

Abstract
Bis(perfluorocatecholato)silane Si(catF)2 was prepared, and stoichiometric binding to Lewis bases was demonstrated with fluoride, triethylphosphine oxide, and N,N′-diisopropylbenzamide. The potent Lewis acidity of Si(catF)2 was suggested from catalytic hydrosilation and silylcyanation reactions with aldehydes. Mechanistic studies of hydrosilation using an optically active silane substrate, R-(+)-methyl(1-napthyl)phenylsilane, proceeded with predominant stereochemical retention at silicon, consistent with a carbonyl activation pathway. The enantiospecificity was dependent on solvent and salt effects, with increasing solvent polarity or addition of NBu4BArF4 leading to a diminished enantiomeric ratio. The medium effects are consistent with an ionic mechanism, wherein hydride transfer occurs prior to silicon–oxygen bond formation.
Graphical Abstract
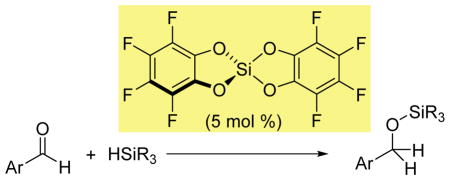
Lewis acidic main group compounds have emerged as broadly applicable reagents. In particular, B(C6F5)3 and other electron-deficient boranes can serve as activators for transition metal compounds,1 and as alternatives to metal-based catalysts.2
In contrast to Group 13 species, silicon Lewis acids remain relatively rare.3 Silicon tetrachloride is the common silicon-based Lewis acid of choice in catalytic applications;4 however, the reactive Si–Cl bonds are readily cleaved by nucleophilic reagents, limiting its utility. Several recent reports have shown that cationic silylium ions promote catalytic imine reduction and Diels-Alder reactions.5 Silylium compounds, in combination with phosphines, engage in frustrated Lewis pair reactions that activate carbon dioxide and dihydrogen.6 Additionally, Leighton and coworkers have demonstrated that chiral silicon complexes can serve as reagents for asymmetric crotylation reactions and as catalysts for Diels-Alder additions.7 Based on these promising results, we were interested in exploring the behavior of neutral silicon compounds as potent Lewis acids. Herein, we report the synthesis and reactivity of a neutral bis(perfluorocatecholato)silane Lewis acid, which represents the first example of a neutral silicon species that catalyzes aldehyde hydrosilation.
The bis(catecholato)silane motif was selected due to its ease of preparation and stability, and fluorinated catechol ligands were employed to enhance the Lewis acidic properties. The novel complex bis(perfluorocatecholato)silane, Si(catF)2 (1), was easily prepared by treatment of silicon tetrachloride with 2 equiv of tetrafluorocatechol in acetonitrile. In the absence of Lewis bases, 1 has very limited solubility in standard organic solvents including benzene, dichloromethane, and acetonitrile.
Reactions with simple anionic and neutral Lewis bases were investigated to probe the binding properties of Si(catF)2. First, addition of tris(dimethylamino)sulfonium difluorotrimethylsilicate (TASF) to Si(catF)2 in tetrahydrofuran lead to the immediate formation of a bis(perfluorocatecholato)fluorosilicate complex (2, eq 1). Single crystal X-ray diffraction confirmed the structure of 2, which exhibits an approximate square pyramidal geometry at silicon (Figure 1). There is π-stacking between the perfluorocatechol rings of two silicate units in the solid state (d = 3.08–3.61 Å).8 The silicon–fluorine bond distance in 2 (1.602(2) Å) is nearly identical to that reported previously for [Si(cat)2F][NEt4].9 This fluoride binding demonstrates that Si(catF)2 can readily accommodate an added Lewis base to form a pentacoordinate species, a general step that is necessary for catalytic applications.
Figure 1.
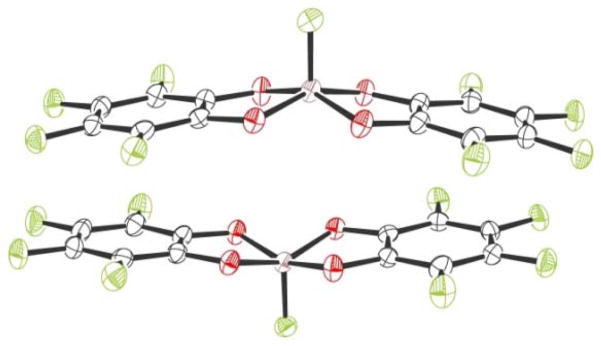
ORTEP diagram of 2, with thermal elipsoids at 50% probability. Tris(dimethylamino)sulfonium cations are omitted for clarity.
