

Abstract
Systemic delivery of a lentiviral vector carrying a therapeutic gene represents a new treatment for monogenic disease. Previously, we have shown that transfer of the adenosine deaminase (ADA) cDNA in vivo rescues the lethal phenotype and reconstitutes immune function in ADA-deficient mice. In order to translate this approach to ADA-deficient severe combined immune deficiency patients, neonatal ADA-deficient mice and newborn rhesus monkeys were treated with species-matched and mismatched vectors and pseudotypes. We compared gene delivery by the HIV-1-based vector to murine γ-retroviral vectors pseudotyped with vesicular stomatitis virus-glycoprotein or murine retroviral envelopes in ADA-deficient mice. The vesicular stomatitis virus-glycoprotein pseudotyped lentiviral vectors had the highest titer and resulted in the highest vector copy number in multiple tissues, particularly liver and lung. In monkeys, HIV-1 or simian immunodeficiency virus vectors resulted in similar biodistribution in most tissues including bone marrow, spleen, liver, and lung. Simian immunodeficiency virus pseudotyped with the gibbon ape leukemia virus envelope produced 10- to 30-fold lower titers than the vesicular stomatitis virus-glycoprotein pseudotype, but had a similar tissue biodistribution and similar copy number in blood cells. The relative copy numbers achieved in mice and monkeys were similar when adjusted to the administered dose per kg. These results suggest that this approach can be scaled-up to clinical levels for treatment of ADA-deficient severe combined immune deficiency subjects with suboptimal hematopoietic stem cell transplantation options.
Introduction
Adenosine deaminase-deficient severe combined immune deficiency (ADA-SCID) is characterized by a severe primary pan-lymphoid immune deficiency and a systemic purine metabolic disorder affecting multiple organs. The disease is fatal if not treated with hematopoietic stem cell (HSC) transplantation, enzyme replacement therapy (ERT), or autologous HSC transplant of gene-corrected cells.1 HSC transplant/gene therapy has resulted in good clinical benefit and immune reconstitution in pediatric patients2; however, in older children, outcomes have been more variable.3,4
ADA-deficient mice are immune deficient but die of a noninfectious pulmonary insufficiency within 3 weeks after birth unless treated with HSC transplant, ERT, or HSC transplant/gene therapy.5,6,7 We have previously shown that intravenous administration of an HIV-1-based, vesicular stomatitis virus-glycoprotein (VSV-G) pseudotyped lentiviral vector with the human ADA cDNA (HIV-ADA/VSV) supported survival and immunological reconstitution in ADA-deficient (ADA−/−) mice. High vector copy numbers and ADA enzyme activity were measured in the liver and lung, and these tissues may have served as a “metabolic sink” to provide systemic detoxification of accumulated adenine metabolites and multisystem correction.8 ADA enzyme production in vivo could provide an additional treatment alternative especially when an appropriate HSC donor is unavailable or for older children or adult late-onset patients where results with HSC transplant/gene therapy may be suboptimal.
Systemic administration of HIV-ADA/VSV was shown to be efficacious in ADA−/− mice, but survival was dose-dependent and required delivery of 5 × 1010 TU/kg. It is not clear if this dosage will be required for clinical benefit in ADA-deficient patients, since in mice the quantity of ADA required for survival appears to be higher than the necessary amount for immune reconstitution.6,9 Regardless, translation of this modality to the clinic requires understanding the feasibility of scaling up the approach from mice to a large animal model. In these studies, we set out to determine the vector biodistribution and pharmacokinetics following systemic administration of integrating gene delivery vectors in neonatal ADA-deficient mice and healthy newborn rhesus monkeys, as well as to understand how vector backbone and pseudotype can impact vector biodistribution. The use of a vector backbone and/or pseudotype derived from a virus related to the host (e.g. murine γ-retrovirus (gRV) in mice or a simian immunodeficiency virus (SIV)-based lentiviral vector in nonhuman primates) may lead to enhanced gene transfer efficiency, potentially lowering the vector dosages necessary to achieve a therapeutic level of transgene expression.
Different combinations of vector backbone and pseudotypes may also reveal potential differences in vector biodistribution observed in mice and humans because mammalian cells have developed a variety of species-specific restriction factors/mechanisms that can block entry or retroviral replication. Some implicated factors include APOBEC proteins (inhibits retroviruses by deaminating cytosine residues), TRIM5α (interferes with uncoating and prevents reverse transcription), tetherin (traps budding virions at the cell surface) and SAMHD1 (hydrolyzes dNTPs to limit retroviral replication).10,11 HIV-1-based lentiviral vectors are blocked from transducing the most primitive rhesus monkey HSC via a TRIM5α-mediated mechanism12,13 and the use of the HIV-1 vector in rhesus monkeys to model the potential biodistribution in humans may provide an inaccurate assessment. Furthermore, while intravenous administration of HIV-ADA/VSV did result in immune reconstitution of rescued ADA−/− mice, we were not able to detect murine HSC transduction,8 which is in contrast to other published reports where intravenous administration of a murine gRV and LV vectors into mice did result in murine HSC transduction.14,15,16
With these issues in mind, we compared the biodistribution of a murine gRV vector, MND-MFG-hADA (MMA), and an HIV-1 lentiviral vector (HIV-ADA) in ADA-deficient neonatal mice. Similarly, we compared the biodistribution of a nonpathogenic SIV molecular clone (SIVmac1A11)-based lentiviral vector to an HIV-1 based lentiviral vector in newborn rhesus monkeys. While VSV-G pseudotyped vectors exhibit a broad tropism, it was unclear if a pseudotype derived from a species-specific virus would result in greater HSC transduction in vivo. Therefore, in these studies, we compared the biodistribution of VSV-G pseudotyped vectors to vectors pseudotyped with the murine ecotropic (Eco) and amphotropic (Ampho) retroviral envelope glycoproteins in mice, and with a gibbon ape leukemia virus (GALV)-derived envelope17 in newborn rhesus monkeys.
We demonstrated that systemic delivery of an HIV-1-based lentiviral vector resulted in the highest tissue vector copy number compared to the murine gRV vector pseudotyped with VSV or murine envelopes in ADA−/− mice. We also demonstrated that the biodistribution of HIV/VSV in rhesus monkeys was comparable to the biodistribution of SIV/VSV, but that only SIV/VSV was detectable in bone marrow CD34+ cells and in some lymphocyte subsets. By comparing the overall biodistribution of ADA expressing lentiviral vectors in mice and monkeys, we found equivalent biodistribution on a dose per kg adjusted basis, demonstrating that this approach is scalable and can be translated to clinical applications for ADA deficiency as well as other monogenic defects. Another important finding was differences observed in the HIV-ADA/VSV vector biodistribution in mice compared to monkeys. These differences may reflect differences in restriction factors but may also reflect differences in the developmental state of newborn mice compared to newborn rhesus monkeys. In either case, these studies emphasize the importance of modeling this approach in a relevant nonhuman primate model.18
Results
ADA knock-out (KO) mouse study 1: systemic administration of lentiviral vectors in ADA−/− and ADA+/− mice
Our previous studies demonstrated that systemic administration of 5.0 × 1010 TU/kg of a lentiviral vector, HIV-ADA/VSV prevented the lethal pulmonary insufficiency associated with ADA deficiency and immune reconstitution, whereas mice treated with one log less vector did not survive. In this study, litters of ADA−/− and ADA+/− newborn mice (days 1–3) were treated with the HIV-ADA/VSV lentiviral vector at a dose of 5.0 × 1010 TU/kg or a twofold lower dose of 2.5 × 1010 TU/kg (Table 1; Figure 1). Even the modest twofold reduction in vector dose resulted in fewer ADA−/− mice surviving out to 3.5 months (2 of 6; 110 days) compared to the ADA−/− mice that received the effective dose of 5.0 × 1010 TU/kg (6 of 8; 110 days) (Figure 2a). However, in the ADA−/− mice surviving to 6 months, there was no dose-dependent difference in the vector copy number nor in the ADA enzyme activity measured in all tissues suggesting a threshold for survival (Figure 2b,c). In our previous studies, we observed a 90% decrease in liver vector copy number from 2 to 4 months.8 In these studies, we did not observe a decrease in liver vector copy number from 3 to 6 months (Figure 2d). Since all littermates were treated identically, we analyzed all of the surviving pups in the litter and found no significant difference in tissue vector copy number in ADA−/− mice compared to ADA+/− mice (Figure 2e). In subsequent studies, when ADA−/− mouse survival was low, the ADA+/− littermates were used as surrogates to gain insight into the biodistribution of each vector tested.
Table 1. Vector construct, pseudotype, titer, and dose of in vivo studies.
