

Abstract
Mast cells (MCs) are tissue-resident immune cells that carry out protective roles against pathogens. In disease states, such as inflammatory bowel disease, these granulocytes release a diverse array of mediators that contribute to inflammatory processes. They also participate in wound repair and tissue remodeling. In this review, the composition of MCs and how their phenotypes can be altered during inflammation of the gastrointestinal tract is detailed. Animal and human clinical studies that have implicated the participation of MCs in inflammatory bowel disease are reviewed, including the contribution of the cell’s mediators to clinical symptoms, stress-triggered inflammation, and fistula and strictures. Studies that have focused on negating the proinflammatory roles of MCs and their mediators in animal models suggest new targets for therapies for patients with inflammatory bowel disease.
Keywords: inflammatory bowel disease, ulcerative colitis, Crohn’s disease, mast cell, tryptase, chymase
Although it has been known for decades that mast cells (MCs) have life-threatening roles in systemic anaphylaxis, asthma, and other immunoglobulin E (IgE)-dependent allergic disorders,1 their participation in inflammatory bowel disease (IBD) is less appreciated. There are many reasons why MCs have received less attention in Crohn’s disease (CD) and ulcerative colitis (UC), although it has been known for sometime that gastrointestinal (GI) manifestations in patients with systemic mastocytosis are frequent and often severe.2 For one thing, routine hematoxylin and eosin histochemistry performed on mucosal biopsies for the diagnosis and monitoring of patients does not detect MCs. Cationic stains (e.g., toluidine blue, safranin, and methylene blue) routinely used to quantitate MCs in the biopsies of skin, and other connective tissues are not particularly effective for identifying these cells in the gastrointestinal tract because of the presence of less sulfated serglycin proteoglycans in the cell’s granules.3,4 Although the chloroacetate esterase cytochemistry approach is a more reliable analytical procedure for enumerating mouse MCs in tissue sections,5 not all human MCs are recognized by this method. Therefore, immunohistochemistry using combinations of antibodies that recognize proteins that are more restricted to human MCs (e.g., the granule proteases hTryptase-β,6–8 hChymase-1,9 and hCarboxypeptidase A310 and the tyrosine-kinase cell surface receptor Kit11) are required to conclusively identify these immune cells in biopsies of human intestine and colon. Another reason for the lack of appreciation of MCs in CD and UC is that researchers who study these effector cells are often interested primarily in their roles in allergic inflammation rather than the non-IgE mechanisms that are operative in IBD. Furthermore, genome wide-association studies performed to date have failed to identify an association between any MC-restricted gene and IBD (reviewed by Cho and Brant12). The latter inability to detect a genetic link between MCs and IBD now seems to be a consequence of the substantial redundancy of many of the cell’s released mediators.13–15
Although increased numbers of activated MCs have been found in involved intestinal segments, their numbers alone do not give insight as to the substantial proinflammatory capacity of these granulocytes, which have been conserved for millions of years of evolution. Just as relevant are the activation status, phenotype, and thereby function of the MCs in the intestine and colon of patients with IBD. The major proteins in human MCs are the tetramer-forming β tryptases derived from the hTPSAB1 and hTPSB2 genes.6–8,16 The mouse orthologs are MC protease (mMCP)-6 and mMCP-7.17–19 The demonstration that dextran sodium sulfate (DSS)-induced and trinitrobenzene sulfonic acid (TNBS)-induced colitis were both markedly diminished in transgenic C57BL/6 (B6) mice that lacked mMCP-6 and mMCP-7 documented for the first time the importance of MCs and their exocytosed tetramer-forming tryptases in experimental IBD.20 In this review, we discuss the human and animal data that implicated MCs and their varied exocytosed mediators in the inflammation that occurs in the intestine and colon of patients with IBD. Although there is still much work to be done, it is now apparent that MCs play central roles in several aspects of IBD. These include regulation of epithelium permeability, transmittance of signals during neuropathologic stress, the initiation and maintenance of inflammatory responses, and the subsequent tissue remodeling that occurs after resolution of the acute inflammatory stage in the GI tract.
DIVERSITY OF MCs LEADS TO DISTINCT FUNCTIONS
MCs are myeloid cells that exhibit substantial plasticity in their development. They exit the bone marrow and fetal liver as poorly granulated CD34+/Kit+ progenitors, and then complete their differentiation and maturation in the GI tract and other tissues where they eventually establish residence.21–23 Unexpectedly, the greatest number of MC-committed progenitors was found in the intestine,24 possibly because of the importance of the MC’s proteases in bacterial and helminth infections.25–28
The trafficking and homing of MC-committed progenitors into the intestine of the mouse is dependent on the integrin α4β7 and the chemokine receptor Cxcr2 on the surface of the MC progenitor and “mucosal addressin cell adhesion molecule-1” and “vascular cell adhesion molecule-1” on the intestinal endothelium.29,30 However, the most important signaling pathway that controls the retention and viability of MC-committed progenitors in the GI tract is that between the tyrosine-kinase receptor Kit/ CD11711 on the outer leaflet of the plasma membrane of the MC and Kit ligand (Kitlg)/stem cell factor31,32 on the outer leaflet of the plasma membranes of fibroblasts, endothelial cells, and other stromal cells.
In humans, the presence of activating and inactivating mutations in the KIT gene are the primary causes of systemic mastocytosis33 and piebalism,34 respectively. Despite the importance of Kit/Kitlg and its downstream transcription factor MITF in controlling MC numbers in tissues, additional cytokines (e.g., interleukin [IL]-3, IL-4, IL-6, IL-9, IL-10, IL-33, nerve growth factor, and transforming growth factor-β) are needed for the development of phenotypically different populations of mouse and human MCs. For example, Levi-Schaffer and Stevens showed in the 1980s that immature IL-3-developed mouse bone marrow-derived MCs (mBMMCs) underwent dramatic differentiation and granule maturation changes when cocultured with fibroblasts for 1 to 4 weeks.35 It subsequently was shown that the primary fibroblast-derived cytokines needed in these developmental changes were Kitlg and IL-33.31,32,36
When activated by their pathogen, complement anaphylatoxins, adenosine, or immunoglobulin receptors, MCs release varied combinations of >50 biologically active factors (Fig. 1). Some of these MC-derived mediators (e.g., histamine, serotonin, and different neutral proteases and serglycin proteoglycans) are preformed and stored in the cell’s secretory granules. Others are newly generated, biologically active lipids (e.g., leukotrienes, prostaglandins, thromboxanes, and platelet activating factor). Several hours after activation, MCs markedly increase their expression of numerous cytokines and chemokines in the delayed phase of MC-dependent inflammation. MCs express numerous activating receptors (e.g., the high-affinity IgE receptor, low-affinity IgG receptors, the complement receptors for C3a and C5a, toll-like receptors, adenosine receptors, and proteinase-activated receptors [PARs]) on their plasma membrane that are counterbalanced by numerous inhibitory receptors (e.g., Lilrb4, CD200, CD300A, FcγRIIB1, and FcγRIIB2). Needless to say, the study of MCs and how they function in the GI tract has remained a formidable challenge due to the many diverse functions of the cell’s membrane receptors and released mediators, which can have contrasting bioactivities.
FIGURE 1.
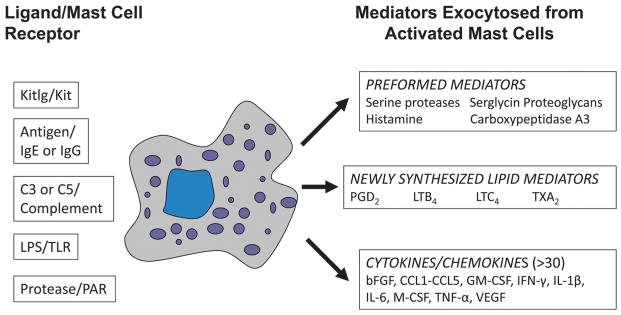
Mediators released from activated MCs. MCs express receptors that recognize different growth factors, immunoglobulins, bacterial components, and complement-derived factors. On a weight basis, ~50% of the protein content of a mature MC in the GI tract consists of varied proteases stored in the cell’s secretory granules ionically bound to serglycin proteoglycans. When MCs are activated (e.g., by their IgG receptors FcγRI and FcγRIII), they exocytose the preformed mediators from their secretory granules. Minutes to hours later, the activated MCs generate and release varied lipid mediators and cytokine/chemokine mediators, respectively. MCs are heterogeneous immune cells in tissues, and the combinations of mediators they produce are dependent on the cell’s phenotype and the activating signaling pathway.
The constitutive MCs in mammals are long-lived cells,37 and studies carried out in mice revealed that the phenotype of a tissue MC is reversibly determined by the complex combinations of factors this cell and its progenitor encounter in different tissue microenvironments. Studies carried out by Stevens, Galli, Kitamura, and others in the 1980s revealed that it is possible to quickly change the phenotype of immature MCs in vitro and in vivo.35,36,38–42 Even mature in vivo-differentiated MCs, such as those in the peritoneal cavity, can reversibly change their phenotype and function when they encounter different cytokine environments.43 Although many showed that a histochemical homogenous population of IL-3-developed mBMMCs express numerous cytokines and chemokines when activated through their high-affinity IgE receptors,44,45 subsequent in situ hybridization studies unexpectedly revealed substantial differences in cytokine expression at the individual cellular level.46 Thus, in mice, there might not be 2 identical MCs in the body.
Relevant to IBD, the MCs in the mouse small intestine increased ~100-fold in number but also reversibly changed their phenotypes in a Trichinella spiralis helminth infection model.5,47 The MCs in the lower submucosa preferentially expressed the chymase mMCP-4, the elastase mMCP-5, and the tryptase mMCP-6 before the animal was infected. Because the expanded MCs made their way from the submucosa to the tips of the mucosa’s villi where the helminth tended to reside, they ceased expressing these 3 granule proteases and began expressing the chymase family members mMCP-1, mMCP-2, and mMCP-10. In the recovery phase of the helminth infection after the adult worms had been successfully expelled, the expanded MCs slowly reverted back to their initial protease phenotype. The induced surplus of MCs then sequentially migrated from the jejunum to the draining lymph nodes, blood, and finally to the spleen, which is the primary graveyard for senescent MCs in mice.48 Crowle and Reed49 noted that the ability of MC-deficient WBB6F1-KitW/KitW-v (W/Wv) mice to expel the gastrointestinal nematode Nippostrongylus brasiliensis was significantly delayed relative to that of their MC-sufficient littermates. Knight et al25 then showed that the ability of naive B6 mice to expel T. spiralis from the infected jejunum was partly dependent on mMCP-1. Thus, what combination of granule pro-teases a MC in the GI tract expresses at any time in its life span can have important biologic consequences.
Although it is likely that a large number of functionally distinct populations of MCs also exist in humans, 3 polarized subsets have been identified by immunohistochemistry50–52 based on their differential expression of hTryptase-β, hChymase-1, and hCarboxypeptidase A3. The functional significance of the immunohistochemical protease data to IBD has not been fully ascertained. Nevertheless, the MCs that express hTryptase-β (but not hChymase-1 or hCarboxypeptidase A3) (MCT) were found predominately in the mucosal layer of the GI tract of those humans who have been evaluated. In the submucosa and serosa of the gut, the MCs tended to express all 3 granule proteases (MCTCA). The third type of polarized human MC that expressed hChymase-1 (but not hCarboxypeptidase A3 or hTryptase-β) (MC) was rarer in the GI tract. In an immunohistochemical investigation of the protease phenotypes of the MCs based on the content of these proteases, Weidner and Austen53 found that the subsets of MCs in the mucosa of the human small intestine were 58% MCT, 35% MCTCA, and 7% MC. In contrast, the sub-mucosa compartment contained 83% MCTCA and 17% MC, but essentially no MCT.
