

Abstract
Major depressive disorder (MDD) is a chronic, recurring, and debilitating mental illness that is the most common mood disorder in the United States. It has been almost 50 years since the monoamine hypothesis of depression was articulated, and just over 50 years since the first pharmacological treatment for MDD was discovered. Several monoamine-based pharmacological drug classes have been developed and approved for the treatment of MDD; however, remission rates are low (often less than 60%) and there is a delayed onset before remission of depressive symptoms is achieved. As a result of a “proof-of-concept” study in 2000 with the noncompetitive NMDA antagonist ketamine, a number of studies have examined the glutamatergic systems as viable targets for the treatment of MDD. This review will provide a brief history on the development of clinically available antidepressant drugs, and then review the possible role of glutamatergic systems in the pathophysiology of MDD. Specifically, the glutamatergic review will focus on the N-methyl-D-aspartate (NMDA) receptor and the efficacy of drugs that target the NMDA receptor for the treatment of MDD. The noncompetitive NMDA receptor antagonist ketamine, which has consistently produced rapid and sustained antidepressant effects in MDD patients in a number of clinical studies, has shown the most promise as a novel glutamatergic-based treatment for MDD. However, compounds that target other glutamatergic mechanisms, such as GLYX-13 (a glycine-site partial agonist at NMDA receptors) appear promising in early clinical trials. Thus, the clinical findings to date are encouraging and support the continued search for and the development of novel compounds that target glutamatergic mechanisms.
Major Depressive Disorder
Major depressive disorder (MDD) is the most common mood disorder in the United States with a lifetime prevalence of 14.4% (Kessler, Petukhova, Sampson, Zaslavsky & Wittchen, 2012). MDD is a chronic, recurring, and debilitating mental disorder that significantly impairs occupational and/or social functioning. Most individuals suffering from MDD have recurring depressive episodes (10.3%) rather than a single lifetime episode (4.1%) (Kessler et al., 2012). It is important to differentiate MDD from major depressive episode (MDE), which includes individuals with bipolar disorder. Because of the inclusion of bipolar disorder, MDE (16.6%) typically has higher prevalence rates as compared to MDD (14.4%) (Kessler et al., 2012). MDD also has been found to have comorbidity with other DSM disorders such as anxiety disorder, substance abuse and impulse control disorder (Kessler et al., 2003).
According to the Diagnostic and Statistical Manual of Mental Disorders (DSM) 5th edition (American Psychiatric Association, 2013), an individual is required to exhibit a minimum of five depressive symptoms every day for a period of at least two weeks, which are newly presented or clearly worsened prior to the onset of the depressive episode, in order to be diagnosed with MDD. One of these five symptoms must include a depressed mood (Criterion A1), which is described as being depressed, or having a loss of interest/pleasure in hobbies/activities that were considered pleasurable (Criterion A2). In addition to one of these two symptoms, an individual must have four other depressive symptoms which may include significant changes in appetite or weight (Criterion A3); sleep (Criterion A4); psychomotor activity (Criterion A5); loss of energy or fatigue (Criterion A6); feelings of worthlessness (Criterion A7); diminished ability to think or concentrate (Criterion A8); or suicidal ideation (Criterion A9). All of these symptoms, with the exception of weight loss/gain and suicidal ideation, need to be present every day for the two week period to meet the DSM-V criterion for MDD. Furthermore, depressive episodes must significantly impair social or occupational functioning (Criterion B). Lastly, episodes must not be attributed to substance abuse (Criterion C) or better explained by other psychological disorders (Criterion D and E) such as schizophrenia, bipolar, etc.
Depressive episodes may appear at any age; however, MDD is most prevalent in adults (18-64 years) with a median age of onset in the 20s. For example, adults are twice as likely to be diagnosed with MDD as compared to both adolescents (13-17 years) and older adults (65+ years) (Kessler et al., 2003; Kessler et al., 2012). This decline of diagnosis in older adults may be attributed to a failure to report previous episodes, memory loss of past episodes, or sampling bias. Females are two-to-three times more likely to be diagnosed with MDD as compared to their male counterparts regardless of age group (Kessler et al., 2012).
Although there are several treatment options (both pharmacological and nonpharmacological) for MDD, 34-46% of MDD patients do not adequately response to treatment (Fava & Davidson, 1996). These patients are categorized as having treatment-resistant depression, which typically is defined as an inadequate response (i.e. fail to achieve full remission) to one or more antidepressant treatments following adequate duration and dose (Fava & Davidson, 1996; Fava, 2003). Treatment-resistant depression is well documented in the literature and is discussed as a subtype of depression; however, there is not a unified definition. For this review, the definition above (i.e. full remission) will be used because the goal for treatment should be to return the patient to the premorbid state of social and occupational functioning. An additional serious issue for the treatment of MDD is that even for the patients that do respond to currently available antidepressant drugs, there is a delayed onset of 4-12 weeks before adequate symptom remission is achieved (Schulberg, Katon, Simon, & Rush, 1998; Uher et al., 2011).
A group of clinicians have developed a strategic plan for treatment-resistant depression called Sequenced Treatment Alternatives to Relieve Depression (STAR*D). STAR*D provides a four step treatment plan, in which a patient proceeds to the next treatment step if they do not achieve full remission under the current treatment step (Fava et al., 2003). SSRIs are the first treatment step and as patients progress through the treatment steps they will be introduced to new antidepressant drugs with different mechanism of actions (e.g. bupropion, tricyclic antidepressants, etc.). Patients that achieve full remission and tolerate treatment at a specific step are then placed on long term treatment with that drug. The results from a large-scale long-term study found a cumulative remission rate of 67% (across all four treatment steps); however, those patients who progressed through more treatment steps had higher relapse rates as compared to those patients that achieved remission in the first treatment step (Rush et al., 2006).
MDD has significant economic and social consequences worldwide, which are driven by prevalence, medical treatment, and unemployment rates. For example, in the United States 36.7% of individuals suffering from MDD are either unemployed or out of labor force and only 59.8% of the MDD population in the workforce is receiving medical treatment for MDD (Greenberg et al., 2003). In 2000, the total economic impact of MDD was $83.1 billion for the United States. The total direct medical treatment costs (which include inpatient, outpatient, and pharmaceutical costs) reached $26.1 billion, and suicide-related costs (i.e. estimated lost lifetime earnings) exceeded $5.4 billion. Interestingly, workplace-related costs (e.g. missed worked days, reduced productivity, and health care expenses) were $51.5 billion (Greenberg et al., 2003). Individuals with severe MDD cost their employers approximately double in health care expenses, are more likely to file for disability and/or be unemployed, and missed approximately 13.7 MORE hours per month as compared to healthy individuals (Birnbaum et al., 2010).
Monoamine Hypothesis and Monoamine Based Pharmacological Treatments
Monoamine Hypothesis
It has been almost 50 years since the monoamine hypothesis of depression was articulated. The monoamine hypothesis proposes that patients with depression have depleted concentrations of serotonin, norepinephrine, and dopamine (Bunney & Davis, 1965; Delgado, 2000; Hirschfeld, 2000; Schildkraut, 1965). Two primary lines of evidence led to the development of the monoamine hypothesis: 1) the effects of reserpine on serotonin and catecholamines; and 2) the pharmacological mechanisms of action of antidepressant drugs. Reserpine, an alkaloid extracted from the Rauwolfia serpentina, was utilized as a treatment for hypertensive vascular disease in the 1950s; however, reserpine was found to precipitate depression in some patients. The depression produced by reserpine was reversed after the treatment was terminated and following either rest or electric shock therapy (Muller, Pryor, Gibbons, & Orgain, 1955). Additionally, reserpine was found to produce depressive-like effects in animals (Hirschfeld, 2000). Reserpine was found to inhibit vesicular monoamine transporter, and as a result, depletes brain monoamines (i.e. serotonin and catecholamines), which provided evidence for the role of serotonin, norepinephrine, and dopamine in depression (Kirshner, 1962; Shore, Pletscher, Tomich, Carlsson, Kuntzman, & Brodie, 1957; Shore, Silver, & Brodie, 1955; Weiner, Cloutier, Bjur, & Pfeffer, 1972). The second line of evidence was based on the underlying pharmacological mechanisms of action of monoamine oxidase (MAO) inhibitors and tricyclic antidepressants (TCA). For example, antidepressant drugs primarily target the monoamine neurotransmitters (i.e. serotonin, norepinephrine, and dopamine) in an attempt to increase the presence of these monoamine neurotransmitters in the synaptic space to activate postsynaptic receptors. The selective serotonin reuptake inhibitors (SSRI), which were developed later, provided additional support for the monoamine hypothesis. More recent clinical studies have provided evidence suggesting that the monoamine hypothesis for MDD needs to be revised and is not as simple as depleted concentrations of serotonin, norepinephrine, and dopamine leading to MDD. For example, monoamine depletion in healthy subjects does not produce depressive symptoms (Salomon, Miller, Krystal, Heninger, & Charney, 1997). Furthermore, monoamine or tryptophan depletion does not increase depressive symptoms in unmediated MDD patients (Berman et al., 2002; Delgado et al., 1994). Thus, the revised monoamine hypothesis suggests that monoamine depletion may play more of a modulatory role such that it influences other neurobiological systems (e.g. intracellular signaling or other neurotransmitter and neuropeptide systems) or must be present in the context of stressors (Charney, 1998; Heninger, Delgado, & Charney, 1996). A brief history of the development and description of the mechanisms of actions for each of the seven major antidepressant drug classes is presented below.
Monoamine oxidase inhibitors
Much like the discovery of the first antipsychotic drug chlorpromazine, serendipity played an important role in the discovery the first pharmacological treatment for depression. Chemists at Hoffmann-La Roche Ltd USA had developed isonicotinyl hydrazide (isoniazid) for the treatment of tuberculosis and isoniazid proved to be a successful antitubercular compound as the mortality rate of tuberculosis significantly decreased after the drug had been on the market for only one year (Lopez-Munoz, Alamo, Juckel, & Assion, 2007; Pletscher, 1991). While developing new antitubercular compounds, Fox and Gibas (1953) synthesized isopropyl-isonicotinyl hydrazide (iproniazid), a monoalkyl derivative of isoniazid, which would later serve as a catalyst for pharmacological treatment of MDD. Clinical observations reported marked “side effects” of iproniazid in patients being treated for tuberculosis, which included euphoria, psychostimulation, increased appetite, and improved sleep. The “side effects” produced by iproniazid were not originally identified as therapeutic effects because these side effects were not consistent across studies (Lopez-Munoz et al., 2007; Pletscher, 1991). Loomer, Saunders, and Kline (1958) conducted a systematic clinical study on patients with depression, in which the patients were treated with iproniazid for several weeks. Loomer and colleagues reported significant improvements in 70% of these patients (Loomer et al., 1958). While iproniazid was marketed as an antitubercular compound under the trade name Marsilid® in 1958, it was used off-label to treat patients suffering from MDD.
Thus, iproniazid became the first successful pharmacological treatment for depression and is classified as a MAO inhibitor. MAO is an enzyme that produces oxidative disamination (or break down) of biogenic amines (e.g. serotonin, dopamine, epinephrine, and norepinephrine) and sympathomimetic amines (e.g. tyramine, benzylamine, etc). There are two isoenzymes, MAOA and MAOB, and the distribution of these isoenzymes varies throughout the body. MAOA is primarily responsible for the enzyme activity for the deaminate of serotonin, melatonin, noradrenaline, and adrenaline (Billett, 2004; Shulman, Herrman, & Walker, 2013). In contrast, MAOB is responsible for enzyme activity for the breakdown of phenethylamine and benzylamine (Campbell, Marangos, Parma, Garrick, & Murphy, 1982; Shulman et al., 2013). Interestingly, both isoenzymes deaminate dopamine, tyramine, and tryptamine. MAOs responsible for the breakdown of biogenic amines are located in the presynaptic terminal. A result of inhibiting MAO is that monoamine neurotransmitter concentrations increase in the presynaptic terminal and are readily available for release when action potentials reach the nerve terminal. Iproniazid is a non-selective irreversible MAO inhibitor, which led to safety concerns (e.g. hypertensive crises), and ultimately led to the removal of iproniazid from the US market. One example of the safety concern was termed the “cheese reaction.” The combination of foods containing high amounts of tyramine, such as cheese or dairy products, and MAO inhibitors, which increase concentrations of tyramine and norepinephrine in the sympathetic nervous system, would lead to increased heart rate, hypertension and sweating. It was later determined that inhibiting MAOA was functionally involved in the antidepressant effects of MAO inhibitors (Lopez-Munoz et al., 2007; Shulman et al., 2013).
In an attempt to improve the safety of MAO inhibitors, drug development has focused on reversible and selective MAOA inhibitors (e.g. moclobemide [Manerix®] and brofaromine [Consonar®]). For example, a meta-analysis revealed that the reversible inhibitor of monoamine oxidase type A (RIMAs) moclobemide was as effective as SSRIs, nonselective MAO inhibitors and TCAs for depressive symptom reduction. Additionally, moclobemide was generally well tolerated with the most common side effect being nausea and insomnia, and only one reported case of hypertensive crisis. It is important to note that moclobemide is available in over 50 countries worldwide, but is not available in the United States, and brofaromine is no longer being developed as an antidepressant drug (Lotufo-Neto, Trivedi, & Thase, 1999).
Tricyclic antidepressants
As a result of the discovery and success of chlorpromazine for the treatment of schizophrenia, the search for more potent antipsychotic drugs intensified. Many of these novel compounds were molecularly modified from the classic antihistamine structure. One compound in particular, G22355 (imipramine), was derived from promethazine by substituting a sulfur bridge of the phenothiazine ring with an ethylene bridge (Domino, 1999; Fangmann, Assion, Juckel, Gonzalez, & Lopez-Munoz, 2008). The Geigy Chemical Corp supplied imipramine, which was synthesized by Hafliger and Schinder, to Dr. Roland Kuhn to test in psychiatric patients. Although imipramine did not exhibit antipsychotic properties in patients with schizophrenia, Kuhn found that imipramine produced marked improvements in patients suffering from severe depression. In an essay written by Kuhn (1958), he states “They commence some activity of their own, again seeking contact with other people, they begin to entertain themselves, take part in the games, become more cheerful and are once again able to laugh… The patients express themselves as feeling much better, fatigue disappears, the feeling of heaviness in the limbs vanish, and the sense of oppression in the chest gives way to a feeling of relief” (p. 459). Kuhn also stated that no serious side effects were recorded in the 500 patients treated with imipramine, which was a vast improvement over MAO inhibitors.
Imipramine (Tofranil®) was approved in 1959 by the Food and Drug Administration (FDA) for the treatment of MDD, which established the class of drugs called tricyclic antidepressants (TCA). The classification of TCAs was based on the three benzene ring molecular core, in part, because the mechanism of action was unknown at the time of discovery. Thus, the classification of TCAs differs from other classes of antidepressant drugs, which are classified based on their mechanism of action. TCAs have a diverse pharmacological profile with significant pharmacological action at two reuptake transporters and three receptor proteins: inhibiting presynaptic norepinephrine reuptake transporters; inhibiting presynaptic serotonin reuptake transporters; blocking postsynaptic adrenergic α1 and α2 receptors; blocking postsynaptic muscarinic receptors; and blocking postsynaptic histamine H1 receptors (Cusack, Nelson, & Richelson, 1994; Owens, Morgan, Plott, & Nemeroff, 1997; Sánchez & Hyttel, 1999: Vaischnavi et al., 2004). The inhibitions of norepinephrine and serotonin reuptake at the transport proteins are thought to be responsible for the therapeutic effects of TCAs and result in increased concentrations of norepinephrine and serotonin in the synaptic cleft, respectively. The selectivity for norepinephrine or serotonin transporters depends on the compounds; however, as most TCAs are more selective for the norepinephrine transporter over the serotonin transporter (Owens et al., 1997; Sánchez & Hyttel, 1999; Thomas, Nelson, & Johnson, 1987). For TCAs, the antagonism of adrenergic, muscarinic, and histaminergic receptors contribute primarily to the side effects of dizziness, memory impairments, and drowsiness, respectively.