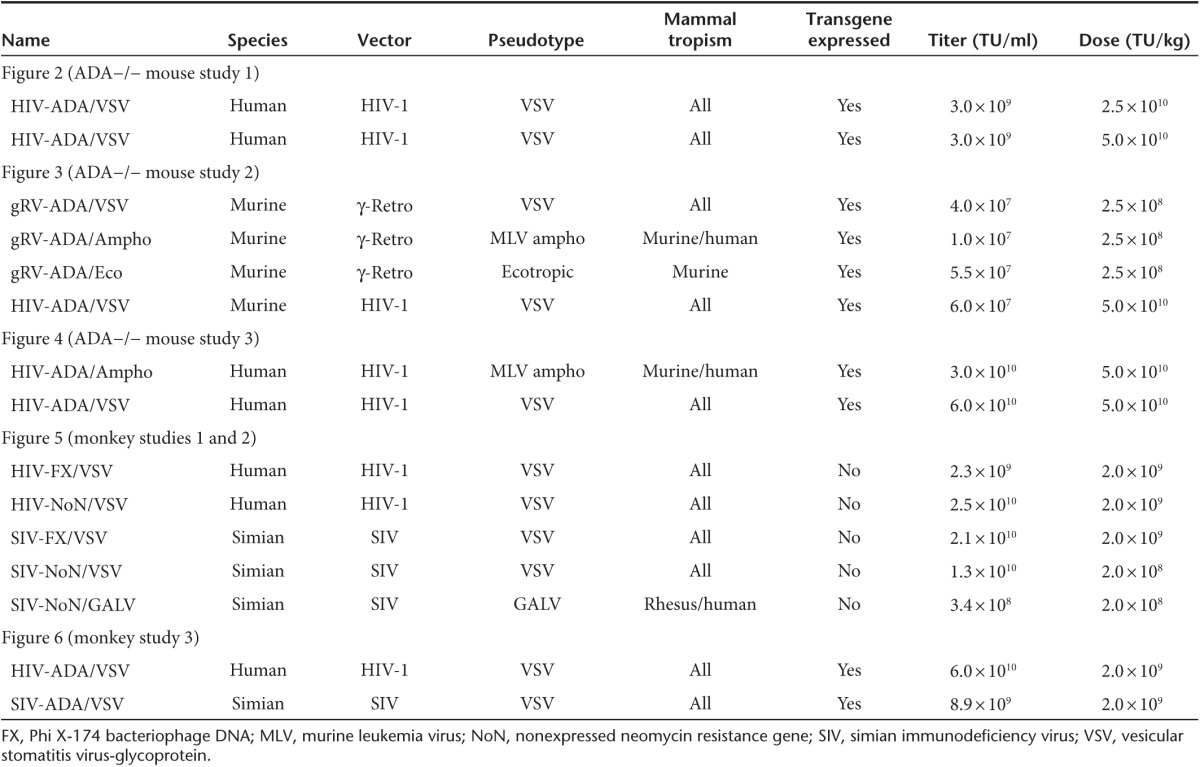
Figure 1.

Vectors. (a) Expressed transgene vectors. MND-MFG-ADA (MMA) γ-retroviral vector with the MND LTR enhancer/promoter driving expression of the human ADA cDNA (HuADA). The Moloney murine leukemia virus packaging region (Ψ), 5′ portion of the gag gene (gag), and env untranslated region (env utr) are indicated. SIN HIV-1 lentiviral vector with the MNDU3 enhancer/promoter driving expression of human ADA cDNA or eGFP and SIN SIV lentiviral vector with the MSCV enhancer/promoter driving expression of the human ADA cDNA or eGFP. The U3, R, and U5 regions of the LTR are shown, with the SIN deletion indicated by the X. HIV-1 or SIVmac1A11 packaging region (Ψ), 5′ portion of the gag gene (gag), rev-responsive element (rre) and central polypurine tract (cPPT) are indicated. (b) Nonexpressed transgene lentiviral vectors. The same HIV-1 and SIVmac1A11 vector backbones described in (a) were used to carry nonexpressed gene sequences derived from truncated PhiX174 DNA (FX) or the bacterial transposon neomycin-resistance gene with the translational start codon eliminated (NoN).
Figure 2.
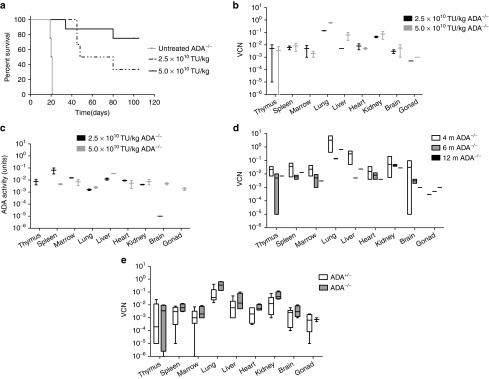
Systemic administration of the lentiviral vector in ADA−/− and ADA+/− mice. (a) Kaplan–Meier survival curves of ADA−/− mice when treated with 5.0 × 1010 TU/kg (N = 8), 2.5 × 1010 TU/kg (N = 6), or untreated (N = 4). (b) Box and whisker (10–90th percentile) plots of tissue vector copy number from ADA−/− mice at 6 months (N = 2 for each dose). (c) Box and whisker (10–90th percentile) plots of ADA enzyme activity at 6 months (N = 2 for each dose). (d). Floating bar (min–max) plots of tissue vector copy number over time (4 months, N = 5; 6 months, N = 2; 12 months, N = 1) in ADA−/− mice treated with 5.0 × 1010 TU/kg. (e) Box and whisker (10–90th percentile) plots of tissue vector copy number from ADA−/− mice (N = 4) and ADA+/− littermates (N = 6) at 6 months (combined data for 2.5 and 5.0 × 1010 TU/kg dose).
ADA KO mouse study 2: comparison of gRV/Ampho, gRV/Eco, and gRV/VSV
Litters of ADA gene KO neonates (ADA−/− and ADA+/−) were treated with the MND-MFG-ADA (MMA) gRV vector pseudotyped with either the VSV-glycoprotein (gRV/VSV), the MLV-amphotropic envelope (gRV/Ampho), or the murine-ecotropic envelope (gRV/Eco) at the best achievable dose of 2.5 × 108 TU/kg, and were compared to mice treated with the HIV-ADA/VSV lentiviral vector at the standard dose of 5.0 × 1010 TU/kg (Table 1). ADA−/− mice did not survive when treated with the gRV/Ampho (0 of 4; 21 days). Survival was reduced with the gRV/Eco (2 of 6; 90 days) and with gRV/VSV (2 of 5; 90 days) compared to the survival of recipients of the HIV-ADA/VSV (5 of 5; 90 days; P <0.0001) (Figure 3a). Despite the lower dose of gRV/VSV administered, there were no differences in the number of vector copies detected in the liver (mean ± SEM: 0.327 ± 0.323) of the two surviving ADA−/− mice treated with the gRV/VSV compared to the recipients of the higher dose of HIV-ADA/VSV (0.067 ± 0.17) (Figure 3b). In contrast, gRV/Eco was detected in most of the ADA−/− tissues analyzed, but the vector copy number was uniformly low and near the detection limit at 10–5 copies or 1 marked cell in 1 × 105. Likewise, in ADA+/− mice treated with the gRV/Ampho or the gRV/Eco, the vector copy number was uniformly low or undetectable (Figure 3c). Also notable was the lack of vector sequences in bone marrow cells isolated from ADA−/− and ADA+/− mice when treated with any gRV vector (total N = 31) regardless of pseudotype, which was significantly lower than the HIV-ADA/VSV vector copy number measured in bone marrow (P < 0.0001 by Fisher's Exact Test).
Figure 3.

Comparison of gRVAmpho, gRV/Eco and gRV/VSV. Litters (ADA−/− and ADA+/−) were injected with the HIV-ADA/VSV lentiviral vector (5.0 × 1010 TU/kg) (N = 5) or with the murine γ-retroviral vector MND-MFG-ADA (gRV; 2.5 × 108 TU/kg) pseudotyped with VSV-G (N = 5), with the murine-amphotropic envelope (gRV/Ampho) (N = 4), or with the murine-ecotropic envelope (gRV/Eco) (N = 6) and were compared to ADA−/− mice (N = 3) on enzyme replacement (ERT). (a) Kaplan–Meier survival curves (P < 0.0001). (b) Floating bar (min–max) plots of vector copy number in surviving ADA−/− mice. (c) Floating bar (min–max) plots of vector copy number in treated ADA+/− littermates injected with HIV-ADA/VSV (N = 4) or with γ-retro- ADA pseudotyped with VSV-G (N = 2), amphotropic (N = 10), or ecotropic (N = 11) envelope.
ADA KO mouse study 3: comparison of HIV-ADA/VSV and HIV-ADA/Ampho
To determine the effect of pseudotype on the HIV-1 lentiviral vector in mice, litters of neonates were treated with HIV-ADA/VSV or with HIV-ADA pseudotyped with the MLV-amphotropic envelope (HIV-ADA/Ampho) each at the effective dose of 5.0 × 1010 TU/kg (Table 1). Survival was not different in ADA−/− mice treated with HIV-ADA/VSV (6 of 7; 90 days) when compared with HIV-ADA/Ampho (4 of 4; 90 days) (Figure 4a). However, HIV-ADA/VSV vector copy number was higher compared to HIV-ADA/Ampho in liver (VSV 3.93 ± 1.50, Ampho 0.186 ± 0.25; P = 0.01) and in lung (VSV 0.373 ± 0.102, Ampho 0.071 ± 0.023; P = 0.03) (Figure 4b). There were no differences detected in the number of vector copies in isolated CD4+ and CD8+ thymocytes, CD19+ splenocytes, or in CD11b+ bone marrow cells; however, the vector copy number in CD11b+ cells was 10-fold higher than in the lymphocyte populations with both vectors (Figure 4c). When compared with historical controls of untreated 16-day-old ADA−/− mice (not aged matched), all treated mice showed immune reconstitution but there were no differences in the absolute numbers of thymocytes or splenocytes in mice treated with either vector, and these numbers were similar to those in ADA-replete mice and ADA-deficient mice treated with ERT (Figure 4d,e). In general, when ADA−/− were rescued from the lethal pulmonary insufficiency, they showed immune reconstitution.6,8
Figure 4.

Comparison of HIV-1/VSV and HIV-1/Ampho. (a) Kaplan–Meier survival curves of ADA−/− treated with HIV/VSV (N = 7) or HIV/Ampho (N = 4) (both at 5.0 × 1010 TU/kg) are compared to historical controls. (b) Box and whisker (10–90th percentile) plots of tissue vector copy number from treated ADA−/− mice. (c) Box and whisker (10–90th percentile) plots of vector copy number measured in isolated cell populations from the thymus (CD4+, CD8+, CD4+CD8+), spleen (CD19+), and bone marrow (CD11b+) (N = 4 for each tissue in each arm). (d,e) Absolute immune cell counts (mean ± SEM) in ADA+/+ (N = 5), ADA−/− on PEG-ADA ERT (N = 3), ADA−/− treated with HIV-ADA/Ampho (N = 4), or ADA−/− treated with HIV-ADA/VSV (N = 4). (d) Thymocyte subpopulations. (e) Splenocyte subpopulations.
