

Abstract
The human heart has a very limited capacity to regenerate lost or damaged cardiomyocytes following cardiac insult. Instead, myocardial injury is characterized by extensive cardiac remodeling by fibroblasts, resulting in the eventual deterioration of cardiac structure and function. Cardiac function would be improved if these fibroblasts could be converted into cardiomyocytes. MicroRNAs (miRNAs), small non-coding RNAs that promote mRNA degradation and inhibit mRNA translation, have been shown to be important in cardiac development. Using this information various researchers have utilized miRNAs to promote the formation of cardiomyocytes through a number of approaches. Several miRNAs acting in combination promote the direct conversion of cardiac fibroblasts into cardiomyocytes. Moreover, a number of miRNAs have been identified that aid the formation of iPS cells and miRNAs also induce these cells to adopt a cardiac fate. MiRNAs have also been implicated in resident cardiac progenitor cell differentiation. In this review we will discuss the current literature as it pertains to these processes as well as discussing the therapeutic implications of these findings.
Keywords: MicroRNA, regeneration, transdifferentiation, stem cell, cardiac myocyte
Introduction
Myocardial infarction leads to significant cardiomyocyte cell death. These specialized cells are not replaced in substantial numbers following injury leading to disproportionate thinning of the heart wall and severely impaired cardiac function. Moreover cardiac fibroblasts, which form a significant proportion of the heart, are expanded further leading to excessive fibrosis and scar formation. Fibrotic remodeling of the injured myocardium negatively impacts contractility and electrical conduction which over time causes a further deterioration in cardiac function1.
A number of strategies are being actively pursued in cardiac regenerative medicine. Replacing lost cardiomyocytes by injecting cardiac progenitors, cardiospheres, or cardiac myocytes derived from inducible pluripotent stem cells and/or embryonic stem cells (iPS/ESCs) has been researched intensively. Others have focused instead on the cardiomyocytes by enabling them to enter cell cycle to replicate and proliferate as a means to generate new muscle cells. However, these approaches, while encouraging, face significant challenges. Recently, much excitement has been turned to the cardiac fibroblast population in the scar tissue with a view to turning these cells into cardiomyocytes 2. Taking cues from heart developmental programs3–5 direct reprogramming of fibroblasts to cardiomyocytes has been achieved recently by specific combinations of transcription factors6–8 and microRNAs (miRNAs)2, 9–11.
In this review we will discuss the role of miRNAs in cardiac development as well as in indirect and direct cardiac reprogramming. Based on this scientific knowledge we will discuss the strategy of targeting miRNAs for cardiac regeneration therapy.
MiRNA biology
MiRNAs are small non-coding RNAs belonging to a class of small silencing RNAs that are critically important in the post-transcriptional regulation of genes12, 13. Indeed, most mammalian mRNAs are targets of miRNAs14–16. MiRNAs have been demonstrated to play an important role in the differentiation and development of many cells and tissues including the heart17–27. Moreover miRNAs are important for stem cell differentiation, as well as indirect and direct reprogramming to multiple lineages9, 28–30.
The RNA polymerase II enzyme transcribes MiRNAs and the resulting transcript is known as a primary-miRNA (pri-miRNA). The pri-miRNA is ~1000 nucleotides in length, possesses a 5′cap, a middle stem loop structure, and a 3′ polyadenylated tail31. Pri-miRNAs are processed further into pre-miRNAs by the Microprocessor complex32. Drosha, a component of the Microprocessor complex, cleaves the 5′cap and 3′ poly (A) tail leaving a ~65bp stem loop structure containing the mature miRNA16. Following cleavage by Drosha, the pre-miRNA is transported from the nucleus into the cytoplasm. Once in the cytoplasm the pre-miRNA is cleaved further by the RNase III-type endonuclease Dicer (Double-Stranded RNA-Specific Endoribonuclease) into a smaller fragment containing the mature miRNA33. In humans, the Dicer generated small dsRNA fragment containing the mature miRNA is loaded onto one of four Argonaute (Ago) proteins, Ago 1-434, an action facilitated a heat shock cognate (HSC70) – heat shock protein 90 (HSP90) complex35. Within this complex, which is named the pre-RNA-induced silencing complex (pre-RISC), processing occurs to generate the final single-stranded mature miRNA36. The single stranded miRNA, associated with Ago1-4, constitutes the mature RISC complex. This is the final functional unit that mediates post-transcriptional repression of a target.
MiRNAs in cardiac development and function
Cardiac specific deletions of Dicer gave the first evidence of a role of miRNAs in cardiac development37–40. Deletion of Dicer in Nkx2-5 (homeobox protein Nkx2-5) cardiac progenitor cells induced development defects in the heart such as cardiac edema and poorly developed ventricular myocardium leading to embryonic lethality at E12.5 due to cardiac failure40. The use of an alternative allele for the Nkx2.5-Cre transgene, which allowed mutant mice to survive beyond E13.5, highlighted additional role for Dicer in cardiac outflow tract alignment and chamber septation 39. Using a αMHC promoter to delete Dicer expression at a later stage of cardiac development caused impairment of cardiac function through dysregulated cardiac contractile protein expression, disrupted sarcomeric structure and disarray of myofibers. Mice quickly developed dilated cardiomyopathy and heart failure with lethality observed within four days of birth37. Similarly, using a tamoxifen-inducible α-MHC-Cre, another group depleted Dicer in the postnatal heart. Loss of Dicer in juveniles resulted in mild cardiac remodeling and premature death within 1 week, whereas deletion in adults led to severe hypertrophy, myofiber disarray, ventricular fibrosis and reactivation of a fetal gene transcription program38.