Selective serotonin reuptake inhibitors
In the late 1960s evidence began to emerge suggesting a significant role of serotonin in MDD. For example, a postmortem study revealed decreased concentrations of serotonin in depressive suicides (Shaw, Eccleston, & Camps, 1967). As a result, the pharmaceutical company Eli Lilly began developing ligands that would selectively inhibit the reuptake of serotonin at serotonin transporters, and as a result would increase serotonin concentrations within the synaptic cleft to further stimulate postsynaptic serotonin receptors. In 1974, the first report on the selective serotonin reuptake inhibitor (SSRI) LY110140 (fluoxetine) was published and in that publication the authors suggested that fluoxetine would be an antidepressant drug (Wong, Horng, Bymaster, Hauser, & Molloy, 1974). The following year Wong and colleagues demonstrated that fluoxetine, an analogue of the phenoxyphenylpropylamine nisoxetine (LY94939), was a potent and selective serotonin reuptake inhibitor with relatively weak affinity for the norepinephrine transporter (Wong, Bymaster, Horng, & Molloy, 1975), which separated fluoxetine pharmacologically from other antidepressant drugs. Fluoxetine was approved by the FDA in December of 1987 and was launched to the market in January 1988 under the trade name Prozac® (for an excellence review on the development of fluoxetine see Wong, Bymaster, & Engleman, 1995; Wong, Perry, & Bymaster, 2005). Since the introduction of fluoxetine to the market, several other SSRIs have been approved by the FDA (e.g. sertraline [Zoloft®], citalopram [Celexa®], paroxetine [Paxil®], and escitalopram [Lexapro®]).
Although fluoxetine was the first SSRI approved and marketed in the United States, the clinical trials (Phase I-Phase III) lasted more than seven years and during that time Astra AB introduced the first SSRI zimeldine (Zelmid®) to the European market in March 1982. Zimeldine, which was derived from pheniramine, was removed from the European market in September 1983 due to severe side effects such as hypersensitivity reactions and Guillain-Barre syndrome, which is acute peripheral neuropathy. The hypersensitivity reactions resembled a flu- like syndrome which included fever, joint/muscle pain, headaches and hepatic effects. The estimated occurrence of the hypersensitivity reaction was 3:1000. Interestingly, zimeldine produced the hypersensitivity reactions in 1.5% of patients during clinical trials, but was still approved in the European market (Montgomery, Gabriel, James, Hawley, & Burkitt, 1989; Nilsson, 1983). During the 16 months that zimelidine was on the market there were 10 confirmed cases of Guillain-Barre syndrome and all of these patients were under zimeldine treatment (Fagius, Osterman, Siden, & Wiholm, 1985).
SSRIs are 20-1500 fold more selective for inhibiting serotonin over norepinephrine at their respective transporter proteins and have minimal binding affinity for other postsynaptic receptors such as adrenergic α1, α2, and β, histamine H1, muscarinic, and dopamine D2 receptors (Owens et al., 1997; Thomas et al., 1987). SSRIs (e.g. fluoxetine and citalopram) also do not stimulate the release of serotonin or norepinephrine presynaptically (Rothman et al., 2001) and have weak or no direct pharmacological action at postsynaptic serotonin receptors (e.g. 5-HT1A, 5-HT2A, and 5-HT2c) (Owens et al., 1997; Sánchez & Hyttel, 1999; Thomas et al., 1987). Therefore, the increase in activity at the postsynaptic serotonin receptors produced by SSRIs is a result of increased concentrations of serotonin in the synaptic cleft via reuptake inhibition rather than direct binding at the postsynaptic receptor. The most common side effects associated with SSRIs are nausea, insomnia, and sexual dysfunction (Papakostas, 2008).
Atypical antidepressant drug bupropion
Several new antidepressant drugs have been developed in the past couple of decades; however, the mechanisms of action for these drugs are similar to established antidepressant drugs in that these drugs target the monoamine neurotransmitter systems. Shortly after the introduction of fluoxetine, the immediate release (IR) form of bupropion (Wellbutrin®) was approved by the FDA in 1989 for the treatment of MDD with the aim to improve efficacy and safety. Bupropion IR requires three daily doses; thus, to reduce daily dosing regimens, the sustained-release (SR) and extended-release (XL) were developed and approved by the FDA in 1996 and 2003, respectively (Fava et al., 2005; Stahl et al., 2004). Bupropion is an “atypical” antidepressant drugs belonging to a unique chemical class (aminoketone) and its binding profile is very different from other antidepressant drugs (i.e. TCAs, SSRI, SNRI). For example, bupropion is primarily a dopamine-norepinephrine reuptake inhibitor. Bupropion displays its highest binding affinity for dopamine transporters and is at least two-fold more selective for dopamine transporters as compared to norepinephrine transporters (Bymaster et al., 2002; Letchworth et al., 2000; Pristupa, Wilson, Hoffman, Kish, & Niznik, 1994). Additionally, bupropion displays minimal or no binding affinity for serotonin transporters or other pre- and postsynaptic receptors (Bymaster et al., 2002; Cusack et al., 1994; Sánchez & Hyttel, 1999). Clinical research has shown that bupropion is as efficacious as other antidepressant drugs for the treatment of MDD and is well tolerated with the three most frequent side effects of bupropion being dry mouth, nausea, and insomnia (Feighner, Hendrickson, Miller, & Stern, 1986; Feighner, Meredith, Stern, Hendrickson, & Miller, 1984; Moreira, 2011). Furthermore, bupropion has the lowest risk of sexual dysfunction (nearly half) as compared to TCAs, MAOIs, SSRIs, and SNRIs (Clayton et al., 2002; Stahl et al., 2004). There are no other selective dopamine-norepinephrine reuptake inhibitors on the market for the treatment of depression.
Serotonin-norepinphrine reuptake inhibitors
The next “atypical” antidepressant drug, venlafaxine, was introduced to the United States market in 1993, and this drug selectively targets the serotonin and norepinephrine transporters. The immediate release form of venlafaxine (Effexor®), a serotonin-norepinephrine reuptake inhibitors (SNRI), was approved by the FDA for the treatment of MDD in 1993 and in 1997 the extended release form of venlafaxine was also approved for MDD (Papakostas, 2009). Since the approval of venlafaxine, several other SNRIs (e.g. duloxetine [Cymbalta®] and milnacipran [Savella®]) have been approved for the treatment of MDD. SNRIs are similar to TCAs in that SNRIs inhibit the reuptake of serotonin and norepinephrine at the serotonin and norepinephrine transporters, respectively. Unlike TCAs, SNRIs have minimal or no pharmacological action at adrenergic (α1, α2, and β), histamine (H1), muscarinic, dopamine, or postsynaptic serotonin receptors (Bymaster et al., 2001; Millan et al., 2001; Owens et al., 1997; Sánchez & Hyttel, 1999; Vaischnavi et al., 2004). There is some evidence that suggests SNRIs may be more effective for the treatment of MDD as compared to SSRIs; however, these differences are relatively modest (Papakostas, Thase, Fava, Nelson, & Shelton, 2007; Stahl, Grady, Moret, & Briley, 2005). The clinical tolerability and the prevalence of sexual dysfunction of SNRIs are comparable with other antidepressant drug treatments (Clayton et al., 2002; Stahl, Grady, Moret, & Briley, 2005).
Atypical antidepressant drug vortioxetine
In September 2013, vortioxetine (Brintellix®) became the most recent antidepressant drug approved by the FDA for the treatment of MDD. Lu AA21004, which was named vortioxetine, was synthesized by H. Lundbeck A/S, but was co-developed by H. Lundbeck A/S and Takeda Pharmaceutical Company Ltd (Gibb & Deeks, 2014). Vortioxetine belongs to the piperazines chemical class and has been marketed as a “multi-modal” drug because vortioxetine displays high binding affinity and complementary mechanisms of action for several serotonin receptors (i.e. 5-HT1A, 5-HT1B, 5-HT3A, 5-HT7, and serotonin transporters). Specifically, vortioxetine is a serotonin 5-HT1A receptor agonist, 5-HT1B receptor partial agonist, 5-HT3A and 5-HT7 receptor antagonist, and a potent serotonin reuptake inhibitor (Bang-Andersen et al., 2011; Mork et al., 2012). Vortioxetine has considerable receptor affinity for dopamine and norepinephrine transporters; however, vortioxetine is approximately 3 to 12 times more selective for serotonin transporters as compared to dopamine and norepinephrine transporters, respectively (Bang-Andersen et al., 2011). The clinical efficacy and tolerability of vortioxetine is comparable to other antidepressants with the most common side effects being nausea and headaches. Vortioxetine appears to have a low risk for sexual dysfunction and weight gain (Alam, Jacobsen, Chen, Serenko, & Mahableshwarkar, 2014; Boulenger, Loft, & Florea, 2012; Boulenger, Loft, & Olsen, 2014; Gibbs & Deeks, 2014; Pearce & Murphy, 2014). It is important to note that clinical (McIntyre, Lophaven, & Olsen, 2014) and preclinical (du Jardin, Jensen, Sanchez, & Pehrson, 2014; Jensen et al., 2014; Mork et al., 2013) studies have shown that vortioxetine may help improve cognitive functioning.
Although there are seven antidepressant drug classes, all of which work to produce an immediate increase in monoamine neurotransmitter concentrations, there is still a population of patients that do not respond to these medications. This lends further support for the revised monoamine hypothesis which suggests that depleted monoamine concentrations may play more of a modulatory role to other neurobiological systems, rather than a major direct role in MDD (Charney, 1998; Heninger, Delgado, & Charney, 1996). Thus, more recent research has focused on finding novel, non-monoaminergic based, receptor targets for treatment-resistant depression (e.g. Lee, Jeong, Kwak, & Park, 2010; Palucha & Pilc 2005). In particular, the glutamatergic system has become a focal point for drug development research.
Glutamatergic Systems and Major Depressive Disorder
Glutamatergic systems
In the central nervous system, glutamate is the major excitatory neurotransmitter and makes functional contributions to more than half of all synapses in the brain. The glutamate system has an integrated tripartite synapse that consists of 1) a presynaptic neuron, 2) a postsynaptic neuron, and 3) an astrocyte. The presynaptic neuron releases glutamate in response to action potentials. The released glutamate then binds to various pre- and postsynaptic receptors, as well as to receptors on the surrounding astrocytes. Synaptic glutamate reuptake is performed primarily by astrocytes, specifically, the excitatory amino acid transporter 2 (EAAT2). Within the astrocyte, glutamate is converted to glutamine (glutamate/glutamine cycle) by glutamine synthetase and then resupplied to the presynaptic neuron where it is used for synthesis of glutamate (Kew & Kemp, 2005; Machado-Vieira, Manji, & Zarate, 2009; Mathews, Henter, & Zarate, 2012). Figure 1 shows the tripartite glutamatergic synapse.
Figure 1.
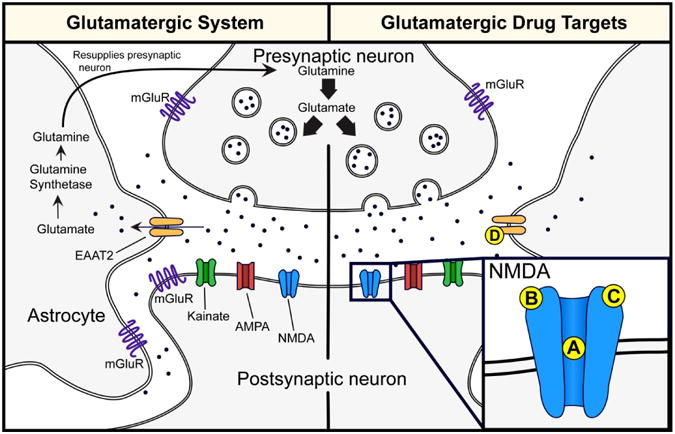
Tripartite glutamatergic synapse and potential drug targets. Left panel: The presynaptic neuron releases glutamate in response to action potentials. Glutamate can bind to ionotropic (i.e. NMDA, AMPA, kainate) and metabotropic (i.e mGluR) receptors located on the presynaptic and postsynaptic neuron as well as on astrocytes. Synaptic glutamate reuptake is performed primarily by the EAAT2 located on astrocytes. Within the astrocyte, glutamate is converted to glutamine (glutamate/glutamine cycle) via glutamine synthetase and then resupplied to the presynaptic neuron where it is used for synthesis of glutamate. Right panel: Potential NMDA and EAAT2 drug targets: (A) Noncompetitive NMDA receptor antagonist (e.g. ketamine and memantine) and low-trapping NMDA receptor channel blockers (lanicemine [AZD6765]); (B) NR2B subunit selective NMDA receptor antagonists (e.g. traxoprodil [CP-101,606] and MK-0657); (C) Partial agonist at the glycine binding site of NMDA receptors (e.g. D-cycloserine, GLYX-13, and NRX- 1074); (D) EAAT2 enhancers (e.g. riluzole). Abbreviations: NMDA, N-methyl-D-aspartate; AMPA, α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid; mGluR, metabotropic glutamate receptors; EAAT2, excitatory amino acid transporter 2.
The glutamatergic system consists of two receptor types, ionotropic and metabotropic. Ionotropic glutamatergic receptors include N-methyl-D-aspartate (NMDA) receptors, α-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA), and kainate receptors. These ionotropic receptors are ion channels that are permeable to cations (i.e. sodium [Na+] and calcium [Ca2+]), which in turn depolarize the neuron and/or promote intracellular signaling cascades (Kew & Kemp, 2005). There are eight G-protein-coupled metabotropic glutamate receptors subtypes (mGluR1-8) that are divided into three distinct groups that are based on their homology and function: Group I (mGluR1 and mGluR5), Group II (mGluR2 and mGluR3), and Group III (mGluR4, mGluR6, mGluR7, and mGluR8). Group I mGluRs are localized on the postsynaptic neuron and are coupled to Gq/G11 subunits; whereas Group II and Group III are localized on the presynaptic neuron and are couple to Gi/Go subunits. mGluRs can mediate intracellular signaling cascades by activating second messenger pathways and/or through βγ subunits. Group I and Group II mGluRs have been investigated in the pathophysiology and treatment of MDD. Specifically, mGluR5 (e.g. AZD2066 and RO4917523) and mGluR2/3 (RO4995819) negative modulators have been tested in Phase II clinical trials for treatment-resistant patients, and some compounds (e.g. RO4917523and RO4995819) have shown promise (for full review see Holly, LaCrosse, & Hillhouse, 2014). Assessing all glutamate receptors and their respective implications in MDD are beyond the scope of this review. Therefore, the present review will primarily focus on NMDA receptors.
N-methyl-D-aspartate (NMDA) receptors
NMDA receptors are a heteromeric complex that has seven subunits, NR1, NR2A-D, and NR3A-B and functional NMDA receptors must be comprised of a NR1 subunit and at least one NR2 subunit. Unlike other ligand-gated ion channels, NMDA receptors require two distinct mechanisms in order to be activated. First, NMDA receptor channels require co-agonist binding at the glycine binding site on the NR1 subunit and at the glutamate binding site on the NR2 subunit (Kew & Kemp, 2005; Marsden, 2011). Thus, if one of these co-agonists (glycine or glutamate) is not bound to their respective binding site, the ion channel will not open. Second, the NMDA receptor channels are blocked by magnesium (Mg2+) ions during the resting state. Depolarization of the neuron is required to dispel the Mg2+ ion from NMDA receptor channels, which is usually achieved by activation of AMPA receptors. The NMDA receptor ion channel is non-selective and will allow both Na+ and Ca2+ to enter. The influx of Ca2+ is associated with the induction of various signaling cascades (Kew & Kemp, 2005; Marsden, 2011).
Glutamatergic dysfunction in MDD
Clinical research has used both indirect and direct measures to evaluate the glutamatergic system in patients suffering from MDD, and have found evidence of glutamatergic dysfunction in MDD. For example, clinical studies that have used indirect measures for analysis, such as plasma, cerebrospinal fluid, and serum concentrations, have found differences in glutamate and glutamine in patients diagnosed with MDD as compared to healthy controls (Table 1). Specifically, several studies have found increased concentrations of glutamate in plasma (Altamura et al., 1993; Küçükibrahimoğlu et al., 2009; Mauri et al., 1998) and increased concentration of glutamine in cerebrospinal fluid (Levine et al., 2000). Furthermore, antidepressant drug treatment has been found to reduce the serum and plasma glutamate concentrations (Küçükibrahimoğlu et al., 2009; Maes, Verkerk, Vandoolaeghe, Lin, & Scharpé, 1998), as well as cerebrospinal fluid glutamine concentrations (Garakani, Martinez, Yehuda, & Gorman, 2013). These findings suggest that these monoamine-based antidepressant drugs are modulating the glutamatergic system. However, there are limitations when using indirect measures, such as the inability to determine central and peripheral substrates and metabolic effects, which make it difficult to interpret these findings. Additionally, peripheral measurements of CNS chemicals/metabolites may be seriously compromised by their inability to easily cross the blood brain barrier.
