations were seeded onto 1.5 × 1.5-cm de-epidermalized porcine dermis and allowed to grow in DMEM and generate extracellular matrix components for 48 hours. A total of 2 × 106 normal human keratinocytes or RDEB keratinocytes untreated or treated with 400 μg/ml genetamicin for 72 hours in culture were then seeded onto the dermal equivalent and grown in combined medium (serum-free keratinocyte growth medium:DMEM in a 60:40 ratio with 10% fetal bovine serum) as described.46 The three-dimensional cultures remained submerged for 72 hours after which the cultures were raised to an air-liquid interface for an additional 2 weeks in the medium with and without gentamicin. At the end of the culture period, the cultures were processed for immunofluorescence staining.
Five-micrometer thick sections from the optimum cutting temperature-embedded SEs were cut on a cryostat, fixed for 5 minutes in cold acetone, and air-dried. SE sections were incubated with a monoclonal antibody against human C7 (clone NP185; Millipore, Billerica, MA) followed by a fluorescein isothiocyanate-conjugated goat anti-mouse IgG. Working dilutions were 1:50 for the primary antibody and 1:40 for the secondary antibody. Immunolabeling of tissue was performed using standard immunofluorescence methods as described previously.47 Representative photographs from stained sections were taken using a Zeiss Axioplan fluorescence microscope equipped with a Zeiss Axiocam MRM digital camera system. All images were photographed using the same camera and at identical exposure times. Mean fluorescence intensity at DEJ was calculated for each sample using Image J (Rasband WS, NIH; http://rsb.info.nih.gov/ij/).
Site-directed mutagenesis and transfection. Site-directed mutagenesis was performed on C7 cDNA in the pRC/CMV vector using a commercial kit (QuikChange II site-directed mutagenesis kit, Stratagene, La Jolla, CA) according to the manufacturer's instructions as described previously.48 Briefly, a pair of complementary primers with 39 bases was designed, and a mutation to change arginine or glutamine residues to a stop codon was placed in the middle. We generated primers for the following 22 mutations associated with RDEB reported in the literature: R20X, R137X, R185X, Q189X, R236X, Q251X, Q281X, R525X, R578X, Q641X, R669X, W796X, R843X, Q906X, Q1211X, R1340X, R1343X, R1630X, R1632X, R1730X, R1933X, R2261X. Parental cDNA inserted in pRC/CMV was amplified using Pyrococcus furiosus DNA polymerase with these primers for 16 cycles in a DNA thermal cycler (Perkin-Elmer, Norwalk, CT). After digestion of the parental DNA with DpnI, the amplified DNA with nucleotide substitution incorporated was transformed into Escherichia coli (XL1-Blue). The mutations were confirmed by automated DNA sequencing.
The expression vector encoding for wild type or mutant C7 cDNA was used to transfect the human embryonal kidney cell line 293 (ATCC, Rockville, MD) using Lipofectamine LTX in combination with Plus Reagent (Life Technologies). Cells were plated and transfected according to the manufacturer's instructions. 24 hours after transfection, media was changed to fresh growth medium containing the various aminoglycosides for 48 hours.
Protein purification and analysis. To purify aminoglycoside-induced C7 from 293 cells, we first generated 293 cells stably transfected with C7 expression constructs using 500 μg/ml G418. Cells were grown to confluence in the absence of G418 and then treated for 48 hours with gentamicin (200 μg/ml). We then purified the secreted C7 protein from serum-free media as described.48 Purified wild-type or aminoglycoside-induced C7 was incubated with chymotrypsin (Sigma) in 50 mmol/l Tris-HCl, pH 7.4, 150 mmol/l NaCl at 14 °C for 3 hours at an enzyme-to-substrate ratio of 1:10 by weight as described previously.48 The digestion products were then analyzed by SDS-PAGE followed by immunoblot analysis with a polyclonal antibody against the triple helical domain of C7 as described.48
SUPPLEMENTARY MATERIAL Figure S1. Aminoglycosides do not significantly affect RDEB cell viability. Figure S2. There is no basal level of C7 readthrough in 293 cells transfected with cDNA constructs coding for RDEB nonsense mutants. Table S1. Summary of PTC sequences and readthrough efficiency of 22 RDEB nonsense mutations in response to aminoglycosides.
Acknowledgments
This work was supported by grants (NIH RO1 AR47981 to M.C., RC4AR060535 and RO1 AR33625 to M.C. and D.T.W.).
