

Abstract
We have previously demonstrated that the small molecule octadecenyl thiophosphate (OTP), a synthetic mimic of the growth factor-like mediator lysophosphatidic acid (LPA), showed radioprotective activity in a mouse model of total-body irradiation (TBI) when given orally or intraperitoneally 30 min before exposure to 9 Gy γ radiation. In the current study, we evaluated the effects of OTP, delivered subcutaneously, for radioprotection or radiomitigation from −24 h before to up to +72 h postirradiation using a mouse TBI model with therapeutic doses at around 1 mg/kg. OTP was injected at 10 mg/kg without observable toxic side effects in mice, providing a comfortable safety margin. Treatment of C57BL/6 mice with a single dose of OTP over the time period from −12 h before to +26 h after a lethal dose of TBI reduced mortality by 50%. When administered at +48 h to +72 h postirradiation (LD50/30 to LD100/30), OTP reduced mortality by ≥34%. OTP administered at +24 h postirradiation significantly elevated peripheral white blood cell and platelet counts, increased crypt survival in the jejunum, enhanced intestinal glucose absorption and reduced endotoxin seepage into the blood. In the 6.4–8.6 Gy TBI range using LD50/10 as the end point, OTP yielded a dose modification factor of 1.2. The current data indicate that OTP is a potent radioprotector and radiomitigator ameliorating the mortality and tissue injury of acute hematopoietic as well as acute gastrointestinal radiation syndrome.
INTRODUCTION
Radiation accidents and the threat of radiation terrorism necessitate the development of radioprotective and radiomitigative treatments for first responders, the military and the public at large. To date, no drug has been approved for the treatment of exposures to high levels of ionizing radiation that lead to the development of hematopoietic (HE) and gastrointestinal (GI) acute radiation syndromes (ARS). Due to obvious limitations in human testing, development of such a medical radiation countermeasure (MCM) is regulated under the U.S. Food and Drug Administration’s Animal Rule (21 CFR 314.600–314.650, 601.90–601.95). An important element of this rule is that in-depth understanding of the mechanism of action of the MCM is required for regulatory approval. Clinical indicators of HE-ARS include pancytopenia, which lasts for weeks after initial exposure. The primary causes of death during the later phases of the HE-ARS are opportunistic infections due to leukopenia, and hemorrhage caused by thrombocytopenia, which may be amenable to currently available treatment interventions (1). GI-ARS is manifested by early onset of vomiting, diarrhea, general malabsorption and the breakdown of the gut barrier function leading to invasion of luminal bacteria into the blood. Concomitant HE-ARS may increase the risk of GI-ARS lethality, whereby potential recovery is limited due to compromised immunity, which subsequently leads to infections. Thus, treatment of GI-ARS is currently the limiting step in treatment of lethal doses of total-body irradiation (TBI). Standard supportive care remains the primary approach in managing radiation casualties. We have developed a novel radioprotectant/radiomitigant octadecenyl thiophosphate (OTP), a synthetic derivative of lysophosphatidic acid (LPA), which activates prosurvival and regenerative pathways similar to those elicited by LPA through the LPA2 receptor subtype (2, 3).
LPA is a growth factor-like lipid mediator generated in biological fluids from lysophospholipids by the action of the lysophospholipase enzyme designated as autotaxin (4, 5). LPA is also an intermediate in the early steps of phospholipid biosynthesis generated by glycerophosphate acyltransferases. Serum is a rich endogenous source of LPA with a concentration of ~10 μM/L (6). Significant amounts of LPA have also been detected in saliva, seminal plasma and tumor cell ascites (7). LPA activates multiple subtypes of G protein coupled receptors (GPCRs) (8, 9). LPA1 is widely expressed in most organs or tissues, however, LPA2~5 have a more restricted expression pattern. The LPA1, LPA2 and LPA3 receptors are encoded by the endothelial differentiation genes (EDG) (8). LPA4, LPA5, LPA6 and other non-EDG family member subtypes are distantly related to the EDG family and their biological functions have yet to be characterized (10–12). Most cells express a combination of at least two of these receptors. Both LPA1 and LPA2 have been identified in the bone marrow as well as gastrointestinal tract (13–15).
Physiologically, LPA accelerates wound healing in vivo (16). Studies have shown that LPA is a potent prosurvival factor that protects cells from a variety of apoptotic stimuli in many cell types including Schwann cells, cardiomyocytes, intestinal epithelial cells, osteoblasts, fibroblasts, renal tubular cells, hepatocytes and lymphocytes (17, 18). It has been shown that LPA2 is unique because it contains a PDZ- and a half Zn-finger domain in its C-terminus (19). The LPA-activated assembly of a molecular complex of LPA2 with the proapoptotic protein Siva-1 (20), the thyroid hormone receptor interacting protein 6 (TRIP-6) mediated through the half Zn-finger C-xx-C motif (19) and the PDZ-motif mediated interaction with the Na+/H+ exchange regulatory cofactor 2 (NHERF2) (21) are required steps for the robust antiapoptotic effect mediated by this receptor subtype (19, 20).
We demonstrated previously that LPA was a survival factor that could prevent and rescue intestinal epithelial cells from radiation-induced apoptosis both in vitro and in vivo (15). However, LPA has a very short half-life in blood that is unsuitable for use as a radioprotective drug candidate (22). Octadecenyl thiophosphate is a fatty alcohol thiophosphate analog of LPA that is a metabolically stabilized agonist of various LPA GPCRs. OTP is a potent full agonist for LPA2 with an EC50 of 124 ± 15 nM (2). We have demonstrated in a mouse TBI model that OTP is a highly effective radiomitigator of GI-ARS and HE-ARS (3). It engages similar prosurvival pathways to LPA through LPA2 receptor subtype. In the current study, we performed further in vitro and in vivo evaluation of the radioprotective and radiomitigative efficacy of OTP.
MATERIALS AND METHODS
Materials
OTP was synthesized as an ammonium salt as described by Durgam et al. (23) and used in all experiments. LPA 18:1 was purchased from Avanti® Polar Lipids, Inc. (Alabaster, AL). IEC-6 and HEK293T cells were acquired from ATCC® (Gaithersburg, MD).
Cell Culture
The culture conditions of rat intestinal crypt derived IEC-6 (3, 24) and HEK293T (19) cells were preformed as described in our previous publications (3, 19, 24).
Detection of LPA2 Macromolecular Complex Assembly
HEK-293T cells transfected with epitope-tagged constructs of FLAG-LPA2, MYC-TRIP6 and GFP-NHERF2 were treated with 2 μM OTP for 10 min. FLAG-LPA2 was pulled down using anti-FLAG beads and bound proteins were analyzed using Western blotting, as described in our earlier publications (19, 20). Briefly, FLAG-tagged LPA2 was pulled down from cell lysates of transfected HEK-293T cells with anti-FLAG beads (Sigma-Aldrich® LLC, St. Louis, MO) and detected using an anti-LPA2 antibody (gift from Dr. A. P. Naren, University of Tennessee Health Science Center, Memphis, TN). TRIP6 was detected with an anti-TRIP6 mouse monoclonal antibody (BD Biosciences, San Jose, CA) and NHERF2 was monitored using an antibody specific to NHERF2, kindly provided by Dr. A. P. Naren.