Despite the above immunohistochemistry data, it is now apparent that the situation is much more complex than initially thought because human MCs can also differ in their expression of hPRSS31 and other granule proteases. In addition, it is now known that 2 genes (designated as hTPSAB1 and hTPSB2) give rise to the enzymatically active β tryptases found in human MCs rather than 1 gene as initially thought.16 These hTPSAB1 and hTPSB2 genes reside on chromosome 16p13.3, which is a region of the human genome that is mutating at a high rate.54 It has been speculated that the extraordinary mutation rate of the locus is a consequence of the need to generate new tryptases in an arms race to help us combat pathogens more effectively versus a need to eliminate those tryptases that have become harmful to our survival. Accordingly, >50 protein isoforms of tetramer-forming tryptases already have been identified by the Human Genome Consortium due to allelic point mutation differences in the hTPSAB1 and hTPSB2 genes (summarized in Prieto-Garcia et al).15,55 Complicating the situation in humans, additional tryptase isoforms have been identified, which are caused by variable splicing of their precursor transcripts.56 The functional significance of these different tryptases has not been evaluated experimentally. Thus, for the purposes of this review, the numerous isoforms of the MC’s tetramer-forming tryptases that originate from the hTPSAB1 and hTPSB2 genes will be collectively referred to as hTryptase-β.
Although MCs are well appreciated for their proinflammatory activity in IgE-dependent allergy, asthma, and anaphylaxis, these granulocytes have been conserved for millions of years in evolution, in part, because of their beneficial roles in innate immunity and adaptive immunity. W/Wv mice constitutively have greatly reduced numbers of MCs in their tissues due to a loss-of-function mutation in the Kit gene.21 W/Wv mice are impaired in their ability to efficiently combat bacterial infections of their peritoneal cavity and lungs.26,57,58 Although it was hypothesized in the 1990s that the primary defect in innate immunity in these MC-deficient mice was due to a loss of MC-derived tumor necrosis factor-α (TNF-α), that conclusion was subsequently shown to be incorrect (reviewed in McNeil et al59). It was later shown that the adoptive transfer of recombinant hTryptase-β or mMCP-6 into the peritoneal cavity of a W/Wv mouse before the treated animal was infected could rescue its defect in bacterial clearance.26 More definitive data were obtained 6 years later when it was shown that transgenic B6 mice lacking mMCP-6 and mMCP-7 were unable to efficiently clear a Klebsiella pneumoniae infection of their peritoneal cavity. Because these infected mice had a delay in the ability to recruit bactericidal neutrophils into the infected tissue sites, the pathogen replicated unchecked early in the infection thereby resulting in sepsis.27
MCs are positioned at mucosal surfaces that interact with the external environment, and these immune cells are able to sense microbes through varied cell surface (e.g., toll-like receptors and complement) and intracellular (e.g., nucleotide-binding oligomerization domain-containing protein [NOD]-1 and NOD-2) receptors. They respond by releasing a diverse array of mediators, some of which attract neutrophils directly (e.g., leukotriene B4 and Cxcl5) or indirectly (e.g., hTryptase-β).60 When challenged with a danger signal or when perturbed in their tissue environments, MCs have the ability to shift from basal beneficial “constitutive” forms to harmful “reactive” forms. This process in the mucosal layer of the intestine is thought to be largely T-cell dependent61 and is driven by Th2 cell-derived cytokines, such as IL-4 and IL-9.62 It is thought that these cytokines induce changes in the protease composition of the developing MCs so that they can more effectively respond to the pathogen. In that regard, IL-9 induces IL-3-developed mBMMCs to markedly increase their expression of mMCP-1,42 which is a chymase needed to combat helminth infections efficiently.25 MCs also are influenced by factors they encounter in their local tissue microenvironments (e.g., Th2 and Th1 cell-derived cytokines). For instance, in vitro studies showed that a Th1 cell stimulation through interferon-γ dampened Kit/Kitlg-dependent MC proliferation and accumulation in tissues. Interferon-γ also induced these MCs to increase their expression of certain IgG receptors.63,64 In the setting of Th2 cell-polarized mucosal inflammation, MCs can express more proinflammatory mediators. They also have a lower threshold for cellular activation by IgE and non-IgE triggering events. The function of a MC in the GI tract may therefore shift from a protective/homeostatic immune cell to one that is proinflammatory to otherwise harmless signals.
The ability of MCs to alter what mediators they express provides clues as to the different inflammatory states of the colon and intestine of patients with IBD. In this regard, a unique MC transcriptome was identified in mucosal biopsies obtained from patients with eosinophilic esophagitis that differed in their response to steroid treatment. The MC protease signatures in these respective samples were hTryptase-β+/hChymase-1−/hCarboxypeptidase A3+ in the eosinophilic esophagitis patients who responded to steroids versus hTryptase-β+/hChymase-1−/hCarboxypeptidase-A3− in the patients who did not respond to steroids and control patients.65 Distinct MC protease profiles have not been evaluated extensively in patients with IBD, but it is expected that this type of assessment will provide useful information pertaining to the extent of inflammation of the GI tract.
CHARACTERIZATION OF THE MCs IN THE INTESTINE OF PATIENTS WITH IBD
Comprehensive studies have not been performed to fully characterize the distribution and phenotype of the MCs within the various inflamed and noninflamed segments of colon and intestine of patients with IBD (Fig. 2). Nevertheless, Nishida et al66 and others67,68 found increased numbers of MCs in the mucosal biopsies of examined UC and CD patients. In contrast, Bischoff et al detected fewer toluidine blue+ MCs in the involved intestinal segments of patients with IBD. When immunohistochemistry was additionally performed for the presence of hTryptase-β and hChymase-1, it was discovered that a large portion of the immunoreactive proteases resided outside the MCs.69 It was therefore concluded that degranulation of the MCs in the setting of active intestinal inflammation accounted for the decreased numbers of MCs observed.
FIGURE 2.

MCs in the colonic mucosa of a patient with IBD. A patient with active UC underwent colonoscopy to determine the extent and severity of the colitis. H&E histochemistry, performed on sections of a biopsy from an inflamed segment, revealed acute and chronic inflammatory changes in the mucosa at low ×20 (A) and higher ×40 power (B). To visualize the Kit+ MCs in the biopsy, replicate sections were stained with an antibody that recognizes this tyrosine kinase receptor. Kit+ MCs were interspersed throughout the lamina propria (black arrows) at low ×20 (C) and higher ×40 power (D). There were no aggregates or sheets of MCs that would be consistent with systemic mastocytosis. Images courtesy of Jason Hornick, MD, Department of Pathology, Brigham and Women’s Hospital.
To obtain additional data to support the contribution of MCs to the inflammation that occurs in the mucosa of patients with UC, Fox et al70 exposed colon biopsies to IgE and the relevant antigen ex vivo, and then evaluated the resulting supernatants for the presence of histamine, prostaglandin D2, and leukotrienes. These investigators discovered that there was an increase in the levels of all 3 mediators in inflamed segments compared with noninflamed segments. The accumulated data suggested increased numbers of MCs in the tissue biopsies and/or MCs that were more susceptible to immunologic activation. In another study to assess the degree of MC activation in patients with IBD, mucosa biopsies were placed in an oxygenation system for 4 hours. The levels of hTryptase-β in the supernatants were then measured by radioimmunoassay.71 In this ex vivo study, there was a significantly enhanced and prolonged increase in hTryptase-β release in the samples obtained from patients with UC compared with those from normal individuals and patients with CD. The exocytosis of the granule protease was more pronounced in inflamed tissue compared with noninflamed tissue. In another ex vivo system, ileum specimens from CD patients with active disease released significantly greater amounts of histamine than specimens taken from patients with inactive disease or from control individuals.72 In support of these data, Raithel et al found that the levels of a histamine metabolite were increased in the urine of patients with active UC and CD compared with that of normal individuals. The level of the histamine metabolite in the urine also correlated with the severity of endoscopic disease in the patients with CD and the extent of disease in the patients with UC.73
Visual evidence of MC degranulation also was observed based on an electron microscopic evaluation of the ultrastructure of the MCs in inflamed segments of patients with IBD (Fig. 3).67,74 Finally, many populations of activated MCs release substantial amounts of TNF-α in vitro. Given the success of anti–TNF-α drugs in the treatment of IBD, it is noteworthy that many of the TNF-α–expressing cells in the lamina propria were MCs.75
FIGURE 3.

Ultrastructure of intestinal MCs in proximity to nerve endings in GI disorders. MCs are granulocytes that can be identified in tissue section by electron microscopy based on their characteristic monolobed nucleus, elongated surface folds (microplicae), and abundant cytoplasmic electron-dense granules (A). In the intestine, MCs (M) frequently reside in close proximity to blood vessels and nerves (N). Depicted are MCs adjacent to nerve endings and in various stages of activation (B–D) in a patient with IBS. White arrowheads highlight granules that are partially or completely empty. Reprinted from Giovanni Barbara et al, Gastroenterology 2007;132:p 30 with permission from Elsevier.
ANIMAL MODELS DEMONSTRATE MC INVOLVEMENT IN EXPERIMENTAL COLITIS
The use of animals in various experimental models that replicate certain aspects of human IBD has been invaluable. The widely used DSS- and TNBS-induced colitis models have given insight into the importance of the barrier function of the colonic epithelium and the acute inflammatory response of the injured lower GI tract.
Several studies have been carried out in which investigators used pharmacologic approaches to inhibit the release of mediators from activated MCs and/or block their activity in the DSS- or TNBS-induce colitis models. In that regard, the serine protease inhibitor nafamostat mesilate dampens the enzymatic activities of tetramer-forming tryptases,76 and this drug reduced the inflammation that occurred in TNBS- and DSS-induced colitis in mice.77,78 In the latter colitis model, it was discovered that the serine protease inhibitor reduced colon inflammation comparable with that obtained with 5-aminosalicylate, a drug commonly used for the treatment of patients with UC. Although chymase activity was somewhat reduced in the colons of the nafamostat mesilate-treated mice, the levels of enzymatically active tryptase were below detection in these studies. Although the latter data raised the possibility of prominent adverse roles for MC tetramer-forming tryptases in colitis, the findings are difficult to interpret because nafamostat mesilate is not a highly specific inhibitor of the MC’s tetramer-forming tryptases. For example, the drug also inhibits the tryptic proteases that participate in the complement pathway.79 In addition, it has not been ruled out that the findings might have been a consequence of inactivation of the tryptase family member mPrss31,80 which also participates in DSS-induced colitis.81
Another study showed a reduction in experimental DSS-induced colitis when mice were simultaneously given the chymase inhibitor NK3201.82 It was concluded in that study that the beneficial effects of NK3201 probably were an indirect consequence of reduced levels of enzymatically active matrix metalloproteinase (MMP)-9. Relevant to this conclusion, the chymase mMCP-4 can activate certain MMP zymogens, as does human and mouse tetramer-forming tryptases.83–85
Genetically manipulated animals also have been used to evaluate the extent of MC involvement in experimental colitis. Araki et al86 used MC-deficient Ws/Ws rats in the DSS-induced colitis model. Wild-type and sibling MC-deficient rats had reduced microscopic and macroscopic colitis with reduced mucosal histamine levels. Although the use of MC-deficient mice and rats has given insight as to the overall importance of MCs and their mediators in different experimental inflammation models, many of the mediators exocytosed from these immune cells have contrasting effector functions (e.g., proinflammatory IL-6 and leukotriene B4 versus anti-inflammatory IL-10 and prostaglandin D2), thereby complicating data interpretation when MC-deficient animals are subjected to different disease models. Thus, to better evaluate the importance of the MC’s tetramer-forming tryptases in colitis and other diseases in the context of the cell’s other mediators, Adachi et al used a homologous recombination approach to create a novel B6 mouse line that cannot express either mMCP-6 or mMCP-7.27 When these transgenic mice, mMCP-5-null B6 mice, and mMCP-7-null B6 mice were subjected to the DSS and TNBS colitis models in a head-to-head comparison, only the mice that were deficient in both tetramer-forming tryptases were significantly protected from the chemically-induced colitis.20 It was therefore determined that both tetramer-forming tryptases must be knocked out in mice to uncover their importance in experimental colitis. Similar conclusions were drawn from studies in experimental arthritis13 and chronic obstructive pulmonary disease.87
As in mice, 2 genes encode MC-restricted tetramer-forming human tryptases. The accumulated mouse data suggest that the reason why the hTPSAB1 and hTPSB2 genes were not identified in genome-wide association studies of patients with CD or UC was due to the redundant proinflammatory activities of the translated enzymes. Although the disease-initiating insults that lead to experimental arthritis (e.g., deposition of complement/IgG complexes in the joint directed against citrullinated proteins) and chronic obstructive pulmonary disease (e.g., exposure of the lungs to cigarette smoke) are very different from those that lead to experimental IBD (e.g., changes in the gut’s microbiota, damage to the gut’s epithelium, and aberrant immune responses), the common feature of these 3 diseases is the activation of tissue MCs and the release of their proinflammatory tryptases.