Table 1. Alterations in glutamatergic substrates in major depressive disorder using indirect measures for analysis.
| Interest | Region of Analysis | Change | Reference |
|---|---|---|---|
| Glutamate | Serum | No Difference | Maes et al., 1998a |
| Plasma | Increased | Altamura et al., 1993 | |
| Mauri et al., 1998 | |||
| Küçükibrahimoğlu et al., 2009 | |||
| Platelet-rich plasmac | Supersensitivity to glutamate | Berk et al., 2001 | |
| Glutamine | Serum | Increased | Maes et al., 1998a |
| Plasma | Decreased | Altamura et al., 1993 | |
| Cerebrospinal fluid | Increased | Levine et al., 2000 | |
| No Difference | Garakani et al., 2013b |
decrease after 5-week antidepressant; however, no difference prior to treatment
decrease after 8-week antidepressant; however, no difference prior to treatment
platelet intracellular calcium response to glutamate stimulation
Clinical studies that have used more direct measures (Table 2), such as proton magnetic resonance spectroscopy (1H-MRS), to evaluate the glutamatergic dysfunction in MDD have found reduced metabolic, typically unresolved, glutamate/glutamine exchange (Glx) in subcortical and cortical brain regions of MDD patients. For example, a reduction of Glx were found the hippocampus (Block et al., 2009) and anterior cingulate cortex (Auer et al., 2000; Mirza et al., 2004; Pfleiderer et al., 2003), which are purported to play an important role in MDD. Further 1H-MRS studies found that cortical regions also had a reduction in Glx which include the dorsomedial and ventromedial prefrontal cortex (Hasler et al., 2007), and left dorsolateral prefrontal cortex (Michael et al., 2003). Following ECT treatment, the anterior cingulate cortex and left dorsolateral prefrontal cortex Glx concentrations were significantly increased and recovered back to healthy control Glx concentrations (Michael et al., 2003; Pfleiderer et al., 2003). Furthermore, these studies found that ECT responders had a great increase of Glx as compared to the non-responders. On the other hand, Sanacora and colleagues (2004) found elevated concentrations of Glx in the occipital cortex, and while this finding is not consistent with the other 1H-MRS, it is consistent with the elevated concentrations of glutamate found in plasma (Altamura et al., 1993; Küçükibrahimoğlu et al., 2009; Mauri et al., 1998) and by recent studies that found decreased concentrations of EAAT2, the main source the glutamate reuptake, in postmortem brain tissue, which would result in the accumulation of extracellular glutamate (Bernard et al., 2001; Choudary et al., 2005). It is important to note that there are limitations when using the magnetic resonance imaging protocol. For example, these studies were measuring glutamate/glutamine cycle rather than glutamate exclusively.
Table 2. Alterations to glutamatergic concentrations measured by proton magnetic resonance spectroscopy (1H-MRS).
| Interest | Region of Analysis | Change | Reference |
|---|---|---|---|
| Glutamate | Occipital cortex | Increased | Sanacora et al., 2004 |
| Glutamate/Glutamine (Glx) | Anterior cingulate cortex | Decreased | Auer et al., 2000 |
| Pfleiderer et al., 2003a | |||
| Mirza et al., 2004 | |||
| Hippocampus | Decreased | Block et al., 2009b | |
| Dorsolateral prefrontal cortex | Decreased | Michael et al., 2003a | |
| Dorsomedial and ventromedial prefrontal cortex | Decreased | Hasler et al., 2007 |
Glx major depressive disorder levels recovered to healthy control following ECT treatment
no change in Glx levels following 8-weeks of citalopram treatment
Several postmortem studies have found changes in the expression of NMDA receptor subunits in MDD patients (Table 3), which are likely compensatory effects to the changes in glutamatergic substrate concentrations, and appear to be brain region specific. For example, the NR2B and NR2C subunits have been shown to have increased expression in the locus coeruleus in postmortem tissue of MDD patients (Chandley et al., 2014; Karolewicz, Stockmeier, & Ordway, 2005). Additionally, the expression of NR2A subunits has been found to be elevated in the lateral amygdala (Karolewicz et al., 2009). Furthermore, MDD patients have shown an increase in glutamate binding in the hippocampus (Beneyto, Kristiansen, Oni-Orisan, McCullumsmith, & Meador-Woodruff, 2007) and a greater sensitivity to glutamate as measured by intracellular calcium influx (Berk, Plein, & Ferreira, 2001). On the other hand, the NR2A and NR2B subunits transcription have been shown to be reduced in the perirhinal and prefrontal cortices in postmortem tissue from MDD patients (Beneyto et al., 2007; Beneyto and Meador-Woodruff, 2008; Feyissa, Chandran, Stockmeier, & Karolewicz, 2009). Moreover, postmortem studies have found decreased levels of the NR1subunit in the superior temporal cortex (Nudmamud-Thanoi and Reynolds, 2004) and prefrontal cortex (Beneyto and Meador-Woodruff, 2008). NR1 and NR2 subunits are required for functional NMDA receptor heteromeric complexes, and thus, increases/decreases in the levels of these NR1/NR2 subunits can be interpreted as changes in total number of functional NMDA receptors. Based on these results, it would be hypothesized that depression may be associated with hyperfunction of NMDA receptors in subcortical regions (i.e. hippocampus, locus coeruleus, and amygdala); whereas, depression may be associated with a hypofunction of NMDA receptors in cortical regions (i.e. prefrontal, perirhinal and temporal cortices). Moreover, the results from the 1H-MRS studies would support the notion of a hypofunction of the glutamatergic system in cortical regions. However, pervious drug treatment is a common limitation when interpreting plasma, 1H-MRS, or postmortem human data. The majority of these studies used patients that were drug free for at least three to four weeks, but there is a possibility that previous pharmacological treatments produced neurobiological changes in the brain that were still evident postmortem.
Table 3. Alterations in N-methyl-D-aspartate (NMDA) receptor subunit expression in postmortem tissue from major depressive disorder patients.
| Receptor Subunit/site | Region of Analysis | Molecular Assay | Change | Reference |
|---|---|---|---|---|
| NR1 | Superior temporal cortex | Western blot | Decreased | Nudmamud-Thanoi et al., 2004 |
| Prefrontal cortex | In situ hybridization | Decreased | Beneyto et al., 2008 | |
| Western blot | No change | Feyissa et al., 2009 | ||
| NR2A | Perirhinal cortex | In situ hybridization | Decreased | Beneyto et al., 2007 |
| Prefrontal cortex | In situ hybridization | Decreased | Beneyto et al., 2008 | |
| Western blot | Decreased | Feyissa et al., 2009 | ||
| Lateral amygdala | Western blot | Increased | Karolewicz et al., 2009 | |
| NR2B | Perirhinal cortex | In situ hybridization | Decreased | Beneyto et al., 2007 |
| Prefrontal cortex | Western blot | Decreased | Feyissa et al., 2009 | |
| Locus coeruleus | Polymerase chain reaction (PCR) | Increased | Chandley et al., 2014 | |
| NR2C | Locus coeruleus | Western blot | Increased | Karolewicz et al., 2005 |
| Polymerase chain reaction (PCR) | Increased | Chandley et al., 2014 | ||
| Glutamate Binding site | Hippocampus | Autoradiography [3H]CGP39653 | Increased binding | Beneyto et al., 2007 |
Collectively, the clinical data suggest the involvement of the glutamatergic system in the pathophysiology of MDD, which includes disruptions in glutamatergic substrate concentrations and NMDA receptor alterations. Although the role of glutamatergic systems have yet to be fully elucidated, a “proof of concept” clinical study reported that the noncompetitive NMDA antagonist ketamine produced rapid and prolonged antidepressant effects in patients suffering from MDD (Berman et al., 2000). This has generated tremendous interest in developing new drugs that target glutamatergic mechanisms for the treatment of MDD. Figure 1 shows potential drug targets on the NMDA receptor and EAAT2.
Ketamine: Noncompetitive NMDA Receptor Antagonist, Drug of Abuse and Antidepressant
Drug of abuse
Ketamine, a dissociative anesthetic with hallucinogenic properties, is a derivative of phencyclidine and was developed in the 1960s by Dr. Calvin Lee Stevens of Wayne State University for the pharmaceutical company Parke-Davis. In 1964, ketamine was experimentally administered to human subjects to test the safety and general anesthetic properties of ketamine, and the subjects reported few side effects and described feelings of floating or having no feeling in their limbs. Ketamine was approved by the FDA in 1970 for use as a short-acting anesthetic in humans and animals and was given to injured American soldiers during the Vietnam War (Domino, 2010). Ketamine, which is distributed under the trade names Ketalar®, Ketaset®, and Vetamine®, is still widely used in human and veterinary medicine with approximately 16,000 dispensed ketamine prescriptions in 2012 (Domino, 2010; Drug Enforcement Administration, 2013).
Ketamine, also known as “Special K,” became popular as a recreational drug in the mid- 1990s. As a result of its increased recreational use, ketamine became a Schedule III non-narcotic substance under the Controlled Substances Act in 1999 (Drug Enforcement Administration, 2013). Although the illicit use of ketamine appears to be relatively low in the United States with less than 2% of the population using hallucinogens (Substance Abuse and Mental Health Services Administration, 2013), which also includes lysergic acid diethylamide, phencyclidine, peyote, mescaline, psilocybin mushrooms, and “ecstasy,” the popularity of ketamine continues to grow in other countries. For example, ketamine (23.2%) is the second most abused substance in Hong Kong with heroin (61.7%) being the most abused substance (Shek, 2007). The use of ketamine also has been on a rise over the past decade, specifically among “dance” or “club” drug users in Europe (McCambridge, Winstock, Hunt, & Mitcheson, 2007, 2007; Shek, 2007; Winstock, Mitcheson, Gillatt, & Cottrell, 2012). At subanesthetic or emergence from anesthetic doses, ketamine produces hallucinations (i.e. distorts perceptions of sight and sounds), mood and body image changes, and make the user feel disconnected (or dissociated) from their body/reality. The hallucinogenic effects of ketamine have been termed the “K-hole” by users, and the duration of these effects are relatively short, 30 to 60 min, as compared to phencyclidine (Drug Enforcement Administration, 2013).
Mechanism of action
Ketamine is a noncompetitive NMDA receptor antagonist (aka channel blocker) that binds to the phencyclidine site inside the ion channel of the NMDA receptor, blocking the channel in a way that is similar to how Mg2+ blocks NMDA receptors, and is non-selective for the NR2A-D subunits of the NMDA receptor channel (Lord et al., 2013; Yamakura, Mori, Masaki, Shimoji, & Mishina, 1993; Yamakura & Shimoji, 1999). While many have categorized ketamine as a high affinity NMDA receptor antagonist, in vivo autoradiography studies suggest that ketamine has relatively low affinity for the NMDA receptor (Ki = 659-1190 nM) (Bresink, Danysz, Parsons, & Mutschler, 1995; Roth et al., 2013). Additionally, selectivity of ketamine for the NMDA receptor appears to be weak as compared to other receptors. For example, ketamine has been shown to have higher receptor affinity for in vitro human cloned dopamine D2 receptors (Ki = 55 nM) than for NMDA receptors (Ki = 659-1190 nM) (Seeman, Ko, & Tallerico, 2005); however, a follow up study found that ketamine did not exert functional activity (i.e. as a full agonist, partial agonist, or antagonists) at in vitro dopamine D2 receptors (Jordan et al., 2006). Ketamine is approximately 12 and 20-fold more selective for NMDA receptors as compared to serotonin 5-HT2A and mu opioid receptors, respectively (Kapur & Seeman, 2002; Smith et al., 1987). Furthermore, ketamine also has weak binding affinity for sigma (Mendelsohn, Kalra, Johnson, & Kerchner, 1985), muscarinic (Hirota, Hashimoto, & Lambert, 2002), and κ and δ opioid receptors (Smith et al., 1987), as well as dopamine, norepinephrine, and serotonin transporters (Nishimura et al., 1998).
Antidepressant clinical studies
Tables 4 provide a summary of the clinical trial results and the current status of ketamine for the treatment of MDD. In 2000, the noncompetitive NMDA receptor antagonist ketamine was first used in a “proof of concept” randomized, double- blind study to assess the effects of ketamine on MDD in seven patients who received both vehicle and ketamine treatment (counterbalanced). A single, subanesthetic dose of ketamine (0.5 mg/kg) was intravenously (i.v.) infused over 40 minutes, and the antidepressant effects of ketamine were assessed using the Hamilton Depression Rating Scale (HDRS) and Beck Depression Inventor (BDI). In comparison, an anesthetic dose for ketamine in humans ranges from 1.0 mg/kg to 4.5 mg/kg intravenous and from 6.5 mg/kg to 13.0 mg/kg intramuscular. In this study, ketamine produced rapid, within four hours, and prolonged antidepressant effects that lasted up to 72 hours as compared to placebo control (Berman et al., 2000). This rapid antidepressant effect of ketamine is far superior to the 4-12 week delay with current antidepressant drugs (Schulberg et al., 1998; Uher et al., 2011). The hallucinogenic (or psychotomimetic) effects (e.g. out of body experience, hallucinations, etc.) of ketamine subsided (within two hours) prior to the onset of the antidepressant effects as measured by the Visual Analog Scales for intoxication “high” (VAS-high) and Brief Psychiatric Rating Scale (BPRS) (Berman et al., 2000). This was the first clinical study to demonstrate that glutamatergic drugs may be effective for the treatment of MDD.
Table 4.
Selected clinical trial results and current status of ketamine for the treatment of major depressive disorder.
| Drug | Primary Mechanism of Action | Study/reference | TRD | N | Study Design | Main Findings | Current Status/Clinical Trials | ||
|---|---|---|---|---|---|---|---|---|---|
| Ketamine | Noncompetitive NMDA receptor antagonist | Breman et al., 2000 | No | 7 | Randomized, double-blind, single infusion (0.5 mg/kg) | Improvement from 4 hours to 3 days | Phase 1: Biomarkers of rapid onset for MDD. NCT02165449 | ||
| Zarate et al., 2006 | Yes | 17 | Randomized, double-blind, single infusion | Improvement from 110 min to 7 days; full remission in 5/17 patients | Phase 2: There are >20 clinical trials evaluating ketamine for MDD. NCT01945047; | ||||
| Ann het Rot et al., 2010 | Yes | 10 | Open-label, repeated infusion (6 infusions over 12 days) | 9/10 met response criteria after first ketamine infusion; mean relapse rate of 19 days after last ketamine infusion | NCT01880593; NCT01920555; NCT01868802; NCT00088699; NCT 01179009; NCT01667926; etc. | ||||
| Murrough et al., 2013 | Yes | 24 | Open-label, repeated infusion (6 infusions over 12 days) | 17/24 met response criteria following 6 ketamine infusions; 4 patients did not relapse after 83 days | |||||
| Murrough et al., 2013 | Yes | 73 | Two-site, randomized, active placebo control (midazolam), single infusion | 30/47 patients met response criteria following ketamine; 7/25 met response criteria following midazolam; no effect of site | |||||
| Ghasemi et al., 2014 | No | 18 | Randomized, active control (ECT), repeated infusion and repeated ECT (3 infusion or ECT treatments) | Improvement with ketamine following first infusion and lasted 1 week; improvement with ECT after second treatment and lasted 1 week | |||||
| Irwin et al., 2010 | No | 2 | Open-label, case study, single oral dose | Both cases showedimprovement from 60 min to 8days | |||||
| McNulty et al., 2012 | No | 1 | Open-label, case study, repeated oral dosing (40 mg/5 mL flavored syrup) | Patient was in remission at 2 month follow up | |||||
| Irwin et al., 2013 | No | 8 | Open-label, repeated nightly oral dosing (0.5 mg/kg; 28 days) | Improvement after 14 days of ketamine dosing and continued until day 28. | |||||
| Lara et al., 2013 | Yes | 26 | Open-label, single sublingual dose (10 mg) | 20/26 achieved remission or improvement 90 min after ketamine dose | |||||
| Chilukuri et al., 2014 | No | 27 | Randomized, open-label parallel group (0.25-0.5 mg/kg IM or 0.5 mg/kg IV) | Improvement after 2 hours: 8/9 following IV; 7/9 follwing 0.25 mg IM; 6/9 following 0.5 mg IM | |||||
| Lapidus et al., 2014 | No | 18 | Randomized, double-blind, crossover study (50 mg intranasal ketamine) | 8/18 met response criteria at 24 hours | |||||
TRD, treatment-resistant depression; N, represents the number of participants to complete the study; MDD, major depressive disorder; NMDA, N-methyl-D-aspartate; IV, intravenous; IM, intramuscular.
One limitation of the Berman et al. (2000) study was the limited three day follow-up period. In that study, the ketamine-treated patients did not return baseline depression scores during the follow-up period, which makes it difficult to determine the duration of the antidepressant effects of ketamine. Thus, Zarate and colleagues conducted a clinical study to assess the antidepressant effects of ketamine in patients with treatment-resistant MDD and to determine a better understanding of the duration of the antidepressant effects. Following a single 0.5 mg/kg infusion of ketamine, treatment-resistant patients showed a significant reduction in depression scores at 110 min that lasted up to seven days as measured by HDRS (Zarate et al., 2006). Specifically, 71% of the patients achieved response criteria one day after the infusion, while 29% achieved full remission. Additionally, 35% maintained response criteria on day seven (Zarate et al., 2006). Again, the hallucinogenic (or psychotomimetic) effects diminished before the onset of the antidepressant effects of ketamine (within two hours). This study confirmed the finding in the Berman et al. (2000) study that ketamine produces rapid and prolonged antidepressant effects in the treatment of depression and extended ketamine's efficacy to treatment-resistant MDD.