Animal Experiments
All animal protocols were reviewed and approved by the Institutional Animal Care and Use Committee of the Memphis VA Medical Center or the University of Tennessee Health Science Center. C57BL/6 mice aged 8–12 weeks were purchased from Harlan® Laboratories (Indianapolis, IN) or Jackson Laboratory (Bar Harbor, ME). Female mice were used for each study except for the pharmacokinetics experiment where both genders were used. Mice were maintained on a 12:12 h light-dark schedule and fed standard laboratory mouse chow and sterilized water ad libitum. Experiments were initiated after 2 weeks of acclimatization.
Pharmacokinetics of OTP
Blood was collected from 4 male and 4 female mice by cardiac puncture at predefined time points relative to test article dosing (0.5, 1, 2, 4, 6, 8, 12, 18, 24 and 48 h) and collected into micro plasma separator BD Microtainer® vials (Becton-Dickinson and Co., Franklin Lakes, NJ) containing lithium heparin. Blood was maintained on ice until centrifugation and subsequent processing. OTP was quantified in the plasma samples using the LC-MS method described by Kosanam et al. (25). The pharmacokinetic parameters were calculated as follows: Cmax = the maximum plasma concentration by visual inspection of the highest mean concentration found on the concentration versus time curve; Tmax = the time at which Cmax is observed; drug exposure (AUCinf) was calculated using the trapezoidal rule with extrapolation to time infinity; clearance (CL) = dose/AUC; volume of distribution (Vd) = dose/C0 where C0 is the plasma concentration at time zero (extrapolated). Pharmacokinetic parameters were estimated using Phoenix WinNonlin software (Certara®, St. Louis, MO). Single-dose subcutaneous pharmacokinetics was investigated in both unirradiated mice and in irradiated mice beginning 24 h after partial exposure with shielding of the legs from the tibiae containing approximately 5% of the bone marrow. Partial-body irradiation was performed in principle as described by Booth et al. (26).
Monitoring Acute Toxicity of OTP
Forty mice were randomized into four equal size groups. Animals were evaluated for 14 days after a single dose of OTP subcutaneous (SQ) injection at 10, 25 or 50 mg/kg. OTP was dissolved in 2% 1, 2-propanediol (Sigma-Aldrich) plus 1% ethanol in phosphate buffered saline (PBS) (pH 7.4) (2). Mortality, abnormal behavior and body weight were recorded for 1 h after injection and daily thereafter. Gross autopsy was performed on all animals, including those found dead on or before day 14 or sacrificed on day 14 without microscopic examination of the bone marrow or the major organs.
Murine Total-Body Irradiation Model
JL Shepherd & Associates (San Fernando, CA) 137Cs irradiators were used for the irradiation experiments. Conscious mice were placed in a plastic container and exposed to gamma radiation at a dose rate of ~3.2 Gy/min based on regular ionization chamber dosimeter calibration and calculated monthly decay correction, as described previously (15). Total radiation doses needed to yield LD90/8–12 to LD100/30 mortality were empirically determined and were applied as specified for each type of experimental design. Animals were randomized into groups of equal numbers. Mice received 0.5–10 mg/kg per dose of OTP (SQ, 0.2 mL; specific doses and schedules are called out in each experiment below and are noted in the figure legends). Animals were monitored daily up to 30 days with two daily observations over the period between days 7–14 with death being the primary end point and mean survival time the secondary end point of the study.
Hematological Evaluation
Mice were exposed to 5.7 Gy TBI with 1 mg/kg OTP administered 24 h postirradiation. Peripheral blood was collected from isofluorane-anesthetized mice by retro-orbital bleeding into EDTA BD Microtainer tubes. Peripheral blood (~400 μL) from the mice was collected on postirradiation day 7 or 18 as a terminal procedure. Total white blood cell (WBC) and platelet (PLT) counts were determined using a Z1 Cell Coulter (Beckman Coulter Inc., Fullerton, CA).
Determination of Survival and Clonogenic Capacity of Irradiated CD34+ Human Hematopoietic Progenitors
Human umbilical cord blood-derived CD34+ cells were purchased from Cincinnati Children’s Hospital Stem Cell Facility of the Ohio State Comprehensive Cancer Center (Cincinnati, OH), an IRB-approved Stem Cell Procurement Facility. The purity of the CD34+ cells based on the CD34 marker was above 90%. CD34+ cells were recovered after thawing in StemSpan™ serum-free media (Stem Cell™ Technologies Inc., Vancouver, Canada) for 3 h. After recovery, cells were washed in serum-free Iscove’s modified Dulbecco’s media (IMDM) and aliquoted in 400 μl in serum-free IMDM (40,000 cells/ml). Cells were irradiated with 6 Gy at 0.86 Gy/min. Thirty minutes after irradiation, cells were treated with 0.1–1.0 μM OTP for 4 h. OTP stock (10 mM) solution was made in distilled water and diluted with complete media to a final concentration of 10 μM. Six thousand cells/plate were plated in triplicate in MethoCult™ H4434 Classic media (Stemcell Technologies, Inc.). Colony-forming units (CFU) for granulocyte/macrophage (CFU-GM) and burst-forming units of the erythroid (BFU-E) lineage were counted based on their unique colony morphology after 14 days of culture.
Determination Crypt Survival in the Jejunum
Intestinal crypt survival was measured using the established clonogenic assay method (27). Briefly, groups of 6 mice were exposed to a high dose of TBI (10.6 Gy, 3.2 Gy/min dose rate). OTP at different doses in the 0.5–5.0 mg/kg range was administered at either 2 h before or up to 24 h postirradiation. Animals were sacrificed on day 4. Proximal jejunum segments (about 5 cm from pylorus) from each mouse were fixed in 10% neutralized formaldehyde buffer (pH 7.4) and subjected to routine histological procedures. Paraffin cross sections (5 μm) were cut perpendicular to the long axis of the small intestine and stained with hematoxylin and eosin (H&E). The number of surviving crypts per jejunum circumference was counted using light microscopy. Two cross sections were scored for each mouse and an average number of surviving crypts were calculated. A surviving crypt was defined as a regenerative crypt that contained a cluster of 10 or more dark H&E-stained cells.
