

Abstract
Galectin-1 (gal-1), an endogenous lectin secreted by a variety of cell types, has pleiotropic immunomodulatory functions, including regulation of lymphocyte survival and cytokine secretion in autoimmune, transplant disease, and parasitic infection models. However, the role of gal-1 in viral infections is unknown. Nipah virus (NiV) is an emerging pathogen that causes severe, often fatal, febrile encephalitis. The primary targets of NiV are endothelial cells. NiV infection of endothelial cells results in cell-cell fusion and syncytia formation triggered by the fusion (F) and attachment (G) envelope glycoproteins of NiV that bear glycan structures recognized by gal-1. In the present study, we report that NiV envelope-mediated cell-cell fusion is blocked by gal-1. This inhibition is specific to the Paramyxoviridae family because gal-1 did not inhibit fusion triggered by envelope glycoproteins of other viruses, including two retroviruses and a pox virus, but inhibited fusion triggered by envelope glycoproteins of the related Hendra virus and another paramyxovirus. The physiologic dimeric form of gal-1 is required for fusion inhibition because a monomeric gal-1 mutant had no inhibitory effect on cell fusion. gal-1 binds to specific N-glycans on NiV glycoproteins and aberrantly oligomerizes NiV-F and NiV-G, indicating a mechanism for fusion inhibition. gal-1 also increases dendritic cell production of proinflammatory cytokines such as IL-6, known to be protective in the setting of other viral diseases such as Ebola infections. Thus, gal-1 may have direct antiviral effects and may also augment the innate immune response against this emerging pathogen.
Emerging viral pathogens pose a threat to global health and the global economy. Nipah virus (NiV)4 emerged in a 1999 outbreak among livestock workers in Malaysia and Singapore, resulting in 40% mortality among infected patients. In humans, NiV causes a severe encephalitic syndrome, with an average time of 9–10 days from fever onset to death (1). Livestock workers appeared to contract the virus from infected pigs; although NiV infection in animals causes respiratory illness and is not fatal, the transmission rate in animals approaches 100%. Thus, the threat to livestock workers (2) resulted in destruction of livestock worth more than $100 million in the 1999 outbreaks (3). From 2001 to 2004, NiV outbreaks occurred in Bangladesh (4); in Bangladesh, pigs were not found to be infected, suggesting that the virus may have jumped directly from its natural host, the flying fox (genus Pteropus) (5), to humans. In more recent NiV outbreaks, mortality rates increased (up to 74% in Bangladesh) (4), similar to or exceeding those seen with other viruses such as Ebola (6). Thus, NiV has potential to be an agent of human and economic bioterrorism (3). In addition, recent outbreaks of viral diseases such as severe acute respiratory syndrome and avian influenza highlight the need for new antiviral strategies that exploit innate immunity (7, 8).
In humans, NiV infects endothelial cells, particularly in the CNS, lung, kidney, and heart. The pathognomonic feature of NiV infection is endothelial cell syncytia formation (1), resulting in endothelial cell destruction, inflammation, and hemorrhage. Coexpression of codon-optimized NiV-F and NiV-G alone results in syncytia formation of permissive cells; furthermore, we found that specific N-glycans in NiV-F and NiV-G are critical to the fusion process (H. C. Aguilar et al., manuscript in preparation).
Glycosylation of envelope glycoproteins of HIV, influenza, West Nile, and Ebola viruses regulates both fusogenicity of the envelope glycoproteins and viral pathogenicity (9, 10). Moreover, direct interaction of N-glycans on viral glycoproteins with cellular receptors has been implicated in Dengue virus entry into cells (11). The viral glycoproteins are recognized by endogenous lectins or carbohydrate-binding proteins, such as dendritic cell (DC)-specific ICAM-3 grabbing nonintegrin, that are expressed on the surface of the target cells and that mediate viral entry or viral transmission to other cell types (12).
Galectin-1 (gal-1) is a member of a family of lectins with a variety of immunomodulatory functions. gal-1 regulates pre-B cell development and thymocyte and T cell survival (13–17). Administration of gal-1 in animal models of autoimmune and transplant-related disease results in altered T cell cytokine production (18–21). However, the effects of gal-1 on innate immune function are not well understood, and no direct antimicrobial effects of gal-1 have been previously described. As both NiV-F and NiV-G bear complex N-glycans that can be recognized by the galectin family of mammalian lectins, we examined the effect of gal-1 on NiV-F/G-mediated cell fusion.
Materials and Methods
NiV and HeV codon optimization and expression plasmids
Codon optimization of F and G genes from NiV and HeV was performed by Geneart according to an in-house proprietary software that addresses codon usage, elimination of cryptic splice sites, as well as the stability of DNA/RNA secondary structures. The codon-optimized NiV and HeV F and G genes were synthesized chemically and subcloned into the pcDNA3.1 mammalian expression vector (Invitrogen Life Technologies). The sequences of the codon-optimized genes have been deposited into GenBank (pcNiV-Fopt, accession no. AY816748; pcNiV-Gopt, accession no. AY816746). The codon-optimized NiV- F and G were tagged at their C terminus with an AU1 sequence (DTYRYI).
Lectin-binding assay
293T cells were transfected with codon-optimized, AU1-tagged NiV-F or NiV-G. Biotinylated lectins (Vector Laboratories) specific for the indicated glycans were added to NiV-F or NiV-G cell lysates. Lectin-bound NiV-F or NiV-G was precipitated with streptavidin beads and subsequently detected by immunoblotting with anti-AU1 mAb and 125I-labeled protein A. The amount of lectin-precipitated protein was quantified by densitometry using a PhosphorImager (445SI; Molecular Dynamics) and ImageQuant (version 5.2).
Fusion assay
Fusogenicity of wild-type (wt) or mutant NiV-F or NiV-G envelope glycoproteins was determined by transfecting 2 μg of a 1:15 ratio of NiV-F:G expression plasmids into Vero cells (6-well plates) and culturing for 16 h (n ≥ 4 for each mutant). After 4′,6′-diamidino-2-phenylindole staining, nuclei inside syncytia per ×100 field were counted by fluorescence microscopy (at least 10 fields/condition). Syncytia were defined as four or more nuclei within a common cell membrane. The fusogenic index was defined by normalizing the number of nuclei in syncytia per field formed by the specified experimental condition or the various N-glycan mutants to that formed by the wt NiV-F and NiV-G proteins, which was set at 100%. The buffer control includes DTT; as gal-1 has unpaired cysteines in the binding pocket that, in the absence of saccharide ligand, can form intramolecular disulfide bonds and prevent saccharide ligand binding, we routinely store and prepare gal-1 in a buffer with DTT, as described in the original gal-1 purification (22). As gal-1 forms noncovalent dimers, reducing agents have no effect on gal-1 dimerization. Buffers with this concentration of DTT have no effect on fusion in the assays.