Besides the 2 tetramer-forming tryptases, mouse and human MCs also store appreciable amounts of Prss31 in their secretory granules.80,88,89 Using a homologous recombination approach, a B6 mouse line was recently created that lacks both mPrss31 and mMCP-7.81 Because DSS-induced colitis also was significantly reduced in this transgenic animal, it is now apparent that all 3 tryptases have adverse roles in experimental colitis and that one needs to knock out at least 2 members of this family of serine proteases to undercover their global importance. In both instances, DSS-treated mMCP-6–null and mPrss31-null mice had significantly reduced levels of neutrophils and neutrophil-responsive chemokines (e.g., Cxcl1) in their colons. These data predict that a synthetic inhibitor designed to target MC tryptase-dependent inflammation in IBD would have to account for both hTryptase-β and hPRSS31 to have optimal therapeutic benefit.
POSSIBLE MECHANISMS OF ACTION OF MCs IN IBD
Although animal studies have been helpful in documenting the importance of MCs in intestinal inflammation, it is still unclear exactly how at the molecular level their exocytosed mediators participate in the inflammation observed in humans with CD or UC (Fig. 4). One mechanism by which MCs seem to contribute to the inflammation of the GI tract is by regulating the permeability of the involved epithelium as occurred in the DSS-induced colitis model in wild-type and mMCP-6-null mice (Fig. 5). Maintenance of the epithelial cell’s tight junctions is needed to prevent the infiltration of harmful bacteria, foreign antigens, and allergens into the mucosa where they can induce MC-dependent inflammatory responses. Perturbations in the integrity and function of the epithelium barrier can initiate or exacerbate inflammation in IBD (reviewed by McGuckin et al90). In an in vitro system using monolayers of T84 epithelial cells and the HMC-1 MC line, it was shown that the latter MC line could be activated by the bacterial component peptidoglycan, which then led to the release of harmful mediators through their bacterial receptors toll-like receptor 2 and NOD-2 (and possibly NOD-1).91 This activation response, in turn, rendered the monolayer of epithelial cells more susceptible to permeability as measured by the passage of horse-radish peroxidase through the epithelial cell monolayer. The trans-epithelial resistance also decreased in a peptidoglycan-dose and MC concentration-dependent manner.
FIGURE 4.

MCs have diverse functions in the intestine in both health and disease. MCs are tissue-resident immune cells that provide protection from invading organisms. However, they can be inappropriately activated in IBD, resulting in the release of mediators that contribute to the pathology of the GI tract. Depicted is a breach of the epithelium barrier, which results in the activation of a mucosal MC by bacterial products that make their way into the mucosa. The diverse arsenal of mediators produced and released by the activated MC leads to the accumulation of neutrophils and other inflammatory cells, further disruption of the epithelial barrier, increased ion/water transport, activation of the enteric nervous system, and ultimately wound repair and tissue remodeling.
FIGURE 5.
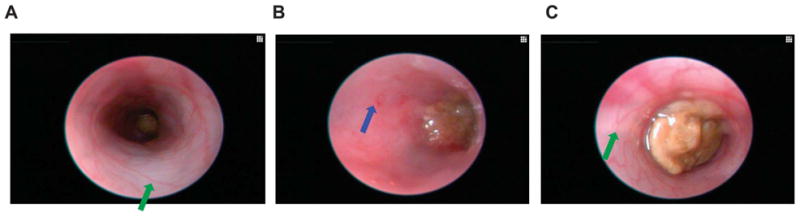
Colonoscopy of an untreated wild-type B6 mouse (A), a DSS-treated wild-type B6 mouse (B), and a DSS-treated MC tryptase knock-out (mMCP-6–null) B6 mouse (C). The mucosal surface of the untreated wild-type B6 mouse is smooth, and its underlining blood vessels can be readily seen (green arrow). In contrast, the blood vessels of the wild-type B6 mouse that received DSS for 5 days were less visible due to the submucosal edema (blue arrow). Ulcers and erosions also were present in this DSS-treated animal. The mucosal surface of the DSS-treated mMCP-6–null B6 mouse was similar to that of the untreated wild-type mouse (green arrow). The brown matter in the center of each colon is feces.
MCs have been shown to carry out important homeostatic roles in the intestine and colon by regulating the baseline permeability of the gut’s epithelium.92 MC-deficient mice had decreased intestinal permeability when given the biomarkers horseradish peroxidase and/or fluorescein isothiocyanate-labeled dextran. It was concluded that the relevant MC protease most likely was mMCP-4 since transgenic mice lacking this chymase had diminished epithelium permeability similar to the treated MC-deficient mice.92 The physiologic role of MC chymases in the intestine in this study was shown to impact mucosal architecture with increased crypt depth, a reduction in crypt-to-villus migration of epithelial cells and decreased expression of the tight-junction protein claudin-3 on the lateral membranes of the crypt epithelium. The accumulated data suggest that the MCs in the mouse and human intestine and colon regulate the architecture and permeability of the epithelium barrier due, in part, to their elaboration and release of different granule proteases.
To examine how serine proteases released from activated MCs may affect the epithelium barrier, several groups studied the G-protein-coupled protease-activated receptor family members, including (PAR)-2/F2RL1, which is expressed in the intestine. The ability of MC tetramer-forming tryptases to activate PAR-2 by tryptic cleavage of its exposed bait region is controversial, possibly because of functionally different allelic isoforms of those tetramer-forming tryptases that have been evaluated to date. Whether a PAR is a preferential target of mouse and/or human Prss31 also has not been investigated. Nevertheless, an undefined MC tryptase cleaves PAR-2 on the basolateral aspect of colonocytes, which then causes a rearrangement of gap-junction proteins and closely associated actin.93 These structural changes result in increased paracellular permeability to macromolecules. In an animal study, intracolonic administration of agonists for PAR-2 resulted in edema of the bowel wall, changes in paracellular permeability, and inflammation characterized by the accumulation of increased numbers of granulocytes in the colon.94
In support of an adverse role for PAR-2 in IBD, the expression of this protease-regulated signaling protein was found to increase in the colons of patients with UC.95 PAR-2 also is expressed on many neurons, including the spinal afferents that are in close proximity to intestinal MCs. A MC tryptase was shown to induce these neurons to release substance P and other inflammatory neuropeptides, thereby initiating neurogenic inflammation.96 Likewise, the neurons of the myenteric plexus of the guinea pig’s intestine express PAR-2, and these neurons can be stimulated by PAR-2 agonists and by supernatants from activated MCs.97
Regarding the MC chymase family members, studies carried out on mMCP-4–null and mMCP-5–null B6 mice revealed that these MC-restricted proteases mediate ischemia-reperfusion injury of skeletal muscle98 and thermal injury of skin,99 in part, through the proteolytic disruption of tight junctions.100 These in vivo mouse data raise the possibility that hChymase-1 might have similar roles in the GI tract of those with IBD. It is not known whether the proteolytic loss of the claudin-4 in the epidermis that occurs in burn-injured mice is a consequence of direct cleavage of this tight-junction protein by mMCP-4 or by the indirect ability of the chymase to activate a pro-MMP zymogen constitutively present in the animal’s skin. However, in support of the latter possibility, hChymase-1 and mMCP-4 can activate certain MMP zymogens in vitro.83
What role, if any, MCs have in intestinal ion transport has not been investigated in-depth but circumstantial evidence has been obtained which implicated adverse roles for these cells and their mediators in the ion and water loss that often occurs in the GI tract of patients with IBD. In that regard, the major source of histamine in the body is the MC. Histamine receptors H1, H2, and H3 are present in the intestine,101 and histamine regulates ion transport in the intestines of animals102 and patients with IBD.103 Other MC mediators that participate in the inflammation that occur in the GI tract of patients with IBD have not been studied extensively. Nevertheless, the MCs in the inflamed segments of patients with UC released more histamine, prostaglandin D2, and leukotriene C4 than uninflamed segments when activated through their high-affinity IgE receptors.70 It also was shown that intraluminal release of leukotriene C4 was higher at baseline and on stimulation than in control patients and those with UC.104 The accumulated data suggested that factors released from activated MCs contribute to the barrier changes that occur in the epithelium of the GI tract of patients with CD.
IRRITABLE BOWEL SYNDROME SYMPTOMS IN IBD ARE MEDIATED BY MC FACTORS
Barbara et al105 showed that patients suffering from irritable bowel syndrome (IBS) had more MCs in the left colon and rectum than those of normal individuals. The MCs at these sites often had degranulated, and many of these activated MCs resided near nerve endings (Fig. 3). These observations could be biologically significant because many patients with IBD have IBS-like symptoms, including abdominal pain and diarrhea. Accordingly, this phenomenon has been observed when patients with IBD are in remission.106,107 It is conceivable that while the inflammatory infiltrate resolves, a subset of activated MCs remain in the mucosa and submucosa, which contribute to gastrointestinal symptoms. This also has been observed after self-limited GI infections. The so-called postinfectious IBS may persist for months and is thought to be due to biologically active factors released from intestinal MCs.108
In an IBD study, UC patients in remission were shown to have evidence of IBS on barostat testing in the rectum.109 Nineteen patients with UC in remission (Mayo score of 0) and 17 control patients undergoing routine screening colonoscopy were tested for rectal compliance and visceroperception. Although the measurements for rectal compliance were not significantly different between the 2 groups, visceroperception was significantly more pronounced in the patients with UC. When the sigmoid biopsy samples were analyzed, the number of MCs was significantly increased in the patients with UC. In addition, more of these cells were degranulated located in close proximity to nerve endings, as observed by electron microscopy. Although these findings suggest MC involvement in the IBS-like symptoms that may follow active IBD, a cause-and-effect direct association has not been shown and the differences in MC numbers correlate only weakly with the visceroperception.
Activation of MCs and their exocytosed mediators have been shown to contribute to IBS through actions on permeability of the epithelium in the GI tract. Wilcz-Villega et al110 showed that a MC tryptase increased permeability to macromolecules and decreased resistance in a Caco-2 epithelial cell layer system. Enhanced MC tryptase activity also has been shown in ex vivo colonic biopsies in patients with diarrhea-predominant IBS.111
RELEVANCE OF STRESS-INDUCED ACTIVATION OF MCs IN IBD
It is well known that stress somehow causes alterations of the epithelium barrier in the intestine,112 and that these stress-induced changes likely contribute to the pathogenesis of IBD.113 Evidence has been obtained that link MC activation and physiologic stress (reviewed by McKay and Bienenstock114). In fact, Ehrlich,115 who first identified tissue MCs in 1878, discovered that many of the MCs in skin and other connective tissues were located adjacent to nerves. In a seminal study that first documented a physiologic link between MCs and nerves in vivo, Bienstock et al116 showed that the Pavlovian conditioning of rats resulted in the activation of mucosal MCs and the release of the granule chymase rMCP-2 into the circulation. In an animal model of chronic stress, MC-deficient Ws/Ws rats and their +/+ sibling controls were subjected to 5 days of water deprivation in a stress model in which the animals could see the water but could not access it for several hours each day.117 Some colon segments from these experiments were placed in Ussing chambers to measure differences in the influx of horseradish peroxidase. The remaining segments were analyzed for MC hyperplasia by histochemistry and for MC activation by electron microscopy. The colons of the stressed, wild-type MC-sufficient rats displayed an increased flux of the reporter, increased MC numbers, and ultrastructural evidence of MC activation that lasted up to 72 hours after the animals were stressed, compared with sham nonstressed rats. These ultra-structural changes were not found in the epithelium of MC-deficient rats that were similarly stressed. Comparable data were obtained by another group which showed that the intestinal epithelium was largely unperturbed without evidence of bacterial adhesion and without associated inflammatory infiltrate in a 10-day model of chronic stress in MC-deficient Ws/Ws rats compared with control animals.112
MC-dependent changes to the epithelium also have been found in acute stress models, such as restraint-induced stress and systemic injection of corticotropin-releasing hormone (CRH).118,119 Finally, a model of early life stress (namely neonatal maternal separation) led to increased spontaneous colitis in IL-10–deficient mice that was characterized by changes in the permeability of the epithelium and increased release of the granule mediator mMCP-6.120
In the clinical setting, patients with IBD often describe a stressful event that precedes the onset of their symptoms. These observations have been supported in the clinical literature.121 There is an abundance of data that support the relationship between the complex network of the central and gut nervous systems with the mucosal immune response that occurs in IBD.113 The MCs positioned in the submucosa and muscle layers of the intestine often are in close proximity to the autonomic and vagal nerve endings of the gut-brain axis, and it has been concluded that they carry out key effector roles in neurogenic inflammation. In this process, it is likely that MCs receive and deliver signals to the nervous system. Release of MC mediators ultimately affects intestinal permeability, motility, secretion, and the mucosal immune response.