Measurement of Carrier-Mediated Glucose Uptake In Situ
For evaluating carrier-mediated glucose uptake, mice were injected with OTP at +24 h after 4.3 Gy TBI (3.2 Gy/min) and sacrificed 48 h later. Apical transport of glucose was measured in vitro by incubating sleeves of intact proximal jejunum segments with 14C D-glucose and 3H L-glucose tracer to correct for carrier-independent D-glucose absorption (28). Briefly, jejunum segments were externalized and two 1 cm sleeves were secured by silk ligatures onto stainless steel rods. The sleeves were kept in cold Ringer’s solution (~4°C) aerated with 95% O2/5% CO2 during and after the mounting. Measurement of glucose uptake began 4 min after the sleeves were exposed to 37°C Ringer’s solution followed by a 2 min incubation of the sleeves in uptake solution (Ringer’s solution with 2 μM solution of the tracers). After the incubation, tissues were rinsed for 20 s in cold Ringer’s solution, blotted, weighed and dissolved using Solvable™ solution (PerkinElmer® Inc., Waltham, MA). Radioactivity taken up into the tissue was counted in Ultima Gold™ scintillation solution (Perkin-Elmer). Rates of glucose uptake (pM/min) were calculated and normalized to wet tissue weight.
Measurement of Plasma Endotoxin Level
Bacterial translocation was monitored by determining plasma endotoxin levels using the Chromogenic Limulus Amebocyte Lysate test kit (Lonza Inc., Wakersville, MD). Mice were treated with 1 mg/ kg OTP 24 h after 7.1 Gy TBI. Peripheral blood was collected on day 6 postirradiation for plasma endotoxin determination. Peripheral blood was collected from isofluorane-anesthetized mice by retro-orbital bleeding into sample tubes with EDTA BD Microtainer tubes. Plasma was separated by centrifugation for 5 min (10,000g) and diluted 50-fold with PBS. The reaction mixtures were measured at 405 nm by a spectrophotometer. A standard curve was generated in parallel to calculate the endotoxin levels of samples.
Clonogenic Survival Assay In Vitro
The effect of OTP on clonogenic survival after irradiation was determined using the method described by Saha et al., with slight modifications (29). Briefly, single cell suspension of IEC-6 cells was obtained with trypsinization for 3 min using 0.05% trypsin EDTA (Corning® Cellgro®, Mediatech Inc., Manassas, VA). Two hundred cells/well were plated on 6-well (Corning® Costar®) plates and the next day the complete growth media was changed for the serum-free media supplemented with 10 μg/ml of insulin for 24 h prior to irradiation. Cells were irradiated with increasing doses of gamma radiation (0, 2, 4, 6 Gy at a dose rate of 4.3 Gy/min). After irradiation the media was removed and replaced with fresh complete growth media supplemented with 10 μM OTP, 3 μM LPA, or vehicle (3 μM BSA + 0.1 % DMSO in complete growth media). Cells were treated with fresh test compound daily on the first three days after exposure, after which they were allowed to grow in complete growth media for seven more days. On day 10 postirradiation the cells were fixed, stained with 0.5% crystal violet and the number of colonies containing at least 50 cells was counted. Surviving fractions for each treatment were determined by normalizing the mean plating efficiency for each dose to the plating efficiency of the unirradiated control plates. Assays were performed in triplicate.
Determination of OTP’s Dose Modification Factor
For determination of the dose modification factor (DMF), groups of 15 mice were exposed to increasing doses of TBI at 6.4, 7.1, 7.8 or 8.6 Gy. OTP (1 mg/kg, SQ) was administered at +24 h postirradiation. Overall survival was tabulated to generate survival curves for OTP-treated and vehicle groups. Comparative probit analysis was used for determining DMF. The DMF was calculated by dividing the estimated LD50/10 of the treated groups by LD50/10 of the control groups (30).
Statistics
The primary end point for the survival/dose-ranging studies was survival at 30 days. Therefore, Fisher’s exact test was selected as an appropriate statistical test to compare control and OTP-treated groups using SAS® software (Cary, NC).
Analysis of variance (ANOVA) with Bonferroni post test was used for analyzing data from crypt microcolony counts and the clonogenic assay after fitting the survival curves with exponential regression using the Prism V.5.0 software (GraphPad Software Inc., San Diego, CA). Student’s t test was used for analyzing glucose uptake and endotoxin level, and for the comparison of crypt survival between vehicle and OTP-treated specimens. The P < 0.05 was considered statistically significant. The DMF was calculated by using a probit analysis to estimate the respective LD50 for control and OTP groups [DMF = LD50(OTP)/LD50(vehicle)] (30). Significance testing of the DMF was conducted using one- and two-sided hypothesis tests. LD50 estimations and significance testing for the DMF were conducted using Rx 64. v.3.1.1 software with MASS v.7.3–34 package.
RESULTS
OTP Recruits NHERF2 and TRIP-6 into an Antiapoptotic Signaling Complex
Within 10 min of OTP treatment MYC-TRIP-6 and GFP-NHERF2 were detectable in complex with the FLAG-LPA2 receptor in the lysates of cells treated with OTP (Fig. 1). When the FLAG-LPA2 construct was omitted from the transfection no TRIP-6 or NHERF2 was detectable in the protein pulled down, although both proteins were expressed in the cell lysates. In contrast to the wild-type FLAG-LPA2 in cell lysates transfected with the FLAG-C314A/L351A-LPA2 mutant, OTP treatment failed to yield detectable MYC-TRIP-6 and GFP-NHERF2 in the protein pulled down by the anti-FLAG beads even though the mutant LPA2 protein was abundantly detectable. These results indicate that OTP, similarly to LPA, recruits MYC-TRIP-6 and GFP-NHERF2 into a ternary complex with LPA2 that has been shown to mediate the antiapoptotic actions of LPA.
FIG. 1.
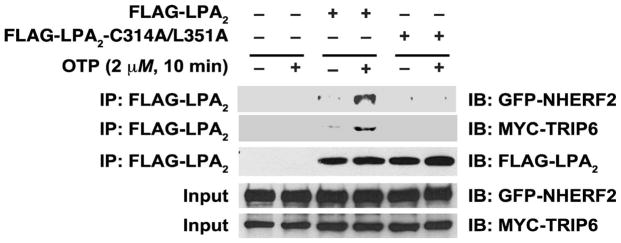
OTP stimulation of LPA2 induces the recruitment of TRIP-6 and NHERF2 into a macromolecular signaling complex. HEK293T cells were transfected with wild-type FLAG-LPA2 or the C314A/L351A mutant defective in Zn-finger- and PDZ-motif-mediated interactions. The cells were co-transfected with MYC-TRIP2 and GFP-NHERF2 constructs. In cells stimulated with 2 μM OTP for 10 min, a ternary complex was formed between LPA2 TRIP6 and NHERF2, however, no such complex was detectable in cells expressing the C314A/L351A LPA2 mutant. The blot shown is representative of the results of three other experiments.