Rhesus monkey studies 1 and 2: experimental design
To assess the potential to translate this approach to clinical applications, studies of in vivo gene delivery were conducted in newborn rhesus monkeys (~500 g at birth, 250-fold higher mass than neonatal mice). Studies in vitro demonstrated that only the SIV vector could transduce the nonadherent fraction of rhesus bone marrow (hematopoietic cells); whereas both the SIV and HIV vector could readily transduce the plastic adherent fraction (stromal cells) (Supplementary Figure S1a,b). Therefore, to determine the best approach for preclinical studies, newborn monkeys were simultaneously coadministered one SIV lentiviral vector and one HIV lentiviral vector, both pseudotyped with VSV (SIV/VSV and HIV/VSV); or one SIV lentiviral vector pseudotyped with VSV (SIV/VSV) and one SIV lentiviral vector pseudotyped with a GALV-derived envelope (SIV/GALV) (Table 2). Each vector contained a different nonexpressed transgene (mutated neomycin resistance gene, NoN; or truncated Phi X-174 bacteriophage DNA, FX) such that the biodistribution of both lentiviral vectors could be quantified (Tables 1 and 2; Figure 1). The dosage for SIV/VSV and HIV/VSV administered at birth was 2.0 × 109 TU/kg, and the dosage for SIV/GALV was 10-fold less at 2.0 × 108 TU/kg because this was the highest dose achievable due to limitations related to production titer (Table 1). All rhesus monkeys had birth weights within the normative range; and after vector administration all showed normal growth rates, complete blood counts, and clinical chemistry panels when compared with historical and concurrent control data for all parameters (data not shown). In monkey study 1, tissue harvests were performed at 1 month postnatal age and in monkey study 2, tissue harvests were performed at 3 months postnatal age. All vector copy number data were analyzed across treatment groups as well as by paired comparison within an individual rhesus monkey.
Table 2. Study design for newborn rhesus monkeys administered lentiviral vectors (HIV or SIV) intravenously at birtha.

Rhesus monkey studies 1 and 2: SIV/VSV compared to SIV/GALV
While transduction by VSV pseudotyped vectors does not appear to be restricted in blood cells, we chose to investigate the use of a nonhuman primate envelope on the transduction of blood cells and, ideally, HSC in vivo. After 1 month, SIV/GALV vector copy number was near the detection limit in CD2+, CD8+, and in CD34+ peripheral blood cells, but there were no differences when compared with the SIV/VSV vector copy number without normalizing for the lower GALV dose (Figure 5a). Furthermore, the SIV/GALV vector copy number was not different when compared with SIV/VSV in isolated bone marrow CD34− and CD34+ cells, suggesting that SIV/GALV may transduce hematopoietic cells more efficiently if given at a dose comparable to the other vectors used (Figure 5b). However, in isolated splenocytes, the SIV/VSV vector copy number was higher than SIV/GALV in paired comparisons (P = 0.0256) (Supplementary Figure S2b), and only the SIV/VSV vector was detected in thymocytes (Figure 5c). Tissue analysis showed that after 1 month (monkey study 1) SIV/GALV was also detectable in the liver, lung, heart, adrenal glands, lymph nodes, and spleen but the vector copy number was not different compared to SIV/VSV (Supplementary Figure S3a,S4c). However, after 3 months (monkey study 2), GALV was still detectable in the liver, lung, heart, adrenal glands, lymph nodes, and spleen, but the SIV/VSV vector copy number was significantly higher in the lateral liver lobe (0.006 ± 0.002; P = 0.035), quadrate liver lobe (0.004 ± 0.001; P = 0.049), and caudal liver lobe (0.005 ± 0.002; P = 0.047), and the VSV:GALV ratio was significantly higher in the right adrenal gland (P = 0.005) even when the GALV vector concentration was normalized for dose (Supplementary Figures S3b,S4d). Thus, because the VSV vector can be produced and concentrated to a higher titer preparation with overall broad tropism, it appears there would be little advantage to using the GALV envelope for human gene transfer in vivo.
Figure 5.

HIV-1 and SIV vector biodistribution in rhesus monkeys (studies 1 and 2). Each monkey received a pair of vectors: HIV-FX/VSV and SIV-NoN/VSV, HIV-NoN/VSV and SIV-FX/VSV, or SIV-NoN/VSV and SIV-FX/GALV. The dose was 2.0 × 109 TU/kg for SIV/VSV and HIV/VSV, and 2.0 × 108 TU/kg for SIV/GALV (see Table 1). (a) Box and whisker plots (10–90th percentile) vector copy number measured in CD34+ cells and CD34- cells isolated from bone marrow at 1 month (study 1). (b) Box and whisker plots (10–90th percentile) vector copy number measured in tissues at 1 month (study 1). (c) Box and whisker plots (10–90th percentile) vector copy number measured in cells isolated from peripheral blood at 1 month (study 1). (d) Vector copy number were measured in liver biopsies collected on days 1, 10, and 30 postvector administration (significant time trend for all vectors; P = 0.001). (e) Box and whisker plots (10–90th percentile) vector copy number from treated rhesus monkeys at 3 months (study 2): SIV/VSV (N = 8), HIV/VSV (N = 4), and SIV/GALV (N = 4). Vector copy number in all tissues analyzed for studies 1 and 2 is in Supplementary Figure S3 and paired comparisons are shown in Supplementary Figure S4. Ax LN, axilary lymph node; BM, bone marrow; CS, cell suspensions; LN, lymph node.
Rhesus monkey studies 1 and 2: HIV compared to SIV
HIV-1 and lentiviral vectors based on HIV-1 have been shown to be restricted in rhesus monkey CD34+ cells. Therefore, we directly compared the biodistribution of an HIV-1-based lentiviral vector to an SIV-based lentiviral vector. After 1 month, HIV/VSV was only detected in CD2+ and CD20+ cells; whereas SIV/VSV was detected in CD2+, CD4+, CD8+, CD20+, and CD34+ cells (Figure 5a). Similarly only SIV/VSV was detected in both bone marrow CD34+ and CD34− cells, whereas HIV/VSV was only detected in CD34− bone marrow cells (Figure 5b). In cell suspensions, both HIV and SIV vectors were detected in liver and spleen, but only SIV was detected in the thymus (Figure 5c). When the vector copy numbers were analyzed as paired comparisons, 80% of the samples had a higher SIV:HIV ratio suggesting there was more SIV/VSV vector detected (Supplementary Figure S2a).
After 1 month, the highest number of HIV and SIV vector copies was detected in liver (0.00085–0.0036), spleen (0.0035–0.002) and adrenal glands (0.0004–0.004) but there were no differences when analyzed across treatment groups or as paired comparisons (Supplementary Figure S3a,S4a). Only the SIV vector, but not HIV, was detected in heart, esophagus, trachea, pancreas, and kidneys, and was significantly higher in whole bone marrow (P = 0.0009), left inguinal lymph nodes (P = 0.0092), right diaphragm (P = 0.0008), and the left middle (P = 0.0001) and the accessory (P = 0.023) lung lobes. When analyzed as paired comparisons, the SIV vector copy number was higher in the right ventricle of the heart (P = 0.0387) and lung accessory lobe (P = 0.045) (Supplementary Figure S4a).
In monkey study 2, liver biopsies were collected under ultrasound guidance at 3, 10, and 30 days postvector administration (Figure 5d). There was a significant time trend (days) with the number of SIV/VSV copies declining from 0.308 ± 0.123 at 3 days postinjection to 0.007 ± 0.002 at 30 days postinjection, and HIV/VSV vector copy number declined from 0.54 ± 0.45 to 0.016 ± 0.009 (P = 0.013). However, there were no differences in the vector copy number when vectors were compared over time (P = 0.258). Interestingly, at 3 months posttreatment, there was a decrease in the number of tissues in which SIV vector copies were detected compared to 1 month posttreatment, and there were no tissues in which only SIV but no HIV vectors were detected. Importantly, both HIV and SIV vector copies were detected equivalently in spleen (0.003–0.006) and bone marrow (0.0002–0.0003). In contrast to study 1 at 1 month posttreatment, the number of HIV vector copies detected was higher compared to SIV in the quadrate liver lobe (P = 0.049) and in the inguinal lymph nodes (P = 0.031) (Figure 5e and Supplementary Figure S3b), but in paired comparisons there were no differences in any tissue nor in the SIV:HIV ratios (Supplementary Figure S4b). In both studies 1 and 2, we observed a relatively high vector copy number in the adrenal glands (0.001) with both HIV and SIV; the implication of this finding is currently uncertain (Supplementary Figure S3a,b).
Monkey study 3: comparison of SIV-ADA to HIV-ADA
In these studies, we compared the biodistribution of an SIV and an HIV vector, each with the human ADA cDNA transgene. Each newborn monkey received an intravenous injection of 2.0 × 109 TU/kg of either HIV-ADA or SIV-ADA (Tables 1 and 2). All monkeys had birth weights within the expected range; and after vector administration all had the anticipated growth rates, with complete blood counts, and chemistry panels within the normative range (data not shown).
To determine the pharmacokinetics of vector clearance from the bloodstream, viral genomes were isolated from the plasma and reverse transcribed to quantitate the absolute number of viral transcripts at 0.5, 2, 4, 10 hours, and then 1, 3, and 7 days postinjection (Figure 6a). There was a significant time trend (P < 0.001) with the number of vector genomes decreasing from 108 to 101 transcripts for both vectors. Noncompartmental pharmacokinetics were determined and there were no differences in the mean maximum concentration (Cmax) of SIV-ADA (mean ± SD) (4.5 × 106 ± 2.0 × 106 vg/μl) or HIV-ADA (4.1 × 106 ± 2.1 × 106 vg/μl) (P = 0.476) (Supplementary Table S1). Similarly, terminal half-life (t1/2) was not different in SIV-ADA (0.971 ± 0.306 hours) compared to HIV-ADA (0.976 ± 0.509 hours) treated monkeys (P = 0.990), nor were there differences in the mean clearance of SIV-ADA (290.9 ± 131.7 μl/hour) or HIV-ADA (228.2 ± 179.0 μl/hour) (P = 0.653). The absolute numbers of vector genomes (RNA transcripts) were also measured in cerebrospinal fluid (Figure 6b) and were detected in 3 of 3 monkeys treated with SIV-ADA and in 0 of 3 monkeys treated with HIV-ADA. A low number of HIV-ADA vector genomes, but not SIV-ADA, were detected in urine 1 day postinjection in two of three monkeys.
