

The development of congestive heart failure is characterized by adaptive responses that lead to maladaptive neurohormonal activation to compensate for the reduction in left ventricular function. These changes are largely characterized by increased sympathetic tone. Neurohormonal activation can alter myocardial loading conditions and lead to myocyte cell death resulting in hypertrophy and further left ventricular dysfunction. In addition, the inhomogeneity of myocardial sympathetic innervation arising from ischemia as well as necrosis along with the sympathovagal imbalance that develops in response to heart failure can lead to a substrate that is arrhythmogenic1–6. Advances in the medical therapy of heart failure have focused on attenuating these physiological and cellular effects through beta adrenergic receptor blockade and RAAS inhibition (angiotensin converting enzyme inhibitors, angiotensin receptor blockers, and aldosterone antagonists). While these therapies attenuate the progression of heart failure, increase survival and improve heart failure symptoms, the long-term prognosis of heart failure patients remains poor.
Cardiac device therapy has provided additional improvement in the care of selected patients with depressed systolic function and a dilated left ventricle. While much of the benefit is derived from preventing sudden death from lethal ventricular arrhythmias using implantable defibrillators, a subset of patients with left bundle branch block and heart failure are clinically improved by reducing dysynchronous left ventricular contraction7, 8. This is accomplished by pacing the left as well as the right ventricular chamber (biventricular pacing) to prevent the temporal and spatial dispersion in mechanical activation of the left ventricle. The mechanisms responsible for the beneficial effects of cardiac resynchronization therapy include immediate improvements in hemodynamics as well as delayed cellular and molecular remodeling of the left ventricle9. Proteomic and transcriptomic studies of the molecular mechanisms altered in response to biventricular pacing have identified multiple biochemical pathways demonstrating favorable reverse remodeling. Among these include enzymes involved in mitochondrial energetics, myocyte calcium handling and contractility10–13. Other mechanisms lead to molecular remodeling throughout the entire left ventricle and include improved beta adrenergic receptor signaling with reductions in neurohormonal activation14.
In this issue of Circulation Research, DeMazumder and colleagues add to the growing body of evidence that adverse cardiac neuronal remodeling in heart failure may underlie some of the long-term therapeutic benefits of resynchronization therapy15. They employed a well-characterized canine model of tachycardia-induced congestive heart failure secondary to rapid atrial pacing with catheter ablation used to produce chronic left bundle branch block. In vivo studies demonstrated that resynchronization with biventricular pacing led to the expected improvement in global myocardial function as well as indices of left ventricular filling pressure and contractility in vivo. In vitro studies evaluating isolated myocytes demonstrated increased cellular contractility as well as improvement in the cellular calcium transients after resynchronization. The cellular responses to adrenergic and muscarinic stimulation in this isolated cellular preparation were quite intriguing. Failing myocytes had an increased sensitivity to cholinergic agonists which led to reduced contractile responses and attenuated contractile responses to beta adrenergic stimulation. This was accompanied by increased M2 muscarinic receptor density. The negative inotropic effects of cholinergic stimulation could be blocked by atropine. While reducing contractility, muscarinic activation protected the myocyte from after transients during isoproterenol in vitro. These effects could be blocked with pertussis toxin suggesting a potential beneficial action of muscarinic activation of inhibitory G proteins on malignant arrhythmias. Cardiac resynchronization therapy also improved cellular calcium transients and myocyte systolic shortening and tissue analyses demonstrated that the upregulation of M2 muscarinic receptors in heart failure was largely normalized. The translational relevance of the findings to humans was supported by additional studies showing an increase in M2 muscarinic receptors using immunofluorescence in explanted cardiac tissue from advanced heart failure patients vs. normal controls. The authors conclude that the beneficial effects of biventricular pacing include post synaptic remodeling of muscarinic as well as beta adrenergic receptors that synergistically act to reverse inhibitory G protein signaling bias in the advanced failing heart. These findings add to the extensive previous work from these investigators regarding molecular remodeling in dysynchronous heart failure and its response to cardiac resynchronization. The upregulation of cardiomyocyte muscarinic receptors in heart failure is novel and appears to be a “two edged sword”. While cholinergic stimulation reduces after depolarizations during adrenergic activation and potentially protects the failing heart from ventricular arrhythmias, it adversely attenuates beta adrenergic mediated increases in myocyte contractility and depresses cardiac function.