OTP Promotes Hematopoiesis of Irradiated Human CD34+ Progenitor Cells In Vitro
In a previous study (3) we showed that OTP reduced mortality in mice exposed to ~9 Gy of 137Cs gamma radiation in a HE-ARS model. The protective effect of OTP in this model of the HE-ARS suggested that OTP directly or indirectly might have a radioprotective effect on hematopoiesis. To test whether OTP has any direct protective/mitigative action in hematopoietic progenitor cells and whether this effect carries over to humans, we obtained cord-derived purified human CD34+ progenitor cells and examined whether overall cell survival and the differentiation of select lineages were affected by the compound after 6 Gy (0.86 Gy/min) gamma irradiation, which produces HE-ARS in vivo. OTP already at ≥0.1 μM significantly increased CFU-GM counts (Fig. 2, P < 0.05), and at 1 μM concentration the increase in BFU-E counts also reached statistical significance. Collectively, OTP at 1 μM increased total colony counts by 64% (P < 0.01).
FIG. 2.

Effect of OTP on the survival and differentiation of CD34+ progenitor cells in vitro. Progenitor cells were 6 Gy γ irradiated and treated for 4 h starting +30 min postirradiation. Note that OTP treatment increased the total number of surviving colonies and significantly enhanced BFU-E and CFU-GM colony growth (n =3, *P< 0.05, **P < 0.01 relative to vehicle control).
Pharmacokinetic and Acute Toxicology Properties of OTP
The concentration versus time profile, clearance, volume of distribution and exposure of OTP in nonirradiated mice are shown in Fig. 3 and Table 1. OTP was rapidly absorbed after SQ administration to nonirradiated mice with a Tmax of 0.5–1.0 h and a corresponding Cmax of 302 ng/mL in females and 476 ng/mL in males. The exposure (AUCinf) in females was 1,152 h*ng/mL and 1,521 ng/mL in males. The Cmax of 407 ng/mL and 622 ng/mL in irradiated females and males, respectively, was slightly higher compared with the corresponding values in nonirradiated females and males. However, the concentration versus time profiles overlapped in nonirradiated and irradiated mice within error, as shown in Fig. 3. There was no change in the Tmax (0.5–1.0 h) or half-life after irradiation.
FIG. 3.

Time-concentration profile of OTP in irradiated (15.6 Gy, 0.14 Gy/min partial-body irradiation) and nonirradiated male and female mice. There was no detectable change in time-concentration profile by gender or as a result of irradiation (n =3–4 mice per group for each time point).
TABLE 1.
Pharmacokinetic Parameters of OTP in Nonirradiated and Irradiated Mice of Both Genders
| Gender | Cmax (ng/mL) | Tmax (h) | T1/2 (h) | AUCinf (h*ng/mL) | CLss_F (mL/h/kg) | Vz_F (mL/kg) |
|---|---|---|---|---|---|---|
| Female | 302 | 1.00 | 1.38 | 1,152 | 868 | 1,728 |
| Male | 476 | 0.50 | 1.41 | 1,521 | 657 | 1,341 |
| Irradiated female | 407 | 1.00 | 1.36 | 1,591 | 629 | 1,230 |
| Irradiated male | 622 | 0.50 | 1.52 | 1,721 | 581 | 1,277 |
OTP by SQ injection in mice at 10, 25 or 50 mg/kg produced no death or observable organ damage at the time of necropsy on day 14. No toxicity was observed at 10 mg/ kg. At 25 mg/kg, we noted mice hunching over for about 10 min after drug injection, however, this response disappeared within 1 h. At 50 mg/kg, we noted a rapid heartbeat, hunching over and “lethargy” around 10 min after drug injection. These symptoms disappeared within 2 h. No animal died in any of the treatment groups. Gross autopsy revealed no sign of macroscopic pathology upon visual observation in all mice treated with OTP. Microscopic analysis was not performed in this study of the organs.
Drug-Dose Ranging of the Radioprotective/Radiomitigating Efficacy of OTP
To determine the effective radioprotective drug-dose range of OTP in a TBI model we administered it as a single dose at 1, 3 and 10 mg/kg subcutaneously 30 min prior to an LD90/30 radiation dose of 6.3 Gy (3.2 Gy/min) in this murine model. At every drug dose in this range OTP yielded a survival advantage that was greater than 50% at day 30 over the vehicle group (Fig. 4A, P < 0.01). Mortalities between a single injection and three daily injections of OTP (1 mg/kg) starting at +24 h postirradiation were compared. Specifically, a three-dose schedule at +24 h plus +48 h plus +72 h, a two-dose schedule at +24 h plus +48 h and a single-dose schedule at +24 h were compared. No significant additional efficacy was observed with repeated daily dosing compared to the single-dose schedule (Fig. 4B).
FIG. 4.

Panel A: OTP (SQ) dose-dependently protects mice from a lethal dose of 6.3 Gy radiation when administered −30 min relative to the time of TBI (n =12 for all groups, *P < 0.01 vs. vehicle). Panel B: A single 1 mg/kg dose of OTP (SQ) is as effective as two (1 mg/kg at + 24 h and +48 h postirradiation) or three doses (1 mg/kg at +24 h, +48 h and +72 h postirradiation) at protecting mice from a lethal dose of radiation (6.3 Gy) (n = 18 per group,*P < 0.03 vs. vehicle).
Using a single 1 mg/kg dose of OTP we next examined the effective time window over a time range that spanned −24 h prior, to +26 h postirradiation in 12 h increments (6.3 Gy, 3.2 Gy/min) (Fig. 5). Using 30 day survival as the primary end point, a single dose delivered between −12 h to +26 h resulted in a >40% survival advantage over the vehicle group (P < 0.02, Fisher’s exact test). Real-life concept of operations in the event of mass casualties may require a delay in the initiation of radiomitigant treatment beyond 26 h post injury. For this reason, we examined how delaying the administration of a single-dose OTP treatment to +48 h and +72 h postirradiation would impact the efficacy, using 30 day survival as our end point (Fig. 6). OTP treatment elicited a 33% survival advantage in mice that first received the drug at +48 or +72 h postirradiation, which at the group sizes used (n = 15 mice/group), was not significantly different compared to the 47% survival advantage found in the group that received OTP treatment at +24 h.
FIG. 5.

OTP protects mice from 6.3 Gy of TBI when administered −24 h to +26 h relative to the time of TBI. (n = 12–24 per group). Panel A shows the radioprotective effects of OTP (1 mg/kg, SQ, *P < 0.02 vs. vehicle) administered at −12 h or −24 h prior to irradiation. Panel B shows the radiomitigative effects of OTP (1 mg/kg, SQ, *P < 0.03 or **P < 0.002 vs. vehicle) administered at +12 h or + 26 h postirradiation.
FIG. 6.

Delayed administration of OTP protects mice from lethal dose of 6.4 Gy TBI. OTP administered (1 mg/kg, SQ) at +24 h, +48 h and +72 h after TBI reduced mortality relative to vehicle treatment (n = 15 mice per group, *P < 0.03 vs. vehicle).