Recombinant dimeric and monomeric gal-1
Recombinant human gal-1 was made exactly as described in Ref. 23. Monomeric gal-1 is the N-Gal-1 mutant (24) and was made as previously described, except that a 3 × 7-cm lactosyl-Sepharose affinity column was used to isolate the recombinant gal-1.
Production of N-glycan mutants
There are five potential N-linked glycosylation sites in NiV-F. The N-glycan consensus sequence, NXS/T, where X is any amino acid except for proline, was altered specifically to produce F1 to F5 glycan mutants. In each case the arginine (N) was mutated conservatively to a glutamine (Q) by changing the AAT/AAC codons to CAG using the QuickChange Site-Directed Mutagenesis kit (Stratagene), according to the manufacturer’s directions. Each mutant was sequenced in its entirety to confirm sequence fidelity.
Oligomerization of NiV envelope
Increasing concentrations of gal-1 or N-Gal-1 were added to 293T cells expressing NiV-F or NiV-G for 30 min. Excess gal-1 was removed by washing with PBS, and the indicated amounts of the membrane impermeant cross-linkers (bis(sulfosuccinimidyl) suberate (BS3) for NiV-F and N-(γ-maleimidobutyryloxy)sulfosuccinimide ester (sulfo-GMBS) for NiV-G (Pierce)) were added to the cells. The reactions were quenched according to the manufacturer’s directions, and cells were lysed in 1% Triton X-100. Lysates were separated by SDS-PAGE and NiV-F or -G detected by immunoblotting with monoclonal anti-AU1. Proteins were visualized and quantified by radiometric densitometry using 125I-labeled protein G to detect the amount of bound anti-AU1.
Binding of NiV-F/G to gal-1
Membrane proteins were isolated from 1 × 107 293T cells expressing NiV-F or NiV-G (25). Isolated membrane proteins were solubilized in 0.1% Triton X-100 and applied to a gal-1 affinity column (22). After washing with wash buffer (PBS, 0.1% Triton X-100, and 0.02% NaN3), bound proteins were eluted with 0.1 M β-lactose or 0.1 M sucrose in wash buffer. Samples were resolved by 10% SDS-PAGE and blotted with anti-AU1 mAb. Proteins were visualized using ECL (Amersham Biosciences).
Coimmunoprecipitation of gal-1 and NiV-F
To coprecipitate NiV-F and gal-1, 20 μM gal-1 was first added to 293T cells expressing equivalent amounts of F or F3 Env proteins for 1 h at 37°C. Cells were washed twice with PBS, lysed, and lysates immunoprecipitated with rabbit anti-gal-1 antiserum (1:100 in 10 mM NaPO4, 150 mM NaCl, 1% Triton X-100, 0.1% SDS, 0.5% sodium deoxycholate, and 1× Complete protease inhibitor mix (Roche)). Precipitated proteins were captured on protein G beads, washed three times with wash buffer 1 (100 mM Tris-HCl, 150 mM NaCl, 2 mM EDTA, and 0.2% Nonidet P-40), and three times with wash buffer 2 (100 mM Tris-HCl, 500 mM NaCl, 2 mM EDTA, and 0.2% Nonidet P-40). Bound material was separated by SDS-PAGE, and precipitated NiV-F was detected with an anti-AU1 mAb.
gal-1 stimulation of DCs
Monocytes were cultured for 5 days with IL-4 (100 ng/ml) and GM-CSF (50 ng/ml) in RPMI 1640 medium. At day 5, recombinant gal-1 (20 μM) or N-Gal-1 (20 μM) was added, and cells were cultured for an additional 48 h. In one well, 100 mM lactose were added 20 min before addition of recombinant gal-1. To control for potential endotoxin contamination in the gal-1 or N-Gal-1 stocks, all cells were preincubated with 10 μg/ml polymyxin B at 37°C for 30 min (26). Supernatant was collected after 48 h; IL-6 and TNF-α levels were measured with the cytometric bead array human inflammation kit (BD Biosciences). Values shown are averages from several independent experiments.
Results
Glycosylation screen of NiV-F and NiV-G suggests the presence of gal-1 binding sites
NiV-F and NiV-G are glycosylated heavily. NiV-F and NiV-G have five and seven N-linked glycosylation sites, respectively (Fig. 1), but the structures of the N-glycans at those sites are not known, nor is it known if NiV-F or NiV-G bear O-glycans, although potential sites of O-glycosylation have been identified in NiV-G (Fig. 1). We used a semiquantitative lectin-binding assay to characterize broad classes of glycans on NiV-F and NiV-G (Table I). To identify O-glycans, we used peanut lectin agglutinin (PNA), which recognizes the Galβ(1,3)GalNAc sequence on core 1 or core 2 O-glycans; the presence of PNA binding indicates that O-glycans are present, whereas the absence of PNA binding can indicate lack of O-glycans or capping of the Galβ(1,3)GalNAc sequence by sialic acid (27). As shown in Table I, NiV-G displayed greater PNA binding compared with NiV-F, indicating that NiV-G has more asialo O-glycans than NiV-F.
FIGURE 1.

Schematic of NiV-G and NiV-F envelope glycoproteins. A, NiV-G is a type II transmembrane protein. NiV-F (F0) is a type I transmembrane protein cleaved into F1 and F2 by a host cell protease. *, Potential N-linked glycosylation sites, F1 to F5 and G1 to G7. ⫯, Potential NiV-G O-linked glycosylation sites. Epitopes of NiV-F and NiV-G antipeptide Abs are indicated. Codon-optimized NiV-G and NiV-F were AU1 epitope tagged at the indicated termini. B, NiV-F and NiV-G bind gal-1. Extracts of cell membranes from 293T cells expressing either NiV-F or NiV-G were applied to a gal-1 affinity column. Bound glycoproteins were eluted with β-lactose and detected with anti-AU1 mAb. No glycoproteins were eluted with sucrose wash.
Table I.
Glycosylation profiling by lectin-immunoprecipitation/immunoblot assaya
| Lectin | Specificity | Binding to G | Binding to F |
|---|---|---|---|
| GNV | α(1,3)mannose | + | ++ |
| PHA-E | β(1,6)GlcNAc | ++ | +++ |
| PNA (peanut) | Gal β(1,3)GalNAc | +++ | +/− |
Relative amounts of NiV-F or -G precipitated by the indicated lectins were quantified by densitometry; arbitrary densitometric units: 1–40, +/−; 41–80, +; 81–120, ++; and 121–160, +++. Background is designated as −.