CRH is a 41-mer peptide enzymatically derived from its 196-mer precursor protein. It is produced in the central nervous system and is thought to play a central role in stress-induced intestinal pathology.122 Some populations of MCs synthesize and secrete CRH and also express receptors that recognize the peptide.123 In an ex vivo intestinal permeability system, it was shown that exposure of CRH to porcine ileum led to increased paracellular permeability.124 Because degranulated MCs were found, various inhibitors were used to show that this CRH-dependent process was mediated, in part, by TNF-α and the MC’s granule proteases, as well as the enteric nervous system. Nerve growth factor and substance P also can induce some populations of MCs to release lipid and cytokine mediators in vitro.125,126
MCs are thought to play a central role in the neural inflammation observed in IBD, and various MC-derived mediators (e.g., histamine, serotonin, and platelet-activating factor) have neuropharmacological activity on the electrical and synaptic behavior of neurons in the enteric nervous system.127 It is thought that the triggering of MCs in the context of inflammation is transmitted to the enteric nervous system, which then leads to a secondary response that promotes protection of the host including mucosal secretion and a coordinated increase in intestinal motility.
EVIDENCE FOR MC INVOLVEMENT EARLY IN CD
It is important to evaluate for the recurrence of CD after ileocolonic resection to prevent future complications and surgeries. This clinical finding also serves as a model to study the natural history of inflammation in these patients. Ferrante et al128 showed that the inflammation the investigators detected around myenteric nerve bundles “plexitis” at the proximal margin of ileal resection specimens in the absence of surrounding inflammation predicted early endoscopic recurrence. Sokol et al129 studied histologic factors that predicted early recurrence of CD in 171 patients. These investigators found that their patients with CD displayed more sub-mucosal plexitis. Moreover, approximately one third of these patients had 3 or more MCs in and around the nerve bundles, as opposed to 2 or less in all of the control cases. In a multivariate analysis to evaluate which of these patients had a clinical recurrence within 2 years, the presence of 3 or more MCs in plexitis at the proximal area of ileal resection was the second greatest predictor after active smoking (see Concluding Remarks).
MC INVOLVEMENT IN INTESTINAL STRICTURES AND FISTULAE
Two of the most important sequelae of chronic intestinal inflammation in patients with CD are fibrotic strictures and fistulae. These irreversible and devastating processes are thought to be related to ineffective wound healing. The process of wound healing may include the urgent need to repair an acute mucosal defect and the remodeling that occurs under chronic inflammatory conditions. MCs and their mediators are key elements in all 3 stages of wound healing, namely inflammation, proliferation, and remodeling.130,131 The involvement of MCs in the repair of the colon after DSS-induced acute colitis was suggested by an increase in tryptase-expressing MCs that persisted up to day 20 at a point when other inflammatory cells had returned to baseline.132
MCs accumulate at the site of wounds, and these tissue-resident immune cells release varied chemotactic factors that attract leucocytes, macrophages, neutrophils, basophils, and/or eosinophils. MC mediators (especially, the cell’s tetramer-forming tryptases) also can induce bystander cells to increase their expression of numerous biologically active factors. The presence of fibroproliferative factors leads to fibroblast proliferation and collagen accumulation130 as occurs when IL-3-derived mBMMCs are cocultured with 3T3 fibroblasts.133 Fibroblasts undergo phenotypic changes in response to MC-derived factors. For example, they can become myofibroblasts that possess more pronounced contractile properties.130,131 The contractile capacity of these cells is critical to the development of stricture formation in CD.134 Garbuzenko et al135 showed that sonicates of hTryptase-β+ human MCs induced monolayers of cultured cutaneous fibroblasts to increase their rate of proliferation and collagen production. Xu et al136 obtained similar data using fibroblasts obtained from biopsies of intestinal segments from patients with CD. Soluble factors from fibroblasts (e.g., IL-33), in turn, can influence the phenotype and function of MCs.35,36 It therefore has been appreciated since the 1980s that complex bilateral interactions occur between MCs and fibroblasts that influence both cell types.
In the setting of chronic intestinal inflammation in several subtypes of CD, the respective intestinal segment can become fibrotic and thickened due to increased expression and deposition of type-III collagen.137,138 Clinically significant strictures often develop at the site of fibrosis. Colonic fibrosis also occurs in TNBS-induced colitis, and Xu et al136 showed that a MC stabilizer was able to decrease histologic evidence of inflammation and fibrosis, secondary to a decrease in MC activation and collagen production. Using an immunohistochemistry approach, Gelbmann et al139 investigated the presence of MCs in surgically resected intestinal strictures in patients with CD. They found a striking accumulation of hTryptase-β+ and/or hChymase-1+ MCs in the hypertrophied and fibrotic areas of the muscularis propria. Because these investigators showed by double-staining approaches that the MCs colocalized with the extracellular matrix protein laminin, this work suggested the significance of MCs in the pathogenesis of fibrotic strictures. Unfortunately, it remains to be determined which MC-derived factor(s) is operative in this setting. Because many types of collagen exist, a more in-depth analysis of the changes of the protein composition of the extra-cellular matrix in the lesions is needed.
Patients with CD who develop a stricturing phenotype that required surgery (or who had a higher rate of inflammatory recurrence after surgery) often expressed allelic variants of the NOD-2/CARD15 gene.140 The intestinal MCs in patients with CD were discovered to have high levels of the latter bacteria receptor; they also could be activated by its ligand muramyl dipeptide.141
Intestinal and perianal fistulae develop in ~50% of patients with CD at least once in their lifetime,142 and therapeutic outcomes using antibiotics, immunosuppressants, or TNF-α inhibitors are poor. In the majority of cases, fistulae develop in the setting of active luminal inflammation. They may also form as a complication of surgery or in response to an abscess.143 The mechanisms that drive fistulae development have not been determined. Nevertheless, recent studies revealed that fistulae are the product of epithelial-to-mesenchymal transition, where highly polarized epithelial cells acquire migratory and invasive features.144–146 This process is likely driven by a combination of environmental (e.g., host microbiota) and genetic factors. In this regard, pathogen-associated molecular patterns (e.g., muramyl dipeptide) induce epithelial-to-mesenchymal transition–related genes in cultured intestinal epithelial cells.147 Mutations have been identified in the human NOD-2 gene that are linked to increased susceptibility to CD, particularly the fistulizing and fibrostenosing type.148 Okumura et al141 showed that intestinal MCs express NOD-2, and that the expression of this bacteria receptor is increased in patients with CD compared with patients with UC and controls. Furthermore, NOD-2 expression in MCs could be augmented by exposure to interferon-γ. It also was associated with factors, such as CXCL10 that result in the recruitment of inflammatory cells. Therefore, inappropriate bacterial sensing leading to epithelial-to-mesenchymal transition and fistulae in genetically susceptible patients could contribute to the dysregulation of intestinal MCs due to their mutated NOD-2 bacteria receptors.
Various studies have been performed on surgically resected fistulae to understand the factors that may lead to their development. Kiregaard et al143 found strong expression of MMP-3 by immunohistochemistry in mononuclear macrophage-like cells around the fistula lumen of CD patients. Recent studies performed on perianal fistulae tissue identified the presence of increased numbers of MCs in and around the fistula tracts by immunohistochemical staining for hTryptase-β and hChymase-1.147 In addition to the MCs, Frei et al discovered that MMP-9 and MMP-13 were highly expressed along the fistula tracts. Because numerous MC proteases can activate MMP zymogens,83–85 it is conceivable that MCs are central in the pathogenesis of CD associated-fistulae. This is an important area of research due to the limited and poorly efficacious medications to treat fistulae in CD. Thus, new therapeutics (e.g., a pharmacologic inhibitor of the relevant MC pro-teases) could significantly advance this field of study.
PHARMACOLOGIC TRIALS DESIGNED TO TARGET MCs FOR THE TREATMENT OF IBD
Regrettably, there have been limited trials to date designed to evaluate the efficacy of drugs that target MCs and their mediators for the treatment of IBD. Aminosalicylates are effective for inducing and maintaining remission in patients with mild-to-moderate UC. Their mechanisms of action are unclear, but 5-aminosalicylate inhibits the release of histamine and prostaglandin D2 in cultured human intestinal MCs.149 In an open-label clinical trial, Tremaine et al150 evaluated daily treatment with a subcutaneous injection of the tryptase inhibitor APC-2059 in 53 patients with mild-to-moderate UC who were symptomatic despite treatment with 5-aminosalicylate medications. Twenty-nine percent of the patients met the primary endpoint of obtaining a clinical response at 1 month with a clinical score of 0 to 3 on a 12-point scale (clinical parameters and endoscopic score). In all, 48% reached the secondary endpoint of a final disease activity score of 0 to 3 or at least a 4-point drop. This clinical trial suggests that the next generation of hTryptase-β inhibitors might be effective in patients with milder forms of colitis or who are not on concomitant 5-aminosalicylate drugs.
In earlier studies, conflicting evidence was obtained regarding the efficacy of disodium cromoglycate to treat ulcerative proctitis or proctosigmoiditis.151,152 Although this drug blocks the release of the mediators from activated MCs, the fact that large amounts were needed to achieve a significant therapeutic effect makes interpretation of the in vivo data difficult. Whatever its mechanism of action, the poor tissue absorption of cromolyn likely hinders its therapeutic value. More recently, naturally occurring flavonoids (e.g., quercetin) have been shown to inhibit the release of inflammatory cytokines from cultured human MCs more efficiently than cromolyn.153 Although quercetin was effective in an open-label trial for atopic dermatitis and photosensitivity, its role at the molecular level in MC-dependent IBD remains to be determined.
The use of corticosteroids is a highly effective treatment for inducing disease remission in some UC and CD patients. Patients with UC who have been given corticosteroids have fewer MCs in their rectal biopsies compared with patients with UC not on steroids. This reduction was independent of the degree of architecture distortion or other signs of inflammation. The findings raise the possibility that the efficacy of steroid treatment in IBD is due, in part, to its effects on MCs.154 In support of this conclusion, glucocorticoids inhibit the IL-3-induced proliferation of mBMMCs, the high-affinity IgE receptor-mediated expression of TNF-α, and the IL-10-induced expression of chymases.155
MCs selectively home to the intestine through the interaction of α4β7 integrin with mucosal addressin cell adhesion molecule-1 and vascular cell adhesion molecule-1.30 Taking advantage of these findings, vedolizumab has been approved for the treatment of UC and CD patients.156–158 Vedolizumab is a humanized monoclonal antibody that binds to α4β7, thereby hindering the homing and accumulation of inflammatory lymphocytes to the intestine. Its clinical efficacy could be partly due to its ability to inhibit the accumulation of MC-committed progenitors in the intestine.