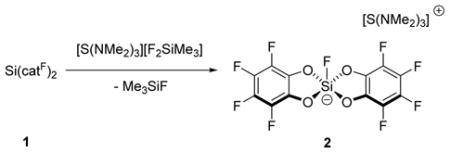 |
(1) |
To apply the Gutmann-Beckett method as a gauge of Lewis acidity, the binding of triethylphosphine oxide to Si(catF)2 was evaluated on the basis of the change in the 31P NMR chemical shift of OPEt3 upon complexation.10 A change of + 35.9 ppm in the 31P NMR chemical shift of OPEt3 was observed upon treatment with 1 equiv of Si(catF)2 in dichloromethane-d2. Only a slightly smaller change in chemical shift was observed using Si(cat)2 (Δδ = + 32.5 ppm). These differences are substantially larger than the change observed upon binding to B(C6F5)3 (Δδ = +26.6 ppm), and suggest that Si(catF)2 is a stronger Lewis acid toward OPEt3 than Si(cat)2 or B(C6F5)3, although both silicon complexes induce large changes in 31P NMR chemical shifts upon complexation.
In contrast to the strong Lewis acidity implied by the Guttmann-Beckett analysis with OPEt3, Si(catF)2 does not readily bind aldehydes or ketones. The combination of 1 equiv of trans-crotonaldehyde and Si(catF)2 resulted in negligible (<2 %) conversion to a Si(catF)2–crotonaldehyde adduct (assessed by 1H NMR spectroscopy in dichloromethane-d2). This suggests that Si(catF)2 displays a substantially diminished affinity for “soft” Lewis bases relative to typical boron Lewis acids, which readily coordinate to carbonyl groups.2a,11
Replacing aldehyde moieties with the strongly coordinating amide functional group promoted coordination, and quantitative adduct formation was observed between N,N′-diisopropylbenzamide and Si(catF)2 (3, eq 2). Upon binding to either Si(catF)2 or B(C6F5)3, the two N-bound isopropyl groups of the benzamide appear as separate, sharp signals, whereas there is coalescence of these signals in the room temperature 1H NMR spectrum of the free benzamide.12 This behavior can be rationalized by invoking strong nitrogen π-donation upon formation of the Lewis acid-base adduct, which slows the exchange of the E and Z isopropyl groups via rotation about the C(amide)–N bond. In contrast to 1, the parent bis(catecholato)silane complex Si(cat)2 does not react with N,N′-bisdiisopropylbenzamide (by NMR spectroscopy in dichloromethane-d2 solvent), indicating that the perfluoro derivative displays an enhanced binding affinity for this substrate.
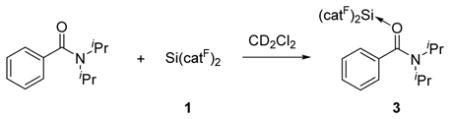 |
(2) |
Single crystals of 3 were grown from a mixture of o-difluorobenzene and toluene at −30 °C (Figure 2). In comparison to the analogous B(C6F5)3 adduct, the C(amide)–O bond of 3 is shorter (1.32(1) vs. 1.304(3) Å) and the C(amide)–N bond is longer (1.28(1) vs. 1.297(3) Å).12 The C=O stretching frequency of 3 (νCO = 1606 cm−1) is intermediate between values for free benzamide and the B(C6F5)3 adduct (1625 and 1570 cm−1, respectively). Thus, both bond length and IR spectroscopic comparisons suggest that B(C6F5)3 is more activating than 1 toward benzamides.
Figure 2.

ORTEP diagram of compound 3 with all thermal elipsoids shown at 50% probability.
Hydrosilation of aldehydes was used to assess the catalytic properties of Si(catF)2. Main–group–catalyzed hydrosilation is well known for both neutral and cationic boron Lewis acids.2a,13 In contrast, only cationic silylium ions have previously been reported as hydrosilation catalysts.2b,14 These silyliumion catalyzed carbonyl reductions often result in over-reduction to the deoxygenated hydrocarbon products, rather than the presumed initial silyl ether complex.