Figure 6.

Pharmacokinetics and biodistribution of lentiviral vectors with the human ADA cDNA in rhesus monkeys (monkey study 3). SIV-ADA/VSV (N = 3) or HIV-ADA/VSV (N = 3) with the human ADA cDNA were administered intravenously to newborns at a dose of 2.0 × 109 TU/kg. (a,b) Vector particles were isolated from plasma, cerebrospinal fluid (CSF), and urine and reverse transcribed, followed by qPCR to quantify vector genomes. (a) Clearance of human ADA vector genomes from peripheral blood plasma (significance time trend P = 0.0001). Additional pharmacokinetics described in Supplementary Table S1. (b) The absolute number of vector genomes measured in CSF at day 1 and in urine at days 1 and 7 postvector administration. (c) Vector copy number (VCN) were measured in liver biopsies collected on day 1, 10, and 30 postvector administration and compared to vector copy number in liver (caudate, quadrate, right and left lateral lobes) harvested at 3 months postnatal age (significant time trend P = 0.004 by repeated measures ANOVA). (d) Box and whisker plots (10–90th percentile) vector copy number from treated rhesus monkeys at 3 months. (e) SIV-ADA/VSV (N = 3) and HIV-ADA/VSV (N = 3) vector copy number detected in rhesus monkey tissues at 3 months compared with HIV-ADA/VSV in ADA−/− mice (N = 4) at 6 months (from Supplementary Figure S5). The dose in the monkeys was 2.0 × 109 TU/kg and the dose in the ADA−/− mice was 25-fold higher at 5.0 × 1010TU/kg (*P = 0.0001, **P = 0.002).
When the number of copies measured in liver biopsies were compared to the number measured in samples from each of the liver lobes collected at tissue harvest (3 months postnatal age), vector copies was shown to decrease significantly (100-fold) from days 1 to 30 postinjection, and remained relatively stable until the tissue harvest time point (P = 0.004) (Figure 6c). There were no significant differences in the number of HIV-ADA and SIV-ADA copies detected at any time point.
Vector copy analysis showed that the number of tissues in which the vector was detected in study 3 was very similar to study 2, in which tissues were also analyzed at 3 months postadministration. In addition to liver, SIV-ADA and HIV-ADA were detected in the heart (0.0004–0.0016), lung lobes (0.0003–0.008), adrenal glands (0.009–0.036), and whole bone marrow (0.0022–0.0026), and were not different when the vectors were compared (Figure 6d, Supplementary Figure S5). The SIV vector copy number was significantly higher in the left adrenal gland (SIV 0.036 ± 0.007, HIV 0.009 ± 0.003; P = 0.017) and the right ventricle of the heart (SIV 0.003 ± 0.0003, HIV 0.001 ± 0.0004; P = 0.026). In contrast to the studies with nonexpressed genes, the number of HIV-ADA vector copies was higher than the number of SIV-ADA copies detected in the spleen (SIV 0.04 ± 0.013, HIV 0.18 ± 0.041; P = 0.029). These data suggest that there are no negative implications for using vectors based on HIV-1 to model the biodistribution of viral vectors after intravenous administration in rhesus monkeys. However, it is possible that if SIV transduced more CD34+ cells similar to study 1, this may not be accurately evaluable within the 3-month time course of studies 2 and 3.
Species comparison: HIV-ADA in mice and monkeys
We compared HIV-ADA biodistribution in both ADA-deficient mice (Figure 3c) and in rhesus monkeys (Figure 6d) since the HIV-ADA vector was species mismatched in both mice and monkeys (Figure 6e). The mice received vector dosages that were ~25-fold higher on a per kg basis than the monkeys, and this was reflected in a concordant 10-fold to 30-fold higher vector copy numbers in the liver, lungs, heart, and bone marrow of the mice. Despite the higher dose administered to the mice, spleen vector copy number was not different than vector copy number in rhesus monkey spleens. Notable were the significantly higher vector copy numbers in the thymus and brain detected in ADA−/− mice compared to monkeys, which far exceeded the differences in vector dose. HIV-ADA was >1000-fold higher in the mouse thymus when compared with the monkey thymus with either HIV-ADA (P < 0.0001) or SIV-ADA (P < 0.0001). Likewise, the HIV-ADA vector copy number was 100-fold higher in the mouse brain when compared with the monkey brain with either HIV-ADA (P < 0.002) or SIV-ADA (P < 0.01).
Discussion
Intravenous injection of the lentiviral vector HIV-ADA/VSV, carrying the human ADA gene, has been shown to provide sufficient active ADA for immune reconstitution and increased survival of ADA-deficient mice that would typically not survive past 3 weeks of age.8 In clinical trials, HSC targeted gene transfer ex vivo has been shown to provide good immune reconstitution, especially in the youngest of ADA deficient patients; however, for older pediatric patients the response has been more variable.2,3,4 Thus, the development of an in vivo enzyme replacement may be a potential alternative to stem cell-based treatments for older ADA-deficient children, including those diagnosed with late onset ADA deficiency.
The goal for these studies was to compare biodistribution when using species-specific vector constructs and/or pseudotypes compared to species mismatched vectors and pseudotypes to accurately model biodistribution in human patients. A second goal was to gain insight into the scalability of the approach, and to determine the relationship between birth weight, dose, and the resultant biodistribution in neonatal mice and rhesus monkeys. In these studies, we used the ADA-deficient mouse model to investigate the efficacy and biodistribution of species-specific murine viral vectors and/or murine envelopes in a murine host, and the rhesus monkey to investigate the scalability and biodistribution of nonhuman primate viral vectors and pseudotypes in a nonhuman primate host.
Studies by others have demonstrated that intravenous injection of murine gRV vectors pseudotyped with murine envelopes resulted in transduction of murine bone marrow cells and HSC.14,19,20 However, we did not detect transduced HSC after intravenous injection of HIV-ADA/VSV in ADA-deficient mice in our previously published studies.8 In this study, we compared the biodistribution of the HIV-ADA/VSV vector to species-specific (murine) vectors or pseudotypes in ADA-deficient mice. Those ADA-deficient mice treated with the murine gRVs either did not survive or had vector copy numbers close to the detection limit across all tissues. In the few surviving mice, the gRV/VSV was only detected in the liver and lung but at similar levels to HIV-ADA/VSV, underscoring the importance of achieving high vector copy numbers in these tissues for survival of the ADA−/− mouse. The murine gRV vector was not detected in the bone marrow, regardless of pseudotype, nor was there any significant increase in vector copy number in any hematopoietic tissue with the murine gRV or murine pseudotypes. In contrast, there were no differences in survival or subsequent vector copy number measured in mononuclear cells isolated from the thymus, spleen, or bone marrow from mice treated with the HIV-ADA vector pseudotyped with VSV compared to mice treated with vector pseudotyped with the murine Ampho envelope. Taken together, these studies demonstrate that the low levels of transduced HSC in ADA-deficient mice treated with an intravenous injection of an HIV-1-based lentiviral vector was likely not solely due to a species mismatch in vector or host restriction factor but may be unique to the ADA-deficient mouse model or due to the route of administration or dose. Others have found that intrafemoral administration of a lentiviral vector did result in transduced HSC in vivo, and that the transduced HSC could repopulate lethally irradiated mice.16
Studies in vitro have shown that nonadherent CD34+ cells isolated from rhesus monkey bone marrow were transduced only by SIV/VSV and not HIV/VSV vectors and similarly our studies in vivo showed that 1 month postintravenous administration, only SIV/VSV was detected in bone marrow CD34+ cells, but both SIV and HIV were detected equivalently in the CD34− fraction. However, tissue analysis revealed few differences across treatment groups in the biodistribution of HIV vectors compared to SIV vectors in all of the rhesus monkey studies performed. Similarly, paired comparisons within individual animals also revealed few differences. In study 1, analysis of SIV:HIV ratios revealed significantly higher SIV copies in splenocytes, and in unfractionated and fractionated bone marrow; however, at 3 months (study 2) there were no differences detected. Despite the lack of significant differences with species-matched and mismatched vectors, the use of a matched vector may still provide a more accurate estimation of an HIV-1-based lentiviral vector in humans, particularly as new host restriction factors of wild-type gRV10,11 are described. In a recent study, differences in HIV lentiviral vector HSC transduction efficiency was correlated with different Trim5α genotypes and levels of expression in rhesus monkeys.21
We also compared the biodistribution of the SIV lentiviral vector pseudotyped with VSV-G to SIV pseudotyped with a species-related GALV pseudotype in rhesus monkey newborns. When comparing treatment groups, monkeys administered SIV/VSV showed the highest vector copy numbers with the widest biodistribution compared to SIV/GALV. However, the blood and marrow cells may have been more permissive to the SIV/GALV vector compared to SIV/VSV vector because even with the lower GALV dose there were very few significant differences between VSV and GALV, whereas other tissues had vector copies commensurate with the lower dose. It should be noted that all of the GALV and VSV titers were determined on HT29 cells; it is not clear if the number of GALV receptors on HT29 is similar or lower on blood cells. If lower, then the titer for the GALV vector would be underestimated. Nonetheless, it appears that the VSV-G pseudotyped vectors packaged much more efficiently than all of the other pseudotypes (GALV, Eco, Ampho), and that this is an important factor in evaluating vectors and pseudotypes for in vivo gene delivery.