N-glycans can be high mannose, complex, or hybrid structures. Terminal mannose residues on high mannose or hybrid structures are recognized by the Galanthus nivalis (GNV) lectin, and both NiV-G and NiV-F bound GNV, although to different extents. Complex or hybrid structures can be modified by the N-acetylglucosaminyltransferase V (GnT V) enzyme to create the β(1,6)Glc-NAc branch; this branch can be elongated with a polylactosamine sequence (28). The β(1,6)GlcNAc branch is recognized by the Phaseolus vulgaris (PHA-E) lectin (29). Both NiV-F and NiV-G bound PHA-E, demonstrating that both NiV-F and NiV-G bear processed N-glycans. Importantly, as the polylactosamine sequences preferentially added to the GnT V branch are recognized by gal-1 (30, 31), this screen suggested that both NiV-G and NiV-F could bind gal-1.
Thus, we sought to determine whether gal-1 could bind directly to NiV-F or NiV-G. The envelope glycoproteins were expressed in 293T cells, cell membrane extracts were applied to a gal-1 affinity matrix, and bound glycoproteins were eluted with lactose. Fig. 1B shows that both NiV-G and NiV-F bound to gal-1.
gal-1 inhibits envelope-mediated fusion
Endothelial cells express several galectins that recognize cell surface saccharides on bacteria, fungi, and parasites (19, 32, 33). As NiV-F and NiV-G bear complex N-glycans that could be recognized by gal-1 (Table I) and endothelial cells are a primary target for NiV infection, we asked whether gal-1 would affect syncytia formation mediated by NiV-F and NiV-G. Because endothelial cell syncytia is a pathognomonic feature of NiV infection (1), we reasoned that a syncytia formation assay would be relevant to test the antiviral activity of gal-1.
Addition of recombinant human gal-1 to Vero cells expressing NiV-F and NiV-G abrogated syncytia formation by ~90% (Fig. 2A). gal-1 also inhibited NiV-F/G-mediated fusion in other permissive cell lines such as human 293T and mouse NIH 3T3 cells (data not shown). The inhibitory effect of gal-1 was specifically antagonized by polyclonal anti-gal-1 antiserum. Importantly, anti-gal-1 antiserum alone in the absence of exogenous gal-1 did not inhibit syncytia formation, indicating that endogenous gal-1 (that can be secreted and bind back to cell surfaces) is not a viral receptor on the cell surface. Moreover, the inhibitory effects of gal-1 on NiV F/G-mediated cell fusion appeared specific for the Paramyxoviridae family (Fig. 2B). gal-1 inhibited fusion mediated by Hendra and human parainfluenza virus type III F and G glycoproteins (both are paramyxoviruses) but had no effect on fusion mediated by two retroviruses, human T lymphotrophic virus II and murine leukemia virus, or by a pox virus, vaccinia (WR strain) (Fig. 2B). In addition, gal-1 inhibition of NiV fusion is fully reversible with lactose, a cognate ligand for gal-1, but not sucrose (Fig. 2C), indicating that the fusion inhibitory effect of gal-1 is dependent on its interaction with carbohydrate moieties on the viral envelope glycoproteins.
FIGURE 2.

gal-1 blocks NiV envelope-mediated fusion. A, gal-1 inhibits NiV-F- and NiV-G-mediated fusion. Vero cells expressing NiV-F and NiV-G were cultured for 16 h in medium with gal-1 (20 μM in 0.32 mM DTT) or buffer control. gal-1 has unpaired cysteines in the binding pocket that can form intra- or intermolecular disulfide binds in the absence of saccharide ligands, which can lead to inactivation of the binding site. Thus, the buffer control includes the same concentration of DTT, a reducing agent, which prevents gal-1 inactivation. To demonstrate the specificity of gal-1 inhibition, rabbit anti-gal-1 serum (in increasing amounts) or preimmune serum (IgG) were added simultaneously with gal-1. Fusion was quantified as described in Materials and Methods. Values of p were calculated using the Student t test (unpaired samples with unequal variance). Mean ± SD of one representative experiment of three is presented. Note that anti-gal-1 sera alone did not inhibit NiV-G/F-mediated fusion. B, Specificity of gal-1 inhibition for Paramyxoviridae family: inhibition was observed only for fusion mediated by Hendra virus (HeV) glycoproteins and human parainfluenza virus type III (HPIV III), whereas no significant inhibition was seen for fusion mediated by glycoproteins from human T lymphotrophic virus II (HTLV II), murine leukemia virus (MLV), or vaccinia (WR strain). Mean ± SE of two determinations. C, Lactose reversal of gal-1 inhibition. A total of 10 mM lactose, a saccharide ligand for gal-1, or 10 mM sucrose, which does not bind gal-1, was added to fusion assays concurrently with gal-1. Fusion was quantified as above. Mean ± SE of two determinations.
The fusion inhibitory activity of gal-1 is dependent on its ability to dimerize. The IC50 of gal-1 in blocking cell fusion induced by NiV-G and NiV-F was in the 7–10 μM range (Fig. 3A). gal-1 forms noncovalent homodimers at this concentration (34, 35), suggesting that dimeric gal-1 was essential for the inhibitory effect on fusion. We confirmed that dimeric gal-1 was required to inhibit NiV-F- and NiV-G-mediated fusion by using a mutant N-Gal-1 that remains monomeric < 250 μM (24). N-Gal-1 had no effect on fusion of cells expressing NiV-F and NiV-G (Fig. 3A), although N-Gal-1 retained specific carbohydrate-binding activity (Fig. 3B). As also shown in Fig. 3C, the N-Gal-1 is monomeric because we observed no agglutination of T cells by the N-Gal-1 at concentrations up to 20 μM, while we see clear T cell agglutination by native gal-1 at the same concentration. Thus, the inhibitory effect of gal-1 on syncytia formation is dependent on both the ability to dimerize and the ability to bind carbohydrate ligand (Fig. 2C).
FIGURE 3.