Initial studies showed that Si(catF)2 is an efficient catalyst for the hydrosilation of 4-nitrobenzaldehyde with triethylsilane at room temperature, to exclusively form the corresponding silyl ether product (Table 1, entry 1). In contrast, we found the previously reported Si(cat)2 and Si(C6F5)4 complexes to be essentially inactive as catalysts (Table 1, entries 2, 3). To our knowledge, Si(catF)2 represents the first neutral silicon Lewis acid to serve as a catalyst for aldehyde hydrosilation.
Table 1.
Hydrosilation trials with neutral silicon catalysts
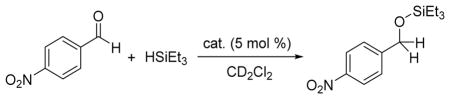
| ||||
|---|---|---|---|---|
| Entry | Catalyst | Time (h) | Temperature (°C) | Yield (%) |
| 1 | Si(catF)2 | 0.5 | 25 | >95 |
| 2 | Si(cat)2 | 48 | 25 | 2 |
| 3 | Si(C6F5)4 | 48 | 45 | 0 |
Reaction conditions: 4-nitrobenzaldehyde (0.30 M), triethylsilane (0.30 M), and catalyst (0.015 M) in dichloromethane-d2 (0.5 mL).
Catalytic amounts of Si(catF)2 also promoted the silylcyanation of 4-nitro-benzaldehyde with trimethylsilylcyanide at 45 °C (eq 3). Previous studies of main group silylcyanation catalysis typically involve (i) combinations of Lewis acid and Lewis base catalysts, such as the Shibasaki bifunctional aluminium and phosphine oxide system,15 or (ii) simple Lewis base activators, including fluoride, phosphines, and amines.16 In the current study, Si(catF)2 behaves as a single component Lewis acid silylcyanation catalyst.
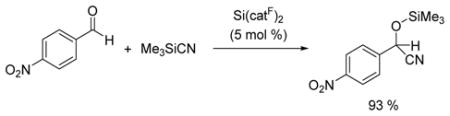 |
(3) |
Hydrosilation catalysis was further evaluated through study of the silane substrate scope (Table 2). Tertiary alkyl and aryl silanes exhibited high activity in hydrosilations of 4-nitrobenzaldehyde (entries 1–4). Bulky silanes such as triisopropylsilane and bis(trimethylsilyl)phenylsilane were also tolerated (entries 5, 6). In contrast, B(C6F5)3 does not react with these sterically demanding substrates, which has been attributed to front strain that prevents silane coordination.2b Silanes incorporating trimethylsiloxy- and dimethylamido- groups were also efficiently transformed (entries 7, 8). Secondary silanes underwent hydrosilation in low conversion (entry 9) and the primary silanes surveyed (H3SiPh and H3SitBu) were completely inactive.
Table 2.
Silane scope for hydrosilation catalysisa
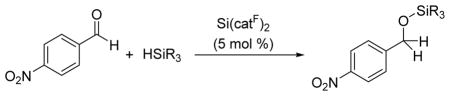
| |||
|---|---|---|---|
| Entry | Silane | Time (h) | Yield (%) |
| 1 | HSiPh3 | 1 | 94 |
| 2 | HSiPh2Me | 0.5 | >95 |
| 3 | HSiPhMe2 | 0.5 | >95 |
| 4 | HSitBuMe2 | 0.5 | 91 |
| 5 | HSiiPr3 | 0.5 | >95 |
| 6 | HSi(SiMe3)2Phb | 1 | 93 |
| 7 | HSi(OSiMe3)2Me | 0.5 | >95 |
| 8 | HSi(NMe2)2Me | 0.5 | 92 |
| 9 | H2SiPhMe | 2 | 42 |
Reaction conditions: 4-nitrobenzaldehyde (0.30 M), silane (0.30 M), and 1 (0.015 M) in CD2Cl2 at 25 °C;
45 °C.
A variety of benzaldehydes were investigated for conversion to the corresponding silyl ethers (Scheme 1). Electron-deficient aldehydes were required for productive catalysis. The nitrile functional group was tolerated (entry c), whereas many Lewis acid catalysts, notably B(C6F5)3, are inactive in the presence of these strongly coordinating groups.2b No inhibitory effect was observed for ortho substituents (entries h, i). Aldehydes were selectively and exclusively hydrosilated in the presence of ketones and esters (entries j, k). Lastly, an electron-deficient cinnemaldehyde derivative underwent exclusive 1,2-addition (entry l).