The feasibility of producing a clinical dose of injectable lentiviral vectors may prove to be a limitation in translating this therapy to the clinic, as large doses would be needed to treat children or young adults. It is not entirely clear how high the titer or concentration will need to be or how the size differences between a neonatal mouse and neonatal human should be addressed. More specific to ADA deficiency, the therapeutic dose needed for untreated ADA−/− mice that do not survive past 3 weeks due to pulmonary complications remains unclear.22 Although ADA-deficient patients exhibit similar pulmonary findings, it is rarely lethal23,24 and thus it is possible that a low dose could still result in clinical benefit in human ADA patients. Nonetheless, if the critical threshold dosage for survival of ADA-deficient mice is considered (e.g. 5 × 1010 TU/kg), the extrapolated pediatric dose would need to be 2 × 1012 TU for a 40-kg child. With a typical initial titer in the range of 1 × 107 TU/ml for an unconcentrated lentiviral vector such as HIV/ADA-VSV, the vector concentrated from >100 l would be needed to produce a single patient dose. This volume is within current production methods but does represent a large amount of material. Methods to increase the efficiency of vector delivery, e.g. by minimizing vector inactivation in the plasma by pegylation25 or by suppressing inflammatory responses to the vector in hepatocytes with dexamethasone,26 may allow a lower vector dose to achieve the needed vector copy numbers. Additionally, vector designs that lead to higher potency of expression of ADA per vector copy, such as strong liver-specific promoters or elements that resist transgene silencing such as the UCOE,27 may allow for lower vector dosages.
Since the same HIV-ADA/VSV vector was used to treat both newborn ADA-deficient mice and rhesus monkeys, we compared the biodistribution in each species. Monkeys received a lower vector dose (~25-fold) than mice and the vector copy numbers in the liver and lung were proportional to these dosages. However, even after adjusting for dose, ADA−/− mice had significantly more HIV-ADA/VSV vector detected (>1000-fold) in the thymus and brain compared to rhesus monkeys. It will be important to accurately assess the potential for ADA production in vivo in the thymus for improved immune reconstitution and in the brain for resolution of significant cognitive and behavioral abnormalities typically seen in some ADA-deficient patients.28 One possible explanation for the difference in biodistribution could be related to selective pressure inherent to ADA-deficiency; however, there was no difference in the biodistribution observed in similarly treated ADA−/− mice compared to ADA+/− mice. Another possibility could be related to a specific restriction of an HIV-based lentiviral vector in some rhesus monkey tissues, but this is highly unlikely as there were no differences between SIV-ADA and HIV-ADA in the brain or thymus. Another possible explanation could be related to the developmental differences when comparing neonatal mice to newborn rhesus monkeys. This is an important consideration because monkeys are developmentally more comparable at birth to humans than mice.18 Others have shown that wild-type HIV-1 and SIV are capable of infecting microvascular endothelial cells found in the brain which may be more accessible in the mouse because of potential differences in the blood–brain–barrier at birth.29,30 Similarly, it has been shown that unlike the adult thymus, the neonatal mouse thymus (days 1–7) represents a distinct period of growth and differentiation that is characterized by a dense, vascular endothelium31 that may be more porous and/or readily transduced when compared with the newborn primate thymus. These results highlight the importance of studying vector biodistribution in an appropriate primate model, one with developmental similarities more relevant to human development for an accurate assessment of therapeutic potential.
The development of gene transfer in vivo using integrating lentiviral vectors has broad applications, particularly when targeting the liver for gene expression for the correction of monogenic disease. Furthermore, systemic delivery during the neonatal period offers the possibility of avoiding postnatal deterioration of disease states; and because of the increased proliferative state of neonatal liver, integrating vectors can ultimately result in more transduced cells and expression.32 However, since the cells are transduced in situ, all of the integration events have oncogenic potential. Therefore, the risk associated with lentiviral vector integration is an important consideration in the development of systemic lentiviral vector administration for gene transfer in vivo.
In HSC directed ex vivo gene therapy, clonal expansion was observed in a β-thalassemia patient after the lentiviral vector integrated in the HMGA2 gene.33 Injection of a nonprimate lentiviral vector, either based on the equine infectious anemia virus or feline immunodeficiency virus, into fetal murine livers resulted in more integrations into gene dense regions near oncogenes or tumor suppressor genes, including several associated with hepatocellular carcinoma, and gave rise to more liver tumors compared to HIV-1-based lentiviral vectors, which gave rise to none.34,35 Indeed, in another study, integrations of a transgene-less lentiviral vector containing highly active hepatic-specific enhancer–promoter sequences was used to identify four new liver cancer genes associated with hepatocellular carcinoma in three different mouse strains after neonatal intravenous injection.36 However, another study using a tumor prone disease model of familial hypercholesterolemia (FAH), FAH−/− mice showed increased survival that was not associated with vector-induced tumors, even within the context of the selective pressure provided by the gene transfer of the FAH gene.37 Likewise, we saw no evidence of tumor outgrowth in any tissue, in any of the ADA KO mice or in monkeys with any of the vectors administered in this study or in a previously published study.8 This has also been reported in rhesus monkeys that have been monitored postgene therapy for long periods of time (≥10 years).38
In conclusion, we have shown that delivery of a lentiviral vector for the treatment of ADA-SCID can be scaled-up to treat newborn monkeys. Initial findings in the ADA-deficient mouse remain relevant for understanding how systemic administration of a lentiviral vector could provide ERT in vivo for ADA-deficient patients, although the studies highlight the importance of considering species differences when attempting to predict biodistribution in human infants. Importantly, the information gained from these studies can also apply to the development of other in vivo therapeutics for the treatment of a variety of genetic disease such as hemophilia (Factor VIII or Factor IX deficiency), Gaucher's Disease (glucocerebrosidase), and Mucopolysaccharidoses (MPSI) (l-alpha-iduronidase), in which systemic delivery and ecotopic expression of a gene in vivo can result in multi-systemic phenotypic correction of a monogenic gene defect.
Materials and Methods
Vector construction and production. The HIV-1-based, self-inactivating (SIN) lentiviral vector pCSO-rc-MNDU3-NeoNT (HIV-NoN) was created by modifying pCSO-rc-MND-ADA (HIV-ADA), described previously to contain the NeoNT fragment in place of the hADA gene. The HIV-1-based, SIN lentiviral vector pCCL-c-MNDU3c-FXM10 vector (HIV-FX) has the FXM10 fragment from bacteriophage PhiX174 in the pCCL-c-MNDU3 backbone.39,40,41 The SIV-based, SIN lentiviral vectors: pCL20c-SM-hADA (SIV-ADA), pCL20c-SM-NeoNT (SIV-NoN), and pCL20c-SM-FXM10 (SIV-FX) were constructed replacing the enhanced green fluorescent protein (eGFP) gene with the corresponding genes (hADA) or nonexpressing fragments (NeoNT and FXM10) in the pCL20cSLFR MSCV-GFP (SIV-eGFP) plasmid.42 The pCCL-MNDU3-eGFP (HIV-eGFP)43 and the retroviral vector pMND-MFG-ADA (MMA) were constructed as previously described.3
The HIV-based vectors (HIV-FX/VSV, HIV-NoN/VSV, HIV-ADA/VSV) were packaged as previously reported.44 The SIV-based vectors (SIV-NoN/VSV, SIV-FX/VSV, SIV-ADA/VSV) were packaged and concentrated by the same method except that the plasmid concentrations for the SIV packaging plasmids were: 90 μg pCAG-SIVgprre, 30 μg pCAG4-RTR-SIV, and 30 μg pCAG-VSV-G.42,44 For SIV-based vectors pseudotyped with a GALV-derived envelope, 150 μg of pGALV-TM was used in place of the VSV-G envelope plasmid.17 For HIV-based vectors pseudotyped with the murine-amphotropic envelope (HIV-ADA/Ampho), 2 µg of the pHit456 plasmid45 was used in place of VSV-G in triple transfection protocol.
The gRV vector, MMA, was made by transfecting the pMND-MFG-ADA (MMA) plasmid with 10 µg of the MLV gag/pol expression plasmid pHIT60 and 2 µg of either the pMD.G plasmid (gRV/VSV), pHit123 (gRV/Eco), or pHit456 (gRV/Ampho)45 into 293T cells (American Type Culture Collection, ATCC; Manassas, VA) in Dulbecco's Modified Eagle Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum, penicillin (100 µg/ml), streptomycin (100 µg/ml), and 2 mmol/l l-glutamine. Viral supernatant was harvested after 48 hours, filtered through a 0.2 µm filter and concentrated ~1000-fold by using Centricon plus 70 ultra-filtration filters (Millipore, Bedford, MA) followed by ultracentrifugation at 25,000 × g for 2 hours.23 Viral pellets were resuspended in phosphate buffered saline and stored in 50 µl aliquots at ≤−80 °C.
Titer determination. Titer of the VSV and GALV pseudotyped vectors was performed by transducing HT29 (human colorectal adenocarcinoma, #HTB-38, ATCC) cells with three dilutions of the lentiviral vector. Similarly, the titer of the murine enveloped vectors was determined on NIH/3T3 (mouse fibroblasts, ATCC CRL-1658) cells. The titer of HIV-ADA/VSV vector was determined on both cells lines and found to be similar. The titer of the HIV vectors was determined by qPCR using primers and probe specific for the HIV psi region using a standard curve constructed with serially diluted DNA extracted from a cellular clone containing two integrated copies of the HIV vector44 (Supplementary Tables S2 andS3). The titer of SIV vectors was determined in a similar manner using an HT29 cellular clone containing one integrated copy of the SIV vector backbone as a standard for qPCR using primers and probes specific for the SIV psi region.13 The titer of the retroviral construct MMA was done as previously reported44 except with qPCR primers/probes to the human ADA gene spanning exons 6 and 7.
Mice. The ADA-deficient mouse was generated and rescued with a two-stage genetic engineering strategy previously described and characterized.46 Due to the accumulation of adenosine and deoxyadenosine, the mice exhibit multisystem abnormalities, including the SCID phenotype prior to death at postnatal days 19–20 from pulmonary insufficiency.5 Mouse husbandry, genotyping procedures, including intravenous administration of lentiviral vectors, was performed as described previously.8 Mice were housed in accordance with the requirements of the Animal Welfare Act and approved by the Institutional Animal Care and Use Committee (IACUC) at the Saban Research Institute at Children's Hospital Los Angeles and the National Institutes of Health (NIH) Guidelines. All animals were housed in microinsulator cages in a pathogen-free colony and all procedures were conducted in laminar flow hoods.