Dimeric gal-1 is required to block NiV-F/G-mediated fusion. A, Monomeric (N-Gal-1) or dimeric gal-1 was added to Vero cells transfected with NiV-F and NiV-G. Fusion was quantified as in Fig. 2. Note that monomeric gal-1 had no effect on fusion even up to 25 μM. Mean ± SD of one representative experiment of three is presented. B, N-Gal-1 binds to T cells in a lactose inhibitable fashion. Increasing concentrations of biotinylated monomeric gal-1 were added to T cells. After binding for 30 min at 4°C, cells were washed once in 1× PBS, and bound gal-1 was detected by addition of FITC-conjugated streptavidin and flow cytometry. The highest amount of N-Gal-1 binding observed was reversed by the addition of 0.1 mM lactose. MFI, mean fluorescence intensity. C, N-Gal-1 is monomeric. gal-1 or N-Gal-1 was added to CEM T cells for 30 or 180 min, and the cells examined by phase-contrast microscopy. Dimeric gal-1 agglutinated T cells (arrows) due to the ability to bind carbohydrate ligands on adjacent cells. N-Gal-1 was unable to agglutinate T cells, indicating that N-Gal-1 is monomeric at this concentration.
gal-1 binds to specific N-glycans on NiV-F
To determine whether gal-1 preferentially binds to specific N-glycan sites on NiV-F, we performed a gal-1 dose-response curve for NiV-F and the NiV-F1, NiV-F2, NiV-F3, NiV-F4, and NiV-F5 mutants (see Materials and Methods). All the N-glycan mutants were similarly sensitive to gal-1 inhibition as wt NiV-F (Fig. 4A), with the exception of the NiV-F3 mutant (Fig. 4B). For example, note that 20 μM gal-1 abrogated fusion for NiV-F and the NiV-F1, F2, F4, and F5 mutants (Fig. 4A), whereas the NiV-F3 mutant still exhibited 50% of control fusion activity at this gal-1 concentration (Fig. 4B) (p < 0.0125, Student’s t test after Bonferroni inequality correction for paired-wise comparison of wt NiV-F against F1, F2, F3, F4, and F5 mutants). Thus, the NiV-F3 mutant was significantly more resistant to gal-1 inhibition, suggesting that the F3 glycan was important for gal-1 binding to NiV-F.
FIGURE 4.

gal-1 binds to a specific N-glycan site in NiV-F and NiV-G. A, F1, F2, F4, and F5 mutants are as sensitive to gal-1 inhibition as wt NiV-F. Vero cells transfected with NiV-G and wt NiV-F or the indicated NiV-F mutants (F1, F2, F4, or F5) were cultured with indicated concentrations of gal-1 and fusogenicity determined as in Fig. 1. Ten fields were counted for each mutant per condition. The number of nuclei in syncytia per field with no gal-1 treatment is normalized to 100% (fusogenic index). Mean ± SD of one representative experiment of three is presented. B, NiV-F3 glycan mutant is resistant to gal-1 inhibition of fusion. For clarity, results are shown only for wt NiV-F and F3 only. The experiment was performed identically as described in A. C, The NiV-F3 mutant reduces gal-1 binding. gal-1 (20 μM) was added to 293T cells expressing equivalent amounts of NiV-F or NiV-F3 protein, cell lysates were immunoprecipitated with rabbit anti-gal-1 antiserum, and NiV-F and NiV-F3 were detected with anti-AU1. D, gal-1 binding to NiV-F is dependent on lectin-glycan interaction. DMNJ (2 mM), which blocks the processing required for formation of complex/hybrid N-glycans, was added to 293T cells expressing NiV-F for 48 h. gal-1 (20 μM) was added to the cells, the cells were lysed, and the lysates were immunoprecipitated with an anti-gal-1 Ab. Precipitated proteins were detected with anti-AU1. CL indicates the input cell lysate while IP indicated precipitated protein. Note that DMNJ treatment itself did not affect levels of NiV-F.
To directly examine gal-1 binding to the NiV-F3 mutant, gal-1 was added to intact 293T cells expressing either NiV-F or NiV-F3. Cell lysates were immunoprecipitated with anti-gal-1 antiserum, and NiV-F or NiV-F3 was detected by immunoblotting. gal-1 precipitated significantly less NiV-F3 compared with wt NiV-F, indicating that gal-1 binds less efficiently to NiV-F3 (Fig. 4C). This result was not due to differential expression because NiV-F and NiV-F3 were expressed equivalently on the cell surface (data not shown). Our results indicate that the F3 N-glycan contributes significantly to gal-1 binding to NiV-F; however, the other NiV-F N-glycans can also contribute to gal-1 binding because a small amount of NiV-F3 is still coimmunoprecipitated with gal-1. To demonstrate that NiV-F binding requires complex N-glycans containing the preferred gal-1 saccharide ligands (see Table I), cells were treated with deoxymannojirimycin (DMNJ), which blocks the Golgi mannosidase II required for complex/hybrid N-glycan formation. As shown in Fig. 4D, DMNJ treatment significantly reduced the ability of gal-1 to coimmunoprecipitate with NiV-F. Next, we investigated the mechanism of gal-1 inhibition of NiV glycoprotein-mediated cell fusion.
Oligomerization of NiV-F/G is aberrantly modulated by gal-1
NiV-F and NiV-G glycoproteins are closely associated on the plasma membrane because the two glycoproteins can be coimmunoprecipitated (Fig. 5A). Because the attachment (G) and fusion (F) proteins of paramyxoviral envelope glycoproteins are oligomeric and the oligomeric structure is required for cell fusion (36), we asked whether gal-1 inhibition of fusion resulted from altered oligomerization of NiV-F and/or NiV-G. As with other paramyxoviruses (37), the NiV-F glycoprotein forms trimers (Fig. 5B). However, in the presence of gal-1, an increased fraction of NiV-F was cross-linked into trimers; the ratio of NiV-F trimers to monomers increased 5-fold, from 0.35 to 1.75, in the presence of increasing amounts of gal-1 (Fig. 5B). gal-1 also markedly augmented the oligomerization of NiV-G. Although we detected tetramers of NiV-G in the absence of gal-1, the addition of gal-1 dramatically enhanced the amount of cell surface NiV-G tetramers; the ratio of NiV-G tetramers to monomers increased >400-fold, from 3.8 to 1665, with increasing amounts of gal-1 (Fig. 5B). Importantly, monomeric N-Gal-1 (10 μM) had no effect on the cross-linking of NiV-F or NiV-G (Fig. 5B), demonstrating that dimeric gal-1 was required for enhanced oligomerization of NiV glycoproteins and indicating that altered oligomerization was likely responsible for the decreased cell fusion caused by dimeric gal-1.
FIGURE 5.