Scheme 1.

Hydrosilation aldehyde scopea
aReaction conditions: aldehyde (0.30 M), triethylsilane (0.30 M), and 1 (0.015 M) in CD2Cl2 at 25 °C; b 0.45 M HSiEt3.
In order to distinguish between possible mechanistic pathways, experiments were performed to determine whether Si(catF)2 binds to and activates the aldehyde or silane substrate during hydrosilation catalysis.17 No changes were observed in the 1H NMR spectrum of HSiEt3 upon addition of Si(catF)2 (in dichloromethane-d2). Previous work by the Piers and Bergman groups has cited loss of JHH coupling between the Si–H and CH2 protons of silicon-bound alkyl groups as evidence for the intervention of a transient Lewis acid adduct.2b,18 Additionally, no scrambling occurred between a 1:1 mixture of HSiPhMe2 and DSiPh2Me upon treatment with 10 mol % Si(catF)2 in dichloromethane-d2. Attempts to observe hydrosilation of alkenes or silation of phenols with catalytic Si(catF)2 were unsuccessful. In contrast, B(C6F5)3, which is known to operate via a silane activation mechanism, is efficient for these catalytic transformations.2d,19 Taken together, these observations suggest that Si(catF)2, unlike B(C6F5)3, does not bind to the silane substrate during catalysis.
An optically active silane substrate was employed to provide further mechanistic insight. Analogous enantiospecificity studies have been reported as evidence for the silane coordination mechanism operative for B(C6F5)3 and transition metal catalysts.20 Reactions were performed with 5 mol % Si(catF)2 for the hydrosilation of 4-nitro-benzaldehyde with enantiopure R-(+)-methyl(1-napthyl)phenylsilane21 to furnish the silyl ether product in 95 % yield (eq 4). The reaction proceeded with predominant stereo-chemical retention; however, the enantiomeric excess was highly dependent on solvent polarity, with increased racemization in more polar media (70 % ee in benzene, 40 % ee in o-dichlorobenzene, and 12 % ee in dichloromethane).
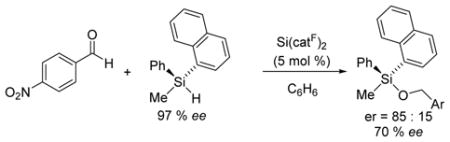 |
(4) |
To investigate the cause of racemization, enantiospecificity studies were performed in the presence of tetrabutylammonium tetrakis(pentafluorophenyl)borate, NBu4BArF4. Salt effects on organic SN1 reactions have provided insight into ionic dissociation steps, typically in polar solvents;22 however, there are fewer examples of salt effects in nonpolar solvents.23 For the hydrosilation shown in eq 4, the addition of NBu4BArF4 had a deleterious effect on the enantiospecificity (26 % ee in the presence of 150.0 mM NBu4BArF4, compared to 70 % ee in the absence of salt).
A proposed mechanism that accounts for the predominant stereochemical retention, as well as the observed solvent and salt effects, is shown in Scheme 2. The aldehyde first coordinates to Si(catF)2 (A), which is followed by hydride transfer from the silane substrate to the activated aldehyde. We suggest that this leads to a silylium alkoxysilicate intimate ion pair intermediate (B) that can undergo rapid silicon–oxygen bond formation, with displacement of the catalyst, within the ion pair prior to silylium rotation, resulting in stereochemical retention. Racemization is caused by the formation of a solvent separated ion pair or ion aggregate (C), which is favored in more polar solvents or upon increasing salt concentration. An alternative mechanism featuring a four-membered cyclic transition state has been proposed for related silane additions to activated carbonyls.24 This concerted, asynchronous addition should afford complete stereochemical retention, and is inconsistent with the observed racemization under more polar conditions. For comparison, a catalytically competent silane–Si(catF)2 adduct should lead to inversion in the major product,20a allowing us to exclude this possible mechanism.
Scheme 2.