All ADA KO mouse studies. Litters were assigned to a treatment group by date of birth in successive order and all litters assigned to a treatment group were treated with the same batch of vector. Vector aliquots were thawed quickly and diluted to a 50-µl injection volume in 0.9% saline (injection, USP; APP, Schaumburg, IL) as described previously.8 Injections were performed on postnatal days 1–3 (1.0–1.9 g) via the superficial temporal vein with a 30-g needle attached to a 1-ml syringe without sedation. Mice were euthanized by carbon dioxide asphyxiation and vector copy numbers were determined in the thymus, spleen, lung, liver, and bone marrow as described previously.8
In study 1, neonatal litters were treated with either 2.5 × 1010 TU/kg of HIV-ADA/VSV or 5.0 × 1010 TU/kg of HIV-ADA/VSV, without prior treatment with ERT (Table 1). Survival of the ADA−/− mice treated with 5.0 × 1010 TU/kg (N = 8) was compared to survival of ADA−/− mice treated with 2.5 × 1010 TU/kg (N = 6) or untreated ADA−/− mice (N = 4). Mice were analysed at 4 (N = 4), 6 (N = 2), and 12 (N = 1) months and were compared to their similarly treated ADA+/− littermates (N = 6).
In study 2, litters were treated with an intravenous injection of 5.0 × 1010 TU/kg of HIV-ADA/VSV LV or with 2.5 × 108 TU/kg of gRV pseudotyped with VSV-G (gRV/VSV), with the murine-amphotropic envelope (gRV/Ampho), or the murine-ecotropic envelope (gRV/Eco) (Table 1). Survival of ADA−/− mice treated with HIV-ADA/VSV was compared to ADA−/− mice treated with gRV/VSV-G (N = 5), gRV/Ampho (N = 4), or gRV/Eco (N = 6). After 3 months, the surviving ADA−/− mice were analysed for tissue vector copy number and compared to the ADA+/− littermates also treated with HIV-ADA/VSV (N = 4), ADA−/−, and/or ADA+/− mice treated with gRV/VSV-G (N = 2), gRV/Ampho (N = 10), or gRV/Eco (N = 11). Absolute cell counts were performed and compared to control ADA−/− mice (N = 3) that received weekly intramuscular injections of ADA ERT.
In study 3, litters were treated with an intravenous injection of 5.0 × 1010 TU/kg of HIV-ADA/VSV lentiviral vector or with the same dose of HIV-ADA pseudotyped with the murine-amphotropic envelope (HIV-ADA/Ampho) (Table 1). ADA−/− treated with HIV/VSV (N = 7) or HIV/Ampho (N = 4) were compared for survival. At 3 months, mice treated with HIV-ADA/VSV (N = 4) or HIV-ADA/Ampho (N = 4) were analyzed for tissue vector copy number and in isolated cell populations from the thymus (CD4+, CD8+, CD4+ CD8+), spleen (CD19+), and bone marrow (CD11b+). Absolute immune cell counts were also determined and compared to ADA+/+ mice (N = 5) and to ADA−/− mice on PEG-ADA ERT (N = 3).
Rhesus monkeys. All animal procedures conformed to the requirements of the Animal Welfare Act and protocols were approved prior to implementation by the IACUC at the University of California, Davis. Normally cycling, adult female rhesus monkeys (Macaca mulatta) (N = 20) with a history of prior pregnancy were bred and identified as pregnant using established methods.47 Activities related to animal care (diet, housing) were performed according to California National Primate Research Center standard operating procedures.
Newborns were delivered by cesarean section at term according to established protocols.47 At birth, umbilical cord blood was collected (0 time point; ~12 ml) and simian Apgar scores assessed.48 Infants were raised in the gene therapy nursery for postnatal studies with daily monitoring of physical signs and food intake. Weights were monitored on a daily or weekly basis following established nursery standard operating procedures. These studies are summarized in Table 2. Newborns were administered the selected lentiviral vectors intravenously (~1 ml volume/injection) via a peripheral vessel within ~30–45 minutes of birth. All newborns were placed in incubators, fed a standard diet based on age, and tissue harvests performed at either 1 or 3 months postnatal age. Blood samples (~1–2 ml) were collected from a peripheral vessel at 1, 3, 7, 14, and 30 days postvector administration (without sedation) for complete blood counts, chemistry panels, and qPCR. Studies 2 and 3 included liver biopsies collected under ultrasound guidance and sterile conditions at 1, 10, and 30 days postvector administration.47 Infants were sedated for these procedures (telazol, 5–8 mg/kg intramuscular). In study 3, blood samples were also collected from a peripheral vessel at 0.5, 2, 4, 10, and 24 hours, then 3 and 7 days postvector administration for pharmacokinetic analysis (~0.7 ml volume/sample). Cerebrospinal fluid was obtained (under telazol, Zoetics, Kalamazoo, MI) 1 day postvector administration using sterile technique (~0.7 ml), urine was collected at 1 and 7 days in infant metabolism cages, and liver biopsies were collected as noted above at 1, 10, and 30 days postvector administration.47
All monkeys were sedated with ketamine (10 mg/kg), final blood samples collected, then euthanized with an overdose of pentobarbital for tissue harvest (study 1 at 1 month, studies 2 and 3 at 3 months postnatal age) according to established protocols. Total body weights and measures were assessed, then all organs were removed including the brain (cerebrum, cerebellum), lung (right and left cranial, middle, caudal, and accessory lobes), trachea, esophagus, heart (right and left ventricle), aorta, pericardium, thymus, spleen, liver (right and left lateral, and quadrate and caudate lobes), lymph nodes (right and left axillary and inguinal, tracheobroncheal, mesenteric), pancreas, right and left adrenals, right and left kidneys, reproductive tract (right and left gonads, uterus or right and left seminal vesicles, and prostate), gastrointestinal tract (stomach, duodenum, jejunum, ileum, colon), right and left diaphragm, omentum, right and left peritoneum, skin, muscle, and bone marrow. Select tissues were weighed (brain, thymus, spleen, liver, adrenals, kidneys, gonads). Cell suspensions were prepared from thymus, spleen, and liver. Cells were immunoselected from blood and bone marrow using established protocols and antibodies (CD3, CD4, CD8, CD20, CD34).49 All tissues collected for qPCR were placed in microcentrifuge tubes and immediately frozen in liquid nitrogen and stored at ≤−80 °C until assay. In addition, sections of tissue were placed in Tissue-Tek optimum cutting temperature compound (Sakura Finetek, Torrance, CA) and quick frozen over liquid nitrogen, and fixed in 10% buffered formalin and embedded in paraffin. Representative 5–6 µm sections were obtained from all tissues and stained with hematoxylin and eosin (H&E) for routine histopathology.
DNA isolation. Genomic DNA from mononuclear cells and positive and negative cell fractions were isolated using the QIAamp DNA Blood Mini kit (Qiagen, Valencia, CA), as recommended by the manufacturer. The positive cell fractions were lysed in 50 mmol/l Tris–HCl, pH = 7.4 (Sigma, St. Louis, MO) containing 0.25 mg/ml of Proteinase K (Invitrogen, Carlsbad, CA) solution by incubating at 65 °C for 1 hour and then for 15 minutes at 95 °C. Genomic DNA was isolated from tissues using the Gentra Puregene Tissue kit (Qiagen, Valencia, CA) as recommended by the manufacturer.
Vector copy number quantification. All vector copy number determinations were performed by qPCR using linear regression of a standard curve constructed from cycle threshold (Ct) values determined during the amplification of DNA extracted from HT29 (colorectal adenocarcinoma, #HTB-38, ATCC) cellular clones which contained known copies of vector as determined by Southern analysis.44 The standard curves were constructed by serially diluting (10-fold) the cellular clone DNA into nontransduced HT29 DNA, and the Ct value was determined at 10 copies down 105 or 106 copies or ~1 marked cell in 100,000–1,000,000 cells. Supplementary Table S2 summarizes the cellular clones used to determine copy number of each gene in cell and tissue samples, and includes their respective copy number as determined by Southern blot analysis. Supplementary Table S3 summarizes the primers and probe sequences used for amplification and detection. Real-time PCR amplifications were performed in 96-well optical plates using the 7900 ABI Sequence Detection System (Applied Biosystems, Foster City, CA) and the TaqMan Universal PCR Master Mix (Applied Biosystems) according to the manufacturer's protocols and as previously described.48 PCR reactions contained 1× TaqMan universal master mix with 400 nmol/l of forward and reverse primers and 100 nmol/l probe in a 50 µl reaction volume. DNA concentration was determined by staining double stranded DNA with Hoechst 33258 (DNA-QF kit; Sigma) according to the manufacturers' instructions. For each sample, 700–1100 ng of DNA (~200,000 cells) was interrogated (350 ng/well in three wells) to achieve an assay sensitivity of 1 marked cell in 100,000. To increase the probability of detecting the lowest amounts of template sequence all of the qPCR reactions were single-plex PCR. DNA quantity was confirmed with a normalizing gene PCR as a separate reaction. The qPCR protocol consisted of one cycle of 2 minutes at 50 °C, 15 minutes at 95 °C, followed by 40 cycles of 15 seconds at 95 °C, and 60 seconds at 60 °C. A valid linear regression of the standard curve had a correlation coefficient (r value) at or above 0.98 and a slope of −3.0 or less. For vector copy numbers less than 1 × 10−7, the vector copy number value was set to 1.0 × 10−7.