gal-1 modulates the oligomeric state of NiV-F and NiV-G. A, Association of NiV-F and NiV-G. Extracts of 293T cells expressing NiV-F and NiV-G (F + G), or NiV-F or NiV-G alone, were immunoprecipitated with anti-F peptide or anti-G peptide antisera. Precipitated proteins were detected with anti-AU1 to detect both NiV-F and NiV-G. Anti-F and anti-G coimmunoprecipitated NiV-G and NiV-F, respectively (lanes 1 and 2). When NiV-F and NiV-G were expressed alone, anti-F and anti-G precipitated only the relevant glycoproteins (lanes 3–6). No precipitated protein was seen in cells transfected with plasmid alone (lanes 7 and 8). B, gal-1 modulates oligomerization of NiV-F and NiV-G. gal-1 was added to 293T cells expressing NiV-F or NiV-G. Cell surface proteins were cross-linked using membrane impermeant cross-linkers (BS3 for NiV-F and sulfo-GMBS (SG) for NiV-G), and NiV-F and NiV-G were detected with anti-AU1. Uncleaved NiV-F monomer (F0) and the estimated NiV-F trimer (3×) are indicated. Molecular mass markers are shown. Molecular mass estimates >180 kDa are not precise, but for NiV-G, the progressive laddering pattern suggests that monomers (1×), dimers (2×), trimers (3×), and tetramers (4×) are formed. M, 10 mM monomeric N-Gal-1, does not enhance oligomerization.
gal-1 induces DC secretion of proinflammatory cytokines
CD45, an established cellular receptor for gal-1 (38, 39), is expressed on DCs (40). A link between CD45 cross-linking and IL-6 signaling has been established in certain cell types, and IL-6 is a proinflammatory cytokine in which increases correlate with increased probability of survival in Ebola-infected patients (41). gal-1 expression is increased at sites of inflammation (21). Thus, as DCs are likely to be exposed to high levels of gal-1 in inflamed tissues, we investigated the effects of gal-1 on the production of proinflammatory mediators by DCs. Monocyte-derived DCs (MDDCs) were matured as described in Fig. 6. gal-1 was added to differentiated MDDCs. As shown in Fig. 6, gal-1 increased MDDC production of IL-6 by at least two orders of magnitude and also stimulated TNF-α secretion. In contrast to gal-1, the monomeric N-Gal-1 mutant failed to induce significant IL-6 or TNF-α secretion, demonstrating that the dimeric form of gal-1 is required to stimulate cytokine secretion by MDDCs. Addition of 100 mM lactose blocked gal-1-induced IL-6 or TNF-α secretion, indicating that induction of proinflammatory cytokines required gal-1 binding to glycan ligands on the MDDCs.
FIGURE 6.
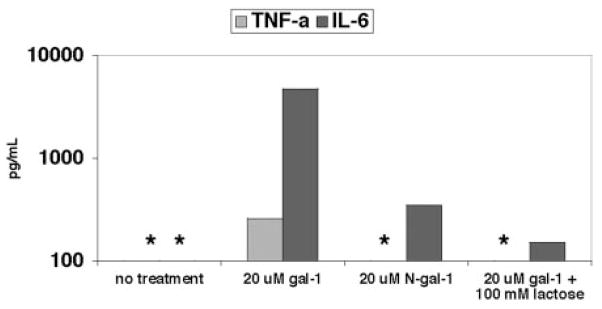
gal-1 induces DC secretion of proinflammatory cytokines. Human PBMCs were cultured for 5 days with IL-4 (100 ng/ml) + GM-CSF (50 ng/ml). At day 5, 20 μM gal-1 (with or without 100 mM lactose) or N-Gal-1 were added, and the culture was continued for an additional 48 h. Cytokine secretion was measured in tissue culture supernatant by cytometric bead array. One experiment of three is shown. *, Lower limit of the linear range.
Discussion
Glycan structures on the Nipah viral F and G envelope glycoproteins play critical roles in viral entry and cell fusion (H. C. Aguilar et al., manuscript in preparation). Some of these glycans bear oligosaccharide structures that can be bound by gal-1, an endogenous lectin expressed by a variety of cell types, including endothelial cells and DCs. The primary targets of NiV infection include endothelial cells and neurons, although the extensive lymphoid necrosis and positive immunostaining for viral Ags seen in lymphoid tissue suggest that this tissue could be similarly involved in primary replication (1). We found that gal-1 can bind directly to NiV-F and NiV-G and that this binding inhibited the cell-cell fusion mediated by NiV-F and NiV-G. It has been reported previously that endothelial cells increase gal-1 synthesis and export to the cell surface in the presence of inflammatory cytokines and that gal-1 is highly expressed by endothelial cells in inflamed lymph nodes (32). In addition, extracellular matrix can concentrate and present enough gal-1 to induce T cell apoptosis (42, 43), suggesting that the amounts of gal-1 that can be found in the extracellular matrix could potentially be sufficient to affect the efficiency of envelope-mediated fusion.
Given the histopathology associated with NiV infection and the elaboration of inflammatory cytokines that participate in the innate immune response to many viral pathogens, our results also suggest that up-regulation of gal-1 expression in the setting of NiV infection could be a mechanism to limit viral spread. The ability of gal-1 to directly block NiV-F/G-mediated cell-cell fusion may result from the ability of gal-1 to perturb NiV-F and NiV-G interactions on the cell surface necessary for membrane fusion. Increased NiV-F and NiV- G homo-oligomerization induced by gal-1 implies that gal-1 segregates NiV-F and NiV-G glycoproteins on the cell surface, perhaps by binding to specific glycans, e.g., the F3 N-glycan. The ability of gal-1 to aggregate cell surface glycoconjugates have been described for the interaction of gal-1 with CD45 on T cells (22). Our report is the first demonstration of a direct antiviral effect for gal-1 or any member of the galectin family. Moreover, the ability of gal-1 to inhibit viral-induced fusion is relatively specific and thus far restricted to Paramyxoviruses.
In addition to endothelial cells, we and others (Ref. 44 and data not shown) have detected gal-1 expression in DCs. Importantly, gal-1 may be an autocrine regulatory factor for endothelial cells and DCs as it is for T cells. For example, Pircher and coworkers (45) found that CD8 cells secrete gal-1 after stimulation and suggested that gal-1 acts as an autocrine factor to control T cell survival after an immune response. Similarly, gal-1 alters T cell secretion of cytokines, including IFN-γ and TNF-α (15, 21, 46). As DCs can express gal-1 and its cognate receptors (such as CD45 and CD43) and DCs adjacent to NiV-infected endothelial cells would be exposed to gal-1 secreted as part of the inflammatory response, we examined the effects of gal-1 on cytokine production by DCs.