Two studies have evaluated the effects of repeated ketamine infusion in patients suffering from treatment-resistant MDD to determine if repeating dosing could extend the antidepressant effects of ketamine beyond seven days. In both studies, patients received six ketamine infusions over approximately 12 days. One study found that the average relapse rate was 19 days after the sixth and final ketamine infusion, with one patient remaining in remission for more than three months (ann het Rot et al., 2010). In the second study, ketamine infusions produced an antidepressant effect in 70.8% of the patients and these antidepressant effects lasted for a median of 18 days (four participants were still in remission at the last follow up visit [83 days]) (Murrough et al., 2013). Both of these studies reported safety and tolerability similar to the single ketamine infusion studies.
In the study by Murrough et al. (2013), they compared the effects of a single low dose of ketamine (0.5 mg/kg, i.v.) to an active placebo, the anesthetic benzodiazepine midazolam (0.045 mg/kg, i.v.), in treatment-resistant patients. Following drug infusion, the ketamine group showed a 64% response rate at 24 hours and 46% response rate on day seven; whereas, the midazolam group showed a response rate of 28% at 24 hours and 18% on day seven (Murrough et al., 2013). These results suggested that the antidepressant effects of ketamine are not a result of its anesthetic properties. Another study compared the effects of ketamine and electroconvulsive therapy (ECT) in patients suffering from MDD. This study found that both ketamine and ECT produced antidepressant effects; however, ketamine produced superior antidepressant effects in terms of response onset. For example, ketamine produced rapid antidepressant effects starting at 24 hours; whereas, the antidepressant effects of ECT were not expressed until day two. The antidepressant effects of both ketamine and ECT lasted until the completion of the study, which was seven days (Ghasemi et al., 2014). These results suggest that ketamine is as efficacious, if not more efficacious, as ECT for treating MDD.
In addition to the previously mentioned studies, several other clinical studies have found that i.v. infusions of low-dose ketamine produce rapid and sustained antidepressant effects in patients with MDD (Machado-Vieira et al., 2009; Mathew et al., 2010; Rasmussen et al., 2013) and a rapid reduction in suicidal ideation/cognition in patients with treatment-resistant MDD (DiazGranados et al., 2010; Price, Nock, Charney, & Mathew, 2009; Price et al., 2014; Zigman & Blier, 2013). An anti-anhedonic effect of ketamine treatment in treatment-resistant bipolar depression was recently demonstrated by Lally et al. (2014). In a randomized, placebo- controlled, double-blind crossover design, 36 treatment-resistant bipolar depression patients were treated with a single, low intravenous dose of 0.5 mg/kg ketamine. They found that ketamine rapidly reduced anhedonia in these patients within 40 minutes and that these effects preceded reductions in other depressive symptoms. Also, the decrease in anhedonic symptoms persisted up to 14 days. The authors concluded that these findings demonstrate the importance of glutamatergic mechanisms for the treatment of treatment-refractory bipolar depression and especially for the treatment of anhedonia symptoms.
Researchers have started to evaluate alternative routes of administration for ketamine to improve safety and possibly reduce the psychotomimetic side effects of ketamine. Furthermore, the hospital visits and the duration associated with the i.v. administration of ketamine may become an issue for patient compliance. Two case studies have demonstrated that orally administered low doses of ketamine were effective in relieving depressive symptoms in several MDD patients (Irwin & Iglewicz, 2010; McNulty & Hahn, 2012). This finding was extended in a 28-day, open-label, proof-of-concept trial of daily, low dose (0.5 mg/kg) oral with 8 patients with symptoms of depression or depression mixed with anxiety (Irwin et al., 2013) and in a study using sublingual administration of low dose ketamine in 26 out-patients with refractory unipolar or bipolar depression (Lara, Bisol, & Munari, 2013). Both of these studies reported minimal to no psychotomimetic effects of ketamine with oral administration. Another small, open-label study found that intramuscular administration of ketamine (0.5 and 0.25 mg/kg) produced rapid (two hours) and sustained (four days) antidepressant effects to a similar magnitude to that of i.v. administration of ketamine (0.5 mg/kg). Psychotomimetic or dissociative effects were not evaluated in this study (Chilukuri et al., 2014). Finally, a double-blind, crossover clinical study has shown that intranasal administration of ketamine (50 mg) produced rapid (40 min) and sustained (two days) antidepressant effects with minimal psychotomimetic and dissociative side effects (Lapidus et al., 2014). These studies demonstrate that there may be effective alternative routes of administration of low-dose ketamine treatment for MDD that have a reduced risk of adverse side effects, which are likely a result of reduced levels of bioavailable ketamine as compared to i.v. administration of ketamine.
Several studies have evaluated potential biomarkers to determine which MDD patients may respond to ketamine treatment. For example, baseline D-serine plasma concentrations and baseline neurocognitive performance (specifically processing speed and attention) prior to initiation of ketamine infusions were significantly lower for treatment-resistant MDD patients that responded to ketamine as compared to non-responders (Moaddel et al., 2014; Murrough et al., 2014; Shiroma et al., 2014). Other potential predictors include higher body mass index (BMI), family history of alcohol abuse (i.e. first degree relatives), and no history of suicide attempts (Niciu et al., 2014). Furthermore, the dissociative side effects produced by ketamine at 40 min (as measured by the Clinician Administered Dissociative States Scale [CADSS]) are negatively correlated to the antidepressant effects of ketamine at 230 min and day seven (i.e. higher CADSS scores were associated with lower HDRS scores) (Luckenbaugh et al., 2014). In contrast, the relationship between peripheral concentrations of brain-derived neurotropic factor (BDNF) and ketamine responders is not fully understood. Several clinical studies have reported a negative correlation between patients that respond to ketamine treatment and BDNF concentrations such that reductions in depressive symptoms are associated with increased concentrations of peripheral BDNF (Duncan et al., 2013; Haile et al., 2014; Laje et al., 2012). Conversely, the plasma concentrations of BDNF were not associated the antidepressant effects of ketamine as there was no difference in BDNF levels between ketamine responders and non- responders (Machado-Vieira et al., 2009). Moreover, a single ketamine infusion did not change the amino acid neurotransmitter (i.e. glutamate, glutamine, and GABA) concentrations in the occipital cortex as measured by 1H-MRS (Valentine et al., 2011), suggesting that changes in the amino acid neurotransmitter concentrations in the occipital cortex are not associated with the antidepressant effects of ketamine. At the time of this review, there are more than 20 clinical trials evaluating the antidepressant effects of ketamine, which include studies focused on treatment-resistant MDD (e.g. NCT01920555 and NCT00088699), suicide risk (e.g. NCT02094898), and the potential biomarkers for ketamine responders (e.g. NCT02165449 and NCT01945047).
Other Glutamatergic Drugs for the Treatment of Major Depressive Disorder
Tables 5 provides a summary of the clinical trial results and the current status of the glutamatergic drugs described below for the treatment of MDD.
Table 5.
Clinical trial results and current status of glutamatergic drugs for the treatment of major depressive disorder.
| Drug | Primary Mechanism of Action | Study/reference | TRD | N | Study Design | Main Findings | Current Status/Clinical Trials |
|---|---|---|---|---|---|---|---|
| Memantine | Noncompetitive NMDA receptor antagonist | Zarate et al., 2006 | No | 26 | Randomized, double-blind, placebo-controlled, repeated dosing (5-20 mg/day) | No significant effects of treatment after 8 weeks | Phase 4: Treatment resistant geriatric depression in primary care. NCT01392287 |
| Lenze et al., 2012 | No | 27 | Randomized, double-blind, placebo-controlled, repeated dosing (10-20 mg/day) | No significant effects of treatment after 8 weeks | |||
| Smith et al., 2013 | Yes | 31 | Randomized, double-blind, placebo-controlled, add-on repeated dosing (5-20 mg/day) | No significant effects of treatment after 8 weeks | |||
| Muhonen et al., 2008 | No | 58 | Randomized, double-blind, active control (escitalopram), repeated dosing (5-20 mg/day); use patients with comorbid alcohol dependence | Improvement from 1 to 6 months; no difference between memantine and escitalopram | |||
| AZD6765 (Lanicemine) | Low trapping NMDA receptor open-channel blocker | Zarate et al., 2013 | Yes | 21 | Randomized, double-blind, crossover study, single infusion (150 mg) | Improvement from 80 to 110 min. No other improvement | AstraZeneca terminated the development of AZD6765 for MDD. |
| Sanacora et al., 2013 | Yes | 34 | Randomized, double-blind, placebo-controlled, single infusion (100 mg) | No significant effects of treatment | |||
| Sanacora et al., 2013 | Yes | 152 | Randomized, double-blind, placebo-controlled, add-on repeated dosing (100-150 mg; 3 infusions per week) | Improvement from week 3 to week 5 (2 weeks post last infusion) | |||
| CP-101,606 (traxoprodil) | NR2B subunit selective NMDA receptor antagonist | Preskorn et al., 2008 | Yes | 30 | Randomized, double-blind, placebo-controlled, add-on single infusion (0.75 mg/kg for 1hour and 0.15 for 6.5 hours) | 60% met response criteria; 33% met remission criteria on day 5 | No clinical trials listed. |
| MK-0657 | NR2B subunit selective NMDA receptor antagonist | Ibrahim et al., 2012 | Yes | 5 | Randomized, double-blind, placebo-controlled, repeated dosing (4-8 mg/day) | Inconsistent improvement from days 5 to 12 | Merck discontinued the manufacturing of MK-0657. Cerecor Inc. purchased the rights from Merck and will continue to develop MK-0657 for MDD. No clinical trials listed. |
| D-cycloserine | Glycine site partial agonist | Heresco-Levy et al., 2006 | Yes | 15 | Randomized, double-blind, placebo-controlled, add-on repeated dosing (250 mg/day) | No significant effects of treatment | Phase 4: Bipolar depression. NCT01833897 |
| Heresco-Levy et al., 2006 | Yes | 22 | Randomized, double-blind, placebo-controlled, add-on repeated dosing (titrated up to 1000 mg/day) | 7/13 met response criteria; 5/13 met remission criteria after 6 weeks of treatment | |||
| GLYX-13 | Glycine site partial agonist | Moskal et al., 2014 | No | 112 | Randomized, double-blind, placebo-controlled, single infusion (1-30 mg/kg) | Improvement from day 1 to day 7 |
Phase 1: Safety, tolerability and pharmacokinetics. NCT01014650 Phase 2: Inadequate/partial response to antidepressant for MDD. NCT01684163; NCT02192099 |
| NRX-1074 | Glycine site partial agonist | No published clinical trials |
Phase 1: Safety and Pharmacokinetics. NCT01856556 Phase 2: IV administration in MDD. NCT02067793 |
||||
| Riluzole | EAAT2 glutamate reuptake enhancer | Zarate et al., 2004 | Yes | 13 | Open-label, repeated oral dosing (50-200 mg/day) | Improvement from week 2 to week 6 | Phase 2: TRD. NCT01204918; NCT01703039; NCT00088699 |
| Sanacora et al., 2007 | Yes | 10 | Open-label, add-on repeated dosing (100 mg/day) | Improvement from week 1 to week 6 | |||
| Mathew et al., 2010 | Yes | 13 | Randomized, double-blind, placebo-controlled, relapse prevention (100-200 mg/day) | No significant effect of treatment for relapse prevention | |||
| Ibrahim et al., 2012 | Yes | 27 | Randomized, double-blind, placebo-controlled, relapse prevention (100-200 mg/day) | No significant effect of treatment for relapse prevention | |||
| Niciu et al., 2014 | Yes | 18 | Randomized, double-blind, placebo-controlled, ketamine non-responder (100-200 mg/day) | No significant effect of treatment in ketamine non-responders |
TRD, treatment-resistant depression; N, represents the number of participants to complete the study; MDD, major depressive disorder; NMDA, N-methyl-D-aspartate; EAAT2, excitatory amino acid transporter 2; IV, intravenous.
Noncompetitive NMDA receptor antagonists
Memantine, which was approved by the FDA for the treatment of Alzheimer's disease in 2013 and marketed by Forest Pharmaceuticals in the United States under the trade name Namenda® (Mount & Downton, 2006), is a selective low-affinity NMDA receptor antagonist that has been evaluated as a treatment for MDD. Unlike ketamine, memantine did not produce rapid or sustained antidepressant effects in clinical research. In fact, three clinical studies found that daily memantine (5-20 mg) was not superior to placebo for the treatment of MDD (Lenze et al., 2012; Smith et al., 2013; Zarate et al., 2006). A single clinical study found that daily memantine produced antidepressant effects in patients suffering from MDD comorbid with alcohol dependence, but it was not superior to the SSRI escitalopram (Muhonen, Lonngvist, Juva, & Alho, 2008). Memantine was generally well tolerated. Although ketamine and memantine possess similar binding affinity for the NMDA receptors, these NMDA antagonists do not produce similar antidepressant effects.
Low trapping NMDA receptor channel blocker
AstraZeneca originally developed the low affinity and low trapping NMDA receptor open-channel blocker AZD6765 (lanicemine) as a neuroprotective agent, but later decided to pursue lanicemine as a treatment for patients with treatment-resistant MDD with the goal of improving safety and tolerability as well as reducing the hallucinogenic (or psychotomimetic) side effects associated with ketamine. There are published results on three different Phase II clinical trials with lanicemine. In two clinical trials (NCT00491686 and NCT00986479), a single i.v. infusion of lanicemine failed to produce any meaningful antidepressant effects in treatment-resistant MDD patients (Sanacora et al., 2013; Zarate et al., 2013). Specifically, in the first clinical trial (NCT00491686) lanicemine (100 mg) failed to produce a statistically significant reduction in depressive symptoms, which was confounded by a large placebo effect (Sanacora et al., 2013). In the second clinical trial (NCT00986479), lanicemine (150 mg) produced rapid (at 80 min) antidepressant effects following a single infusion, but these antidepressant effects dissipated by 230 mins (Zarate et al., 2013). In a third clinical trial (NCT00781742), three weeks of repeated lanicemine treatment (100 or 150 mg) was tested as an adjunctive treatment to ongoing antidepressant treatment. Lanicemine (100 and 150 mg) produced antidepressant effects after two weeks of repeated dosing, and these antidepressant effects continued until week five of the study, which was two weeks after the last lanicemine i.v. infusion (Sanacora et al., 2013). In these three clinical trials, lanicemine was well tolerated and did not produce a significant change in psychotomimetic effects (Sanacora et al., 2013; Zarate et al., 2013). However, by the end of 2013, AstraZeneca discontinued projects for lanicemine as a treatment for treatment-resistant MDD citing lack of efficacy in Phase II clinical trials.
NR2B subunit selective NMDA receptor antagonists
Pfizer developed the potent and NR2B subunit selective NMDA receptor antagonist CP-101,606 (traxoprodil) as a neuroprotectant for head injury and stroke, but later it was evaluated as an adjunctive treatment for patients with treatment-resistant MDD (Butler et al., 1998). The selectivity of traxoprodil for NR2B subunits of the NMDA receptor complex was believed to reduce the psychotomimetic effects that have been associated with the nonspecific NMDA receptor antagonist ketamine. A single eight hour infusion of traxoprodil (0.75 mg/kg per hour for 1.5 hours, then 0.05 mg/kg per hour for 6.5 hours) was evaluated as an adjunctive treatment to paroxetine (40.0 mg/day) in a double-blind between subjects design clinical study. Traxoprodil produced rapid (five days) antidepressant effects with 60% of the patients meeting response criteria. However, traxoprodil produced psychotomimetic effects in four of the nine patients that met response criteria (Preskorn et al., 2008). Although a phase II clinical trial was conducted in 2005-2006 to evaluate the effects of monotherapy traxoprodil in patients with treatment-resistant depression (NCT00163059), to date, there are no published results from these clinical trials.
Recently, the NR2B subunit selective NMDA receptor antagonist MK-0657, which was developed by Merck for the treatment of Parkinson's disease, was the first oral formulation NMDA receptor antagonist to be tested in treatment-resistant MDD patients. This was a double- blind, placebo-controlled study in which the patients received either MK-0657 (4.0-8.0 mg/d) or placebo for 12 days. MK-0657 produced inconsistent antidepressant effects from day 5 to day 12 as measured by HDRS and BDI, receptively; however, MK-0657 failed to produce a significant reduction in depression symptoms as measured by Montgomery-Asberg Depression Rating Scale (MADRS). MK-0657 did not produce psychotomimetic or adverse side effects. One possible explanation for the inconsistent results is that the study was terminated after only five patients completed both phases of the study. Early termination of the study was due to recruitment challenges and Merck discontinued the manufacturing of the compound (Ibrahim et al., 2012). In April 2013, the neuroscience biotech company Cerecor Inc. announced that it had acquired exclusive rights from Merck to develop and commercialize MK-0657 for all human indications. Cerecor Inc. will continue to pursue MK-0657 as a rapid treatment for depression as well as other central nervous system disorders.