OTP Increases Leukocyte and Platelet Counts during the HE-ARS
Radiation injury to the bone marrow results in loss of hematopoietic cells and consequent leukopenia and thrombocytopenia. Based on the in vitro observations for the radioprotective effect of OTP in purified human CD34+ hematopoietic progenitor cells we examined whether OTP-treated mice suffering from the HE-ARS would show higher leukocyte and thrombocyte counts compared to vehicle-treated mice. TBI of C57BL/6 mice to 5.7 Gy reduced the total WBC counts in peripheral blood from above 1.8 ×103/ μl (normal level) to about 0.4 × 103/μl on day 7 postirradiation. By day 18, which marks the recovery phase of leukopenia after radiation exposure in mice, the WBC counts in the vehicle group remained low at 0.55 × 103/μl. In contrast, in the group of mice treated with a single dose of OTP, 1 mg/kg at 24 h postirradiation, significantly increased WBC counts were detected (Fig. 7A) (~0.9 × 103/μl, P < 0.01 vs. vehicle control). Peripheral PLT counts decreased from above 600 × 103/μl to 200 ×103/μl by day 7 postirradiation and this reduction persisted up to day 18. In contrast, in groups treated with 1 mg/kg OTP at 24 h postirradiation, the PLT counts were close to normal levels on day 7 and 18 postirradiation (Fig. 7B) (P < 0.01 vs. vehicle control). One mouse in the control group died by day 18 postirradiation, while no deaths occurred in the OTP-treated group. These data together indicate that OTP treatment administered at +24 h postirradiation attenuates cell loss due to the HE-ARS.
FIG. 7.

Effect of OTP on WBC and PLT counts on day 7 and 18 postirradiation. Mice were exposed to 5.7 Gy TBI and OTP (1 mg/kg, SQ, open bars) was administered at +24 h postirradiation. WBC (panel A) and PLT (panel B) were counted on day 7 or 18 postirradiation (n = 7–8 mice per group, *P < 0.01 vs. vehicle, filled bars).
OTP Increases Clonogenic Survival of Irradiated IEC-6 Cells
OTP and LPA caused a statistically significant increase in colony survival indicated by the slope of the survival curves compared to vector treated cells (regression coefficients R2 were ≥0.97 in each case) (Fig. 8). This finding extended our earlier observations that OTP is effective in inhibiting the progression of the apoptotic cascade by showing that treated cells actually survive in higher numbers after radiation injury.
FIG. 8.

IEC-6 cells were exposed to increasing doses of gamma radiation and were treated with either vehicle, 3 μM LPA 18:1 or 10 μM OTP starting +30 min postirradiation. Colonies were counted on day 10 postirradiation. Both LPA- and OTP-treated cultures showed increased colony survival compared to vehicle-treated cultures at all radiation doses, as demonstrated by the slopes of the survival curves (regression fit R2 > 0.97 of colony survival, ANOVA for comparison of the slopes with Bonferroni post test, *P < 0.01).
Effect of OTP on Radiation Injury to the GI Tract
Exposure of mice to gamma radiation doses higher than ~8 Gy leads to the development of the GI-ARS that manifests morphologically in a loss of the intestinal stem cells and the ensuing decrease in crypt number. We investigated the effect of OTP on radiation-associated GI crypt losses using clonogenic crypt survival assay. To evaluate the radioprotective effect on crypt survival, OTP was administered by SQ injection −2 h with single doses of 0.5, 2.0 and 5.0 mg/kg prior to 10.6 Gy (3.2 Gy/min) of TBI and the proximal jejunum was sampled at day 4 postirradiation (Fig. 9A). OTP at and above 0.5 mg/kg significantly increased the number of surviving crypts per cross section. We also administered OTP at +24 h after TBI. In this paradigm, a single 2.5 mg/kg dose of OTP significantly increased the number of regenerating crypts in the jejunum compared to the vehicle group (P =0.034) (Fig. 9B).
FIG. 9.

Effect of OTP on crypt survival. Mice were exposed to 10.6 Gy TBI (3.2 Gy/min) and crypt survival in the proximal jejunum was evaluated on day 4 postirradiation. Panel A: OTP (SQ) dose dependently and significantly reduced crypt loss when administered at −2 h pre-exposure. Panel B: In this experiment the administration of OTP (2.5 mg/kg) was delayed to +24 h postirradiation. Samples were harvested on day 4 postirradiation (n = 6 mice per group, 2 jejunum sections/mouse, *P < 0.05 vs. vehicle Student’s t test).
OTP Treatment Improves Glucose Uptake and Bacterial Barrier Function in the Irradiated Gut
Apical transport of glucose by the sodium-dependent transporter SCLT-1 is a sensitive indicator of absorptive functions of enterocytes. To evaluate the effect of OTP on enterocyte function after a 4.3 Gy (3.2 Gy/min) TBI, we measured the subsequent uptake of radiolabeled glucose from excised jejunum. Radiation exposure significantly reduced carrier-mediated glucose uptake by more than 50% (Fig. 10A). OTP (1 mg/kg) administered at +24 h postirradiation almost completely reversed the reduction seen in enterocytes from vehicle-treated mice (P < 0.003 relative to sham irradiation; P < 0.03 relative to irradiation with vehicle).
FIG. 10.

Panel A: 14C-glucose uptake was monitored in jejuni isolated from mice treated with 1 mg/kg OTP (SQ) at +24 h after 4.3 Gy TBI (*P < 0.003 relative to sham irradiation, **P < 0.03 relative to irradiation with vehicle). Panel B: Plasma endotoxin level in mice on day 6 postirradiation after treatment with 1 mg/kg OTP at 24 h postirradiation (n = 8 mice per group, *P < 0.01 relative to sham irradiation, #P < 0.01 relative to irradiation + vehicle, 7.1 Gy TBI).
Plasma endotoxin level is an indirect measure of bacterial invasion into blood from the GI system due to a breakdown of the gut barrier. Plasma endotoxin level was quantified on day 6 postirradiation after a 7.1 Gy (3.2 Gy/min) dose of gamma radiation, which was determined to cause endotoxemia in preliminary experiments (data not shown). As shown in Fig. 10B, in the vehicle-treated mice, a tenfold increase in endotoxin level was observed on day 7 postirradiation. In contrast, 1 mg/kg OTP treatment at 24 h postirradiation significantly reduced the plasma endotoxin level to near control level (P < 0.01). These data indicate that OTP protects enterocyte barrier function after radiation injury.
OTP Treatment Shows a Dose Modification Factor of 1.2
Dose modification factor (DMF) is a quantitative measurement of radiation countermeasures that assesses the change in radiation dose needed to produce the same estimated LD50 after test agent treatment compared to vehicle control. We determined the DMF of OTP treatment over the 6.4–8.6 Gy TBI dose range. OTP treatment delayed death on an average of 4 days after exposure to 8.6 Gy of gamma radiation. Probit analysis of the dose-mortality relationship after a single dose of OTP administered at 24 h after irradiation yielded a DMF of 1.2 (Fig. 11) (P < 0.0001). The LD50/10 for the control was 10.08 Gy, while the LD50/10 for OTP treatment was 11.92 Gy.