This study adds to the growing body of evidence demonstrating surprising plasticity in adrenergic and cholinergic neurotransmitters in heart failure as well as other pathophysiological states. While providing important new insight regarding muscarinic receptor plasticity, several issues are not directly addressed in the present study. First, while the isolated myocyte responses show convincing effects during pharmacological stimulation in vitro, studies were not conducted in the intact heart to assess the quantitative impact of parasympathetic activation or administration of muscarinic agonists or antagonists on global left ventricular function in vivo. Second, it isn’t clear whether muscarinic receptor effects were regional or occurred throughout the left ventricle of the dysynchronous heart since all myocytes were obtained from the mid myocardial lateral wall between the anterior and circumflex arteries. Finally, the studies were conducted in the absence of the usual medical care afforded to heart failure patients which include chronic beta blockade and RAAS inhibition. It remains unclear whether upregulation of muscarinic receptors might be less prominent when pharmacological therapy to attenuate neurohormonal activation was present. Further studies addressing these aspects would be welcome since they would provide insight into the in vivo role of muscarinic receptor modulation in heart failure.
An important remaining question relates to how the upregulated muscarinic receptors would be activated to bias Gi signaling in vivo. There is relatively high vagal tone and low sympathetic tone in the normal heart at rest but as heart failure develops, sympathetic tone increases and vagal tone diminishes6. In addition, the effects of electrical vagal stimulation on contractility are variable and perhaps dependent on species as well as the experimental preparation16. Some studies in dogs have demonstrated no effect of vagal stimulation on contractility in the normal heart when the effects of heart rate and afterload are controlled17, 18. Based upon these considerations, one might anticipate that there could be little parasympathetic tone to activate the upregulated muscarinic receptors on cardiac myocytes in advanced heart failure.
While there is a withdrawal of vagal tone in heart failure, a potential source of muscarinic activation may arise from the known plasticity of neurotransmitters expressed in presynaptic cardiac sympathetic nerves19, 20. Adrenergic nerves have their cell bodies in the stellate ganglion and they normally contain tyrosine hydroxylase as well as other enzymes required to produce the norepinephrine released from presynaptic sympathetic nerves. Kanazawa et al. have demonstrated that congestive heart failure can transition sympathetic nerves from an adrenergic to a cholinergic subtype with acetylcholinesterase replacing tyrosine hydroxylase19. Thus, a subpopulation of sympathetic nerves with their cell bodies in the stellate ganglion could shift to a cholinergic subtype and activate muscarinic receptors independent of the usual vagal innervation pathway (Figure). The net effect of this presynaptic neuronal plasticity in the failing heart would be to increase muscarinic responses while reducing adrenergic activation thus biasing myocyte intracellular signaling towards inhibitory G protein activation. This would protect the failing heart from lethal arrhythmias during sympathetic activation but enhanced muscarinic activation would reduce contractility during exercise. While speculative, the upregulation of muscarinic receptors could also be a factor responsible for shifting the mechanism of cardiovascular death in advanced heart failure from ventricular fibrillation to electromechanical dissociation as well as asystole21. If this proves to be correct, novel approaches to restore the normal neurotransmitter composition of sympathetic nerves along with approaches to reduce muscarinic receptor upregulation in cardiac myocytes may become important new targets to treat patients with advanced heart failure. Further study to determine whether cardiac resynchronization normalizes the plasticity of presynaptic sympathetic neurotransmitters in dysynchronous heart failure would also be informative.
Figure. Cholinergic remodeling in heart failure.