Partial agonist at the glycine binding site of NMDA receptors
D-cycloserine, which is a broad spectrum antibiotic used to treat tuberculosis and marketed by Eli Lilly under the trade name Seromycin®, has been evaluated in patients with treatment-resistant MDD because of the pharmacological actions produced by D-cycloserine at the NMDA receptor. D-cycloserine binds to the glycine binding site on the NMDA receptor complex and pharmacological functions as a partial agonist. Typically, partial agonists will produce agonist effects at low doses, but will produce antagonist effects at high doses. In 2006, the antidepressant effects of adjunctive D- cycloserine (250 mg/day) treatment was evaluated in a double-blind 6 week crossover study. While D-cycloserine was well tolerated and did not produce psychotomimetic effects, D- cycloserine failed to produce antidepressant effects (Heresco-Levy et al., 2006). In a recent study, a high dose (titrated up to 1000 mg/day) adjunctive treatment of D-cycloserine produced antidepressant effects after six weeks of treatment (or two week after the final 1000 mg/day was reached) (Heresco-Levy et al., 2013). To date, there are no published studies evaluating D- cycloserine as a monotherapy for the treatment of MDD. At the time of this review, participants with bipolar depression are being recruited for a Phase IV clinical trial to evaluate D-cycloserine for relapse prevention in patients that response to ketamine infusions (NCT01833897).
Naurex Inc. developed a novel compound, GLYX-13, which is a tetrapeptide (TPPT- amide), for the treatment of MDD with the goal of producing rapid antidepressant effects without producing psychotomimetic side effects. Unlike the NMDA receptor antagonists and channel blockers, GLYX-13 binds to the glycine-site of the NMDA receptor and acts as a functional partial agonist and this difference in pharmacological action is believed to reduce psychotomimetic side effects (Burgdorf et al., 2013; Moskal et al., 2005). In a Phase II clinical study comprised of 112 MDD patients across 12 United States centers, GLYX-13 produced rapid and sustained antidepressant effects following a single infusion (5.0-10.0 mg/kg; 3-15 min infusion), and, most importantly, did not produce psychotomimetic effects. Specifically, the antidepressant effects of GLYX-13 were apparent at the end of day one and persisted until day seven following the single infusion (Moskal et al., 2014). At the time of this review, a Phase II double-blind, placebo control, multi-dose clinical trial is underway (NCT01684163).
In addition to GLYX-13, Naurex Inc. has developed another novel compound NRX- 1074, which is similar to GLYX-13; however, NRX-1074 is an orally bioavailable compound and is more potent than GLYX-13. In Phase I clinical trials, NRX-1074 was well tolerated (Naurex Inc., 2014). At the time of this review, Naurex Inc. is recruiting for Phase I safety and pharmacokinetics (NCT01856556) and Phase II multi-dose single infusion for patients with MDD (NCT02067793) clinical trials.
Excitatory amino acid transporter 2 (EAAT2) enhancer
The EAAT2 glutamate reuptake enhancer riluzole, which is approved by the FDA for the treatment of amyotrophic lateral sclerosis (ALS) and marketed by Sanofi-Aventis under the trade name (Rilutek®), has been evaluated under a number of conditions for the treatment of MDD including monotherapy, adjunctive therapy, and ketamine relapse prevention. Riluzole was evaluated as a treatment for MDD because of its duel pharmacological in the glutamatergic system. Specifically, riluzole increases the reuptake of glutamate into astrocytes via EAAT2 (Azbill, Mu, & Springer, 2000) and inhibits glutamate release (Wang, Wang, & Wang, 2004), which produces pharmacological effects similar to the effects NMDA receptor antagonists such that riluzole can reduce NMDA receptor activation by decreasing the concentrations of glutamate available to bind to postsynaptic NMDA receptors. The antidepressant effects of riluzole were first evaluated in an open-label clinical study in patients with treatment-resistant MDD. In the open-label clinical study, daily riluzole (mean dose of 169 mg/day) produced antidepressant effects on weeks three through week six as compared to baseline MADRS score. There was not a placebo control in this study (Zarate et al., 2004). In a small scale clinical study (n = 10), adjunctive riluzole (100 mg/day) treatment produced a rapid decrease in depressive symptoms from week one through week six as compared to baseline HDRS scores. There was not a placebo control in this study (Sanacora et al., 2007). Two double-blind clinical studies evaluated riluzole as relapse prevention for patients that response to a single infusion of ketamine; however, both studies found that riluzole was no more effective than placebo for ketamine relapse prevention (Ibrahim et al., 2012; Mathew et al., 2010). Moreover, riluzole did not produce antidepressant effects in patients that did not response to ketamine infusions (i.e. ketamine non-responders) (Niciu et al., 2014). In general, riluzole was well tolerated in these studies and psychotomimetic effects were not observed. At the time of this review, two Phase II double-blind, placebo control, adjunctive treatment clinical trial are underway for patients with treatment-resistant MDD (NCT01204918 and NCT01703039).
Conclusion
While a majority of MDD patients respond to monoamine based pharmacological treatments, there is a large subgroup of patients that do not response to these treatments (i.e. treatment-resistant patients). Since the first proof-of-concept study in 2000, researchers have evaluated the glutamatergic system and pursued glutamatergic-based drugs as novel mechanism for the treatment of these patients with some success. Specifically, the noncompetitive NMDA receptor antagonist ketamine produces rapid and sustained antidepressant effects in open-label, double-blind, and active-placebo clinical trials, but it also produces psychotomimetic effects (albeit, of short duration). Several potential biomarkers, such as baseline D-serine concentrations and neurocognitive baseline performance, have been identified as predictors for the patients that respond to ketamine treatment. Additionally, the glycine-site partial agonist GLYX-13 has produced rapid and sustained antidepressant effects in Phase II clinical trials with no significant psychotomimetic effects. Other glutamatergic-based drugs (e.g. memantine and MK-0657) have failed to produce meaningful antidepressant effects in MDD patients. Tables 4 and 5 provide a summary of the clinical trial results and the current status of ketamine and other glutamatergic drugs for the treatment of MDD. Currently, preclinical research is evaluating the pharmacological and intracellular effects that are responsible for the antidepressant effects of ketamine (e.g. Li et al., 2010; Autry et al., 2011), which will aid the development of novel glutamatergic drugs (for an excellent review on the preclinical findings see Browne & Lucki, 2013). In conclusion, the clinical findings to date are encouraging and support the continued search for and the development of novel compounds, such as ketamine and GLYX-13, which target glutamatergic mechanisms for the treatment of MDD and for treatment-resistant depression.
Acknowledgments
Todd M. Hillhouse received funding support by the National Institute of Drug Abuse of the National Institutes of Health under Award Number T32DA07268 while working on this publication. Joseph H. Porter has previously received grant support from Eli Lilly and Company, H. Lundbeck A/S, Pfizer, Inc., Orexigen Therapeutics, Inc., and Roche Palo Alto LLC and currently has a research contract from Lundbeck A/S; however, Lundbeck A/S had no financial or intellectual role in the present review. We would like to thank Drs. Steve Negus, Adam Prus, and James McCullough Jr. for their guidance on portions of this review. Additionally, we thank Kevin McKee for his graphical/illustrative contribution to the glutamatergic system and glutamatergic drug targets image (Figure 1).
Footnotes
All authors made substantial contributions and approved the final version of the manuscript.
Disclosures: There are no conflicts of interest to declare.
Contributor Information
Todd M. Hillhouse, the Department of Psychology at Virginia Commonwealth University at the time this review was written and is now at the University of Michigan in the Department of Pharmacology
Joseph H. Porter, the Department of Psychology at Virginia Commonwealth University
References
- aan het Rot M, Collins KA, Murrough JW, Perez AM, Reich DL, Charney DS, Mathew SJ. Safety and efficacy of repeated-dose intravenous ketamine for treatment-resistant depression. Biological Psychiatry. 2010;67:139–145. doi: 10.1016/j.biopsych.2009.08.038. [DOI] [PubMed] [Google Scholar]
- Alam MY, Jacobsen PL, Chen Y, Serenko M, Mahableshwarkar AR. Safety, tolerability, and efficacy of vortioxetine (Lu AA21004) in major depressive disorder: Results of an open-label, flexible-dose, 52-week extension study. International Clinical Psychopharmacology. 2014;29:36–44. doi: 10.1097/YIC.0000000000000010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altamura AC, Mauri CM, Ferrara A, Moro RA, D'Andrea G, Zamberlan F. Plasma and platelet excitatory amino acids in psychiatric disorders. American Journal of Psychiatry. 1993;150:1731–1733. doi: 10.1176/ajp.150.11.1731. [DOI] [PubMed] [Google Scholar]
- American Psychiatric Association. Diagnostic and statistical manual of mental disorders. 5th. Arlington, VA: American Psychiatric Publishing; 2013. [Google Scholar]
- Auer DP, Pütz B, Kraft E, Lipinski B, Schill J, Holsboer F. Reduced glutamate in the anterior cingulate cortex in depression: an in vivo proton magnetic resonance spectroscopy study. Biological Psychiatry. 2000;47:305–313. doi: 10.1016/s0006-3223(99)00159-6. [DOI] [PubMed] [Google Scholar]
- Autry AE, Adachi M, Nosyreva E, Na ES, Los MF, Peng-fei C, et al. Monteggia LM. NMDA receptor blockade at rest triggers rapid behavioural antidepressant responses. Nature. 2011;475:91–95. doi: 10.1038/nature10130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Azbill RD, Mu X, Springer JE. Riluzole increases high-affinity glutamate uptake in rat spinal cord synaptosomes. Brain Research. 2000;871:175–180. doi: 10.1016/s0006-8993(00)02430-6. doi: http://dx.doi.org/10.1016/S0006-8993(00)02430-6. [DOI] [PubMed] [Google Scholar]
- Bang-Andersen B, Ruhland T, Jørgensen M, Smith G, Frederiksen K, Jensen KG, et al. Stensbøl TB. Discovery of 1-[2-(2,4- Dimethylphenylsulfanyl)phenyl]piperazine (Lu AA21004): A novel multimodal compound for the treatment of major depressive disorder. Journal of Medicinal Chemistry. 2011;54:3206–3221. doi: 10.1021/jm101459g. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Kristiansen LV, Oni-Orisan A, McCullumsmith RE, Meador-Woodruff JH. Abnormal glutamate receptor expression in the medial temporal lobe in schizophrenia and mood disorders. Neuropsychopharmacology. 2007;32:1888–1902. doi: 10.1038/sj.npp.1301312. [DOI] [PubMed] [Google Scholar]
- Beneyto M, Meador-Woodruff JH. Lamina-Specific Abnormalities of NMDA Receptor-Associated Postsynaptic Protein Transcripts in the Prefrontal Cortex in Schizophrenia and Bipolar Disorder. Neuropsychopharmacology. 2008;33(9):2175–2186. doi: 10.1038/sj.npp.1301604. doi: http://www.nature.com/npp/journal/v33/n9/suppinfo/1301604s1.html. [DOI] [PubMed] [Google Scholar]
- Berman RM, Cappiello A, Anand A, Oren DA, Heninger GR, Charney DS, Krystal JH. Antidepressant effects of ketamine in depressed patients. Biological Psychiatry. 2000;47:351–354. doi: 10.1016/s0006-3223(99)00230-9. [DOI] [PubMed] [Google Scholar]
- Berman RM, Sanacora G, Anand A, Roach LM, Fasula MK, Finkelstein CO, et al. Charney DS. Monoamine depletion in unmedicated depressed subjects. Biological Psychiatry. 2002;51:469–473. doi: 10.1016/s0006-3223(01)01285-9. doi: http://dx.doi.org/10.1016/S0006-3223(01)01285-9. [DOI] [PubMed] [Google Scholar]
- Bernard R, Kerman IA, Thompson RC, Jones EG, Bunney WE, Barchas JD, et al. Watson SJ. Altered expression of glutamate signaling, growth factor, and glia genes in the locus coeruleus of patients with major depression. Molecular Psychiatry. 2011;16:634–646. doi: 10.1038/mp.2010.44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Berk M, Plein H, Ferreira D. Platelet glutamate receptor supersensitivity in major depressive disorder. Clinical Neuropharmacology. 2001;24:129–132. doi: 10.1097/00002826-200105000-00002. [DOI] [PubMed] [Google Scholar]
- Billett EE. Monoamine oxidase (MAO) in human peripheral tissues. Neurotoxicology. 2004;25(1–2):139–148. doi: 10.1016/S0161-813X(03)00094-9. doi: http://dx.doi.org/10.1016/S0161-813X(03)00094-9. [DOI] [PubMed] [Google Scholar]
- Birnbaum HG, Kessler RC, Kelley D, Ben-Hamadi R, Joish VN, Greenberg PE. Employer burden of mild, moderate, and severe major depressive disorder: mental health services utilization and costs, and work performance. Depression & Anxiety. 2010;27:78–89. doi: 10.1002/da.20580. [DOI] [PubMed] [Google Scholar]
- Block W, Träber F, von Widdern O, Metten M, Schild H, Maier W, et al. Jessen F. Proton MR spectroscopy of the hippocampus at 3 T in patients with unipolar major depressive disorder: correlates and predictors of treatment response. The International Journal of Neuropsychopharmacology. 2009;12:415–422. doi: 10.1017/S1461145708009516. [DOI] [PubMed] [Google Scholar]
- Boulenger JP, Loft H, Florea I. A randomized clinical study of Lu AA21004 in the prevention of relapse in patients with major depressive disorder. Journal of Psychopharmacology. 2012;26:1408–1416. doi: 10.1177/0269881112441866. [DOI] [PubMed] [Google Scholar]
- Boulenger JP, Loft H, Olsen CK. Efficacy and safety of vortioxetine (Lu AA21004), 15 and 20 mg/day: A randomized, double-blind, placebo-controlled, duloxetine-referenced study in the acute treatment of adult patients with major depressive disorder. International Clinical Psychopharmacology. 2014;29:138–149. doi: 10.1097/YIC.0000000000000018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bresink I, Danysz W, Parsons CG, Mutschler E. Different binding affinities of NMDA receptor channel blockers in various brain regions—Indication of NMDA receptor heterogeneity. Neuropharmacology. 1995;34:533–540. doi: 10.1016/0028-3908(95)00017-z. [DOI] [PubMed] [Google Scholar]
- Browne CA, Lucki I. Antidepressant effects of ketamine: Mechanisms underlying fast-acting novel antidepressants. Frontiers in Pharmacology. 2013;4:1–18. doi: 10.3389/fphar.2013.00161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bunney WE, Jr, Davis JM. Norepinephrine in depressive reactions: A review. Archives of General Psychiatry. 1965;13:483–494. doi: 10.1001/archpsyc.1965.01730060001001. [DOI] [PubMed] [Google Scholar]