Supplementary Material
References
- Lin AN, Carter DM. Epidermolysis Bullosa: Basic and Clinical Aspects. Springer-Verlag New York, Inc; New York; 1992. [Google Scholar]
- Uitto J, Christiano AM. Molecular basis for the dystrophic forms of epidermolysis bullosa: mutations in the type VII collagen gene. Arch Dermatol Res. 1994;287:16–22. doi: 10.1007/BF00370713. [DOI] [PubMed] [Google Scholar]
- Uitto J, Christiano AM. Molecular genetics of the cutaneous basement membrane zone. Perspectives on epidermolysis bullosa and other blistering skin diseases. J Clin Invest. 1992;90:687–692. doi: 10.1172/JCI115938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parente MG, Chung LC, Ryynänen J, Woodley DT, Wynn KC, Bauer EA, et al. Human type VII collagen: cDNA cloning and chromosomal mapping of the gene. Proc Natl Acad Sci USA. 1991;88:6931–6935. doi: 10.1073/pnas.88.16.6931. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Burgeson RE. Type VII collagen, anchoring fibrils, and epidermolysis bullosa. J Invest Dermatol. 1993;101:252–255. doi: 10.1111/1523-1747.ep12365129. [DOI] [PubMed] [Google Scholar]
- Sakai LY, Keene DR, Morris NP, Burgeson RE. Type VII collagen is a major structural component of anchoring fibrils. J Cell Biol. 1986;103:1577–1586. doi: 10.1083/jcb.103.4.1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dang N, Murrell DF. Mutation analysis and characterization of COL7A1 mutations in dystrophic epidermolysis bullosa. Exp Dermatol. 2008;17:553–568. doi: 10.1111/j.1600-0625.2008.00723.x. [DOI] [PubMed] [Google Scholar]
- Wertheim-Tysarowska K, Sobczyńska-Tomaszewska A, Kowalewski C, Skroński M, Swiȩćkowski G, Kutkowska-Kaźmierczak A, et al. The COL7A1 mutation database. Hum Mutat. 2012;33:327–331. doi: 10.1002/humu.21651. [DOI] [PubMed] [Google Scholar]
- Zingman LV, Park S, Olson TM, Alekseev AE, Terzic A. Aminoglycoside-induced translational read-through in disease: overcoming nonsense mutations by pharmacogenetic therapy. Clin Pharmacol Ther. 2007;81:99–103. doi: 10.1038/sj.clpt.6100012. [DOI] [PubMed] [Google Scholar]
- Barton-Davis ER, Cordier L, Shoturma DI, Leland SE, Sweeney HL. Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice. J Clin Invest. 1999;104:375–381. doi: 10.1172/JCI7866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedwell DM, Kaenjak A, Benos DJ, Bebok Z, Bubien JK, Hong J, et al. Suppression of a CFTR premature stop mutation in a bronchial epithelial cell line. Nat Med. 1997;3:1280–1284. doi: 10.1038/nm1197-1280. [DOI] [PubMed] [Google Scholar]
- Linde L, Kerem B. Introducing sense into nonsense in treatments of human genetic diseases. Trends Genet. 2008;24:552–563. doi: 10.1016/j.tig.2008.08.010. [DOI] [PubMed] [Google Scholar]
- Wagner KR, Hamed S, Hadley DW, Gropman AL, Burstein AH, Escolar DM, et al. Gentamicin treatment of Duchenne and Becker muscular dystrophy due to nonsense mutations. Ann Neurol. 2001;49:706–711. [PubMed] [Google Scholar]
- Kuschal C, DiGiovanna JJ, Khan SG, Gatti RA, Kraemer KH. Repair of UV photolesions in xeroderma pigmentosum group C cells induced by translational readthrough of premature termination codons. Proc Natl Acad Sci USA. 2013;110:19483–19488. doi: 10.1073/pnas.1312088110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholson P, Yepiskoposyan H, Metze S, Zamudio Orozco R, Kleinschmidt N, Mühlemann O. Nonsense-mediated mRNA decay in human cells: mechanistic insights, functions beyond quality control and the double-life of NMD factors. Cell Mol Life Sci. 2010;67:677–700. doi: 10.1007/s00018-009-0177-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bidou L, Allamand V, Rousset JP, Namy O. Sense from nonsense: therapies for premature stop codon diseases. Trends Mol Med. 2012;18:679–688. doi: 10.1016/j.molmed.2012.09.008. [DOI] [PubMed] [Google Scholar]
- Chen M, Kasahara N, Keene DR, Chan L, Hoeffler WK, Finlay D, et al. Restoration of type VII collagen expression and function in dystrophic epidermolysis bullosa. Nat Genet. 2002;32:670–675. doi: 10.1038/ng1041. [DOI] [PubMed] [Google Scholar]