FIG. 11.

OTP (1 mg/kg) or vehicle was administered to mice at +24 h after 6.4–8.6 Gy TBI. The graph shows the survival probit curves as a function of radiation dose with or without OTP treatment that yielded a DMF of 1.2.(horizontal bars = 95% confidence intervals, n = 15 mice per group, P < 0.0001).
DISCUSSION
In the current study we demonstrate that OTP, an LPA receptor agonist, is not only a potent radioprotectant but also a radiomitigator over the time window that spans from −12 h pre-exposure to +72 h postirradiation to otherwise lethal levels of gamma radiation in mice. The GI tract abundantly expresses LPA1 and LPA2 receptors (3, 14). Luminal and systemic LPA play an important role in maintaining GI barrier function and integrity. Both LPA and OTP are potent antiapoptotic agents in intestinal epithelial cells both in vitro and in vivo (3, 15, 24, 31–33). We observed that in LPA2 receptor knockout (KO) mice (15) and cells (34) the radiation sensitivity increased compared to wild-type mice or cells, and LPA or OTP fail to protect against radiation-induced apoptosis. It has been shown that activation of the LPA2 GPCR subtype is required for mediation of the robust and long-lasting activation of the ERK1/2 and AKT-NFκB prosurvival kinase pathways (19, 20, 35). This signaling response is dependent on the LPA-elicited assembly of a ternary macromolecular signaling complex consisting of LPA2, TRIP-6 and NHERF2 (19). Here we showed that stimulation of LPA2-expressing cells by OTP mimics this effect of LPA and recruits TRIP-6 and NHERF2 into a signaling complex. Thus, this common mechanism is shared between OTP and LPA in attenuating programmed cell death. This macromolecular complex is unique to LPA2, since no other LPA GPCR contains both the PDZ and the Zn-finger motif in their C-termini that are the docking sites for the NHERF2 and TRIP6, respectively. The ternary complex has been shown to mediate robust prosurvival signals through the ERK1/2 and AKT kinase pathways (19, 35, 36), which are also required for the prosurvival actions of OTP (24).
These data support the hypothesis that LPA2 plays a unique role in the radioprotective and radiomitigating machinery under either physiological or pathological conditions. Downstream events involve: reduction of cytochrome c release from mitochondria; prevention of caspase-9 activation by preventing the translocation of Bax from cytosol to mitochondria; and increase of the expression of the antiapoptotic Bcl-2 and Bcl-Xl proteins leading to inhibition of caspase-3 activity and reduced cleavage of PARP-1 (3, 15, 24, 31). We found that OTP significantly enhanced clonogenic survival of the intestinal crypt-derived IEC-6 cell line irradiated in vitro, reduced endotoxemia and increased intestinal glucose transport in in vivo. OTP also reduced damage to irradiated human hematopoietic progenitor cells and increased WBC and PLT counts during the HE-ARS. These results expand on our previous observations that OTP treatment reduced activation of the caspase cascade and reduced DNA fragmentation in irradiated IEC-6 cells (3). When combined, these findings may explain the reduction of apoptosis and increased clonogenic crypt survival, as well as the reduction in the number of apoptotic bodies and caspase 9 positive enterocytes after exposure of mice to gamma radiation, which we previously reported (3).
Although, we have previously shown that pre- and postirradiation OTP treatment augmented the survival of C57BL/6 mice exposed to gamma radiation that elicited the HE-ARS (3), in our current investigation we sought evidence for a direct radioprotective/radiomitigative effect of OTP on CD34+ hematopoietic progenitor cells, which represent an important cell population in the reconstitution of hematopoiesis after radiation injury. Our current data confirmed this hypothesis by showing that CD34+ progenitors demonstrate increased survival and lineage-specific clonogenicity when treated with OTP. It is known that LPA1 and LPA2 receptors are expressed in primitive Lin–Sca+Kit+ hematopoietic stem cells. LPA2 expression is more abundant in Kit+ cells than in their less primitive counterparts, Kit– cells. LPA2, but not LPA1 is also expressed in less primitive Kit– cells (37). It has been observed that LPA induces migration of both the Kit+ and Kit– hematopoietic stem cells in vitro and enhances the chemotactic migratory response of the Kit+ cells to stromal-derived factor 1. Based on these findings and our current data, it is reasonable to hypothesize that OTP, either through receptor-mediated direct action or indirectly by regulation of hematopoietic cytokine production, plays an important role in hematopoiesis.
Further experiments will be needed to dissect the stem cell responses in OTP-treated gamma-irradiated mice, especially in the model where the tibiae are shielded and may contain intact progenitor cells that might respond to OTP with clonal expansion. Our data indirectly support this hypothesis, since we detected significant increases in WBC and PLT counts in OTP-treated gamma-irradiated mice over vehicle controls. Alternatively, increases in circulating WBC and PLT can also be due to cell OTP-activated mobilization of these cells from stores. OTP-treated CD34+ progenitor cells showed a significant increase in the granulocyte/macrophage lineage that coincides with the increased WBC count found in OTP-treated irradiated mice. This contrasts with the lack of increased megakaryocytic response in vitro even though we found higher platelet counts. One plausible explanation for this dichotomy could be that OTP mobilizes stored platelets rather than increases their production. This alternative explanation will have to be addressed in future studies.
Our previous reports detailed the radioprotective effect of OTP and LPA in mice TBI with 12 Gy γ rays, by demonstrating its protective effect in reducing the number of apoptotic bodies, caspase 9 positive cells, Bax positive enterocytes and increasing the number of surviving crypts on day 4 postirradiation (3, 15). In the current study we extended these experiments to evaluate the OTP dose-response relationship on crypt protection and found that this action plateaued above 2 mg/kg. Furthermore, we have tested whether OTP could mitigate crypt loss when administered after radiation exposure. These experiments showed that OTP caused an increase in the number of surviving crypts when administered up to 24 h postirradiation. Thus, OTP appears to mitigate the mechanisms of the ARS that manifest beyond the first few hours of radiation injury.
The gastrointestinal epithelial barrier allows efficient absorption of nutrients and exchange of electrolytes while it prevents the entry of luminal pathogens. Here we showed that OTP restored glucose absorption and inhibited endotoxemia (Fig. 9). These data indicate that subcutaneous OTP administration not only attenuates radiation-induced cell injury but also enhances enterocyte function and barrier integrity, two key steps in the pathogenesis of the GI-ARS leading to malabsorption and sepsis. LPA inhibits cholera toxin-induced secretory diarrhea through CFTR- and NHERF2-dependent protein interactions involving LPA2 receptor (21). We showed that OTP recruited NHERF2 into a macromolecular complex with LPA2, which provides the scaffold for interaction with CFTR (Fig. 1). Diarrhea is also a symptom of the GI-ARS and in this context further experiments will be needed in a more suitable model to evaluate the effect of OTP on radiation-induced diarrhea.