Inhibitors of TNF-α are now a mainstay in the treatment of moderate-to-severe CD and UC. Although intestinal MCs have small amounts of TNF-α prepackaged in the secretory granules, they have the ability to synthesize and release much more TNF-α when exposed to an inflammatory stimulus. Nevertheless, there is no evidence that the success of anti–TNF-α therapy in IBD is due to the inhibition of expression of this cytokine from activated MCs because macrophages and other cell types in the intestine and colon also produce substantial amounts of TNF-α.
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Herein, we reviewed the ability of MCs to carry out diverse functions in the GI tract by their varied cell surface receptors and released mediators. MCs may transition from protective immune cells that contribute to intestinal homeostasis to potent proinflammatory cells that exacerbate the many features of IBD. This shift in function is likely due to the activation status and phenotypic changes of the MCs in the intestine and colon that are driven by cues from the altered tissue microenvironments. Furthermore, it is possible that the phenotype of MCs may be altered by environmental influences, such as the dramatic shifts in the composition of the microbiota159 that have been observed in Western societies and are thought to contribute to the increased prevalence of IBD in the last decade.160,161 The release of proinflammatory mediators from the activated MCs in the intestine and colon of patients with IBD likely contributes to the inability of the epithelium to act as an effective barrier to pathogens and immune-activating antigens in the GI tract. We therefore need to better understand how factors released from activated MCs in the GI tract contribute to IBD inflammation and wound healing, as we search for more effective and safe therapies for our patients.
Despite major advances in our understanding of the causes of IBD at the genetic level, it is now apparent that environmental factors play a substantial role in disease susceptibility. In that regard, exposure to cigarette smoke has been the one factor consistently shown to exacerbate CD. It therefore is imperative that we understand how cigarette smoke and other environmental factors affect some of the key cells in the intestine and colon that cause the substantial pathology that is observed in patients with IBD. Interestingly, cigarette smoke-induced chronic obstructive pulmonary disease was significantly reduced in tryptase-deficient B6 mice relative to similarly treated wild-type B6 and BALB/c mice.87 It therefore will be interesting to compare the consequences of cigarette smoke exposure in the GI tract of wild-type and tryptase-null B6 mice. Finally, the ongoing creation of transgenic mouse lines are needed to help us better understand the complex interplay between genetic and environmental risk factors that ultimately predispose one’s susceptibility to developing IBD.
Acknowledgments
Supported in part by grants from the National Institutes of Health DK094971 (MJH) and AI059746 (RLS) and the Harvard Digestive Diseases Center.
The authors thank Jason Hornick, MD (Brigham and Women’s Hospital, Boston, MA) for the histochemistry images shown in Figure 2 from colon biopsies of a patient with IBD.
Footnotes
The authors have no conflicts of interest to disclose.
References
- 1.Kinet JP. The high-affinity IgE receptor (FcεRI): from physiology to pathology. Annu Rev Immunol. 1999;17:931–972. doi: 10.1146/annurev.immunol.17.1.931. [DOI] [PubMed] [Google Scholar]
- 2.Sokol H, Georgin-Lavialle S, Canioni D, et al. Gastrointestinal manifestations in mastocytosis: a study of 83 patients. J Allergy Clin Immunol. 2013;132:866–873. doi: 10.1016/j.jaci.2013.05.026. [DOI] [PubMed] [Google Scholar]
- 3.Stevens RL, Lee TD, Seldin DC, et al. Intestinal mucosal mast cells from rats infected with Nippostrongylus brasiliensis contain protease-resistant chondroitin sulfate diB proteoglycans. J Immunol. 1986;137:291–295. [PubMed] [Google Scholar]
- 4.Enerbäck L, Kolset SO, Kusche M, et al. Glycosaminoglycans in rat mucosal mast cells. Biochem J. 1985;227:661–668. doi: 10.1042/bj2270661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Friend DS, Ghildyal N, Austen KF, et al. Mast cells that reside at different locations in the jejunum of mice infected with Trichinella spiralis exhibit sequential changes in their granule ultrastructure and chymase phenotype. J Cell Biol. 1996;135:279–290. doi: 10.1083/jcb.135.1.279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schwartz LB, Lewis RA, Austen KF. Tryptase from human pulmonary mast cells: purification and characterization. J Biol Chem. 1981;256:11939–11943. [PubMed] [Google Scholar]
- 7.Miller JS, Moxley G, Schwartz LB. Cloning and characterization of a second complementary DNA for human tryptase. J Clin Invest. 1990;86:864–870. doi: 10.1172/JCI114786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Vanderslice P, Ballinger SM, Tam EK, et al. Human mast cell tryptase: multiple cDNAs and genes reveal a multi-gene serine protease family. Proc Natl Acad Sci U S A. 1990;87:3811–3815. doi: 10.1073/pnas.87.10.3811. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Caughey GH, Zerweck EH, Vanderslice P. Structure, chromosomal assignment, and deduced amino acid sequence of a human gene for mast cell chymase. J Biol Chem. 1991;266:12956–12963. [PubMed] [Google Scholar]
- 10.Reynolds DS, Gurley DS, Stevens RL, et al. Cloning of cDNAs that encode human mast cell carboxypeptidase A, and comparison of the protein with mouse mast cell carboxypeptidase A and rat pancreatic carboxypeptidases. Proc Natl Acad Sci U S A. 1989;86:9480–9484. doi: 10.1073/pnas.86.23.9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Geissler EN, Ryan MA, Housman DE. The dominant-white spotting (W) locus of the mouse encodes the c-kit proto-oncogene. Cell. 1988;55:185–192. doi: 10.1016/0092-8674(88)90020-7. [DOI] [PubMed] [Google Scholar]
- 12.Cho JH, Brant SR. Recent insights into the genetics of inflammatory bowel disease. Gastroenterology. 2011;140:1704–1712. doi: 10.1053/j.gastro.2011.02.046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.McNeil HP, Shin K, Campbell IK, et al. The mouse mast cell-restricted tetramer-forming tryptases mouse mast cell protease 6 and mouse mast cell protease 7 are critical mediators in inflammatory arthritis. Arthritis Rheum. 2008;58:2338–2346. doi: 10.1002/art.23639. [DOI] [PubMed] [Google Scholar]
- 14.Huang C, Wong GW, Ghildyal N, et al. The tryptase, mouse mast cell protease 7, exhibits anticoagulant activity in vivo and in vitro due to its ability to degrade fibrinogen in the presence of the diverse array of protease inhibitors in plasma. J Biol Chem. 1997;272:31885–31893. doi: 10.1074/jbc.272.50.31885. [DOI] [PubMed] [Google Scholar]
- 15.Prieto-Garcia A, Zheng D, Adachi R, et al. Mast cell restricted mouse and human tryptase-heparin complexes hinder thrombin-induced coagulation of plasma and the generation of fibrin by proteolytically destroying fibrinogen. J Biol Chem. 2012;287:7834–7844. doi: 10.1074/jbc.M111.325712. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Pallaoro M, Fejzo MS, Shayesteh L, et al. Characterization of genes encoding known and novel human mast cell tryptases on chromosome 16p13.3. J Biol Chem. 1999;274:3355–3362. doi: 10.1074/jbc.274.6.3355. [DOI] [PubMed] [Google Scholar]
- 17.Reynolds DS, Stevens RL, Lane WS, et al. Different mouse mast cell populations express various combinations of at least six distinct mast cell serine proteases. Proc Natl Acad Sci U S A. 1990;87:3230–3234. doi: 10.1073/pnas.87.8.3230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Reynolds DS, Gurley DS, Austen KF, et al. Cloning of the cDNA and gene of mouse mast cell protease 6: transcription by progenitor mast cells and mast cells of the connective tissue subclass. J Biol Chem. 1991;266:3847–3853. [PubMed] [Google Scholar]
- 19.McNeil HP, Reynolds DS, Schiller V, et al. Isolation, characterization, and transcription of the gene encoding mouse mast cell protease 7. Proc Natl Acad Sci U S A. 1992;89:11174–11178. doi: 10.1073/pnas.89.23.11174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Hamilton MJ, Sinnamon MJ, Lyng GD, et al. Essential role for mast cell tryptase in acute experimental colitis. Proc Natl Acad Sci U S A. 2011;108:290–295. doi: 10.1073/pnas.1005758108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kitamura Y, Go S, Hatanaka K. Decrease of mast cells in W/Wv mice and their increase by bone marrow transplantation. Blood. 1978;52:447–452. [PubMed] [Google Scholar]
- 22.Gurish MF, Pear WS, Stevens RL, et al. Tissue-regulated differentiation and maturation of a v-abl-immortalized mast cell-committed progenitor. Immunity. 1995;3:175–186. doi: 10.1016/1074-7613(95)90087-x. [DOI] [PubMed] [Google Scholar]
- 23.Kirshenbaum AS, Kessler SW, Goff JP, et al. Demonstration of the origin of human mast cells from CD34+ bone marrow progenitor cells. J Immunol. 1991;146:1410–1415. [PubMed] [Google Scholar]
- 24.Schrader JW, Scollay R, Battye F. Intramucosal lymphocytes of the gut: Lyt-2 and thy-1 phenotype of the granulated cells and evidence for the presence of both T cells and mast cell precursors. J Immunol. 1983;130:558–564. [PubMed] [Google Scholar]
- 25.Knight PA, Wright SH, Lawrence CE, et al. Delayed expulsion of the nematode Trichinella spiralis in mice lacking the mucosal mast cell-specific granule chymase, mouse mast cell protease-1. J Exp Med. 2000;192:1849–1856. doi: 10.1084/jem.192.12.1849. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Huang C, De Sanctis GT, O’Brien PJ, et al. Evaluation of the substrate specificity of human mast cell tryptase β1 and demonstration of its importance in bacterial infections of the lung. J Biol Chem. 2001;276:26276–26284. doi: 10.1074/jbc.M102356200. [DOI] [PubMed] [Google Scholar]
- 27.Adachi R, Melicoff E, Sansores-Garcia L, et al. The mast cell-restricted tryptase mMCP-6 has a critical immunoprotective role in bacterial infections. J Biol Chem. 2007;282:20809–20815. doi: 10.1074/jbc.M611842200. [DOI] [PubMed] [Google Scholar]
- 28.Shin K, Watts GF, Oettgen HC, et al. Mouse mast cell tryptase mMCP-6 is a critical link between adaptive and innate immunity in the chronic phase of Trichinella spiralis infection. J Immunol. 2008;180:4885–4891. doi: 10.4049/jimmunol.180.7.4885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Abonia JP, Austen KF, Rollins BJ, et al. Constitutive homing of mast cell progenitors to the intestine depends on autologous expression of the chemokine receptor CXCR2. Blood. 2005;105:4308–4313. doi: 10.1182/blood-2004-09-3578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gurish MF, Tao H, Abonia JP, et al. Intestinal mast cell progenitors require CD49dβ7 (α4β7 integrin) for tissue-specific homing. J Exp Med. 2001;194:1243–1252. doi: 10.1084/jem.194.9.1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Flanagan JG, Leder P. The kit ligand: a cell surface molecule altered in steel mutant fibroblasts. Cell. 1990;63:185–194. doi: 10.1016/0092-8674(90)90299-t. [DOI] [PubMed] [Google Scholar]