- Contard P, Bartel RL, Jacobs L, 2nd, Perlish JS, MacDonald ED, 2nd, Handler L, et al. Culturing keratinocytes and fibroblasts in a three-dimensional mesh results in epidermal differentiation and formation of a basal lamina-anchoring zone. J Invest Dermatol. 1993;100:35–39. doi: 10.1111/1523-1747.ep12349952. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Keene DR, Atha T, Huang Y, Lipman K, Li W, et al. Injection of recombinant human type VII collagen restores collagen function in dystrophic epidermolysis bullosa. Nat Med. 2004;10:693–695. doi: 10.1038/nm1063. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Krueger GG, Jorgensen CM, Fairley JA, Atha T, Huang Y, et al. Normal and gene-corrected dystrophic epidermolysis bullosa fibroblasts alone can produce type VII collagen at the basement membrane zone. J Invest Dermatol. 2003;121:1021–1028. doi: 10.1046/j.1523-1747.2003.12571.x. [DOI] [PubMed] [Google Scholar]
- Wong T, Gammon L, Liu L, Mellerio JE, Dopping-Hepenstal PJ, Pacy J, et al. Potential of fibroblast cell therapy for recessive dystrophic epidermolysis bullosa. J Invest Dermatol. 2008;128:2179–2189. doi: 10.1038/jid.2008.78. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Keene DR, Atha T, Huang Y, Ram R, Kasahara N, et al. Intradermal injection of lentiviral vectors corrects regenerated human dystrophic epidermolysis bullosa skin tissue in vivo. Mol Ther. 2004;10:318–326. doi: 10.1016/j.ymthe.2004.05.016. [DOI] [PubMed] [Google Scholar]
- Remington J, Wang X, Hou Y, Zhou H, Burnett J, Muirhead T, et al. Injection of recombinant human type VII collagen corrects the disease phenotype in a murine model of dystrophic epidermolysis bullosa. Mol Ther. 2009;17:26–33. doi: 10.1038/mt.2008.234. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Urda S, Thyagarajan B, Keene DR, Lin Q, Fang M, Calos MP, et al. Stable nonviral genetic correction of inherited human skin disease. Nat Med. 2002;8:1166–1170. doi: 10.1038/nm766. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Wang X, Amir M, Hwang B, Remington J, Hou Y, et al. Intravenously injected recombinant human type VII collagen homes to skin wounds and restores skin integrity of dystrophic epidermolysis bullosa. J Invest Dermatol. 2013;133:1910–1913. doi: 10.1038/jid.2013.10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Woodley DT, Remington J, Huang Y, Hou Y, Li W, Keene DR, et al. Intravenously injected human fibroblasts home to skin wounds, deliver type VII collagen, and promote wound healing. Mol Ther. 2007;15:628–635. doi: 10.1038/sj.mt.6300041. [DOI] [PubMed] [Google Scholar]
- Wagner JE, Ishida-Yamamoto A, McGrath JA, Hordinsky M, Keene DR, Woodley DT, et al. Bone marrow transplantation for recessive dystrophic epidermolysis bullosa. N Engl J Med. 2010;363:629–639. doi: 10.1056/NEJMoa0910501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tidman MJ, Eady RA. Evaluation of anchoring fibrils and other components of the dermal-epidermal junction in dystrophic epidermolysis bullosa by a quantitative ultrastructural technique. J Invest Dermatol. 1985;84:374–377. doi: 10.1111/1523-1747.ep12265460. [DOI] [PubMed] [Google Scholar]
- Fritsch A, Loeckermann S, Kern JS, Braun A, Bösl MR, Bley TA, et al. A hypomorphic mouse model of dystrophic epidermolysis bullosa reveals mechanisms of disease and response to fibroblast therapy. J Clin Invest. 2008;118:1669–1679. doi: 10.1172/JCI34292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swan SK. Aminoglycoside nephrotoxicity. Semin Nephrol. 1997;17:27–33. [PubMed] [Google Scholar]
- Forge A, Schacht J. Aminoglycoside antibiotics. Audiol Neurootol. 2000;5:3–22. doi: 10.1159/000013861. [DOI] [PubMed] [Google Scholar]
- Kellermayer R, Szigeti R, Keeling KM, Bedekovics T, Bedwell DM. Aminoglycosides as potential pharmacogenetic agents in the treatment of Hailey-Hailey disease. J Invest Dermatol. 2006;126:229–231. doi: 10.1038/sj.jid.5700031. [DOI] [PubMed] [Google Scholar]
- Shiozuka M, Wagatsuma A, Kawamoto T, Sasaki H, Shimada K, Takahashi Y, et al. Transdermal delivery of a readthrough-inducing drug: a new approach of gentamicin administration for the treatment of nonsense mutation-mediated disorders. J Biochem. 2010;147:463–470. doi: 10.1093/jb/mvp185. [DOI] [PubMed] [Google Scholar]