Proposed hydrosilation mechanism
In conclusion, bis(perfluorocatecholato)silane (1) was prepared, and reactions to assess its Lewis acidity were investigated. Coordination of fluoride, triethylphosphine oxide, and N,N′-diisopropylbenzamide demonstrate the ability of Si(catF)2 to bind several common classes of Lewis bases. Additionally, hydrosilation and silylcyanation of electron-deficient aldehydes were catalyzed by Si(catF)2 under mild conditions. A stereogenic silicon substrate was employed in combination with solvent and salt effect studies to provide evidence for a carbonyl activation mechanism involving an ionic intermediate. We hope that future work will expand upon the use of neutral, yet potent, silicon Lewis acids in catalytic transformations.
Supplementary Material
Acknowledgments
This work was supported by the Director, Office of Science, Office of Basic Energy Sciences of the US Department of Energy under contract no. DE-AC02-05CH11231 and the National Science Foundation under award no. CHE-0841786. We also acknowledge the National Institutes of Health for funding of the ChexRay X-ray crystallographic facility (College of Chemistry, University of California, Berkeley) under grant no. S10-RR027172. We thank Michael Lipschutz for assistance with X-ray diffraction and Jigar Patel for Chiral HPLC expertise.
Footnotes
Notes
The authors declare no competing financial interests.
Additional experimental information, characterization, X-ray crystallographic details, and CIF files. This material is available free of charge via the Internet at http://pubs.acs.org.
References
- 1.(a) Piers WE, Chivers T. Chem Soc Rev. 1997;26:345. [Google Scholar]; (b) Chen EYX, Marks TJ. Chem Rev. 2000;100:1391. doi: 10.1021/cr980462j. [DOI] [PubMed] [Google Scholar]; (c) Stern CL, Marks TJ. J Am Chem Soc. 1994;116:10015. [Google Scholar]; (d) Bochmann M, Lancaster SJ, Hursthouse MB, Malik KMA. Organometallics. 1994;13:2235. [Google Scholar]; (e) Bochmann M, Dawson DM. Angew Chem Int Ed Engl. 1996;35:2226. [Google Scholar]
- 2.(a) Parks DJ, Piers WE. J Am Chem Soc. 1996;118:9440. [Google Scholar]; (b) Parks DJ, Blackwell JM, Piers WE. J Org Chem. 2000;65:3090. doi: 10.1021/jo991828a. [DOI] [PubMed] [Google Scholar]; (c) Chandrasekhar S, Reddy CR, Babu BN. J Org Chem. 2002;67:9080. doi: 10.1021/jo0204045. [DOI] [PubMed] [Google Scholar]; (d) Rubin M, Schwier T, Gevorgyan V. J Org Chem. 2002;67:1936. doi: 10.1021/jo016279z. [DOI] [PubMed] [Google Scholar]; (e) Ishihara K, Hanaki N, Yamamoto H. Synlett. 1993:577. [Google Scholar]
- 3.Revunova K, Nikonov GI. Dalton Trans. 2015;44:840. doi: 10.1039/c4dt02024c. [DOI] [PubMed] [Google Scholar]
- 4.(a) Denmark SE, Beutner GL, Wynn T, Eastgate MD. J Am Chem Soc. 2005;127:3774. doi: 10.1021/ja047339w. [DOI] [PubMed] [Google Scholar]; (b) Denmark SE, Chung W. J Org Chem. 2008;73:4582. doi: 10.1021/jo8006539. [DOI] [PubMed] [Google Scholar]; (c) Azizi N, Baghi R, Ghafuri H, Boloutchian M, Hashemi M. Synlett. 2010:379. [Google Scholar]
- 5.(a) Müther K, Mohr J, Oestreich M. Organometallics. 2013;32:6643. [Google Scholar]; (b) Nödling AR, Müther K, Rohde VHG, Hilt G, Oestreich M. Organometallics. 2014;33:302. [Google Scholar]
- 6.(a) Reißmann M, Schäfer A, Jung S, Müller T. Organometallics. 2013;32:6736. [Google Scholar]; (b) Herrington TJ, Ward BJ, Doyle LR, McDermott J, White AJP, Hunt PA, Ashley AE. Chem Commun. 2014;50:12753. doi: 10.1039/c4cc05905k. [DOI] [PubMed] [Google Scholar]
- 7.(a) Suen LM, Steigerwald ML, Leighton JL. Chem Sci. 2013;4:2413. doi: 10.1039/C3SC50714A. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Kubota K, Hamblett CL, Wang X, Leighton JL. Tetrahedron. 2006;62:11397. [Google Scholar]
- 8.This range represents the shortest and longest distances measured between the least-squares mean plane defined by the catecholate ligands of the molecule containing Si1 to individual atoms of the Si2-containing silicate complex.