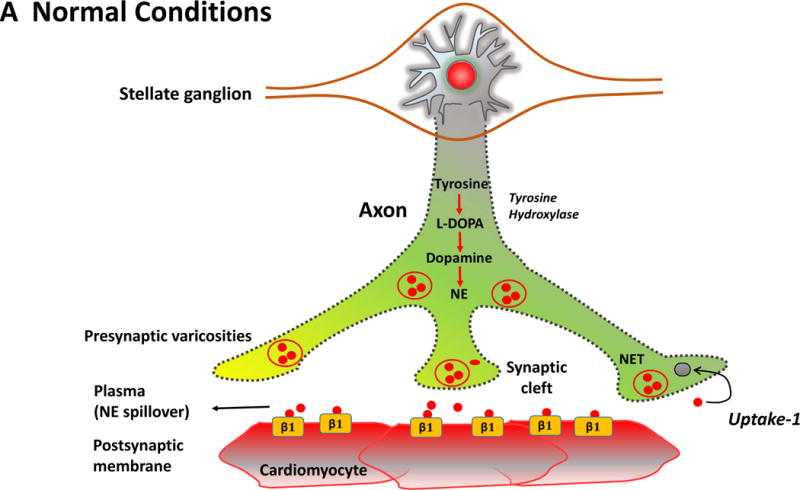
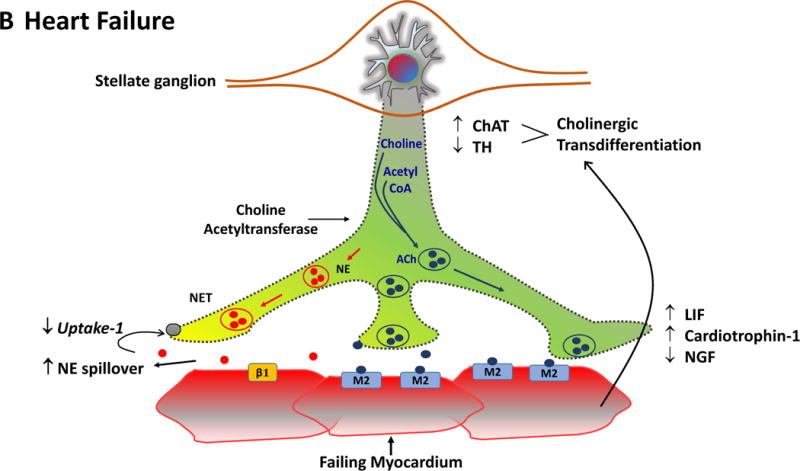
The Figure summarizes the plasticity of neurotransmitters in the sympathetic nerve ganglion in conjunction with postsynaptic receptors on cardiac myocytes. In the normal heart (Panel A), myocardial sympathetic nerves originating in the stellate ganglion secrete norepinephrine (NE, red) to increase heart rate and myocardial contractility through β1-adrenergic receptor signaling. The interstitial NE level at the receptor is dependent upon release as well as the presynaptic reuptake (Uptake 1) mechanism for recycling it into the presynaptic terminal. Parasympathetic control of ventricular contractility (not illustrated) is generally felt to play a minor role. In heart failure (Panel B), plasticity of neurons in the sympathetic ganglia leads to transdifferentiation of some cell bodies to a cholinergic phenotype (blue). Some sympathetic nerves now express Choline Acetyltransferase (AChAT) and tyrosine hydroxylase (TH) expression decreases. This transdifferentiation is promoted by Leukemia Inhibitory Factor (LIF), Cardiotrophin-1 expression and Nerve Growth Factor (NGF) released from the myocardium. Reductions in NE reuptake as well as increased circulating NE levels elevate interstitial NE and lead to post-synaptic down regulation of β1 receptor density, uncoupling of β1 receptors with G proteins and desensitization of β adrenergic signaling. In heart failure, cardiomyocytes paradoxically increase the expression of muscarinic (M2) receptors. The result of cholinergic transdifferentiation of sympathetic neurons and the upregulation of M2 muscarinic receptors leads to diminished beta adrenergic responses and could paradoxically potentiate cholinergic myocardial responses (paradoxic bradycardia and reduced contractility) during sympathetic nerve activation. ACh: Acetylcholine, NET: Norepinephrine transporter.
In summary, this study provides new insight into the diverse pathophysiological alterations in cardiac neurohormonal control in heart failure. There is currently great clinical interest in developing devices that employ neural stimulation to favorably modulate sympathovagal balance6. While small studies have demonstrated that neural stimulation can improve left ventricular function and reduce arrhythmias in heart failure, the most recent prospective randomized trial (DEFEAT-HF) of spinal stimulation has been negative22. This may reflect the complexity of postsynaptic and presynaptic neural plasticity which varies in relation to the underlying disease process as well as its severity. The combination of in vivo large animal models of heart failure with in vitro approaches to identify cellular mechanisms as employed in the study the study of DeMazumder et al. is likely to help us unravel the effects of other types of cardiac neural modulation. Information such as this may help direct these therapeutic interventions to circumstances where they would most likely improve symptoms, prevent death and retard the progression of heart failure.
Acknowledgments
Sources of Funding
Supported by the National Heart Lung and Blood Institute (HL-055324, HL-061610) and the Albert and Elizabeth Rekate Fund in Cardiovascular Medicine.