In the present study, we report the novel observation that gal-1 dramatically increased IL-6 and TNF-α secretion by MDDCs. This indicates that, in addition to directly blocking NiV F- and G-mediated cell fusion, gal-1 may also promote an innate inflammatory response that would facilitate a successful host response to viral infection. Significantly, two groups have documented human cytokine responses that predict survival vs mortality in patients infected with Ebola virus. Georges and coworkers (41) found that several proinflammatory cytokines, including IL-6 and TNF-α, are markedly elevated in survivors of Ebola infection, whereas both the Georges group and the Peters group (47) found that patients who succumbed to infection typically had increased serum IFN-γ and IL-2 but little or no increase in IL-6 response (41, 47–49). Importantly, we and others (21, 50) have demonstrated that gal-1 decreases IL-2 and IFN-γ production when administered in murine models of immune-mediated disease. The proinflammatory response triggered by gal-1 binding to MDDCs is a novel function for gal-1 and may be particularly important as DCs may be an early target of NiV during lymph node infection (1). Significantly, IL-6 is a prototypical proinflammatory mediator known to induce DC maturation and enhance Ag presentation function or allostimulatory capacity and has been used to generate clinical grade DCs for immunotherapy applications (51–54). Therefore, we hypothesize that gal-1 induction of IL-6 can be further protective during NiV infection by enhancing the Ag presentation function of cognate DCs and acting as an immune adjuvant. Thus, gal-1 may have multiple direct and indirect mechanisms, including inhibition of viral-mediated cell fusion and promotion of cytokine production, to enhance the innate immune system’s response to emerging viral pathogens such as NiV.
Acknowledgments
We thank Irvin Chen for comments, and Jessica Zellhoefer for technical assistance.
Footnotes
This work is supported by National Institutes of Health Grants AI059051 (to B.L.), GM63281 (to L.G.B.), AI06094 (to B.L. and L.G.B.), AI07323 (to L.K.), and AI61824 (to E.L.L.). B.L. is a Charles E. Culpepper Medical Scholar supported by the Rockefeller Brothers Fund and a recipient of the Burroughs Wellcome Fund Career Development Award.
Abbreviations used in this paper: NiV, Nipah virus; DC, dendritic cell; gal-1, galectin-1; wt, wild type; PNA, peanut lectin agglutinin; DMNJ, deoxymannojirimycin; MDDC, monocyte-derived DC; BS3, bis(sulfosuccinimidyl) suberate; sulfo-GMBS, N-(γ-maleimidobutyryloxy)sulfosuccinimide ester; GNV, Galanthus nivalis; GnT V, N-acetylglucosaminyltransferase V; PHA-E, Phaseolus vulgaris.
Disclosures
The authors have no financial conflict of interest.
References
- 1.Wong KT, Shieh WJ, Kumar S, Norain K, Abdullah W, Guarner J, Goldsmith CS, Chua KB, Lam SK, Tan CT, et al. Nipah virus infection: pathology and pathogenesis of an emerging paramyxoviral zoonosis. Am J Pathol. 2002;161:2153–2167. doi: 10.1016/S0002-9440(10)64493-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chua KB, Bellini WJ, Rota PA, Harcourt BH, Tamin A, Lam SK, Ksiazek TG, Rollin PE, Zaki SR, Shieh W, et al. Nipah virus: a recently emergent deadly paramyxovirus. Science. 2000;288:1432–1435. doi: 10.1126/science.288.5470.1432. [DOI] [PubMed] [Google Scholar]
- 3.Lam SK. Nipah virus: a potential agent of bioterrorism? Antiviral Res. 2003;57:113–119. doi: 10.1016/s0166-3542(02)00204-8. [DOI] [PubMed] [Google Scholar]
- 4.Nipah virus outbreak(s) in Bangladesh, January–April 2004. Wkly Epidemiol Rec. 2004;79:168. [PubMed] [Google Scholar]
- 5.Mackenzie JS, Chua KB, Daniels PW, Eaton BT, Field HE, Hall RA, Halpin K, Johansen CA, Kirkland PD, Lam SK, et al. Emerging viral diseases of Southeast Asia and the Western Pacific. Emerg Infect Dis. 2001;7(3 Suppl):497–504. doi: 10.3201/eid0707.017703. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Polesky A, Bhatia G. Ebola hemorrhagic fever in the era of bioterrorism. Semin Respir Infect. 2003;18:206–215. [PubMed] [Google Scholar]
- 7.Hackett CJ. Innate immune activation as a broad-spectrum biodefense strategy: prospects and research challenges. J Allergy Clin Immunol. 2003;112:686–694. doi: 10.1016/S0091-6749(03)02025-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Hsu VP, Hossain MJ, Parashar UD, Ali MM, Ksiazek TG, Kuzmin I, Niezgoda M, Rupprecht C, Bresee J, Breiman RF. Nipah virus encephalitis reemergence, Bangladesh. Emerg Infect Dis. 2004;10:2082–2087. doi: 10.3201/eid1012.040701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Olofsson S, Hansen JE. Host cell glycosylation of viral glycoproteins: a battlefield for host defence and viral resistance. Scand J Infect Dis. 1998;30:435–440. doi: 10.1080/00365549850161386. [DOI] [PubMed] [Google Scholar]
- 10.Wei X, Decker JM, Wang S, Hui H, Kappes JC, Wu X, Salazar-Gonzalez JF, Salazar MG, Kilby JM, Saag MS, et al. Antibody neutralization and escape by HIV-1. Nature. 2003;422:307–312. doi: 10.1038/nature01470. [DOI] [PubMed] [Google Scholar]
- 11.Navarro-Sanchez E, Altmeyer R, Amara A, Schwartz O, Fieschi F, Virelizier JL, Arenzana-Seisdedos F, Despres P. Dendritic-cell-specific ICAM3-grabbing non-integrin is essential for the productive infection of human dendritic cells by mosquito-cell-derived dengue viruses. EMBO Rep. 2003;4:723–728. doi: 10.1038/sj.embor.embor866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Van Kooyk Y, Geijtenbeek TB. DC-SIGN: escape mechanism for pathogens. Nat Rev Immunol. 2003;3:697–709. doi: 10.1038/nri1182. [DOI] [PubMed] [Google Scholar]