We evaluated OTP pharmacokinetics in nonirradiated and irradiated mice. Importantly, we found no significant effect of radiation exposure on the rate of elimination and consequent plasma half-life of the compound (Fig. 3 and Table 1). Our current data with radiation doses that are known to elicit the HE-ARS yielded significant survival advantage (Figs. 4, 5 and 6) in experimental paradigms using pre- and post-exposure administration of OTP. We established that OTP has a DMF of 1.2 over the radiation dose range from 6.4–8.6 Gy (Fig. 11).
In summary, we demonstrated that OTP is a potent radioprotector and radiomitigator. It is unique in that: 1. It is a small molecule that is stable with a shelf life of more than two years at room temperature; 2. OTP represents a prototype of a novel class of radioprotectants acting through modulating LPA receptors; 3. OTP is a potent radiomigitator, effective even when administered by parenteral injection up to 72 h postirradiation; 4. OTP is also protective when administered by oral route (3); and 5. OTP ameliorates radiation-associated hematopoietic suppression as well as gastrointestinal injury and augments enterocyte function. Further studies will be needed to address the radiomitigative properties of OTP at higher radiation doses using a well controlled shielded murine model and in nonhuman primates.
Acknowledgments
The authors thank Dr. Fang-Tsyr Lin for her help with the pull-down assays. We thank Drs. Richard Trout and Fridtjof Thomas for their advice on the statistical methods. These studies were supported by NIAID grants RC1AI78512 (WD), 3RC1AI078512 (WD), AI080405 (GT), NCI grant R43CA130424 (WD) and by grant I01BX007080 from the Biomedical Laboratory Research & Development Service of the VA Office of Research and Development (GT).
References
- 1.Dainiak N, Gent RN, Carr Z, Schneider R, Bader J, Buglova E, et al. Literature review and global consensus on management of acute radiation syndrome affecting nonhematopoietic organ systems. Disaster Med Public Health Prep. 2011;5:183–201. doi: 10.1001/dmp.2011.73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Durgam GG, Virag T, Walker MD, Tsukahara R, Yasuda S, Liliom K, et al. Synthesis, structure-activity relationships, and biological evaluation of fatty alcohol phosphates as lysophosphatidic acid receptor ligands, activators of PPARgamma, and inhibitors of autotaxin. J Med Chem. 2005;48:4919–30. doi: 10.1021/jm049609r. [DOI] [PubMed] [Google Scholar]
- 3.Deng W, Shuyu E, Tsukahara R, Valentine WJ, Durgam G, Gududuru V, et al. The lysophosphatidic acid type 2 receptor is required for protection against radiation-induced intestinal injury. Gastroenterology. 2007;132:1834–51. doi: 10.1053/j.gastro.2007.03.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Tokumura A, Harada K, Fukuzawa K, Tsukatani H. Involvement of lysophospholipase D in the production of lysophosphatidic acid in rat plasma. Biochim Biophys Acta. 1986;875:31–8. [PubMed] [Google Scholar]
- 5.Jalink K, Hordijk PL, Moolenaar WH. Growth factor-like effects of lysophosphatidic acid, a novel lipid mediator. Biochim Biophys Acta. 1994;1198:185–96. doi: 10.1016/0304-419x(94)90013-2. [DOI] [PubMed] [Google Scholar]
- 6.Baker DL, Desiderio DM, Miller DD, Tolley B, Tigyi GJ. Direct quantitative analysis of lysophosphatidic acid molecular species by stable isotope dilution electrospray ionization liquid chromatography-mass spectrometry. Anal Biochem. 2001;292:287–95. doi: 10.1006/abio.2001.5063. [DOI] [PubMed] [Google Scholar]
- 7.Baker DL, Morrison P, Miller B, Riely CA, Tolley B, Westermann AM, et al. Plasma lysophosphatidic acid concentration and ovarian cancer. JAMA. 2002;287:3081–2. doi: 10.1001/jama.287.23.3081. [DOI] [PubMed] [Google Scholar]
- 8.Ishii I, Fukushima N, Ye X, Chun J. Lysophospholipid receptors: signaling and biology. Annu Rev Biochem. 2004;73:321–54. doi: 10.1146/annurev.biochem.73.011303.073731. [DOI] [PubMed] [Google Scholar]
- 9.Mutoh T, Chun J. Lysophospholipid activation of G protein-coupled receptors. Subcell Biochem. 2008;49:269–97. doi: 10.1007/978-1-4020-8831-5_10. [DOI] [PubMed] [Google Scholar]
- 10.Noguchi K, Ishii S, Shimizu T. Identification of p2y9/GPR23 as a novel G protein-coupled receptor for lysophosphatidic acid, structurally distant from the Edg family. J Biol Chem. 2003;278:25600–6. doi: 10.1074/jbc.M302648200. [DOI] [PubMed] [Google Scholar]
- 11.Kotarsky K, Boketoft A, Bristulf J, Nilsson NE, Norberg A, Hansson S, et al. Lysophosphatidic acid binds to and activates GPR92, a G protein-coupled receptor highly expressed in gastrointestinal lymphocytes. J Pharmacol Exp Ther. 2006;318:619–28. doi: 10.1124/jpet.105.098848. [DOI] [PubMed] [Google Scholar]
- 12.Lee CW, Rivera R, Gardell S, Dubin AE, Chun J. GPR92 as a new G12/13- and Gq-coupled lysophosphatidic acid receptor that increases cAMP, LPA5. J Biol Chem. 2006;281:23589–97. doi: 10.1074/jbc.M603670200. [DOI] [PubMed] [Google Scholar]
- 13.Fukushima N, Ishii I, Contos JJ, Weiner JA, Chun J. Lysophospholipid receptors. Annu Rev Pharmacol Toxicol. 2001;41:507–34. doi: 10.1146/annurev.pharmtox.41.1.507. [DOI] [PubMed] [Google Scholar]
- 14.Yun CC, Sun H, Wang D, Rusovici R, Castleberry A, Hall RA, Shim H. LPA2 receptor mediates mitogenic signals in human colon cancer cells. Am J Physiol Cell Physiol. 2005;289:C2–11. doi: 10.1152/ajpcell.00610.2004. [DOI] [PubMed] [Google Scholar]
- 15.Deng W, Balazs L, Wang DA, Van Middlesworth L, Tigyi G, Johnson LR. Lysophosphatidic acid protects and rescues intestinal epithelial cells from radiation- and chemotherapy-induced apoptosis. Gastroenterology. 2002;123:206–16. doi: 10.1053/gast.2002.34209. [DOI] [PubMed] [Google Scholar]