Viral genome quantification. In study 3, reverse transcribed (RT)-qPCR was used to detect viral RNA in body fluids. HIV-ADA and SIV-ADA viral genomes were isolated from plasma, urine, and cerebrospinal fluid using the PureLink Viral RNA/DNA Mini Kit (Invitrogen/Life Technologies, Carlsbad, CA) according to the manufacturer's instructions. For each sample, 10 µl of RNA were DNAse treated using Purelink DNAse (Invitrogen/Life Technologies) and then reverse transcribed with 10 units of M-MLV reverse transcriptase, 5 mmol/l dithiothreitol, 500 mmol/l dNTP mix (PCR grade), 7.5 ng/ul of Random Primer mix, and 0.1 U/µl of RNase OUT Recombinant Ribonuclease Inhibitor (Invitrogen/Life Technologies). For each sample, no-RT controls were performed by omission of the M-MLV RT. Reverse transcription was performed at 37 °C for 1 hour followed by denaturation at 94 oC for 10 minutes. Following cDNA synthesis, qPCR was performed on 1 µl of cDNA from the RT reaction using human ADA primers (400 mmol/l) and probe (100 nmol/l) (Supplementary Table S3) that span exons 6/7 in a 25-µl reaction volume containing 2× Universal Master Mix (Applied Biosystems/Life Technologies). Serial dilutions of plasmid DNA containing the ADA cDNA (10,000,000 to 100 molecules) were used to construct the standard curve. Amplification was performed under default conditions for the Via 7 Sequence Detector (Applied Biosystems/Life Technologies).
Statistical analysis. Descriptive statistics of continuous outcome variables, such as the mean and SEM by experimental groups, are presented in the figures. For time-to-event outcome, Kaplan–Meier method50 and log-rank test51 were used to summarize and compare the survival experience of mice across different groups. For continuous outcome measurements, group differences were assessed by unpaired t-test (for two experimental groups) or one-way analysis of variance (for more than two groups)52 followed by pairwise comparisons. For longitudinal outcomes, repeated measure analysis of variance53 was performed to evaluate the effects of group differences, time-trend, as well as group-by-time interactions. Log-transformation was performed on vector copy number. Noncompartmental analysis was used for the pharmacokinetics of vector clearance.54 Vector-associated pharmacokinetic parameters, such as Cmax, tmax, area under the curve (AUC), area under moment curve (AUMC), mean residence time (MRT), terminal half-life (t1/2), clearance, and volume of distribution at steady state, were calculated. For all statistical investigations, tests for significance were two-tailed unless otherwise specified. A P-value less than the 0.05 significance level was considered to be statistically significant. Pharmacokinetics analysis was performed using the “PK” package in statistical software R Version 3.0.1.55 All other statistical analyses were carried out using SAS version 9.3.56
SUPPLEMENTARY MATERIAL Figure S1. HIV-1 and SIV lentiviral mediated transduction in rhesus monkey bone marrow cells in vitro Figure S2. The SIV:HIV ratio and VSV:GALV ratio within each monkey. Figure S3. Vector Copy Number by vector group for all tissues analyzed in Study 1 and Study 2. Figure S4. Paired comparisons of vector biodistribution in individual monkeys (Monkey Study 1 and Study 2) Figure S5. Biodistribution of HIV-ADA/VSV and SIV-ADA/VSV. Table S1. Non-compartmental pharmacokinetics in young rhesus monkeys after intravenous injection. Table S2. HT29 cellular clones used as standards for copy number determination. Table S3. Sequence detection primers and probes used for qPCR.
Acknowledgments
These studies were supported by grants from the National Institute of Allergy and Infectious Diseases (#R01-AI074043, D.B.K.), the National Heart, Lung, and Blood Institute (NHLBI) Center for Fetal Monkey Gene Transfer for Heart, Lung, and Blood Diseases #HL085794 (A.F.T.), the Primate Center base operating grant (#OD011107), and the Cores for Animal Care, Research Vectors, and Cell & Tissue Analysis from P01 #HL073104 (D.B.K.). The Flow Cytometry Core of the Eli and Edythe Broad Center for Regenerative Medicine & Stem Cell Research also supported these studies. Sigma Tau, Gaithersburg MD kindly provided ADA-GEN, and the authors thank Kristine David of Sigma Tau for her helpful assistance. The authors declare no conflict of interest.
Supplementary Material
References
- Gaspar HB, Aiuti A, Porta F, Candotti F, Hershfield MS, Notarangelo LD. How I treat ADA deficiency. Blood. 2009;114:3524–3532. doi: 10.1182/blood-2009-06-189209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aiuti A, Cattaneo F, Galimberti S, Benninghoff U, Cassani B, Callegaro L, et al. Gene therapy for immunodeficiency due to adenosine deaminase deficiency. N Engl J Med. 2009;360:447–458. doi: 10.1056/NEJMoa0805817. [DOI] [PubMed] [Google Scholar]
- Candotti F, Shaw KL, Muul L, Carbonaro D, Sokolic R, Choi C, et al. Gene therapy for adenosine deaminase-deficient severe combined immune deficiency: clinical comparison of retroviral vectors and treatment plans. Blood. 2012;120:3635–3646. doi: 10.1182/blood-2012-02-400937. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaspar HB, Cooray S, Gilmour KC, Parsley KL, Zhang F, Adams S, et al. Hematopoietic stem cell gene therapy for adenosine deaminase-deficient severe combined immunodeficiency leads to long-term immunological recovery and metabolic correction. Sci Transl Med. 2011;3:97ra80. doi: 10.1126/scitranslmed.3002716. [DOI] [PubMed] [Google Scholar]
- Blackburn MR, Datta SK, Wakamiya M, Vartabedian BS, Kellems RE. Metabolic and immunologic consequences of limited adenosine deaminase expression in mice. J Biol Chem. 1996;271:15203–15210. doi: 10.1074/jbc.271.25.15203. [DOI] [PubMed] [Google Scholar]
- Carbonaro DA, Jin X, Cotoi D, Mi T, Yu XJ, Skelton DC, et al. Neonatal bone marrow transplantation of ADA-deficient SCID mice results in immunologic reconstitution despite low levels of engraftment and an absence of selective donor T lymphoid expansion. Blood. 2008;111:5745–5754. doi: 10.1182/blood-2007-08-103663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mortellaro A, Hernandez RJ, Guerrini MM, Carlucci F, Tabucchi A, Ponzoni M, et al. Ex vivo gene therapy with lentiviral vectors rescues adenosine deaminase (ADA)-deficient mice and corrects their immune and metabolic defects. Blood. 2006;108:2979–2988. doi: 10.1182/blood-2006-05-023507. [DOI] [PubMed] [Google Scholar]
- Carbonaro DA, Jin X, Petersen D, Wang X, Dorey F, Kil KS, et al. In vivo transduction by intravenous injection of a lentiviral vector expressing human ADA into neonatal ADA gene knockout mice: a novel form of enzyme replacement therapy for ADA deficiency. Mol Ther. 2006;13:1110–1120. doi: 10.1016/j.ymthe.2006.02.013. [DOI] [PubMed] [Google Scholar]
- Carbonaro DA, Jin X, Wang X, Yu XJ, Rozengurt N, Kaufman ML, et al. Gene therapy/bone marrow transplantation in ADA-deficient mice: roles of enzyme-replacement therapy and cytoreduction. Blood. 2012;120:3677–3687. doi: 10.1182/blood-2012-02-408591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wolf D, Goff SP. Host restriction factors blocking retroviral replication. Annu Rev Genet. 2008;42:143–163. doi: 10.1146/annurev.genet.42.110807.091704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayinde D, Casartelli N, Schwartz O. Restricting HIV the SAMHD1 way: through nucleotide starvation. Nat Rev Microbiol. 2012;10:675–680. doi: 10.1038/nrmicro2862. [DOI] [PubMed] [Google Scholar]
- Nakayama EE, Shioda T. TRIM5α and Species Tropism of HIV/SIV. Front Microbiol. 2012;3:13. doi: 10.3389/fmicb.2012.00013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kahl CA, Cannon PM, Oldenburg J, Tarantal AF, Kohn DB. Tissue-specific restriction of cyclophilin A-independent HIV-1- and SIV-derived lentiviral vectors. Gene Ther. 2008;15:1079–1089. doi: 10.1038/gt.2008.50. [DOI] [PubMed] [Google Scholar]
- Xu L, O'Malley T, Sands MS, Wang B, Meyerrose T, Haskins ME, et al. In vivo transduction of hematopoietic stem cells after neonatal intravenous injection of an amphotropic retroviral vector in mice. Mol Ther. 2004;10:37–44. doi: 10.1016/j.ymthe.2004.04.010. [DOI] [PubMed] [Google Scholar]
- Pan D, Gunther R, Duan W, Wendell S, Kaemmerer W, Kafri T, et al. Biodistribution and toxicity studies of VSVG-pseudotyped lentiviral vector after intravenous administration in mice with the observation of in vivo transduction of bone marrow. Mol Ther. 2002;6:19–29. doi: 10.1006/mthe.2002.0630. [DOI] [PubMed] [Google Scholar]
- Worsham DN, Schuesler T.et al. (2006In vivo gene transfer into adult stem cells in unconditioned mice by in situ delivery of a lentiviral vector Mol Ther 14514–. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christodoulopoulos I, Cannon PM. Sequences in the cytoplasmic tail of the gibbon ape leukemia virus envelope protein that prevent its incorporation into lentivirus vectors. J Virol. 2001;75:4129–4138. doi: 10.1128/JVI.75.9.4129-4138.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Batchelder CA, Keyser JL, Lee CC, Tarantal AF. Characterization of growth, glomerular number, and tubular proteins in the developing rhesus monkey kidney. Anat Rec (Hoboken) 2013;296:1747–1757. doi: 10.1002/ar.22756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang B, O'Malley TM, Xu L, Vite C, Wang P, O'Donnell PA, et al. Expression in blood cells may contribute to biochemical and pathological improvements after neonatal intravenous gene therapy for mucopolysaccharidosis VII in dogs. Mol Genet Metab. 2006;87:8–21. doi: 10.1016/j.ymgme.2005.08.014. [DOI] [PubMed] [Google Scholar]