Footnotes
Disclosures
None.
References
- 1.Cao JM, Fishbein MC, Han JB, Lai WW, Lai AC, Wu TJ, Czer L, Wolf PL, Denton TA, Shintaku IP, Chen PS, Chen LS. Relationship between regional cardiac hyperinnervation and ventricular arrhythmia. Circulation. 2000;101:1960–1969. doi: 10.1161/01.cir.101.16.1960. [DOI] [PubMed] [Google Scholar]
- 2.Canty JM, Jr, Suzuki G, Banas MD, Verheyen F, Borgers M, Fallavollita JA. Hibernating myocardium: Chronically adapted to ischemia but vulnerable to sudden death. Circ Res. 2004;94:1142–1149. doi: 10.1161/01.RES.0000125628.57672.CF. [DOI] [PubMed] [Google Scholar]
- 3.Han S, Kobayashi K, Joung B, Piccirillo G, Maruyama M, Vinters HV, March K, Lin SF, Shen C, Fishbein MC, Chen PS, Chen LS. Electroanatomic remodeling of the left stellate ganglion after myocardial infarction. J Am Coll Cardiol. 2012;59:954–61. doi: 10.1016/j.jacc.2011.11.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fernandez SF, Ovchinnikov V, Canty JM, Jr, Fallavollita JA. Hibernating myocardium results in partial sympathetic denervation and nerve sprouting. Am J Physiol Heart Circ Physiol. 2013;304:H318–27. doi: 10.1152/ajpheart.00810.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Fallavollita JA, Heavey BM, Luisi J AJ, Michalek SM, Baldwa S, Mashtare J TL, Hutson AD, deKemp RA, Haka MS, Sajjad M, Cimato TR, Curtis AB, Cain ME, Canty J JM. Regional myocardial sympathetic denervation predicts the risk of sudden cardiac arrest in ischemic cardiomyopathy. J Am Coll Cardiol. 2014;63:141–149. doi: 10.1016/j.jacc.2013.07.096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shen MJ, Zipes DP. Role of the autonomic nervous system in modulating cardiac arrhythmias. Circ Res. 2014;114:1004–21. doi: 10.1161/CIRCRESAHA.113.302549. [DOI] [PubMed] [Google Scholar]
- 7.Bristow MR, Saxon LA, Boehmer J, Krueger S, Kass DA, De Marco T, Carson P, DiCarlo L, DeMets D, White BG, DeVries DW, Feldman AM. Cardiac-resynchronization therapy with or without an implantable defibrillator in advanced chronic heart failure. The New England Journal of Medicine. 2004;350:2140–2150. doi: 10.1056/NEJMoa032423. [DOI] [PubMed] [Google Scholar]
- 8.Moss AJ, Hall WJ, Cannom DS, Klein H, Brown MW, Daubert JP, Estes NA, 3rd, Foster E, Greenberg H, Higgins SL, Pfeffer MA, Solomon SD, Wilber D, Zareba W. Cardiac-Resynchronization Therapy for the Prevention of Heart-Failure Events. N Engl J Med. 2009;361:1329–1338. doi: 10.1056/NEJMoa0906431. [DOI] [PubMed] [Google Scholar]
- 9.Spragg DD, Kass DA. Pathobiology of left ventricular dyssynchrony and resynchronization. Prog Cardiovasc Dis. 2006;49:26–41. doi: 10.1016/j.pcad.2006.05.001. [DOI] [PubMed] [Google Scholar]
- 10.Spragg DD, Leclercq C, Loghmani M, Faris OP, Tunin RS, DiSilvestre D, McVeigh ER, Tomaselli GF, Kass DA. Regional alterations in protein expression in the dyssynchronous failing heart. Circulation. 2003;108:929–932. doi: 10.1161/01.CIR.0000088782.99568.CA. [DOI] [PubMed] [Google Scholar]