- Burgdorf J, Zhang Xl, Nicholson KL, Balster RL, David Leander J, Stanton PK, et al. Moskal JR. GLYX-13, a NMDA receptor glycine-site functional partial agonist, induces antidepressant-like effects without ketamine-like side effects. Neuropsychopharmacology. 2013;38:729–742. doi: 10.1038/npp.2012.246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butler TW, Blake JF, Bordner J, Butler P, Chenard BL, Collins MA, et al. Zhao D. (3R,4S)-3-[4-(4-Fluorophenyl)-4-hydroxypiperidin-1-yl]chroman-4,7-diol: A conformationally restricted analogue of the NR2B subtype-selective NMDA antagonist (1S,2S)-1-(4-Hydroxyphenyl)-2- (4-hydroxy-4-phenylpiperidino)-1-propanol. Journal of Medicinal Chemistry. 1998;41:1172–1184. doi: 10.1021/jm9707986. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Dreshfield-Ahmad LJ, Threlkeld PG, Shaw JL, Thompson L, Nelson DL, et al. Wong DT. Comparative affinity of duloxetine and venlafaxine for serotonin and norepinephrine transporters in vitro and in vivo, human serotonin receptor subtypes, and other neuronal receptors. Neuropsychopharmacology. 2001;25:871–880. doi: 10.1016/S0893-133X(01)00298-6. doi: http://dx.doi.org/10.1016/S0893-133X(01)00298-6. [DOI] [PubMed] [Google Scholar]
- Bymaster FP, Katner JS, Nelson DL, Hemrick-Luecke SK, Threlkeld PG, Heiligenstein JH, et al. Perry KW. Atomoxetine increases extracellular levels of norepinephrine and dopamine in prefrontal cortex of rat: A potential mechanism for efficacy in attention deficit/hyperactivity disorder. Neuropsychopharmacology. 2002;27:699–711. doi: 10.1016/S0893-133X(02)00346-9. [DOI] [PubMed] [Google Scholar]
- Campbell I, Marangos P, Parma A, Garrick N, Murphy D. Localization of monoamine oxidases A and B in primate brains relative to neuron-specific and non- neuronal enolases. Neurochemical Research. 1982;7:657–666. doi: 10.1007/bf00965519. [DOI] [PubMed] [Google Scholar]
- Chen TY, Tzeng NS. Aripiprazole: A dopamine modulator that mimics methylphenidate in producing faster antidepressant effects. Medical Hypotheses. 2013;81:183–185. doi: 10.1016/j.mehy.2013.05.009. doi: http://dx.doi.org/10.1016/j.mehy.2013.05.009. [DOI] [PubMed] [Google Scholar]
- Chilukuri H, Pothula Reddy N, Mohan Pathapati R, Nagesha Manu A, Jollu S, Basha Shaik A. Acute antidepressant effects of intramuscular versus intravenous ketamine. Indian Journal of Psychological Medicine. 2014;36:71–76. doi: 10.4103/0253-7176.127258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choudary PV, Molnar M, Evans SJ, Tomita H, Li JZ, Vawter MP, et al. Jones EG. Altered cortical glutamatergic and GABAergic signal transmission with glial involvement in depression. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:15653–15658. doi: 10.1073/pnas.0507901102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chandley MJ, Szebeni A, Szebeni K, Crawford JD, Stockmeier CA, Turecki G, et al. Ordway GA. Elevated gene expression of glutamate receptors in noradrenergic neurons from the locus coeruleus in major depression. The International Journal of Neuropsychopharmacology. 2014:1–10. doi: 10.1017/S1461145714000662. [DOI] [PubMed] [Google Scholar]
- Charney DS. Monoamine dysfunction and the pathophysiology and treatment of depression. Journal of Clinical Psychiatry. 1998;59(Suppl):11–14. [PubMed] [Google Scholar]
- Clayton AH, Pradko JF, Croft HA, Montano CB, Leadbetter RA, Bolden-Watson C, et al. Metz A. Prevalence of sexual dysfunction among newer antidepressants. Journal of Clinical Psychiatry. 2002;63:357–366. doi: 10.4088/jcp.v63n0414. [DOI] [PubMed] [Google Scholar]
- Cusack B, Nelson A, Richelson E. Binding of antidepressants to human brain receptors: focus on newer generation compounds. Psychopharmacology. 1994;114:559–565. doi: 10.1007/bf02244985. [DOI] [PubMed] [Google Scholar]
- Delgado PL. Depression: The case for a monoamine deficiency. Journal of Clinical Psychiatry. 2000;61(Supp 6):7–11. [PubMed] [Google Scholar]
- Delgado PL, Price LH, Miller HL, et al. Serotonin and the neurobiology of depression: Effects of tryptophan depletion in drug-free depressed patients. Archives of General Psychiatry. 1994;51:865–874. doi: 10.1001/archpsyc.1994.03950110025005. [DOI] [PubMed] [Google Scholar]
- DiazGranados N, Ibrahim L, Brutsche NE, Ameli R, Henter ID, Luckenbaugh DA, et al. Zarate CA., Jr A randomized add-on trial of an n-methyl-d-aspartate antagonist in treatment-resistant bipolar depression. Archives of General Psychiatry. 2010;67:793–802. doi: 10.1001/archgenpsychiatry.2010.90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Domino EF. History of modern psychopharmacology: A personal view with an emphasis on antidepressants. Psychosomatic Medicine. 1999;61:591–598. doi: 10.1097/00006842-199909000-00002. [DOI] [PubMed] [Google Scholar]
- Domino EF. Taming the Ketamine Tiger. Anesthesiology. 2010;113:678–684. doi: 10.1097/ALN.0b013e3181ed09a2. [DOI] [PubMed] [Google Scholar]
- Drug Enforcement Administration. Ketamine. Springfield, VA: US Department of Justice, Drug Enforcement Administration; 2013. [Google Scholar]
- du Jardin KG, Jensen JB, Sanchez C, Pehrson AL. Vortioxetine dose- dependently reverses 5-HT depletion-induced deficits in spatial working and object recognition memory: A potential role for 5-HT1A receptor agonism and 5-HT3 receptor antagonism. European Neuropsychopharmacology. 2014;24:160–171. doi: 10.1016/j.euroneuro.2013.07.001. doi: http://dx.doi.org/10.1016/j.euroneuro.2013.07.001. [DOI] [PubMed] [Google Scholar]
- Duncan WCJ, Sarasso S, Ferrarelli F, Selter J, Riedner BA, Hejazi NS, et al. Zarate CAJ. Concomitant BDNF and sleep slow wave changes indicate ketamine- induced plasticity in major depressive disorder. The International Journal of Neuropsychopharmacology. 2013;16:301–311. doi: 10.1017/S1461145712000545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faguis J, Osterman PO, Siden A, Wiholm BE. Guillain-Barre syndrome following zimeldine treatment. Journal of Neurology, Neurosurgery, and Psychiatry. 1985;48:65–69. doi: 10.1136/jnnp.48.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fangmann P, Assion HJ, Juckel G, Gonzalez CA, Lopez-Munoz F. Half a century of antidepressant drugs: On the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part II: Tricyclics, and tetracyclics. Journal of Clinical Psychopharmacology. 2008;28:1–4. doi: 10.1097/jcp.0b013e3181627b60. [DOI] [PubMed] [Google Scholar]
- Fava M. Diagnosis and definition of treatment-resistant depression. Biological Psychiatry. 2003;53:649–659. doi: 10.1016/s0006-3223(03)00231-2. [DOI] [PubMed] [Google Scholar]
- Fava M, Davidson KG. Definition and epidemiology of treatment-resistant depression. The Psychiatric Clinics of North America. 1996;19:179–200. doi: 10.1016/s0193-953x(05)70283-5. [DOI] [PubMed] [Google Scholar]
- Fava M, Rush JA, Thase ME, Clayton A, Stahl SM, Pradko JF, Johnston AJ. 15 years of clinical experience with bupropion HCl: From bupropion to bupropion SR to bupropion XL. Primary Care Companion to The Journal of Clinical Psychiatry. 2005;7:106–113. doi: 10.4088/pcc.v07n0305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fava M, Rush AJ, Trivedi MH, Nierenberg AA, Thase ME, Sackeim HA, et al. Kupfer DJ. Background and rationale for the sequenced treatment alternatives to relieve depression (STAR*D) study. The Psychiatric Clinics of North America. 2003;26:457–494. doi: 10.1016/s0193-953x(02)00107-7. [DOI] [PubMed] [Google Scholar]
- Feighner JP, Hendrickson G, Miller LL, Stern WC. Double-blind comparison of doxepin versus bupropion in outpatients with a major depressive disorder. Journal of Clinical Psychopharmacology. 1986;6:27–32. [PubMed] [Google Scholar]
- Feighner JP, Meredith CH, Stern WC, Hendrickson G, Miller LL. A double-blind study of bupropion and placebo in depression. American Journal of Psychiatry. 1984;141:525–529. doi: 10.1176/ajp.141.4.525. [DOI] [PubMed] [Google Scholar]
- Feyissa AM, Chandran A, Stockmeier CA, Karolewicz B. Reduced levels of NR2A and NR2B subunits of NMDA receptor and PSD-95 in the prefrontal cortex in major depression. Progress in Neuro-Psychopharmacology and Biological Psychiatry. 2009;33:70–75. doi: 10.1016/j.pnpbp.2008.10.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fox HH, Gibas JT. Synthetic tuberculostats. VII monoalkyl derivatives of isonicotinylhydrazine. Journal of Organic Chemistry. 1953;18:994–1002. [Google Scholar]
- Garakani A, Martinez JM, Yehuda R, Gorman JM. Cerebrospinal fluid levels of glutamate and corticotropin releasing hormone in major depression before and after treatment. Journal of Affective Disorders. 2013;146:262–265. doi: 10.1016/j.jad.2012.06.037. doi: http://dx.doi.org/10.1016/j.jad.2012.06.037. [DOI] [PubMed] [Google Scholar]
- Ghasemi M, Kazemi MH, Yoosefi A, Ghasemi A, Paragomi P, Amini H, Afzali MH. Rapid antidepressant effects of repeated doses of ketamine compared with electroconvulsive therapy in hospitalized patients with major depressive disorder. Psychiatry Research. 2014;215:355–361. doi: 10.1016/j.psychres.2013.12.008. doi: http://dx.doi.org/10.1016/j.psychres.2013.12.008. [DOI] [PubMed] [Google Scholar]
- Gibb A, Deeks E. Vortioxetine: First global approval. Drugs. 2014;74:135–145. doi: 10.1007/s40265-013-0161-9. [DOI] [PubMed] [Google Scholar]
- Greenberg P, Kessler R, Birnbaum H, Leong S, Lowe S, Berglund P, Corey-Lisle P. The economic burden of depression in the United States: How did it change between 1990 and 2000? Journal of Clinical Psychiatry. 2003;64:1465–1475. doi: 10.4088/jcp.v64n1211. [DOI] [PubMed] [Google Scholar]
- Haile CN, Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Foulkes A, et al. Mathew SJ. Plasma brain derived neurotrophic factor (BDNF) and response to ketamine in treatment-resistant depression. The International Journal of Neuropsychopharmacology. 2014;17:331–336. doi: 10.1017/S1461145713001119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hashimoto K, Sawa A, Iyo M. Increased levels of glutamate in brains from patients with mood disorders. Biological Psychiatry. 2007;62:1310–1316. doi: 10.1016/j.biopsych.2007.03.017. [DOI] [PubMed] [Google Scholar]
- Hasler G, van der Veen J, Tumonis T, Meyers N, Shen J, Drevets WC. Reduced prefrontal glutamate/glutamine and γ-aminobutyric acid levels in major depression determined using proton magnetic resonance spectroscopy. Archives of General Psychiatry. 2007;64:193–200. doi: 10.1001/archpsyc.64.2.193. [DOI] [PubMed] [Google Scholar]
- Heninger GR, Delgado PL, Charney DS. The revised monoamine theory of depression: a modulatory role for monoamines, based on new findings from monoamine depletion experiments in humans. Pharmacopsychiatry. 1996;29:2–11. doi: 10.1055/s-2007-979535. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Gelfin G, Bloch B, Levin R, Edelman S, Javitt DC, Kremer I. A randomized add-on trial of high-dose d-cycloserine for treatment-resistant depression. The International Journal of Neuropsychopharmacology. 2013;16:501–506. doi: 10.1017/S1461145712000910. [DOI] [PubMed] [Google Scholar]
- Heresco-Levy U, Javitt DC, Gelfin Y, Gorelik E, Bar M, Blanaru M, Kremer I. Controlled trial of d-cycloserine adjuvant therapy for treatment-resistant major depressive disorder. Journal of Affective Disorders. 2006;93(1–3):239–243. doi: 10.1016/j.jad.2006.03.004. doi: http://dx.doi.org/10.1016/j.jad.2006.03.004. [DOI] [PubMed] [Google Scholar]
- Hirota K, Hashimoto Y, Lambert DG. Interaction of intravenous anesthetics with recombinant human M1-M3 muscarinic receptors expressed in Chinese hamster ovary cells. Anesthesia & Analgesia. 2002;95:1607–1610. doi: 10.1213/01.ane.0000037146.52925.d5. [DOI] [PubMed] [Google Scholar]
- Hirschfeld RMA. History and evolution of the monoamine hypothesis depression. Journal of Clinical Psychiatry. 2000;61(Supp 6):4–6. [PubMed] [Google Scholar]
- Holly EN, LaCrosse AL, Hillhouse TM. Review of Group I and Group II metabotropic glutamate receptors, and their role in the pathophysiology of major depressive disorder. In: Foster OM, editor. Metabotropic Glutamate Receptors: Molecular Mechanisms, Role in Neurological Disorders, and Pharmacological Effects. Hauppauge, NY: Nova Science Publishers; 2014. [Google Scholar]
- Ibrahim L, DiazGranados N, Franco-Chaves J, Brutsche N, Henter ID, Kronstein P, et al. Zarate CA., Jr Course of improvement in depressive symptoms to a single intravenous infusion of ketamine vs add-on riluzole: Results from a 4-week, double- blind, placebo-controlled study. Neuropsychopharmacology. 2012;37:1526–1533. doi: 10.1038/npp.2011.338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim L, DiazGranados N, Jolkovsky L, Brutsche N, Luckenbaugh DA, Herring WJ, et al. Zarate CA., Jr A randomized, placebo-controlled, crossover pilot trial of the oral selective NR2B antagonist MK-0657 in patients with treatment-resistant major depressive disorder. Journal of Clinical Psychopharmacology. 2012;32:551–557. doi: 10.1097/JCP.0b013e31825d70d6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Iglewicz A. Oral ketamine for the rapid treatment of depression and anxiety in patients receiving hospice care. Journal of Palliative Medicine. 2010;13:903–908. doi: 10.1089/jpm.2010.9808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irwin SA, Iglewicz A, Nelesen RA, Lo JY, Carr CH, Romero SD, Lloyd LS. Daily oral ketamine for the treatment of depression and anxiety in patients receiving hospice care: A 28-day open-label proof-of-concept trial. Journal of Palliative Medicine. 2013;16:958–965. doi: 10.1089/jpm.2012.0617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jensen JB, du Jardin KG, Song D, Budac D, Smagin G, Sanchez C, Pehrson AL. Vortioxetine, but not escitalopram or duloxetine, reverses memory impairment induced by central 5-HT depletion in rats: Evidence for direct 5-HT receptor modulation. European Neuropsychopharmacology. 2014;24:148–159. doi: 10.1016/j.euroneuro.2013.10.011. doi: http://dx.doi.org/10.1016/j.euroneuro.2013.10.011. [DOI] [PubMed] [Google Scholar]
- Jordan S, Chen R, Fernalld R, Johnson J, Regardie K, Kambayashi J, et al. Kikuchi T. In vitro biochemical evidence that the psychotomimetics phencyclidine, ketamine and dizocilpine (MK-801) are inactive at cloned human and rat dopamine D2 receptors. European Journal of Pharmacology. 2006;540(1–3):53–56. doi: 10.1016/j.ejphar.2006.04.026. [DOI] [PubMed] [Google Scholar]
- Kapur S, Seeman P. NMDA receptor antagonists ketamine and PCP have direct effects on the dopamine D2 and serotonin 5-HT2 receptors implications for models of schizophrenia. Molecular Psychiatry. 2002;7:837–844. doi: 10.1038/sj.mp.4001093. [DOI] [PubMed] [Google Scholar]