- 9.Harland JJ, Day RO, Vollano JF, Sau AC, Holmes RR. J Am Chem Soc. 1981;103:5269. [Google Scholar]
- 10.(a) Mayer U, Gutmann V, Gerger W. Monatsh Chem. 1975;106:1235. [Google Scholar]; (b) Beckett MA, Strickland GC, Holland JR, Varma KS. Polymer. 1996;37:4629. [Google Scholar]
- 11.Childs RF, Mulholland DL, Nixon A. Can J Chem. 1982;60:801. [Google Scholar]
- 12.Parks DJ, Piers WE, Parvez M, Atencio R, Zaworotko MJ. Organometallics. 1998;17:1369. [Google Scholar]
- 13.Denmark SE, Ueki Y. Organometallics. 2013;32:6631. doi: 10.1021/om400582k. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.(a) Kira M, Hino T, Sakurai H. Chem Lett. 1992:555. [Google Scholar]; (b) Müther K, Oestreich M. Chem Commun. 2011;47:334. doi: 10.1039/c0cc02139c. [DOI] [PubMed] [Google Scholar]
- 15.Hamashima Y, Sawada D, Kanai M, Shibasaki M. J Am Chem Soc. 1999;121:2641. [Google Scholar]
- 16.(a) Kim SS, Rajagopal G, Song DH. J Organomet Chem. 2004;689:1734. [Google Scholar]; (b) Denmark SE, Chung W. J Org Chem. 2006;71:4002. doi: 10.1021/jo060153q. [DOI] [PubMed] [Google Scholar]
- 17.NMR kinetics experiments were attempted for the hydrosilation of 2-trifluoromethylbenzaldehyde with a 10-fold excess of triethylsilane at 30 °C. Unfortunately, the kinetics data were complex and could not be fit to a standard rate law, in part due to complications from catalyst decomposition.
- 18.Koller J, Bergman RG. Organometallics. 2012;31:2530. [Google Scholar]
- 19.Blackwell JM, Foster KL, Beck VH, Piers WE. J Org Chem. 1999;64:4887. doi: 10.1021/jo9903003. [DOI] [PubMed] [Google Scholar]
- 20.(a) Rendler S, Oestreich M. Angew Chem Int Ed. 2008;47:5997. doi: 10.1002/anie.200801675. [DOI] [PubMed] [Google Scholar]; (b) Metsänen TT, Hrobárik P, Klare HFT, Kaupp M, Oestreich M. J Am Chem Soc. 2014;136:6912. doi: 10.1021/ja503254f. [DOI] [PubMed] [Google Scholar]; (c) Shinke S, Tsuchimoto T, Kawakami Y. Silicon Chem. 2007;3:243. [Google Scholar]
- 21.(a) Sommer LH, Frye CL, Parker GA, Michael KW. J Am Chem Soc. 1964;86:3271. [Google Scholar]; (b) Ojima Y, Yamaguchi K, Mizuno N. Adv Synth Catal. 2009;351:1405. [Google Scholar]
- 22.(a) Winstein S, Klinedinst PE, Robinson GC. J Am Chem Soc. 1961;83:885. [Google Scholar]; (b) Allen AD, Tidwell TT, Tee OS. J Am Chem Soc. 1993;115:10091. [Google Scholar]; (c) Loupy A, Tchoubar B. Salt Effects in Organic and Organometallic Chemistry. VCH; Weinheim, Germany: 1992. [Google Scholar]
- 23.(a) Winstein S, Friedrich EC, Smith S. J Am Chem Soc. 1964;86:305. [Google Scholar]; (b) Perrin CL, Pressing J. J Am Chem Soc. 1971;93:5705. [Google Scholar]; (c) Tellers DM, Yung CM, Arndtsen BA, Adamson DR, Bergman RG. J Am Chem Soc. 2002;124:1400. doi: 10.1021/ja011809u. [DOI] [PubMed] [Google Scholar]
- 24.(a) Doyle MP, West CT. J Org Chem. 1975;40:3835. [Google Scholar]; (b) Doyle MP, West CT, Donnelly SJ, McOsker CC. J Organomet Chem. 1976;117:129. [Google Scholar]; (c) Sakata K, Fujimoto H. J Org Chem. 2013;78:12505. doi: 10.1021/jo402195x. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.