- 13.Gauthier L, Rossi B, Roux F, Termine E, Schiff C. Galectin-1 is a stromal cell ligand of the pre-B cell receptor (BCR) implicated in synapse formation between pre-B and stromal cells and in pre-BCR triggering. Proc Natl Acad Sci USA. 2002;99:13014–13019. doi: 10.1073/pnas.202323999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Perillo NL, Uittenbogaart CH, Nguyen JT, Baum LG. Galectin-1, an endogenous lectin produced by thymic epithelial cells, induces apoptosis of human thymocytes. J Exp Med. 1997;185:1851–1858. doi: 10.1084/jem.185.10.1851. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Vespa GN, Lewis LA, Kozak KR, Moran M, Nguyen JT, Baum LG, Miceli MC. Galectin-1 specifically modulates TCR signals to enhance TCR apoptosis but inhibit IL-2 production and proliferation. J Immunol. 1999;162:799–806. [PubMed] [Google Scholar]
- 16.Endharti AT, Zhou YW, Nakashima I, Suzuki H. Galectin-1 supports survival of naive T cells without promoting cell proliferation. Eur J Immunol. 2005;35:86–97. doi: 10.1002/eji.200425340. [DOI] [PubMed] [Google Scholar]
- 17.Rubinstein N, Alvarez M, Zwirner NW, Toscano MA, Ilarregui JM, Bravo A, Mordoh J, Fainboim L, Podhajcer OL, Rabinovich GA. Targeted inhibition of galectin-1 gene expression in tumor cells results in heightened T cell-mediated rejection: a potential mechanism of tumor-immune privilege. Cancer Cell. 2004;5:241–251. doi: 10.1016/s1535-6108(04)00024-8. [DOI] [PubMed] [Google Scholar]
- 18.Hernandez JD, Baum LG. Ah, sweet mystery of death! Galectins and control of cell fate. Glycobiology. 2002;12:127R–136R. doi: 10.1093/glycob/cwf081. [DOI] [PubMed] [Google Scholar]
- 19.Almkvist J, Karlsson A. Galectins as inflammatory mediators. Glycoconj J. 2004;19:575–581. doi: 10.1023/B:GLYC.0000014088.21242.e0. [DOI] [PubMed] [Google Scholar]
- 20.Hsu DK, Liu FT. Regulation of cellular homeostasis by galectins. Glycoconj J. 2004;19:507–515. doi: 10.1023/B:GLYC.0000014080.95829.52. [DOI] [PubMed] [Google Scholar]
- 21.Rabinovich GA, Baum LG, Tinari N, Paganelli R, Natoli C, Liu FT, Iacobelli S. Galectins and their ligands: amplifiers, silencers or tuners of the inflammatory response? Trends Immunol. 2002;23:313–320. doi: 10.1016/s1471-4906(02)02232-9. [DOI] [PubMed] [Google Scholar]
- 22.Pace KE, Lee C, Stewart PL, Baum LG. Restricted receptor segregation into membrane microdomains occurs on human T cells during apoptosis induced by galectin-1. J Immunol. 1999;163:3801–3811. [PubMed] [Google Scholar]
- 23.Pace KE, Hahn HP, Baum LG. Preparation of recombinant human galectin-1 and use in T cell death assays. Methods Enzymol. 2003;363:499–518. doi: 10.1016/S0076-6879(03)01075-9. [DOI] [PubMed] [Google Scholar]
- 24.Cho M, Cummings RD. Characterization of monomeric forms of galectin-1 generated by site-directed mutagenesis. Biochemistry. 1996;35:13081–13088. doi: 10.1021/bi961181d. [DOI] [PubMed] [Google Scholar]
- 25.De Maio A, Lis H, Gershoni JM, Sharon N. Identification of glycoproteins that are receptors for peanut agglutinin on immature (cortical) mouse thymocytes. FEBS Lett. 1986;194:28–32. doi: 10.1016/0014-5793(86)80045-x. [DOI] [PubMed] [Google Scholar]
- 26.Jacobs DM, Morrison DC. Inhibition of the mitogenic response to lipopolysaccharide (LPS) in mouse spleen cells by polymyxin B. J Immunol. 1977;118:21–27. [PubMed] [Google Scholar]
- 27.Gillespie W, Paulson JC, Kelm S, Pang M, Baum LG. Regulation of α2,3-sialyltransferase expression correlates with conversion of peanut agglutinin (PNA)+ to PNA− phenotype in developing thymocytes. J Biol Chem. 1993;268:3801–3804. [PubMed] [Google Scholar]
- 28.Partridge EA, Le Roy C, Di Guglielmo GM, Pawling J, Cheung P, Granovsky M, Nabi IR, Wrana JL, Dennis JW. Regulation of cytokine receptors by Golgi N-glycan processing and endocytosis. Science. 2004;306:120–124. doi: 10.1126/science.1102109. [DOI] [PubMed] [Google Scholar]
- 29.Cummings RD, Kornfeld S. Characterization of the structural determinants required for the high affinity interaction of asparagine-linked oligosaccharides with immobilized Phaseolus vulgaris leukoagglutinating and erythroagglutinating lectins. J Biol Chem. 1982;257:11230–11234. [PubMed] [Google Scholar]
- 30.Di Virgilio S, Glushka J, Moremen K, Pierce M. Enzymatic synthesis of natural and 13C enriched linear poly-N-acetyllactosamines as ligands for galectin-1. Glycobiology. 1999;9:353–364. doi: 10.1093/glycob/9.4.353. [DOI] [PubMed] [Google Scholar]
- 31.Amano M, Galvan M, He J, Baum LG. The ST6Gal I sialyltransferase selectively modifies N-glycans on CD45 to negatively regulate galectin-1-induced CD45 clustering, phosphatase modulation, and T cell death. J Biol Chem. 2003;278:7469–7475. doi: 10.1074/jbc.M209595200. [DOI] [PubMed] [Google Scholar]
- 32.Baum LG, Seilhamer JJ, Pang M, Levine WB, Beynon D, Berliner JA. Synthesis of an endogeneous lectin, galectin-1, by human endothelial cells is up-regulated by endothelial cell activation. Glycoconj J. 1995;12:63–68. doi: 10.1007/BF00731870. [DOI] [PubMed] [Google Scholar]
- 33.Zuniga E, Gruppi A, Hirabayashi J, Kasai KI, Rabinovich GA. Regulated expression and effect of galectin-1 on Trypanosoma cruzi-infected macrophages: modulation of microbicidal activity and survival. Infect Immun. 2001;69:6804–6812. doi: 10.1128/IAI.69.11.6804-6812.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Cho M, Cummings RD. Galectin-1, a β-galactoside-binding lectin in Chinese hamster ovary cells. I. Physical and chemical characterization. J Biol Chem. 1995;270:5198–5206. doi: 10.1074/jbc.270.10.5198. [DOI] [PubMed] [Google Scholar]