- Karolewicz B, Stockmeier CA, Ordway GA. Elevated levels of the NR2C subunit of the NMDA receptor in the locus coeruleus in depression. Neuropsychopharmacology. 2005;30:1557–1567. doi: 10.1038/sj.npp.1300781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Karolewicz B, Szebeni K, Gilmore T, Maciag D, Stockmeier CA, Ordway GA. Elevated levels of NR2A and PSD-95 in the lateral amygdala in depression. The International Journal of Neuropsychopharmacology. 2009;12:143–153. doi: 10.1017/S1461145708008985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kessler RC, Berglund P, Demler O, Jin R, Koretz D, Merikangas KR, et al. Wang PS. The epidemiology of major depressive disorder: Results from the national comorbidity survey replication (NCS-R) JAMA: The Journal of the American Medical Association. 2003;289:3095–3105. doi: 10.1001/jama.289.23.3095. [DOI] [PubMed] [Google Scholar]
- Kessler RC, Petukhova M, Sampson NA, Zaslavsky AM, Wittchen HU. Twelve-month and lifetime prevalence and lifetime morbid risk of anxiety and mood disorders in the United States. International Journal of Methods in Psychiatric Research. 2012;21:169–184. doi: 10.1002/mpr.1359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kew JC, Kemp J. Ionotropic and metabotropic glutamate receptor structure and pharmacology. Psychopharmacology (Berl) 2005;179:4–29. doi: 10.1007/s00213-005-2200-z. [DOI] [PubMed] [Google Scholar]
- Kirshner N. Uptake of Catecholamines by a Particulate Fraction of the Adrenal Medulla. Journal of Biological Chemistry. 1962;237(7):2311–2317. [PubMed] [Google Scholar]
- Küçükibrahimoğlu E, Saygın M, Çalışkan M, Kaplan O, Ünsal C, Gören MZ. The change in plasma GABA, glutamine and glutamate levels in fluoxetine- or S- citalopram-treated female patients with major depression. European Journal of Clinical Pharmacology. 2009;65:571–577. doi: 10.1007/s00228-009-0650-7. [DOI] [PubMed] [Google Scholar]
- Laje G, Lally N, Mathews D, Brutsche N, Chemerinski A, Akula N, et al. Zarate C., Jr Brain-Derived Neurotrophic Factor Val66Met Polymorphism and Antidepressant Efficacy of Ketamine in Depressed Patients. Biological Psychiatry. 2012;72:e27–e28. doi: 10.1016/j.biopsych.2012.05.031. doi: http://dx.doi.org/10.1016/j.biopsych.2012.05.031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lally N, Nugent AC, Luckenbaugh DA, Ameli R, Roiser JP, Zarate CA. Anti-anhedonic effect of ketamine and its neural correlates in treatment-resistant bipolar depression. Translational Psychiatry. 2014;4:e469. doi: 10.1038/tp.2014.105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lapidus KAB, Levitch CF, Perez AM, Brallier JW, Parides MK, Soleimani L, et al. Murrough JW. A randomized controlled trial of intranasal ketamine in major depressive disorder. Biological Psychiatry. 2014:1–7. doi: 10.1016/j.biopsych.2014.03.026. doi: http://dx.doi.org/10.1016/j.biopsych.2014.03.026. [DOI] [PMC free article] [PubMed]
- Lara DR, Bisol LW, Munari LR. Antidepressant, mood stabilizing and procognitive effects of very low dose sublingual ketamine in refractory unipolar and bipolar depression. The International Journal of Neuropsychopharmacology. 2013;16:2111–2117. doi: 10.1017/S1461145713000485. [DOI] [PubMed] [Google Scholar]
- Lee S, Jeong J, Kwak Y, Park SK. Depression research: where are we now? Molecular Brain. 2010;3:1–10. doi: 10.1186/1756-6606-3-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lenze EJ, Skidmore ER, Begley AE, Newcomer JW, Butters MA, Whyte EM. Memantine for late-life depression and apathy after a disabling medical event: a 12-week, double-blind placebo-controlled pilot study. International Journal of Geriatric Psychiatry. 2012;27:974–980. doi: 10.1002/gps.2813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letchworth SR, Smith HR, Porrino LJ, Bennett BA, Davies HML, Sexton T, Childers SR. Characterization of a tropane radioligand, [3H]2β-Propanoyl-3β- (4-tolyl) Tropane ([3H]PTT), for dopamine transport sites in rat brain. Journal of Pharmacology and Experimental Therapeutics. 2000;293:686–696. [PubMed] [Google Scholar]
- Levine J, Panchalingam K, Rapoport A, Gershon S, McClure RJ, Pettegrew JW. Increased cerebrospinal fluid glutamine levels in depressed patients. Biological Psychiatry. 2000;47:586–593. doi: 10.1016/s0006-3223(99)00284-x. [DOI] [PubMed] [Google Scholar]
- Li N, Lee B, Liu RJ, Banasr M, Dwyer JM, Iwata M, et al. Duman RS. mTOR-Dependent Synapse Formation Underlies the Rapid Antidepressant Effects of NMDA Antagonists. Science. 2010;329:959–964. doi: 10.1126/science.1190287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lopez-Munoz F, Alamo C, Juckel G, Assion HJ. Half a century of antidepressant drugs: On the clinical introduction of monoamine oxidase inhibitors, tricyclics, and tetracyclics. Part I: Monoamine oxidase inhibitors. Journal of Clinical Psychopharmacology. 2007;27:555–559. doi: 10.1097/jcp.0b013e3181bb617. [DOI] [PubMed] [Google Scholar]
- Lord B, Wintmolders C, Langlois X, Nguyen L, Lovenberg T, Bonaventure P. Comparison of the ex vivo receptor occupancy profile of ketamine to several NMDA receptor antagonists in mouse hippocampus. European Journal of Phamacology. 2013;715(1-3):21–25. doi: 10.1016/j.ejphar.2013.06.028. doi: http://dx.doi.org/10.1016/j.ejphar.2013.06.028. [DOI] [PubMed] [Google Scholar]
- Luckenbaugh DA, Niciu MJ, Ionescu DF, Nolan NM, Richards EM, Brutsche NE, et al. Zarate CA., Jr Do the dissociative side effects of ketamine mediate its antidepressant effects? Journal of Affective Disorders. 2014;159:56–61. doi: 10.1016/j.jad.2014.02.017. doi: http://dx.doi.org/10.1016/j.jad.2014.02.017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lotufo-Neto F, Trivedi M, Thase ME. Meta-Analysis of the Reversible Inhibitors of Monoamine Oxidase Type A Moclobemide and Brofaromine for the Treatment of Depression. Neuropsychopharmacology. 1999;20:226–247. doi: 10.1016/S0893-133X(98)00075-X. [DOI] [PubMed] [Google Scholar]
- Machado-Vieira R, Manji HK, Zarate CA. The role of the tripartite glutamatergic synapse in the pathophysiology and therapeutics of mood disorders. The Neuroscientist. 2009;15:525–539. doi: 10.1177/1073858409336093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Machado-Vieira R, Yuan P, Brutsche N, DiazGranados N, Luckenbaugh D, Manji HK, Zarate CA., Jr Brain-derived neurotrophic factor and initial antidepressant response to an N-Methyl-D-Aspartate antagonist. Journal of Clinical Psychiatry. 2009;70:1662–1666. doi: 10.4088/JCP.08m04659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maes M, Verkerk R, Vandoolaeghe E, Lin A, Scharpé S. Serum levels of excitatory amino acids, serine, glycine, histidine, threonine, taurine, alanine and arginine in treatment-resistant depression: modulation by treatment with antidepressants and prediction of clinical responsivity. Acta Psychiatrica Scandinavica. 1998;97(Suppl):302–308. doi: 10.1111/j.1600-0447.1998.tb10004.x. [DOI] [PubMed] [Google Scholar]
- Marsden WN. Stressor-induced NMDAR dysfunction as a unifying hypothesis for the aetiology, pathogenesis and comorbidity of clinical depression. Medical Hypotheses. 2011;77:508–528. doi: 10.1016/j.mehy.2011.06.021. doi: http://dx.doi.org/10.1016/j.mehy.2011.06.021. [DOI] [PubMed] [Google Scholar]
- Mathew SJ, Murrough JW, aan het Rot M, Collins KA, Reich DL, Charney DS. Riluzole for relapse prevention following intravenous ketamine in treatment- resistant depression: a pilot randomized, placebo-controlled continuation trial. The International Journal of Neuropsychopharmacology. 2010;13:71–82. doi: 10.1017/S1461145709000169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mathews DC, Henter ID, Zarate CAJ. Targeting the glutamatergic system to treat major depressive disorder: Rationale and progress to date. Drugs. 2012;72:1313–1333. doi: 10.2165/11633130-000000000-00000. 1310.2165/11633130-000000000-000000000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mauri MC, Ferrara A, Boscati L, Bravin S, Zamberlan F, Alecci M, Invernizzi G. Plasma and platelet amino acid concentrations in patients affected by major depression and under fluvoxamine treatment. Neuropsychobiology. 1998;37:124–129. doi: 10.1159/000026491. [DOI] [PubMed] [Google Scholar]
- McCambridge J, Winstock A, Hunt N, Mitcheson L. 5-Year trends in use of hallucinogens and other adjunct drugs among UK dance drug users. European Addiction Research. 2007;13:57–64. doi: 10.1159/000095816. [DOI] [PubMed] [Google Scholar]
- McIntyre RS, Lophaven S, Olsen CK. A randomized, double-blind, placebo- controlled study of vortioxetine on cognitive function in depressed adults. The International Journal of Neuropsychopharmacology. 2014:1–11. doi: 10.1017/S1461145714000546. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McNulty JP, Hahn K. Compounded oral ketamine. The International Journal of Pharmaceutical Compounding. 2012;16:364–368. [PubMed] [Google Scholar]
- Mendelsohn LG, Kalra V, Johnson BG, Kerchner GA. Sigma opioid receptor: characterization and co-identity with the phencyclidine receptor. Journal of Pharmacology and Experimental Therapeutics. 1985;233:597–602. [PubMed] [Google Scholar]
- Michael N, Erfurth A, Ohrmann P, Arolt V, Heindel W, Pfleiderer B. Metabolic changes within the left dorsolateral prefrontal cortex occurring with electroconvulsive therapy in patients with treatment resistant unipolar depression. Psychological Medicine. 2003;33:1277–1284. doi: 10.1017/S0033291703007931. [DOI] [PubMed] [Google Scholar]
- Millan MJ, Gobert A, Lejeune F, Newman-Tancredi A, Rivet JM, Auclair A, Peglion JL. S33005, a novel ligand at both serotonin and norepinephrine transporters: I. receptor binding, electrophysiological, and neurochemical profile in comparison with venlafaxine, reboxetine, citalopram, and clomipramine. Journal of Pharmacology and Experimental Therapeutics. 2001;298:565–580. [PubMed] [Google Scholar]
- Mirza Y, Tang J, Russell A, Banerjee SP, Bhandari R, Ivey J, et al. Rosenberg DR. Reduced anterior cingulate cortex glutamatergic concentrations in childhood major depression. Journal of the American Academy of Child & Adolescent Psychiatry. 2004;43:341–348. doi: 10.1097/00004583-200403000-00017. [DOI] [PubMed] [Google Scholar]
- Moaddel R, Luckenbaugh D, Xie Y, Villaseñor A, Brutsche N, Machado-Vieira R, et al. Wainer I. D-serine plasma concentration is a potential biomarker of (R,S)- ketamine antidepressant response in subjects with treatment-resistant depression. Psychopharmacology. 2014:1–11. doi: 10.1007/s00213-014-3669-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Montgomery SA, Gabriel R, James D, Hawley C, Burkitt P. The specificity of the zimelidine reaction. International Clinical Psychopharmacology. 1989;4:19–23. doi: 10.1097/00004850-198901000-00003. [DOI] [PubMed] [Google Scholar]
- Moreira R. The efficacy and tolerability of bupropion in the treatment of major depressive disorder. Clinical Drug Investigation. 2011;31:5–17. doi: 10.2165/1159616-s0-000000000-00000. [DOI] [PubMed] [Google Scholar]
- Mørk A, Pehrson A, Brennum LT, Nielsen SM, Zhong H, Lassen AB, et al. Stensbøl TB. Pharmacological effects of Lu AA21004: A novel multimodal compound for the treatment of major depressive disorder. Journal of Pharmacology and Experimental Therapeutics. 2012;340:666–675. doi: 10.1124/jpet.111.189068. [DOI] [PubMed] [Google Scholar]
- Mørk A, Montezinho LP, Miller S, Trippodi-Murphy C, Plath N, Li Y, et al. Sanchez C. Vortioxetine (Lu AA21004), a novel multimodal antidepressant, enhances memory in rats. Pharmacology Biochemistry and Behavior. 2013;105:41–50. doi: 10.1016/j.pbb.2013.01.019. doi: http://dx.doi.org/10.1016/j.pbb.2013.01.019. [DOI] [PubMed] [Google Scholar]
- Moskal JR, Burch R, Burgdorf JS, Kroes RA, Stanton PK, Disterhoft JF, Leander JD. GLYX-13, an NMDA receptor glycine site functional partial agonist enhances cognition and produces antidepressant effects without the psychotomimetic side effects of NMDA receptor antagonists. Expert Opinion on Investigational Drugs. 2014;23:243–254. doi: 10.1517/13543784.2014.852536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moskal JR, Kuo AG, Weiss C, Wood PL, O'Connor Hanson A, Kelso S, et al. Disterhoft JF. GLYX-13: A monoclonal antibody-derived peptide that acts as an N-methyl-d-aspartate receptor modulator. Neuropharmacology. 2005;49:1077–1087. doi: 10.1016/j.neuropharm.2005.06.006. doi: http://dx.doi.org/10.1016/j.neuropharm.2005.06.006. [DOI] [PubMed] [Google Scholar]
- Mount C, Downton C. Alzheimer disease: progress or profit? Nature Medicine. 2006;12:780–784. doi: 10.1038/nm0706-780. [DOI] [PubMed] [Google Scholar]
- Muhonen LH, Lonngvist J, Juva K, Alho H. Double-blind, randomized comparison of memantine and escitalopram for the treatment of major depressive disorder comorbid with alcohol dependence. Journal of Clinical Psychiatry. 2008;69:392–399. doi: 10.4088/jcp.v69n0308. [DOI] [PubMed] [Google Scholar]
- Muller JC, Pryor WW, Gibbons JE, Orgain ES. Depression and anxiety occurring during rauwolfia therapy. Journal of the American Medical Association. 1955;159:836–839. doi: 10.1001/jama.1955.02960260006002. [DOI] [PubMed] [Google Scholar]
- Murrough JW, Iosifescu DV, Chang LC, Al Jurdi RK, Green CE, Perez AM, et al. Mathew SJ. Antidepressant efficacy of ketamine in treatment-resistant major depression: A two-site randomized controlled trial. American Journal of Psychiatry. 2013;170:1134–1142. doi: 10.1176/appi.ajp.2013.13030392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough JW, Perez AM, Pillemer S, Stern J, Parides MK, aan het Rot M, et al. Iosifescu DV. Rapid and longer-term antidepressant effects of repeated ketamine infusions in treatment-resistant major depression. Biological Psychiatry. 2013;74:250–256. doi: 10.1016/j.biopsych.2012.06.022. doi: http://dx.doi.org/10.1016/j.biopsych.2012.06.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Murrough J, Wan LB, Iacoviello B, Collins K, Solon C, Glicksberg B, et al. Burdick K. Neurocognitive effects of ketamine in treatment-resistant major depression: association with antidepressant response. Psychopharmacology. 2014;231:481–488. doi: 10.1007/s00213-013-3255-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- National Institute of Mental Health's Psychoactive Drug Screening Program. 2014 Contract # HHSN-271-2013-00017-C (NIMH PDSP). Available from http://pdsp.med.unc.edu/
- Naurex Inc. Naurex reports positive top-line phase 2b results for novel antidepressant GLYX-13 and advances NRX-1074 into phase 2 depression study. 2014 Retrieved from http://naurex.com/wp-content/uploads/2014/05/Naurex_Clinical_Advances-2014.05.06.pdf.