- 32.Martin FH, Suggs SV, Langley KE, et al. Primary structure and functional expression of rat and human stem cell factor DNAs. Cell. 1990;63:203–211. doi: 10.1016/0092-8674(90)90301-t. [DOI] [PubMed] [Google Scholar]
- 33.Akin C, Metcalfe DD. Systemic mastocytosis. Annu Rev Med. 2004;55:419–432. doi: 10.1146/annurev.med.55.091902.103822. [DOI] [PubMed] [Google Scholar]
- 34.Giebel LB, Spritz RA. Mutation of the KIT (mast/stem cell growth factor receptor) protooncogene in human piebaldism. Proc Natl Acad Sci U S A. 1991;88:8696–8699. doi: 10.1073/pnas.88.19.8696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Levi-Schaffer F, Austen KF, Gravallese PM, et al. Coculture of interleukin-3-dependent mouse mast cells with fibroblasts results in a phenotypic change of the mast cells. Proc Natl Acad Sci U S A. 1986;83:6485–6488. doi: 10.1073/pnas.83.17.6485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kaieda S, Shin K, Nigrovic PA, et al. Synovial fibroblasts promote the expression and granule accumulation of tryptase via interleukin-33 and its receptor ST-2 (IL1RL1) J Biol Chem. 2010;285:21478–21486. doi: 10.1074/jbc.M110.114991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Padawer J. Mast cells: extended lifespan and lack of granule turnover under normal in vivo conditions. Exp Mol Pathol. 1974;20:269–280. doi: 10.1016/0014-4800(74)90059-8. [DOI] [PubMed] [Google Scholar]
- 38.Nakano T, Sonoda T, Hayashi C, et al. Fate of bone marrow-derived cultured mast cells after intracutaneous, intraperitoneal, and intravenous transfer into genetically mast cell-deficient W/Wv mice: evidence that cultured mast cells can give rise to both connective tissue and muosal mast cells. J Exp Med. 1985;162:1025–1043. doi: 10.1084/jem.162.3.1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Otsu K, Nakano T, Kanakura Y, et al. Phenotypic changes of bone marrow-derived mast cells after intraperitoneal transfer into W/Wv mice that are genetically deficient in mast cells. J Exp Med. 1987;165:615–627. doi: 10.1084/jem.165.3.615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Gurish MF, Ghildyal N, McNeil HP, et al. Differential expression of secretory granule proteases in mouse mast cells exposed to interleukin-3 and c-kit ligand. J Exp Med. 1992;175:1003–1012. doi: 10.1084/jem.175.4.1003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ghildyal N, Friend DS, Nicodemus CF, et al. Reversible expression of mouse mast cell protease 2 mRNA and protein in cultured mast cells exposed to IL-10. J Immunol. 1993;151:3206–3214. [PubMed] [Google Scholar]
- 42.Eklund KK, Ghildyal N, Austen KF, et al. Induction by IL-9 and suppression by IL-3 and IL-4 of the levels of chromosome 14-derived transcripts that encode late-expressed mouse mast cell proteases. J Immunol. 1993;151:4266–4273. [PubMed] [Google Scholar]
- 43.Sonoda S, Sonoda T, Nakano T, et al. Development of mucosal mast cells after injection of a single connective tissue-type mast cell in the stomach mucosa of genetically mast cell-deficient W/Wv mice. J Immunol. 1986;137:1319–1322. [PubMed] [Google Scholar]
- 44.Plaut M, Pierce JH, Watson CJ, et al. Mast cell lines produce lymphokines in response to cross-linkage of FcεRI or to calcium ionophores. Nature. 1989;339:64–67. doi: 10.1038/339064a0. [DOI] [PubMed] [Google Scholar]
- 45.Burd PR, Rogers HW, Gordon JR, et al. Interleukin 3-dependent and -independent mast cells stimulated with IgE and antigen express multiple cytokines. J Exp Med. 1989;170:245–257. doi: 10.1084/jem.170.1.245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Gurish MF, Ghildyal N, Arm J, et al. Cytokine mRNAs are preferentially increased relative to secretory granule protein mRNAs in mouse bone marrow-derived mast cells that have undergone IgE-mediated activation and degranulation. J Immunol. 1991;146:1527–1533. [PubMed] [Google Scholar]
- 47.Friend DS, Ghildyal N, Gurish MF, et al. Reversible expression of tryptases and chymases in the jejunal mast cells of mice infected with Trichinella spiralis. J Immunol. 1998;160:5537–5545. [PubMed] [Google Scholar]
- 48.Friend DS, Gurish MF, Austen KF, et al. Senescent jejunal mast cells and eosinophils in the mouse preferentially translocate to the spleen and draining lymph node, respectively, during the recovery phase of helminth infection. J Immunol. 2000;165:344–352. doi: 10.4049/jimmunol.165.1.344. [DOI] [PubMed] [Google Scholar]
- 49.Crowle PK, Reed ND. Rejection of the intestinal parasite Nippostrongylus brasiliensis by mast cell-deficient W/Wv anemic mice. Infect Immun. 1981;33:54–58. doi: 10.1128/iai.33.1.54-58.1981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Irani AA, Schechter NM, Craig SS, et al. Two types of human mast cells that have distinct neutral protease compositions. Proc Natl Acad Sci U S A. 1986;83:4464–4468. doi: 10.1073/pnas.83.12.4464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Irani AM, Goldstein SM, Wintroub BU, et al. Human mast cell carboxypeptidase: selective localization to MCTC cells. J Immunol. 1991;147:247–253. [PubMed] [Google Scholar]
- 52.Schechter NM, Irani AM, Sprows JL, et al. Identification of a cathepsin G-like proteinase in the MCTC type of human mast cell. J Immunol. 1990;145:2652–2661. [PubMed] [Google Scholar]
- 53.Weidner N, Austen KF. Heterogeneity of mast cells at multiple body sites: fluorescent determination of avidin binding and immunofluorescent determination of chymase, tryptase, and carboxypeptidase content. Pathol Res Pract. 1993;189:156–162. doi: 10.1016/S0344-0338(11)80086-5. [DOI] [PubMed] [Google Scholar]
- 54.Badge RM, Yardley J, Jeffreys AJ, et al. Crossover breakpoint mapping identifies a subtelomeric hotspot for male meiotic recombination. Hum Mol Genet. 2000;9:1239–1244. doi: 10.1093/hmg/9.8.1239. [DOI] [PubMed] [Google Scholar]
- 55.Prieto-Garcia A, Castells MC, Hansbro PM, et al. Mast cell-restricted tetramer-forming tryptases and their beneficial roles in hemostasis and blood coagulation. Immunol Allergy Clin North Am. 2014;34:263–281. doi: 10.1016/j.iac.2014.01.001. [DOI] [PubMed] [Google Scholar]
- 56.Jackson NE, Wang HW, Bryant KJ, et al. Alternate mRNA splicing in multiple human tryptase genes is predicted to regulate tetramer formation. J Biol Chem. 2008;283:34178–34187. doi: 10.1074/jbc.M807553200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Echtenacher B, Männel DN, Hültner L. Critical protective role of mast cells in a model of acute septic peritonitis. Nature. 1996;381:75–77. doi: 10.1038/381075a0. [DOI] [PubMed] [Google Scholar]
- 58.Malaviya R, Ikeda T, Ross E, et al. Mast cell modulation of neutrophil influx and bacterial clearance at sites of infection through TNF-α. Nature. 1996;381:77–80. doi: 10.1038/381077a0. [DOI] [PubMed] [Google Scholar]
- 59.McNeil HP, Adachi R, Stevens RL. Mast cell-restricted tryptases: structure and function in inflammation and pathogen defense. J Biol Chem. 2007;282:20785–20789. doi: 10.1074/jbc.R700017200. [DOI] [PubMed] [Google Scholar]
- 60.Bischoff SC. Physiological and pathophysiological functions of intestinal mast cells. Semin Immunopathol. 2009;31:185–205. doi: 10.1007/s00281-009-0165-4. [DOI] [PubMed] [Google Scholar]
- 61.Ruitenberg EJ, Elgersma A. Absence of intestinal mast cell response in congenitally athymic mice during Trichinella spiralis infection. Nature. 1976;264:258–260. doi: 10.1038/264258a0. [DOI] [PubMed] [Google Scholar]
- 62.Finkelman FD, Shea-Donohue T, Goldhill J, et al. Cytokine regulation of host defense against parasitic gastrointestinal nematodes: lessons from studies with rodent models. Annu Rev Immunol. 1997;15:505–533. doi: 10.1146/annurev.immunol.15.1.505. [DOI] [PubMed] [Google Scholar]
- 63.Kirshenbaum AS, Worobec AS, Davis TA, et al. Inhibition of human mast cell growth and differentiation by interferon gamma-1b. Exp Hematol. 1998;26:245–251. [PubMed] [Google Scholar]
- 64.Okayama Y, Kirshenbaum AS, Metcalfe DD. Expression of a functional high-affinity IgG receptor, FcγRI, on human mast cells: up-regulation by IFN-γ. J Immunol. 2000;164:4332–4339. doi: 10.4049/jimmunol.164.8.4332. [DOI] [PubMed] [Google Scholar]
- 65.Abonia JP, Blanchard C, Butz BB, et al. Involvement of mast cells in eosinophilic esophagitis. J Allergy Clin Immunol. 2010;126:140–149. doi: 10.1016/j.jaci.2010.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Nishida Y, Murase K, Isomoto H, et al. Different distribution of mast cells and macrophages in colonic mucosa of patients with collagenous colitis and inflammatory bowel disease. Hepatogastroenterology. 2002;49:678–682. [PubMed] [Google Scholar]
- 67.Dvorak AM, Monahan RA, Osage JE, et al. Crohn’s disease: transmission electron microscopic studies. II. Immunologic inflammatory response. Alterations of mast cells, basophils, eosinophils, and the microvasculature. Hum Pathol. 1980;11:606–619. doi: 10.1016/s0046-8177(80)80072-4. [DOI] [PubMed] [Google Scholar]
- 68.Nolte H, Spjeldnaes N, Kruse A, et al. Histamine release from gut mast cells from patients with inflammatory bowel diseases. Gut. 1990;31:791–794. doi: 10.1136/gut.31.7.791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Bischoff SC, Wedemeyer J, Herrmann A, et al. Quantitative assessment of intestinal eosinophils and mast cells in inflammatory bowel disease. Histopathology. 1996;28:1–13. doi: 10.1046/j.1365-2559.1996.262309.x. [DOI] [PubMed] [Google Scholar]
- 70.Fox CC, Lazenby AJ, Moore WC, et al. Enhancement of human intestinal mast cell mediator release in active ulcerative colitis. Gastroenterology. 1990;99:119–124. doi: 10.1016/0016-5085(90)91238-2. [DOI] [PubMed] [Google Scholar]
- 71.Raithel M, Winterkamp S, Pacurar A, et al. Release of mast cell tryptase from human colorectal mucosa in inflammatory bowel disease. Scand J Gastroenterol. 2001;36:174–179. doi: 10.1080/003655201750065933. [DOI] [PubMed] [Google Scholar]
- 72.Knutson L, Ahrenstedt O, Odlind B, et al. The jejunal secretion of histamine is increased in active Crohn’s disease. Gastroenterology. 1990;98:849–854. doi: 10.1016/0016-5085(90)90006-m. [DOI] [PubMed] [Google Scholar]
- 73.Winterkamp S, Weidenhiller M, Otte P, et al. Urinary excretion of N-methylhistamine as a marker of disease activity in inflammatory bowel disease. Am J Gastroenterol. 2002;97:3071–3077. doi: 10.1111/j.1572-0241.2002.07028.x. [DOI] [PubMed] [Google Scholar]
- 74.Dvorak AM, McLeod RS, Onderdonk A, et al. Ultrastructural evidence for piecemeal and anaphylactic degranulation of human gut mucosal mast cells in vivo. Int Arch Allergy Immunol. 1992;99:74–83. doi: 10.1159/000236338. [DOI] [PubMed] [Google Scholar]