- 35.Perillo NL, Pace KE, Seilhamer JJ, Baum LG. Apoptosis of T cells mediated by galectin-1. Nature. 1995;378:736–739. doi: 10.1038/378736a0. [DOI] [PubMed] [Google Scholar]
- 36.Lamb RA, Kolakofsky D. Paramyxoviridae: The Viruses and Their Replication. In: Knipe DM, Howley PM, editors. Fundamental Virology. Lippincott, Williams & Wilkins; Philadelphia: 2001. pp. 689–724. [Google Scholar]
- 37.Russell R, Paterson RG, Lamb RA. Studies with cross-linking reagents on the oligomeric form of the paramyxovirus fusion protein. Virology. 1994;199:160–168. doi: 10.1006/viro.1994.1108. [DOI] [PubMed] [Google Scholar]
- 38.Walzel H, Schulz U, Neels P, Brock J. Galectin-1, a natural ligand for the receptor-type protein tyrosine phosphatase CD45. Immunol Lett. 1999;67:193–202. doi: 10.1016/s0165-2478(99)00012-7. [DOI] [PubMed] [Google Scholar]
- 39.Nguyen JT, Evans DP, Galvan M, Pace KE, Leitenberg D, Bui TN, Baum LG. CD45 modulates galectin-1-induced T cell death: regulation by expression of core 2 O-glycans. J Immunol. 2001;167:5697–5707. doi: 10.4049/jimmunol.167.10.5697. [DOI] [PubMed] [Google Scholar]
- 40.Hart DNJ, MacDonald K, Vuckovic S, Ckark GJ. Phenotypic characterization of dendritic cells. In: Lotze MT, Thomson AW, editors. Dendritic Cells. Academic Press; San Diego: 2001. pp. 97–117. [Google Scholar]
- 41.Baize S, Leroy EM, Georges AJ, Georges-Courbot MC, Capron M, Bedjabaga I, Lansoud-Soukate J, Mavoungou E. Inflammatory responses in Ebola virus-infected patients. Clin Exp Immunol. 2002;128:163–168. doi: 10.1046/j.1365-2249.2002.01800.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Collins BE, Paulson JC. Cell surface biology mediated by low affinity multivalent protein-glycan interactions. Curr Opin Chem Biol. 2004;8:617–625. doi: 10.1016/j.cbpa.2004.10.004. [DOI] [PubMed] [Google Scholar]
- 43.He J, Baum LG. Presentation of galectin-1 by extracellular matrix triggers T cell death. J Biol Chem. 2004;279:4705–4712. doi: 10.1074/jbc.M311183200. [DOI] [PubMed] [Google Scholar]
- 44.Dietz AB, Bulur PA, Knutson GJ, Matasic R, Vuk-Pavlovic S. Maturation of human monocyte-derived dendritic cells studied by microarray hybridization. Biochem Biophys Res Commun. 2000;275:731–738. doi: 10.1006/bbrc.2000.3372. [DOI] [PubMed] [Google Scholar]
- 45.Blaser C, Kaufmann M, Muller C, Zimmermann C, Wells V, Mallucci L, Pircher H. β-Galactoside-binding protein secreted by activated T cells inhibits antigen-induced proliferation of T cells. Eur J Immunol. 1998;28:2311–2319. doi: 10.1002/(SICI)1521-4141(199808)28:08<2311::AID-IMMU2311>3.0.CO;2-G. [DOI] [PubMed] [Google Scholar]
- 46.Rabinovich GA, Ariel A, Hershkoviz R, Hirabayashi J, Kasai KI, Lider O. Specific inhibition of T cell adhesion to extracellular matrix and proinflammatory cytokine secretion by human recombinant galectin-1. Immunology. 1999;97:100–106. doi: 10.1046/j.1365-2567.1999.00746.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Villinger F, Rollin PE, Brar SS, Chikkala NF, Winter J, Sundstrom JB, Zaki SR, Swanepoel R, Ansari AA, Peters CJ. Markedly elevated levels of interferon (IFN)-γ, IFN-α, interleukin (IL)-2, IL-10, and tumor necrosis factor α associated with fatal Ebola virus infection. J Infect Dis. 1999;179(Suppl 1):S188–S191. doi: 10.1086/514283. [DOI] [PubMed] [Google Scholar]
- 48.Baize S, Leroy EM, Georges-Courbot MC, Capron M, Lansoud-Soukate J, Debre P, Fisher-Hoch SP, McCormick JB, Georges AJ. Defective humoral responses and extensive intravascular apoptosis are associated with fatal outcome in Ebola virus-infected patients. Nat Med. 1999;5:423–426. doi: 10.1038/7422. [DOI] [PubMed] [Google Scholar]
- 49.Leroy EM, Baize S, Volchkov VE, Fisher-Hoch SP, Georges-Courbot MC, Lansoud-Soukate J, Capron M, Debre P, McCormick JB, Georges AJ. Human asymptomatic Ebola infection and strong inflammatory response. Lancet. 2000;355:2210–2215. doi: 10.1016/s0140-6736(00)02405-3. [DOI] [PubMed] [Google Scholar]
- 50.Baum LG, Blackall DP, Arias-Magallano S, Nanigian D, Uh SY, Browne JM, Hoffmann D, Emmanouilides CE, Territo MC, Baldwin GC. Amelioration of graft versus host disease by galectin-1. Clin Immunol. 2003;109:295–307. doi: 10.1016/j.clim.2003.08.003. [DOI] [PubMed] [Google Scholar]
- 51.Nieda M, Tomiyama M, Egawa K. Ex vivo enhancement of antigen-presenting function of dendritic cells and its application for DC-based immunotherapy. Hum Cell. 2003;16:199–204. doi: 10.1111/j.1749-0774.2003.tb00154.x. [DOI] [PubMed] [Google Scholar]
- 52.Sorg RV, Ozcan Z, Brefort T, Fischer J, Ackermann R, Muller M, Wernet P. Clinical-scale generation of dendritic cells in a closed system. J Immunother. 2003;26:374–383. doi: 10.1097/00002371-200307000-00010. [DOI] [PubMed] [Google Scholar]
- 53.Luft T, Jefford M, Luetjens P, Hochrein H, Masterman KA, Maliszewski C, Shortman K, Cebon J, Maraskovsky E. IL-1β enhances CD40 ligand-mediated cytokine secretion by human dendritic cells (DC): a mechanism for T cell-independent DC activation. J Immunol. 2002;168:713–722. doi: 10.4049/jimmunol.168.2.713. [DOI] [PubMed] [Google Scholar]
- 54.Jonuleit H, Kuhn U, Muller G, Steinbrink K, Paragnik L, Schmitt E, Knop J, Enk AH. Pro-inflammatory cytokines and prostaglandins induce maturation of potent immunostimulatory dendritic cells under fetal calf serum-free conditions. Eur J Immunol. 1997;27:3135–3142. doi: 10.1002/eji.1830271209. [DOI] [PubMed] [Google Scholar]