- 11.Barth AS, Aiba T, Halperin V, DiSilvestre D, Chakir K, Colantuoni C, Tunin RS, Dimaano VL, Yu W, Abraham TP, Kass DA, Tomaselli GF. Cardiac resynchronization therapy corrects dyssynchrony-induced regional gene expression changes on a genomic level. Circ Cardiovasc Genet. 2009;2:371–8. doi: 10.1161/CIRCGENETICS.108.832345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Agnetti G, Kaludercic N, Kane LA, Elliott ST, Guo Y, Chakir K, Samantapudi D, Paolocci N, Tomaselli GF, Kass DA, Van Eyk JE. Modulation of mitochondrial proteome and improved mitochondrial function by biventricular pacing of dyssynchronous failing hearts. Circ Cardiovasc Genet. 2010;3:78–87. doi: 10.1161/CIRCGENETICS.109.871236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Lichter JG, Carruth E, Mitchell C, Barth AS, Aiba T, Kass DA, Tomaselli GF, Bridge JH, Sachse FB. Remodeling of the sarcomeric cytoskeleton in cardiac ventricular myocytes during heart failure and after cardiac resynchronization therapy. J Mol Cell Cardiol. 2014;72:186–95. doi: 10.1016/j.yjmcc.2014.03.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Chakir K, Daya SK, Aiba T, Tunin RS, Dimaano VL, Abraham TP, Jaques-Robinson KM, Lai EW, Pacak K, Zhu WZ, Xiao RP, Tomaselli GF, Kass DA. Mechanisms of enhanced beta-adrenergic reserve from cardiac resynchronization therapy. Circulation. 2009;119:1231–40. doi: 10.1161/CIRCULATIONAHA.108.774752. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.DeMazumder D, Kass DA, O’Rourke B, Tomaselli GF. Cardiac resynchronization therapy restores sympathovagal balance in the failing heart by differential remodeling of cholinergic signaling. Circ Res. 2015 doi: 10.1161/CIRCRESAHA.116.305268. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Degeest H, Levy MN, Zieske H, Lipman RI. Depression of Ventricular Contractility by Stimulation of the Vagus Nerves. Circ Res. 1965;17:222–35. doi: 10.1161/01.res.17.3.222. [DOI] [PubMed] [Google Scholar]
- 17.Sarnoff SJ, Brockman SK, Gilmore JP, Linden RJ, Mitchell JH. Regulation of ventricular contraction. Influence of cardiac sympathetic and vagal nerve stimulation on atrial and ventricular dynamics. Circ Res. 1960;8:1108–22. doi: 10.1161/01.res.8.5.1108. [DOI] [PubMed] [Google Scholar]
- 18.Matsuura W, Sugimachi M, Kawada T, Sato T, Shishido T, Miyano H, Nakahara T, Ikeda Y, Alexander J, Jr, Sunagawa K. Vagal stimulation decreases left ventricular contractility mainly through negative chronotropic effect. Am J Physiol. 1997;273:H534–9. doi: 10.1152/ajpheart.1997.273.2.H534. [DOI] [PubMed] [Google Scholar]
- 19.Kanazawa H, Ieda M, Kimura K, Arai T, Kawaguchi-Manabe H, Matsuhashi T, Endo J, Sano M, Kawakami T, Kimura T, Monkawa T, Hayashi M, Iwanami A, Okano H, Okada Y, Ishibashi-Ueda H, Ogawa S, Fukuda K. Heart failure causes cholinergic transdifferentiation of cardiac sympathetic nerves via gp130-signaling cytokines in rodents. J Clin Invest. 2010;120:408–21. doi: 10.1172/JCI39778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Shivkumar K, Fukuda K, Kanazawa H, Aizawa Y, Ardell JL. Cardiac innervation and sudden cardiac death. Circ Res. 2015 doi: 10.1161/CIRCRESAHA.116.304679. (In Press) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Goldberger JJ, Basu A, Boineau R, Buxton AE, Cain ME, Canty JM, Jr, Chen PS, Chugh SS, Costantini O, Exner DV, Kadish AH, Lee B, Lloyd-Jones D, Moss A, Myerburg R, Olgin J, Passman R, Stevenson W, Tomaselli GF, Zareba W, Zoloth L. Risk stratification for sudden cardiac death: A plan for the future. Circulation. 2014;129:516–526. doi: 10.1161/CIRCULATIONAHA.113.007149. [DOI] [PubMed] [Google Scholar]
- 22.Zipes DP, Neuzil P, Theres H, Caraway D, Mann DL, Mannheimer C, Van Buren P, Linde C, Linderoth B, Kueffer F, Sarazin SA, DeJongste MJ. Ventricular functional response to spinal cord stimuation for advanced heart failure: Primary results of the randomized Defeat-HF trial. Circulation. 2014;130:2114. (Abstract) [Google Scholar]