- Worsham DN, Schuesler T, von Kalle C, Pan D. In vivo gene transfer into adult stem cells in unconditioned mice by in situ delivery of a lentiviral vector. Mol Ther. 2006;14:514–524. doi: 10.1016/j.ymthe.2006.05.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Evans ME, Kumkhaek C, Hsieh MM, Donahue RE, Tisdale JF, Uchida N. TRIM5α variations influence transduction efficiency with lentiviral vectors in both human and rhesus CD34(+) cells in vitro and in vivo. Mol Ther. 2014;22:348–358. doi: 10.1038/mt.2013.256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Volmer JB, Thrasher JL, Zhong H, Crosby JR, Lee JJ, et al. Metabolic consequences of adenosine deaminase deficiency in mice are associated with defects in alveogenesis, pulmonary inflammation, and airway obstruction. J Exp Med. 2000;192:159–170. doi: 10.1084/jem.192.2.159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Booth C, Algar VE, Xu-Bayford J, Fairbanks L, Owens C, Gaspar HB. Non-infectious lung disease in patients with adenosine deaminase deficient severe combined immunodeficiency. J Clin Immunol. 2012;32:449–453. doi: 10.1007/s10875-012-9658-3. [DOI] [PubMed] [Google Scholar]
- Grunebaum E, Cutz E, Roifman CM. Pulmonary alveolar proteinosis in patients with adenosine deaminase deficiency. J Allergy Clin Immunol. 2012;129:1588–1593. doi: 10.1016/j.jaci.2012.02.003. [DOI] [PubMed] [Google Scholar]
- Croyle MA, Callahan SM, Auricchio A, Schumer G, Linse KD, Wilson JM, et al. PEGylation of a vesicular stomatitis virus G pseudotyped lentivirus vector prevents inactivation in serum. J Virol. 2004;78:912–921. doi: 10.1128/JVI.78.2.912-921.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Agudo J, Ruzo A, Kitur K, Sachidanandam R, Blander JM, Brown BD. A TLR and non-TLR mediated innate response to lentiviruses restricts hepatocyte entry and can be ameliorated by pharmacological blockade. Mol Ther. 2012;20:2257–2267. doi: 10.1038/mt.2012.150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang F, Frost AR, Blundell MP, Bales O, Antoniou MN, Thrasher AJ. A ubiquitous chromatin opening element (UCOE) confers resistance to DNA methylation-mediated silencing of lentiviral vectors. Mol Ther. 2010;18:1640–1649. doi: 10.1038/mt.2010.132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Titman P, Pink E, Skucek E, O'Hanlon K, Cole TJ, Gaspar J, et al. Cognitive and behavioral abnormalities in children after hematopoietic stem cell transplantation for severe congenital immunodeficiencies. Blood. 2008;112:3907–3913. doi: 10.1182/blood-2008-04-151332. [DOI] [PubMed] [Google Scholar]
- Mankowski JL, Spelman JP, Ressetar HG, Strandberg JD, Laterra J, Carter DL, et al. Neurovirulent simian immunodeficiency virus replicates productively in endothelial cells of the central nervous system in vivo and in vitro. J Virol. 1994;68:8202–8208. doi: 10.1128/jvi.68.12.8202-8208.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moses AV, Stenglein SG, Strussenberg JG, Wehrly K, Chesebro B, Nelson JA. Sequences regulating tropism of human immunodeficiency virus type 1 for brain capillary endothelial cells map to a unique region on the viral genome. J Virol. 1996;70:3401–3406. doi: 10.1128/jvi.70.6.3401-3406.1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuddihy AR, Suterwala BT, Ge S, Kohn LA, Jang J, Andrade J, et al. Rapid thymic reconstitution following bone marrow transplantation in neonatal mice is VEGF-dependent. Biol Blood Marrow Transplant. 2012;18:683–689. doi: 10.1016/j.bbmt.2012.01.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park F, Ohashi K, Chiu W, Naldini L, Kay MA. Efficient lentiviral transduction of liver requires cell cycling in vivo. Nat Genet. 2000;24:49–52. doi: 10.1038/71673. [DOI] [PubMed] [Google Scholar]
- Cavazzana-Calvo M, Payen E, Negre O, Wang G, Hehir K, Fusil F, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassaemia. Nature. 2010;467:318–322. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Themis M, Waddington SN, Schmidt M, von Kalle C, Wang Y, Al-Allaf F, et al. Oncogenesis following delivery of a nonprimate lentiviral gene therapy vector to fetal and neonatal mice. Mol Ther. 2005;12:763–771. doi: 10.1016/j.ymthe.2005.07.358. [DOI] [PubMed] [Google Scholar]
- Nowrouzi A, Cheung WT, Li T, Zhang X, Arens A, Paruzynski A, et al. The fetal mouse is a sensitive genotoxicity model that exposes lentiviral-associated mutagenesis resulting in liver oncogenesis. Mol Ther. 2013;21:324–337. doi: 10.1038/mt.2012.224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ranzani M, Cesana D, Bartholomae CC, Sanvito F, Pala M, Benedicenti F, et al. Lentiviral vector-based insertional mutagenesis identifies genes associated with liver cancer. Nat Methods. 2013;10:155–161. doi: 10.1038/nmeth.2331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rittelmeyer I, Rothe M, Brugman MH, Iken M, Schambach A, Manns MP, et al. Hepatic lentiviral gene transfer is associated with clonal selection, but not with tumor formation in serially transplanted rodents. Hepatology. 2013;58:397–408. doi: 10.1002/hep.26204. [DOI] [PubMed] [Google Scholar]
- Tarantal AF, Skarlatos SI. Center for fetal monkey gene transfer for heart, lung, and blood diseases: an NHLBI resource for the gene therapy community. Hum Gene Ther. 2012;23:1130–1135. doi: 10.1089/hum.2012.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanazono Y, Brown KE, Handa A, Metzger ME, Heim D, Kurtzman GJ, et al. In vivo marking of rhesus monkey lymphocytes by adeno-associated viral vectors: direct comparison with retroviral vectors. Blood. 1999;94:2263–2270. [PubMed] [Google Scholar]
- Kahl CA, Tarantal AF, Lee CI, Jimenez DF, Choi C, Pepper K, et al. Effects of busulfan dose escalation on engraftment of infant rhesus monkey hematopoietic stem cells after gene marking by a lentiviral vector. Exp Hematol. 2006;34:369–381. doi: 10.1016/j.exphem.2005.12.005. [DOI] [PubMed] [Google Scholar]
- Podsakoff GM, Engel BC, Carbonaro DA, Choi C, Smogorzewska EM, Bauer G, et al. Selective survival of peripheral blood lymphocytes in children with HIV-1 following delivery of an anti-HIV gene to bone marrow CD34(+) cells. Mol Ther. 2005;12:77–86. doi: 10.1016/j.ymthe.2005.02.024. [DOI] [PubMed] [Google Scholar]
- Hanawa H, Hematti P, Keyvanfar K, Metzger ME, Krouse A, Donahue RE, et al. Efficient gene transfer into rhesus repopulating hematopoietic stem cells using a simian immunodeficiency virus-based lentiviral vector system. Blood. 2004;103:4062–4069. doi: 10.1182/blood-2004-01-0045. [DOI] [PubMed] [Google Scholar]
- Haas DL, Case SS, Crooks GM, Kohn DB. Critical factors influencing stable transduction of human CD34(+) cells with HIV-1-derived lentiviral vectors. Mol Ther. 2000;2:71–80. doi: 10.1006/mthe.2000.0094. [DOI] [PubMed] [Google Scholar]
- Cooper AR, Patel S, Senadheera S, Plath K, Kohn DB, Hollis RP. Highly efficient large-scale lentiviral vector concentration by tandem tangential flow filtration. J Virol Methods. 2011;177:1–9. doi: 10.1016/j.jviromet.2011.06.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soneoka Y, Cannon PM, Ramsdale EE, Griffiths JC, Romano G, Kingsman SM, et al. A transient three-plasmid expression system for the production of high titer retroviral vectors. Nucleic Acids Res. 1995;23:628–633. doi: 10.1093/nar/23.4.628. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn MR, Datta SK, Kellems RE. Adenosine deaminase-deficient mice generated using a two-stage genetic engineering strategy exhibit a combined immunodeficiency. J Biol Chem. 1998;273:5093–5100. doi: 10.1074/jbc.273.9.5093. [DOI] [PubMed] [Google Scholar]
- Tarantal A.2005Ultrasound imaging in rhesus and long-tailed macaques: Reproductive and research applications. Wolfe-Coote S.ed.). The Laboratory Primate Elsevier Academic Press; Amsterdam; Boston; 317–351. [Google Scholar]
- Tarantal AF, McDonald RJ, Jimenez DF, Lee CC, O'Shea CE, Leapley AC, et al. Intrapulmonary and intramyocardial gene transfer in rhesus monkeys (Macaca mulatta): safety and efficiency of HIV-1-derived lentiviral vectors for fetal gene delivery. Mol Ther. 2005;12:87–98. doi: 10.1016/j.ymthe.2005.01.019. [DOI] [PubMed] [Google Scholar]
- Lee CC, Jimenez DF, Kohn DB, Tarantal AF. Fetal gene transfer using lentiviral vectors and the potential for germ cell transduction in rhesus monkeys (Macaca mulatta) Hum Gene Ther. 2005;16:417–425. doi: 10.1089/hum.2005.16.417. [DOI] [PubMed] [Google Scholar]
- Kaplan EL, Meier P. Nonparametric-estimation from incomplete observations. J Am Statis Assoc. 457. 1958;53 [Google Scholar]
- Mantel N. Evaluation of survival data and two new rank order statistics arising in its consideration. Cancer Chemother Rep. 1966;50:163–170. [PubMed] [Google Scholar]
- Tukey JW. Variances of variance-components .2. The unbalanced single classification. Ann Math Statis. 43. 1957;28 [Google Scholar]
- Fitzmaurice G, Davidian M, Verbeke G, Molenberghs G. Longitudinal Data Analysis. Chapman and Hall/CRC; Boca Raton, FL; 2008. [Google Scholar]
- Gabrielsson JL, Weiner DL. Methodology for pharmacokinetic/pharmacodynamic data analysis. Pharm Sci Technol Today. 1999;2:244–252. doi: 10.1016/s1461-5347(99)00162-5. [DOI] [PubMed] [Google Scholar]
- Jaki T, Wolfsegger MJ. Estimation of pharmacokinetic parameters with the R package PK. Pharm Stat. 2011;10:284–288. [Google Scholar]
- SAS Institute . SAS Institute; Cary, NC; 2012. SAS/STAT user's guide, Version 9.3. 9.3 ed. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.