- 75.Bischoff SC, Lorentz A, Schwengberg S, et al. Mast cells are an important cellular source of tumour necrosis factor α in human intestinal tissue. Gut. 1999;44:643–652. doi: 10.1136/gut.44.5.643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mori S, Itoh Y, Shinohata R, et al. Nafamostat mesilate is an extremely potent inhibitor of human tryptase. J Pharmacol Sci. 2003;92:420–423. doi: 10.1254/jphs.92.420. [DOI] [PubMed] [Google Scholar]
- 77.Isozaki Y, Yoshida N, Kuroda M, et al. Anti-tryptase treatment using nafamostat mesilate has a therapeutic effect on experimental colitis. Scand J Gastroenterol. 2006;41:944–953. doi: 10.1080/00365520500529470. [DOI] [PubMed] [Google Scholar]
- 78.Cho EY, Choi SC, Lee SH, et al. Nafamostat mesilate attenuates colonic inflammation and mast cell infiltration in the experimental colitis. Int Immunopharmacol. 2011;11:412–417. doi: 10.1016/j.intimp.2010.12.008. [DOI] [PubMed] [Google Scholar]
- 79.Issekutz AC, Roland DM, Patrick RA. The effect of FUT-175 (nafamstat mesilate) on C3a, C4a, and C5a generation in vitro and inflammatory reactions in vivo. Int J Immunopharmacol. 1990;12:1–9. doi: 10.1016/0192-0561(90)90062-r. [DOI] [PubMed] [Google Scholar]
- 80.Wong GW, Tang Y, Feyfant E, et al. Identification of a new member of the tryptase family of mouse and human mast cell proteases that possesses a novel C-terminal hydrophobic extension. J Biol Chem. 1999;274:30784–30793. doi: 10.1074/jbc.274.43.30784. [DOI] [PubMed] [Google Scholar]
- 81.Hansbro PM, Hamilton MJ, Fricker M, et al. Importance of mast cell Prss31/transmembrane tryptase/tryptase–gamma in lung function and experimental chronic obstructive pulmonary disease and colitis. J Biol Chem. 2014;289:18214–18277. doi: 10.1074/jbc.M114.548594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ishida K, Takai S, Murano M, et al. Role of chymase-dependent matrix metalloproteinase-9 activation in mice with dextran sodium sulfate-induced colitis. J Pharmacol Exp Ther. 2008;324:422–426. doi: 10.1124/jpet.107.131946. [DOI] [PubMed] [Google Scholar]
- 83.Tchougounova E, Lundequist A, Fajardo I, et al. A key role for mast cell chymase in the activation of pro-matrix metalloprotease-9 and pro-matrix metalloprotease-2. J Biol Chem. 2005;280:9291–9296. doi: 10.1074/jbc.M410396200. [DOI] [PubMed] [Google Scholar]
- 84.Gruber BL, Schwartz LB, Ramamurthy NS, et al. Activation of latent rheumatoid synovial collagenase by human mast cell tryptase. J Immunol. 1988;140:3936–3942. [PubMed] [Google Scholar]
- 85.Magarinos NJ, Bryant KJ, Fosang AJ, et al. Mast cell-restricted, tetramer-forming tryptases induce aggrecanolysis in articular cartilage by activating matrix metalloproteinase-3 and -13 zymogens. J Immunol. 2013;191:1404–1412. doi: 10.4049/jimmunol.1300856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Araki Y, Andoh A, Fujiyama Y, et al. Development of dextran sulphate sodium-induced experimental colitis is suppressed in genetically mast cell-deficient Ws/Ws rats. Clin Exp Immunol. 2000;119:264–269. doi: 10.1046/j.1365-2249.2000.01094.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Beckett EL, Stevens RL, Jarnicki AG, et al. A new short-term mouse model of chronic obstructive pulmonary disease identifies a role for mast cell tryptase in pathogenesis. J Allergy Clin Immunol. 2013;131:752–762. doi: 10.1016/j.jaci.2012.11.053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Caughey GH, Raymond WW, Blount JL, et al. Characterization of human γ tryptases, novel members of the chromosome 16p mast cell tryptase and prostasin gene families. J Immunol. 2000;164:6566–6575. doi: 10.4049/jimmunol.164.12.6566. [DOI] [PubMed] [Google Scholar]
- 89.Wong GW, Foster PS, Yasuda S, et al. Biochemical and functional characterization of human transmembrane tryptase (TMT)/tryptase γ: TMT is an exocytosed mast cell protease that induces airway hyperresponsiveness in vivo via an IL-13/IL-4Rα/STAT6-dependent pathway. J Biol Chem. 2002;277:41906–41915. doi: 10.1074/jbc.M205868200. [DOI] [PubMed] [Google Scholar]
- 90.McGuckin MA, Eri R, Simms LA, et al. Intestinal barrier dysfunction in inflammatory bowel diseases. Inflamm Bowel Dis. 2009;15:100–113. doi: 10.1002/ibd.20539. [DOI] [PubMed] [Google Scholar]
- 91.Wu L, Feng BS, He SH, et al. Bacterial peptidoglycan breaks down intestinal tolerance via mast cell activation: the role of TLR2 and NOD2. Immunol Cell Biol. 2007;85:538–545. doi: 10.1038/sj.icb.7100079. [DOI] [PubMed] [Google Scholar]
- 92.Groschwitz KR, Ahrens R, Osterfeld H, et al. Mast cells regulate homeostatic intestinal epithelial migration and barrier function by a chymase/ Mcpt4-dependent mechanism. Proc Natl Acad Sci U S A. 2009;106:22381–22386. doi: 10.1073/pnas.0906372106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Jacob C, Yang PC, Darmoul D, et al. Mast cell tryptase controls paracellular permeability of the intestine: role of protease-activated receptor 2 and β-arrestins. J Biol Chem. 2005;280:31936–31948. doi: 10.1074/jbc.M506338200. [DOI] [PubMed] [Google Scholar]
- 94.Cenac N, Coelho AM, Nguyen C, et al. Induction of intestinal inflammation in mouse by activation of proteinase-activated receptor-2. Am J Pathol. 2002;161:1903–1915. doi: 10.1016/S0002-9440(10)64466-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Kim JA, Choi SC, Yun KJ, et al. Expression of protease-activated receptor 2 in ulcerative colitis. Inflamm Bowel Dis. 2003;9:224–229. doi: 10.1097/00054725-200307000-00002. [DOI] [PubMed] [Google Scholar]
- 96.Steinhoff M, Vergnolle N, Young SH, et al. Agonists of proteinase-activated receptor-2 induce inflammation by a neurogenic mechanism. Nat Med. 2000;6:151–158. doi: 10.1038/72247. [DOI] [PubMed] [Google Scholar]
- 97.Corvera CU, Dery O, McConalogue K, et al. Thrombin and mast cell tryptase regulate guinea-pig myenteric neurons through proteinase-activated receptors-1 and -2. J Physiol. 1999;517:741–756. doi: 10.1111/j.1469-7793.1999.0741s.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Abonia JP, Friend DS, Austen WG, et al. Mast cell protease 5 mediates ischemia-reperfusion injury of mouse skeletal muscle. J Immunol. 2005;174:7285–7291. doi: 10.4049/jimmunol.174.11.7285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Younan G, Suber F, Xing W, et al. The inflammatory response after an epidermal burn depends on the activities of mouse mast cell proteases 4 and 5. J Immunol. 2010;185:7681–7690. doi: 10.4049/jimmunol.1002803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Bankova LG, Lezcano C, Pejler G, et al. Mouse mast cell proteases 4 and 5 mediate epidermal injury through disruption of tight junctions. J Immunol. 2014;192:2812–2820. doi: 10.4049/jimmunol.1301794. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bertaccini G, Coruzzi G. An update on histamine H3 receptors and gastrointestinal functions. Dig Dis Sci. 1995;40:2052–2063. doi: 10.1007/BF02208678. [DOI] [PubMed] [Google Scholar]
- 102.Homaidan FR, Tripodi J, Zhao L, et al. Regulation of ion transport by histamine in mouse cecum. Eur J Pharmacol. 1997;331:199–204. doi: 10.1016/s0014-2999(97)00184-2. [DOI] [PubMed] [Google Scholar]
- 103.Crowe SE, Luthra GK, Perdue MH. Mast cell mediated ion transport in intestine from patients with and without inflammatory bowel disease. Gut. 1997;41:785–792. doi: 10.1136/gut.41.6.785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Casellas F, Guarner F, Antolin M, et al. Abnormal leukotriene C4 released by unaffected jejunal mucosa in patients with inactive Crohn’s disease. Gut. 1994;35:517–522. doi: 10.1136/gut.35.4.517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Barbara G, Stanghellini V, De GR, et al. Activated mast cells in proximity to colonic nerves correlate with abdominal pain in irritable bowel syndrome. Gastroenterology. 2004;126:693–702. doi: 10.1053/j.gastro.2003.11.055. [DOI] [PubMed] [Google Scholar]
- 106.Chang L, Munakata J, Mayer EA, et al. Perceptual responses in patients with inflammatory and functional bowel disease. Gut. 2000;47:497–505. doi: 10.1136/gut.47.4.497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Minderhoud IM, Oldenburg B, Wismeijer JA, et al. IBS-like symptoms in patients with inflammatory bowel disease in remission; relationships with quality of life and coping behavior. Dig Dis Sci. 2004;49:469–474. doi: 10.1023/b:ddas.0000020506.84248.f9. [DOI] [PubMed] [Google Scholar]
- 108.Spiller RC. Postinfectious irritable bowel syndrome. Gastroenterology. 2003;124:1662–1671. doi: 10.1016/s0016-5085(03)00324-x. [DOI] [PubMed] [Google Scholar]
- 109.van Hoboken EA, Thijssen AY, Verhaaren R, et al. Symptoms in patients with ulcerative colitis in remission are associated with visceral hypersensitivity and mast cell activity. Scand J Gastroenterol. 2011;46:981–987. doi: 10.3109/00365521.2011.579156. [DOI] [PubMed] [Google Scholar]
- 110.Wilcz-Villega EM, McClean S, O’Sullivan MA. Mast cell tryptase reduces junctional adhesion molecule-A (JAM-A) expression in intestinal epithelial cells: implications for the mechanisms of barrier dysfunction in irritable bowel syndrome. Am J Gastroenterol. 2013;108:1140–1151. doi: 10.1038/ajg.2013.92. [DOI] [PubMed] [Google Scholar]
- 111.Lee JW, Park JH, Park DI, et al. Subjects with diarrhea-predominant IBS have increased rectal permeability responsive to tryptase. Dig Dis Sci. 2010;55:2922–2928. doi: 10.1007/s10620-009-1094-8. [DOI] [PubMed] [Google Scholar]
- 112.Soderholm JD, Yang PC, Ceponis P, et al. Chronic stress induces mast cell-dependent bacterial adherence and initiates mucosal inflammation in rat intestine. Gastroenterology. 2002;123:1099–1108. doi: 10.1053/gast.2002.36019. [DOI] [PubMed] [Google Scholar]
- 113.Bonaz BL, Bernstein CN. Brain-gut interactions in inflammatory bowel disease. Gastroenterology. 2013;144:36–49. doi: 10.1053/j.gastro.2012.10.003. [DOI] [PubMed] [Google Scholar]
- 114.McKay DM, Bienenstock J. The interaction between mast cells and nerves in the gastrointestinal tract. Immunol Today. 1994;15:533–538. doi: 10.1016/0167-5699(94)90210-0. [DOI] [PubMed] [Google Scholar]
- 115.Ehrlich P. doctoral thesis. Germany: University of Leipzig; 1878. Beitrage zur Theorie und Praxis der Histologischen Farbung. [Google Scholar]
- 116.MacQueen G, Marshall J, Perdue M, et al. Pavlovian conditioning of rat mucosal mast cells to secrete rat mast cell protease II. Science. 1989;243:83–85. doi: 10.1126/science.2911721. [DOI] [PubMed] [Google Scholar]
- 117.Santos J, Yang PC, Soderholm JD, et al. Role of mast cells in chronic stress induced colonic epithelial barrier dysfunction in the rat. Gut. 2001;48:630–636. doi: 10.1136/gut.48.5.630. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Santos J, Saunders PR, Hanssen NP, et al. Corticotropin-releasing hormone mimics stress-induced colonic epithelial pathophysiology in the rat. Am J Physiol. 1999;277:G391–G399. doi: 10.1152/ajpgi.1999.277.2.G391. [DOI] [PubMed] [Google Scholar]