- Niciu MJ, Luckenbaugh D, Ionescu DF, Guevara S, Machado-Vieira R, Richards EM, et al. Zarate CA., Jr Clinical predictors of ketamine response in treatment- resistant major depression. Journal of Clinical Psychiatry. 2014;75:417–423. doi: 10.4088/JCP.13m08698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niciu MJ, Luckenbaugh DA, Ionescu DF, Richards Erica M, Vande Voort JL, Ballard ED, et al. Zarate CA., Jr Riluzole likely lacks antidepressant efficacy in ketamine non-responders. Journal of Psychiatric Research. 2014:1–3. doi: 10.1016/j.jpsychires.2014.07.022. doi: http://dx.doi.org/10.1016/j.jpsychires.2014.07.022. [DOI] [PMC free article] [PubMed]
- Nilsson BS. Adverse reactions in connection with zimeldine treatment--a review. Acta Psychiatrica Scandinavica. 1983;308(Suppl):115–119. doi: 10.1111/j.1600-0447.1983.tb11110.x. [DOI] [PubMed] [Google Scholar]
- Nishimura M, Sato K, Okada T, Yoshiya I, Schloss P, Shimada S, Tohyama M. Ketamine inhibits monoamine transporters expressed in human embryonic kidney 293 cells. Anesthesiology. 1998;88:768–774. doi: 10.1097/00000542-199803000-00029. [DOI] [PubMed] [Google Scholar]
- Nudmamud-Thanoi S, Reynolds GP. The NR1 subunit of the glutamate/NMDA receptor in the superior temporal cortex in schizophrenia and affective disorders. Neuroscience Letters. 2004;372(1–2):173–177. doi: 10.1016/j.neulet.2004.09.035. doi: http://dx.doi.org/10.1016/j.neulet.2004.09.035. [DOI] [PubMed] [Google Scholar]
- Owens MJ, Morgan WN, Plott SJ, Nemeroff CB. Neurotransmitter receptor and transporter binding profile of antidepressants and their metabolites. Journal of Pharmacology and Experimental Therapeutics. 1997;283:1305–1322. [PubMed] [Google Scholar]
- Papakostas GI. Tolerability of modern antidepressants. Journal of Clinical Psychiatry. 2008;69(suppl E1):8–13. [PubMed] [Google Scholar]
- Palucha A, Pilc A. The involvement of glutamate in the pathology of depression. Drug News Perspective. 2005;18:262–268. doi: 10.1358/dnp.2005.18.4.908661. doi:101358/dmp.2005.18.4.908661. [DOI] [PubMed] [Google Scholar]
- Papakostas GI. Serotonin norepinephrine reuptake inhibitors: Spectrum of efficacy in major depressive disorder. Primary Psychiatry. 2009;16(Suppl 4):16–24. [Google Scholar]
- Papakostas GI, Thase ME, Fava M, Nelson JC, Shelton RC. Are antidepressant drugs that combine serotonergic and noradrenergic mechanisms of action more effective than the selective serotonin reuptake inhibitors in treating major depressive disorder? A meta-analysis of studies of newer agents. Biological Psychiatry. 2007;62:1217–1227. doi: 10.1016/j.biopsych.2007.03.027. doi: http://dx.doi.org/10.1016/j.biopsych.2007.03.027. [DOI] [PubMed] [Google Scholar]
- Pearce EF, Murphy JA. Vortioxetine for the treatment of depression. Annals of Pharmacotherapy. 2014;48:758–765. doi: 10.1177/1060028014528305. [DOI] [PubMed] [Google Scholar]
- Pfleiderer B, Michael N, Erfurth A, Ohrmann P, Hohmann U, Wolgast M, et al. Heindel W. Effective electroconvulsive therapy reverses glutamate/glutamine deficit in the left anterior cingulum of unipolar depressed patients. Psychiatry Research: Neuroimaging. 2003;122:185–192. doi: 10.1016/s0925-4927(03)00003-9. doi: http://dx.doi.org/10.1016/S0925-4927(03)00003-9. [DOI] [PubMed] [Google Scholar]
- Pin JP, Duvoisin R. The metabotropic glutamate receptors: Structure and functions. Neuropharmacology. 1995;34:1–26. doi: 10.1016/0028-3908(94)00129-g. doi: http://dx.doi.org/10.1016/0028-3908(94)00129-G. [DOI] [PubMed] [Google Scholar]
- Pletscher A. The discovery of antidepressants: A winding path. Experientia. 1991;47:4–8. doi: 10.1007/bf02041242. [DOI] [PubMed] [Google Scholar]
- Preskorn SH, Baker B, Kolluri S, Menniti FS, Krams M, Landen JW. An innovative design to establish proof of concept of the antidepressant effects of the NR2B subunit selective N-Methyl-D-Aspartate antagonist, CP-101,606, in patients with treatment-refractory major depressive disorder. Journal of Clinical Psychopharmacology. 2008;28:631–637. doi: 10.1097/JCP.0b013e31818a6cea. [DOI] [PubMed] [Google Scholar]
- Price RB, Iosifescu DV, Murrough JW, Chang LC, Al Jurdi RK, Iqbal SZ, et al. Mathew SJ. Effects of ketamine on explicit and implicit suicidal cognition: A randomized controlled trial in treatment-resistant depression. Depression and Anxiety. 2014;31:335–343. doi: 10.1002/da.22253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price RB, Nock MK, Charney DS, Mathew SJ. Effects of intravenous ketamine on explicit and implicit measures of suicidality in treatment-resistant depression. Biological Psychiatry. 2009;66:522–526. doi: 10.1016/j.biopsych.2009.04.029. doi: http://dx.doi.org/10.1016/j.biopsych.2009.04.029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pristupa ZB, Wilson JM, Hoffman BJ, Kish SJ, Niznik HB. Pharmacological heterogeneity of the cloned and native human dopamine transporter: disassociation of [3H]WIN 35,428 and [3H]GBR 12,935 binding. Molecular Pharmacology. 1994;45:125–135. [PubMed] [Google Scholar]
- Rasmussen KG, Lineberry TW, Galardy CW, Kung S, Lapid MI, Palmer BA, et al. Frye MA. Serial infusions of low-dose ketamine for major depression. Journal of Psychopharmacology. 2013;27:444–450. doi: 10.1177/0269881113478283. [DOI] [PubMed] [Google Scholar]
- Rothman RB, Baumann MH, Dersch CM, Romero DV, Rice KC, Carroll FI, Partilla JS. Amphetamine-type central nervous system stimulants release norepinephrine more potently than they release dopamine and serotonin. Synapse. 2001;39:32–41. doi: 10.1002/1098-2396(20010101)39:1<32∷aid-syn5>3.0.co;2-3. [DOI] [PubMed] [Google Scholar]
- Roth BL, Gibbons S, Arunotayanun W, Huang XP, Setola V, Treble R, Iversen L. The ketamine analogue methoxetamine and 3- and 4-Methoxy analogues of phencyclidine are high affinity and selective ligands for the glutamate NMDA receptor. PLoS ONE. 2013;8:e59334. doi: 10.1371/journal.pone.0059334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rush A, Trivedi M, Wisniewski S, Nierenberg A, Stewart J, Warden D, et al. Fava M. Acute and longer-term outcomes in depressed outpatients requiring one or several treatment steps: A STAR*D report. American Journal of Psychiatry. 2006;163:1905–1917. doi: 10.1176/ajp.2006.163.11.1905. [DOI] [PubMed] [Google Scholar]
- Salomon RM, Miller HL, Krystal JH, Heninger GR, Charney DS. Lack of behavioral effects of monoamine depletion in healthy subjects. Biological Psychiatry. 1997;41:58–64. doi: 10.1016/0006-3223(95)00670-2. doi: http://dx.doi.org/10.1016/0006-3223(95)00670-2. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Gueorguieva R, Epperson C, Wu YT, Appel M, Rothman DL, et al. Mason GF. Subtype-specific alterations of γ-aminobutyric acid and glutamatein patients with major depression. Archives of General Psychiatry. 2004;61:705–713. doi: 10.1001/archpsyc.61.7.705. [DOI] [PubMed] [Google Scholar]
- Sanacora G, Kendell SF, Levin Y, Simen AA, Fenton LR, Coric V, Krystal JH. Preliminary evidence of riluzole efficacy in antidepressant-treated patients with residual depressive symptoms. Biological Psychiatry. 2007;61:822–825. doi: 10.1016/j.biopsych.2006.08.037. doi: http://dx.doi.org/10.1016/j.biopsych.2006.08.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanacora G, Smith MA, Pathak S, Su HL, Boeijinga PH, McCarthy DJ, Quirk MC. Lanicemine: A low-trapping NMDA channel blocker produces sustained antidepressant efficacy with minimal psychotomimetic adverse effects. Molecular Psychiatry. 2013:1–8. doi: 10.1038/mp.2013.130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sánchez C, Hyttel J. Comparison of the effects of antidepressants and their metabolites on reuptake of biogenic amines and on receptor binding. Cellular and Molecular Neurobiology. 1999;19:467–489. doi: 10.1023/a:1006986824213. [DOI] [PubMed] [Google Scholar]
- Schildkraut JJ. The catecholamine hypothesis of affective disorders: A review of supporting evidence. American Journal of Psychiatry. 1965;122:509–522. doi: 10.1176/ajp.122.5.509. [DOI] [PubMed] [Google Scholar]
- Schulberg HC, Katon W, Simon GE, Rush A. Treating major depression in primary care practice: An update of the agency for health care policy and research practice guidelines. Archives of General Psychiatry. 1998;55:1121–1127. doi: 10.1001/archpsyc.55.12.1121. [DOI] [PubMed] [Google Scholar]
- Seeman P, Ko F, Tallerico T. Dopamine receptor contribution to the action of PCP, LSD and ketamine psychotomimetics. Molecular Psychiatry. 2005;10:877–883. doi: 10.1038/sj.mp.4001682. [DOI] [PubMed] [Google Scholar]
- Shaw DM, Eccleston EG, Camps FE. 5-Hydroxytryptamine in the hind-brain of depressive suicides. The British Journal of Psychiatry. 1967;113:1407–1411. doi: 10.1192/bjp.113.505.1407. [DOI] [PubMed] [Google Scholar]
- Shek DTL. Tackling adolescent substance abuse in Hong Kong: Where we should and should not go. The Scientific World Journal. 2007;7:2021–2030. doi: 10.1100/tsw.2007.315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shiroma PR, Albott CS, Johns B, Thuras P, Wels J, Lim KO. Neurocognitive performance and serial intravenous subanesthetic ketamine in treatment- resistant depression. The International Journal of Neuropsychopharmacology. 2014:1–9. doi: 10.1017/S1461145714001011. [DOI] [PubMed] [Google Scholar]
- Shore PA, Silver SL, Brodie BB. Interaction of reserpine, serotonin, and lysergic acid diethylamide in brain. Science. 1955;122:284–285. doi: 10.2307/1751305. [DOI] [PubMed] [Google Scholar]
- Shore PA, Pletscher A, Tomich EG, Carlsson A, Kuntzman R, Brodie BB. Role of brain serotonin in reserpine action. Annals of the New York Academy of Sciences. 1957;66:609–617. doi: 10.1111/j.1749-6632.1957.tb40751.x. [DOI] [PubMed] [Google Scholar]
- Shulman K, Herrmann N, Walker S. Current place of monoamine oxidase inhibitors in the treatment of depression. CNS Drugs. 2013;27:789–797. doi: 10.1007/s40263-013-0097-3. [DOI] [PubMed] [Google Scholar]
- Smith DJ, Bouchal RL, DeSanctis CA, Monroe PJ, Amedro JB, Perrotti JM, Crisp T. Properties of the interaction between ketamine and opiate binding sites in vivo and in vitro. Neuropharmacology. 1987;26:1253–1260. doi: 10.1016/0028-3908(87)90084-0. [DOI] [PubMed] [Google Scholar]
- Smith ER, Deligiannidis KM, Ulbricht CM, Landolin CS, Patel JK, Rothschild AJ. Antidepressant augmentation using the N-Methyl-D-Aspartate antagonist memantine: A randomized, double-blind, placebo-controlled trial. Journal of Clinical Psychiatry. 2013;4:966–973. doi: 10.4088/JCP.12m08252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stahl SM, Grady MM, Moret C, Briley M. SNRIs: Their pharmacology, clinical efficacy, and tolerability in comparison with other classes of antidepressants. CNS Spectrums. 2005;10:732–747. doi: 10.1017/s1092852900019726. [DOI] [PubMed] [Google Scholar]
- Stahl SM, Pradko JF, Haight BR, Modell JG, Rockett CB, Learned-Coughlin S. A review of the neuropharmacology of bupropion, a dual norepinephrine and dopamine reuptake inhibitor. Primary Care Companion to The Journal of Clinical Psychiatry. 2004;6:159–166. doi: 10.4088/pcc.v06n0403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Substance Abuse and Mental Health Services Administration. Results from the 2012 National Survey on Drug Use and Health: Summary of National Findings, NSDUH Series H-46, HHS Publication No (SMA) 13-4795. Rockville, MD: Substance Abuse and Mental Health Services Administration; 2013. [Google Scholar]
- Thomas DR, Nelson DR, Johnson AM. Biochemical effects of the antidepressant paroxetine, a specific 5-hydroxytryptamine uptake inhibitor. Psychopharmacology. 1987;93:193–200. doi: 10.1007/bf00179933. [DOI] [PubMed] [Google Scholar]
- Uher R, Mors O, Rietschel M, Rajewska-Rager A, Petrovic A, Zobel A, et al. McGuffin P. Early and delayed onset of response to antidepressants in individual trajectories of change during treatment of major depression: a secondary analysis of data from the genome-based therapeutic drugs for depression (GENDEP) study. Journal of Clinical Psychiatry. 2011;72:1478–1484. doi: 10.4088/JCP.10m06419. [DOI] [PubMed] [Google Scholar]
- Vaishnavi SN, Nemeroff CB, Plott SJ, Rao SG, Kranzler J, Owens MJ. Milnacipran: a comparative analysis of human monoamine uptake and transporter binding affinity. Biological Psychiatry. 2004;55:320–322. doi: 10.1016/j.biopsych.2003.07.006. [DOI] [PubMed] [Google Scholar]
- Valentine GW, Mason GF, Gomez R, Fasula M, Watzl J, Pittman B, et al. Sanacora G. The antidepressant effect of ketamine is not associated with changes in occipital amino acid neurotransmitter content as measured by [1H]-MRS. Psychiatry Research: Neuroimaging. 2011;191:122–127. doi: 10.1016/j.pscychresns.2010.10.009. doi: http://dx.doi.org/10.1016/j.pscychresns.2010.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang SJ, Wang KY, Wang WC. Mechanisms underlying the riluzole inhibition of glutamate release from rat cerebral cortex nerve terminals (synaptosomes) Neuroscience. 2004;125:191–201. doi: 10.1016/j.neuroscience.2004.01.019. doi: http://dx.doi.org/10.1016/j.neuroscience.2004.01.019. [DOI] [PubMed] [Google Scholar]
- Weiner N, Cloutier G, Bjur R, Pfeffer RI. Modification of Norepinephrine Synthesis in Intact Tissue by Drugs and during Shor-term Adrenergic Nerve Stimulation. Pharmacological Reviews. 1972;24:203–221. [PubMed] [Google Scholar]
- Winstock AR, Mitcheson L, Gillatt DA, Cottrell AM. The prevalence and natural history of urinary symptoms among recreational ketamine users. BJU International. 2012;110:1762–1766. doi: 10.1111/j.1464-410X.2012.11028.x. [DOI] [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Horng JS, Molloy BB. A new selective inhibitor for uptake of serotonin into synaptosomes of rat brain: 3-(p-trifluoromethylphenoxy). N- methyl-3-phenylpropylamine. Journal of Pharmacology and Experimental Therapeutics. 1975;193:804–811. [PubMed] [Google Scholar]
- Wong DT, Bymaster FP, Engleman EA. Prozac (fluoxetine, lilly 110140), the first selective serotonin uptake inhibitor and an antidepressant drug: Twenty years since its first publication. Life Sciences. 1995;57:411–441. doi: 10.1016/0024-3205(95)00209-o. doi: http://dx.doi.org/10.1016/0024-3205(95)00209-O. [DOI] [PubMed] [Google Scholar]
- Wong DT, Horng JS, Bymaster FP, Hauser KL, Molloy BB. A selective inhibitor of serotonin uptake: Lilly 110140, 3-(p-Trifluoromethylphenoxy)-n-methyl-3- phenylpropylamine. Life Sciences. 1974;15:471–479. doi: 10.1016/0024-3205(74)90345-2. doi: http://dx.doi.org/10.1016/0024-3205(74)90345-2. [DOI] [PubMed] [Google Scholar]
- Wong DT, Perry KW, Bymaster FP. The discovery of fluoxetine hydrochloride (Prozac) Nature Reviews Drug Discovery. 2005;4:764–774. doi: 10.1038/nrd1821. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Mori H, Masaki H, Shimoji K, Mishina M. Different sensitivities of NMDA receptor channel subtypes to non-competitive antagonists. NeuroReport. 1993;4:687–690. doi: 10.1097/00001756-199306000-00021. [DOI] [PubMed] [Google Scholar]
- Yamakura T, Shimoji K. Subunit- and site-specific pharmacology of the NMDA receptor channel. Progress in Neurobiology. 1999;59:279–298. doi: 10.1016/s0301-0082(99)00007-6. doi: http://dx.doi.org/10.1016/S0301-0082(99)00007-6. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Mathews D, Ibrahim L, Chaves JF, Marquardt C, Ukoh I, et al. Luckenbaugh DA. A randomized trial of a low-trapping nonselective N- Methyl-D-Aspartate channel blocker in major depression. Biological Psychiatry. 2013;74:257–264. doi: 10.1016/j.biopsych.2012.10.019. doi: http://dx.doi.org/10.1016/j.biopsych.2012.10.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate CA, Jr, Payne JL, Quiroz J, Sporn J, Denicoff KK, Luckenbaugh D, et al. Manji HK. An open-label trial of riluzole in patients with treatment-resistant major depression. American Journal of Psychiatry. 2004;161:171–174. doi: 10.1176/appi.ajp.161.1.171. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Carlson PJ, Brutsche NE, Ameli R, Luckenbaugh DA, et al. Manji HK. A randomized trial of an n-methyl-d-aspartate antagonist in treatment-resistant major depression. Archives of General Psychiatry. 2006;63:856–864. doi: 10.1001/archpsyc.63.8.856. [DOI] [PubMed] [Google Scholar]
- Zarate CA, Jr, Singh JB, Quiroz JA, De Jesus G, Denicoff KK, Luckenbaugh DA, et al. Charney DS. A double-blind, placebo-controlled study of memantine in the treatment of major depression. American Journal of Psychiatry. 2006;163:153–155. doi: 10.1176/appi.ajp.163.1.153. [DOI] [PubMed] [Google Scholar]
- Zigman D, Blier P. Urgent ketamine infusion rapidly eliminated suicidal ideation for a patient with major depressive disorder: A case report. Journal of Clinical Psychopharmacology. 2013;33:270–272. doi: 10.1097/JCP.0b013e3182856865. [DOI] [PubMed] [Google Scholar]