- 16.Balazs L, Okolicany J, Ferrebee M, Tolley B, Tigyi G. Topical application of the phospholipid growth factor lysophosphatidic acid promotes wound healing in vivo. Am J Physiol Regul Integr Comp Physiol. 2001;280:R466–72. doi: 10.1152/ajpregu.2001.280.2.R466. [DOI] [PubMed] [Google Scholar]
- 17.Moolenaar WH, van Meeteren LA, Giepmans BN. The ins and outs of lysophosphatidic acid signaling. Bioessays. 2004;26:870–81. doi: 10.1002/bies.20081. [DOI] [PubMed] [Google Scholar]
- 18.Radeff-Huang J, Seasholtz TM, Matteo RG, Brown JH. G protein mediated signaling pathways in lysophospholipid induced cell proliferation and survival. J Cell Biochem. 2004;92:949–66. doi: 10.1002/jcb.20094. [DOI] [PubMed] [Google Scholar]
- 19.Shuyu E, Lai Y-J, Tsukahara R, Chen CS, Fujiwara Y, Yue J, et al. Lysophosphatidic acid 2 receptor-mediated supramolecular complex formation regulates its antiapoptotic effect. J Biol Chem. 2009;284:14558–71. doi: 10.1074/jbc.M900185200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lin FT, Lai YJ, Makarova N, Tigyi G, Lin WC. The lysophosphatidic acid 2 receptor mediates down-regulation of Siva-1 to promote cell survival. J Biol Chem. 2007;282:37759–69. doi: 10.1074/jbc.M705025200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Li C, Dandridge KS, Di A, Marrs KL, Harris EL, Roy K, et al. Lysophosphatidic acid inhibits cholera toxin-induced secretory diarrhea through CFTR-dependent protein interactions. J Exp Med. 2005;202:975–86. doi: 10.1084/jem.20050421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Albers HM, Dong A, van Meeteren LA, Egan DA, Sunkara M, van Tilburg EW, et al. Boronic acid-based inhibitor of autotaxin reveals rapid turnover of LPA in the circulation. Proc Natl Acad Sci U S A. 2010;107:7257–62. doi: 10.1073/pnas.1001529107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Durgam GG, Tsukahara R, Makarova N, Walker MD, Fujiwara Y, Pigg KR, et al. Synthesis and pharmacological evaluation of second-generation phosphatidic acid derivatives as lysophosphatidic acid receptor ligands. Bioorg Med Chem Lett. 2006;16:633–40. doi: 10.1016/j.bmcl.2005.10.031. [DOI] [PubMed] [Google Scholar]
- 24.Deng W, Wang DA, Gosmanova E, Johnson LR, Tigyi G. LPA protects intestinal epithelial cells from apoptosis by inhibiting the mitochondrial pathway. Am J Physiol Gastrointest Liver Physiol. 2003;284:G821–9. doi: 10.1152/ajpgi.00406.2002. [DOI] [PubMed] [Google Scholar]
- 25.Kosanam H, Ma F, He H, Ramagiri S, Gududuru V, Tigyi GJ, et al. Development of an LC-MS/MS assay to determine plasma pharmacokinetics of the radioprotectant octadecenyl thiophosphate (OTP) in monkeys. J Chromatogr B Analyt Technol Biomed Life Sci. 2010;878:2379–83. doi: 10.1016/j.jchromb.2010.07.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Booth C, Tudor G, Tudor J, Katz BP, MacVittie TJ. Acute gastrointestinal syndrome in high-dose irradiated mice. Health Phys. 2012;103:383–99. doi: 10.1097/hp.0b013e318266ee13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Withers HR, Elkind MM. Microcolony survival assay for cells of mouse intestinal mucosa exposed to radiation. Int J Radiat Biol Relat Stud Phys Chem Med. 1970;17:261–7. doi: 10.1080/09553007014550291. [DOI] [PubMed] [Google Scholar]
- 28.Kimura Y, Turner JR, Braasch DA, Buddington RK. Lumenal adenosine and AMP rapidly increase glucose transport by intact small intestine. Am J Physiol Gastrointest Liver Physiol. 2005;289:G1007–14. doi: 10.1152/ajpgi.00085.2005. [DOI] [PubMed] [Google Scholar]
- 29.Saha S, Bhanja P, Liu L, Alfieri AA, Yu D, Kandimalla ER, et al. TLR9 agonist protects mice from radiation-induced gastrointestinal syndrome. PLoS One. 2012;7:e29357. doi: 10.1371/journal.pone.0029357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Finney DJ. Probit analysis. Cambridge: Cambridge University Press; 1971. [Google Scholar]
- 31.Deng W, Poppleton H, Yasuda S, Makarova N, Shinozuka Y, Wang DA, et al. Optimal lysophosphatidic acid-induced DNA synthesis and cell migration but not survival require intact autophosphorylation sites of the epidermal growth factor receptor. J Biol Chem. 2004;279:47871–80. doi: 10.1074/jbc.M405443200. [DOI] [PubMed] [Google Scholar]
- 32.Funk-Archuleta MA, Foehr MW, Tomei LD, Hennebold KL, Bathurst IC. A soy-derived antiapoptotic fraction decreases methotrexate toxicity in the gastrointestinal tract of the rat. Nutr Cancer. 1997;29:217–21. doi: 10.1080/01635589709514627. [DOI] [PubMed] [Google Scholar]
- 33.Sturm A, Sudermann T, Schulte KM, Goebell H, Dignass AU. Modulation of intestinal epithelial wound healing in vitro and in vivo by lysophosphatidic acid. Gastroenterology. 1999;117:368–77. doi: 10.1053/gast.1999.0029900368. [DOI] [PubMed] [Google Scholar]
- 34.Kiss GN, Lee SC, Fells JI, Liu J, Valentine WJ, Fujiwara Y, et al. Mitigation of radiation injury by selective stimulation of the LPA(2) receptor. Biochimica Biophys Acta. 2013;1831:117–25. doi: 10.1016/j.bbalip.2012.08.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lai YJ, Chen CS, Lin WC, Lin FT. c-Src-mediated phosphorylation of TRIP6 regulates its function in lysophosphatidic acid-induced cell migration. Mol Cell Biol. 2005;25:5859–68. doi: 10.1128/MCB.25.14.5859-5868.2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lai YJ, Lin VT, Zheng Y, Benveniste EN, Lin FT. The adaptor protein TRIP6 antagonizes Fas-induced apoptosis but promotes its effect on cell migration. Mol Cell Biol. 2010;30:5582–96. doi: 10.1128/MCB.00134-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Whetton AD, Lu Y, Pierce A, Carney L, Spooncer E. Lysophospholipids synergistically promote primitive hematopoietic cell chemotaxis via a mechanism involving Vav 1. Blood. 2003;102:2798–802. doi: 10.1182/blood-2002-12-3635. [DOI] [PubMed] [Google Scholar]