- 119.Castagliuolo I, Wershil BK, Karalis K, et al. Colonic mucin release in response to immobilization stress is mast cell dependent. Am J Physiol. 1998;274:G1094–G1100. doi: 10.1152/ajpgi.1998.274.6.G1094. [DOI] [PubMed] [Google Scholar]
- 120.Lennon EM, Maharshak N, Elloumi H, et al. Early life stress triggers persistent colonic barrier dysfunction and exacerbates colitis in adult IL-10−/− mice. Inflamm Bowel Dis. 2013;19:712–719. doi: 10.1097/MIB.0b013e3182802a4e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Triantafillidis JK, Merikas E, Gikas A. Psychological factors and stress in inflammatory bowel disease. Expert Rev Gastroenterol Hepatol. 2013;7:225–238. doi: 10.1586/egh.13.4. [DOI] [PubMed] [Google Scholar]
- 122.Larauche M, Bradesi S, Million M, et al. Corticotropin-releasing factor type 1 receptors mediate the visceral hyperalgesia induced by repeated psychological stress in rats. Am J Physiol Gastrointest Liver Physiol. 2008;294:G1033–G1040. doi: 10.1152/ajpgi.00507.2007. [DOI] [PubMed] [Google Scholar]
- 123.Theoharides TC, Cochrane DE. Critical role of mast cells in inflammatory diseases and the effect of acute stress. J Neuroimmunol. 2004;146:1–12. doi: 10.1016/j.jneuroim.2003.10.041. [DOI] [PubMed] [Google Scholar]
- 124.Overman EL, Rivier JE, Moeser AJ. CRF induces intestinal epithelial barrier injury via the release of mast cell proteases and TNF-α. PLoS One. 2012;7:e39935. doi: 10.1371/journal.pone.0039935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Ansel JC, Brown JR, Payan DG, et al. Substance P selectively activates TNF-α gene expression in murine mast cells. J Immunol. 1993;150:4478–4485. [PubMed] [Google Scholar]
- 126.Marshall JS, Gomi K, Blennerhassett MG, et al. Nerve growth factor modifies the expression of inflammatory cytokines by mast cells via a prostanoid-dependent mechanism. J Immunol. 1999;162:4271–4276. [PubMed] [Google Scholar]
- 127.Wood JD. Effects of bacteria on the enteric nervous system: implications for the irritable bowel syndrome. J Clin Gastroenterol. 2007;41(suppl 1):S7–S19. doi: 10.1097/MCG.0b013e31802f1331. [DOI] [PubMed] [Google Scholar]
- 128.Ferrante M, de HG, Hlavaty T, et al. The value of myenteric plexitis to predict early postoperative Crohn’s disease recurrence. Gastroenterology. 2006;130:1595–1606. doi: 10.1053/j.gastro.2006.02.025. [DOI] [PubMed] [Google Scholar]
- 129.Sokol H, Polin V, Lavergne-Slove A, et al. Plexitis as a predictive factor of early postoperative clinical recurrence in Crohn’s disease. Gut. 2009;58:1218–1225. doi: 10.1136/gut.2009.177782. [DOI] [PubMed] [Google Scholar]
- 130.Noli C, Miolo A. The mast cell in wound healing. Vet Dermatol. 2001;12:303–313. doi: 10.1046/j.0959-4493.2001.00272.x. [DOI] [PubMed] [Google Scholar]
- 131.Douaiher J, Succar J, Lancerotto L, et al. Development of mast cells and importance of their tryptase and chymase serine proteases in inflammation and wound healing. Adv Immunol. 2014;122:211–252. doi: 10.1016/B978-0-12-800267-4.00006-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Iba Y, Shibata A, Kato M, et al. Possible involvement of mast cells in collagen remodeling in the late phase of cutaneous wound healing in mice. Int Immunopharmacol. 2004;4:1873–1880. doi: 10.1016/j.intimp.2004.08.009. [DOI] [PubMed] [Google Scholar]
- 133.Dayton ET, Caulfield JP, Hein A, et al. Regulation of the growth rate of mouse fibroblasts by IL-3-activated mouse bone marrow-derived mast cells. J Immunol. 1989;142:4307–4313. [PubMed] [Google Scholar]
- 134.Regan MC, Flavin BM, Fitzpatrick JM, et al. Stricture formation in Crohn’s disease: the role of intestinal fibroblasts. Ann Surg. 2000;231:46–50. doi: 10.1097/00000658-200001000-00007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Garbuzenko E, Nagler A, Pickholtz D, et al. Human mast cells stimulate fibroblast proliferation, collagen synthesis and lattice contraction: a direct role for mast cells in skin fibrosis. Clin Exp Allergy. 2002;32:237–246. doi: 10.1046/j.1365-2222.2002.01293.x. [DOI] [PubMed] [Google Scholar]
- 136.Xu X, Rivkind A, Pikarsky A, et al. Mast cells and eosinophils have a potential profibrogenic role in Crohn disease. Scand J Gastroenterol. 2004;39:440–447. doi: 10.1080/00365520310008566. [DOI] [PubMed] [Google Scholar]
- 137.Stallmach A, Schuppan D, Riese HH, et al. Increased collagen type III synthesis by fibroblasts isolated from strictures of patients with Crohn’s disease. Gastroenterology. 1992;102:1920–1929. doi: 10.1016/0016-5085(92)90314-o. [DOI] [PubMed] [Google Scholar]
- 138.Graham MF, Diegelmann RF, Elson CO, et al. Collagen content and types in the intestinal strictures of Crohn’s disease. Gastroenterology. 1988;94:257–265. doi: 10.1016/0016-5085(88)90411-8. [DOI] [PubMed] [Google Scholar]
- 139.Gelbmann CM, Mestermann S, Gross V, et al. Strictures in Crohn’s disease are characterised by an accumulation of mast cells colocalised with laminin but not with fibronectin or vitronectin. Gut. 1999;45:210–217. doi: 10.1136/gut.45.2.210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Alvarez-Lobos M, Arostegui JI, Sans M, et al. Crohn’s disease patients carrying Nod2/CARD15 gene variants have an increased and early need for first surgery due to stricturing disease and higher rate of surgical recurrence. Ann Surg. 2005;242:693–700. doi: 10.1097/01.sla.0000186173.14696.ea. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Okumura S, Yuki K, Kobayashi R, et al. Hyperexpression of NOD2 in intestinal mast cells of Crohn’s disease patients: preferential expression of inflammatory cell-recruiting molecules via NOD2 in mast cells. Clin Immunol. 2009;130:175–185. doi: 10.1016/j.clim.2008.08.027. [DOI] [PubMed] [Google Scholar]
- 142.Schwartz DA, Loftus EV, Jr, Tremaine WJ, et al. The natural history of fistulizing Crohn’s disease in Olmsted County, Minnesota. Gastroenterology. 2002;122:875–880. doi: 10.1053/gast.2002.32362. [DOI] [PubMed] [Google Scholar]
- 143.Kirkegaard T, Hansen A, Bruun E, et al. Expression and localisation of matrix metalloproteinases and their natural inhibitors in fistulae of patients with Crohn’s disease. Gut. 2004;53:701–709. doi: 10.1136/gut.2003.017442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Bataille F, Rohrmeier C, Bates R, et al. Evidence for a role of epithelial mesenchymal transition during pathogenesis of fistulae in Crohn’s disease. Inflamm Bowel Dis. 2008;14:1514–1527. doi: 10.1002/ibd.20590. [DOI] [PubMed] [Google Scholar]
- 145.Scharl M, Weber A, Furst A, et al. Potential role for SNAIL family transcription factors in the etiology of Crohn’s disease-associated fistulae. Inflamm Bowel Dis. 2011;17:1907–1916. doi: 10.1002/ibd.21555. [DOI] [PubMed] [Google Scholar]
- 146.Scharl M, Frei S, Pesch T, et al. Interleukin-13 and transforming growth factor β synergise in the pathogenesis of human intestinal fistulae. Gut. 2013;62:63–72. doi: 10.1136/gutjnl-2011-300498. [DOI] [PubMed] [Google Scholar]
- 147.Frei SM, Pesch T, Lang S, et al. A role for tumor necrosis factor and bacterial antigens in the pathogenesis of Crohn’s disease-associated fistulae. Inflamm Bowel Dis. 2013;19:2878–2887. doi: 10.1097/01.MIB.0000435760.82705.23. [DOI] [PubMed] [Google Scholar]
- 148.Radlmayr M, Torok HP, Martin K, et al. The c-insertion mutation of the NOD2 gene is associated with fistulizing and fibrostenotic phenotypes in Crohn’s disease. Gastroenterology. 2002;122:2091–2092. doi: 10.1053/gast.2002.34020. [DOI] [PubMed] [Google Scholar]
- 149.Fox CC, Moore WC, Lichtenstein LM. Modulation of mediator release from human intestinal mast cells by sulfasalazine and 5-aminosalicylic acid. Dig Dis Sci. 1991;36:179–184. doi: 10.1007/BF01300753. [DOI] [PubMed] [Google Scholar]
- 150.Tremaine WJ, Brzezinski A, Katz JA, et al. Treatment of mildly to moderately active ulcerative colitis with a tryptase inhibitor (APC 2059): an open-label pilot study. Aliment Pharmacol Ther. 2002;16:407–413. doi: 10.1046/j.1365-2036.2002.01194.x. [DOI] [PubMed] [Google Scholar]
- 151.Heatley RV, Calcraft BJ, Rhodes J, et al. Disodium cromoglycate in the treatment of chronic proctitis. Gut. 1975;16:559–563. doi: 10.1136/gut.16.7.559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Dronfield MW, Langman MJ. Comparative trial of sulphasalazine and oral sodium cromoglycate in the maintenance of remission in ulcerative colitis. Gut. 1978;19:1136–1139. doi: 10.1136/gut.19.12.1136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Weng Z, Zhang B, Asadi S, et al. Quercetin is more effective than cromolyn in blocking human mast cell cytokine release and inhibits contact dermatitis and photosensitivity in humans. PLoS One. 2012;7:e33805. doi: 10.1371/journal.pone.0033805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Goldsmith P, McGarity B, Walls AF, et al. Corticosteroid treatment reduces mast cell numbers in inflammatory bowel disease. Dig Dis Sci. 1990;35:1409–1413. doi: 10.1007/BF01536749. [DOI] [PubMed] [Google Scholar]
- 155.Eklund KK, Humphries DE, Xia Z, et al. Glucocorticoids inhibit the cytokine-induced proliferation of mast cells, the high-affinity IgE receptor-mediated expression of TNF-α, and the IL-10-induced expression of chymases. J Immunol. 1997;158:4373–4380. [PubMed] [Google Scholar]
- 156.Soler D, Chapman T, Yang LL, et al. The binding specificity and selective antagonism of vedolizumab, an anti-α4β7 integrin therapeutic antibody in development for inflammatory bowel diseases. J Pharmacol Exp Ther. 2009;330:864–875. doi: 10.1124/jpet.109.153973. [DOI] [PubMed] [Google Scholar]
- 157.Sandborn WJ, Feagan BG, Rutgeerts P, et al. Vedolizumab as induction and maintenance therapy for Crohn’s disease. N Engl J Med. 2013;369:711–721. doi: 10.1056/NEJMoa1215739. [DOI] [PubMed] [Google Scholar]
- 158.Feagan BG, Rutgeerts P, Sands BE, et al. Vedolizumab as induction and maintenance therapy for ulcerative colitis. N Engl J Med. 2013;369:699–710. doi: 10.1056/NEJMoa1215734. [DOI] [PubMed] [Google Scholar]
- 159.Takahashi K. Interaction between the intestinal immune system and commensal bacteria and its effect on the regulation of allergic reactions. Biosci Biotechnol Biochem. 2010;74:691–695. doi: 10.1271/bbb.90962. [DOI] [PubMed] [Google Scholar]
- 160.Peterson DA, McNulty NP, Guruge JL, et al. IgA response to symbiotic bacteria as a mediator of gut homeostasis. Cell Host Microbe. 2007;2:328–339. doi: 10.1016/j.chom.2007.09.013. [DOI] [PubMed] [Google Scholar]
- 161.Frank DN, St Amand AL, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A. 2007;104:13780–13785. doi: 10.1073/pnas.0706625104. [DOI] [PMC free article] [PubMed] [Google Scholar]