- Brendel C, Belakhov V, Werner H, Wegener E, Gärtner J, Nudelman I, et al. Readthrough of nonsense mutations in Rett syndrome: evaluation of novel aminoglycosides and generation of a new mouse model. J Mol Med (Berl) 2011;89:389–398. doi: 10.1007/s00109-010-0704-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Houghton JL, Green KD, Chen W, Garneau-Tsodikova S. The future of aminoglycosides: the end or renaissance. Chembiochem. 2010;11:880–902. doi: 10.1002/cbic.200900779. [DOI] [PubMed] [Google Scholar]
- Welch EM, Barton ER, Zhuo J, Tomizawa Y, Friesen WJ, Trifillis P, et al. PTC124 targets genetic disorders caused by nonsense mutations. Nature. 2007;447:87–91. doi: 10.1038/nature05756. [DOI] [PubMed] [Google Scholar]
- Bordeira-Carriço R, Pêgo AP, Santos M, Oliveira C. Cancer syndromes and therapy by stop-codon readthrough. Trends Mol Med. 2012;18:667–678. doi: 10.1016/j.molmed.2012.09.004. [DOI] [PubMed] [Google Scholar]
- Du M, Liu X, Welch EM, Hirawat S, Peltz SW, Bedwell DM. PTC124 is an orally bioavailable compound that promotes suppression of the human CFTR-G542X nonsense allele in a CF mouse model. Proc Natl Acad Sci USA. 2008;105:2064–2069. doi: 10.1073/pnas.0711795105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McElroy SP, Nomura T, Torrie LS, Warbrick E, Gartner U, Wood G, et al. A lack of premature termination codon read-through efficacy of PTC124 (Ataluren) in a diverse array of reporter assays. PLoS Biol. 2013;11:e1001593. doi: 10.1371/journal.pbio.1001593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keeling KM, Bedwell DM. Clinically relevant aminoglycosides can suppress disease-associated premature stop mutations in the IDUA and P53 cDNAs in a mammalian translation system. J Mol Med (Berl) 2002;80:367–376. doi: 10.1007/s00109-001-0317-z. [DOI] [PubMed] [Google Scholar]
- Floquet C, Hatin I, Rousset JP, Bidou L. Statistical analysis of readthrough levels for nonsense mutations in mammalian cells reveals a major determinant of response to gentamicin. PLoS Genet. 2012;8:e1002608. doi: 10.1371/journal.pgen.1002608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boyce ST, Ham RG. Calcium-regulated differentiation of normal human epidermal keratinocytes in chemically defined clonal culture and serum-free serial culture. J Invest Dermatol. 1983;81 suppl. 1:33s–40s. doi: 10.1111/1523-1747.ep12540422. [DOI] [PubMed] [Google Scholar]
- O'Keefe EJ, Chiu ML. Stimulation of thymidine incorporation in keratinocytes by insulin, epidermal growth factor, and placental extract: comparison with cell number to assess growth. J Invest Dermatol. 1988;90:2–7. doi: 10.1111/1523-1747.ep12462409. [DOI] [PubMed] [Google Scholar]
- Normand J, Karasek MA. A method for the isolation and serial propagation of keratinocytes, endothelial cells, and fibroblasts from a single punch biopsy of human skin. In Vitro Cell Dev Biol Anim. 1995;31:447–455. doi: 10.1007/BF02634257. [DOI] [PubMed] [Google Scholar]
- Woodley DT, Bachmann PM, O'Keefe EJ. Laminin inhibits human keratinocyte migration. J Cell Physiol. 1988;136:140–146. doi: 10.1002/jcp.1041360118. [DOI] [PubMed] [Google Scholar]
- Krueger GG, Jorgensen CM. Defined system to assess the in vitro induction of a psoriasis phenotype on normal keratinocytes by fibroblasts from psoritic subjects. J Cutan Med Surg. 1997;2:20–25. [Google Scholar]
- Gammon WR, Briggaman RA, Inman AO, 3rd, Queen LL, Wheeler CE. Differentiating anti-lamina lucida and anti-sublamina densa anti-BMZ antibodies by indirect immunofluorescence on 1.0 M sodium chloride-separated skin. J Invest Dermatol. 1984;82:139–144. doi: 10.1111/1523-1747.ep12259692. [DOI] [PubMed] [Google Scholar]
- Chen M, Costa FK, Lindvay CR, Han YP, Woodley DT. The recombinant expression of full-length type VII collagen and characterization of molecular mechanisms underlying dystrophic epidermolysis bullosa. J Biol Chem. 2002;277:2118–2124. doi: 10.1074/jbc.M108779200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.