The study of individual mature miRNAs has demonstrated their importance in cardiac development.
MiRNA-1 is produced from two loci. The two mature miRNAs are called miRNA-1-1 and miRNA-1-2 to distinguish them from each other. MiRNA-1-1 and miRNA-1-2, co-transcribed with miRNA-133-a2 and miRNA-133-a1 respectively, are expressed specifically in cardiac and skeletal muscle with expression increasing substantially from E8.5 to adulthood28, 41, 42. Transcription of miRNA-1/miRNA-133 in heart and skeletal muscle is regulated by Serum Response Factor (SRF), Myocardin, Myogenic Differentiation 1 (MyoD) and Myocyte Enhancer Factor (Mef2) transcription factors41, 43. Expression of SRF is repressed by miRNA-133 in myoblasts suggesting a negative regulatory loop42.
Overexpression of miRNA-1 in the developing mouse heart inhibits proliferation of ventricular cardiomyocytes. Developmental arrest occurs at E13.5 secondary to thinning of ventricle walls and heart failure. Hand2, a direct target of miRNA-1, may mediate these effects41. Similar findings have been observed in Xenopus42 suggesting miRNA-1 is important for the development of the vertebrate heart.
MiRNA-1-2 deletion in mice induces a range of phenotypes40. Approximately half of the miRNA-1-2 null mice died between E15.5 and birth from severe ventricular septation defects. The surviving miRNA-1-2 null mice possessed thickened cardiac walls arising from increased cardiomyocyte proliferation 40. Moreover, conduction defects were observed due to de-repression of Irx5 and a number of the null mice died within 2–3 months from dilatation of the heart and ventricular dysfunction40. MiRNA-1-1 null mice are similar, with partial lethality, mild ventricular dilation and conduction defects all being observed44.
Partially penetrant lethality in single miRNA-1-1 or miRNA-1-2 null mice may reflect compensation by the remaining miRNA-1. To elucidate the role of miRNA-1, double mutant mice for miRNA-1-1 and miRNA-1-2 (miRNA-1 dKO) were recently engineered by two groups44, 45. Heidersbach et al.44 found that miRNA-1dKO mice died uniformly before P10 due to severe cardiac dysfunction including ventricular septal defects, heart chamber dilatation, abnormal conduction and sarcomere disruption. In the miRNA-1dKO mice Telokin, the smooth muscle-restricted inhibitor of MLC2 (myosin light chain-2) phosphorylation, was ectopically expressed in the myocardium. Telokin was found to be a direct target of miRNA-1 suggesting that miRNA-1 promotes cardiac development and sarcomeric organization by repressing the smooth muscle gene program44. Similarly miRNA-1dKO mice generated by Wei et al.45 died by P17 from heart dilatation, an increase in cardiac mass arising from elevated cardiomyocyte proliferation, and sarcomeric defects. However, ventricular septal defects were not observed. In the miRNA-1 dKO mice increased expression of the nuclear receptor ERRβ (estrogen related receptor 2), a direct miRNA-1 target, activated a fetal gene program which included the expression of fetal sarcomere-associated genes. Expressing ERRβ in heart tissue mimicked the miRNA-1 dKO phenotype; further validating the model45.
The differences between these two studies may reflect that miRNA-1 controls heart development through multiple pathways, the methods used to generate the null mice, or genetic background.
MiRNA-133 is co-transcribed with miRNA-1 and shares a commonality of function during heart development. Overexpression of miRNA-133 in Xenopus induces defects in cardiac looping and chamber formation42. Zebrafish regenerate their hearts following severe injury by increasing cardiomyocyte proliferation. Interestingly, transgenic expression of miRNA-133 inhibited this process in part by targeting the cell junction protein connexin-43 and the cell cycle regulator monopolar spindle 1 46. To gain further insights into the role of miRNA-133 in the development of the mammalian heart, both miRNA-133a1 and miRNA-133a2 were ablated in mice47 (miRNA-133 dKO). The phenotype of the miRNA-133 dKO mice was found to be somewhat similar to that of the miRNA-1 dKO. Approximately half of miRNA-133 dKO mice died post-natally due to ventricular septal defects, increased cardiomyocyte proliferation, and aberrant expression of smooth muscle genes47. Surviving mutants displayed severe cardiac dysfunction with death from heart failure within 6 months. The miRNA-133 dKO phenotype was attributed in part to the increased expression of cyclinD2, a negative regulator of cell cycle, and SRF, a co-activator of smooth muscle genes47. The high level of miRNA-133 expression in the adult heart may also be important for homeostasis of the organ as knockdown of miRNA-133 promoted hypertrophy in one report48 and overexpression of miRNA-133 inhibits hypertrophic stimuli48, 49. Calcineurin levels increase during cardiac hypertrophy and this protein may act in a reciprocal fashion with miRNA-133. Overexpression of miRNA-133 inhibited calcineurin expression and inhibition of calcineurin by cyclosporin-A prevented miRNA-133 downregulation in a hypertrophic model50.
Mice with a combined deletion of the two miRNA-1/miRNA-133a clusters (miRNA-1/133 dKO) have also been generated51. Genetic ablation of either cluster was not detrimental to mice development and survival. However, deletion of both miRNA-1/133a clusters was embryonically lethal. MiRNA-1/133 dKO mice displayed severe cardiac defects with thinning of ventricular walls and decreased cardiomyocyte proliferation. These effects were associated with increased smooth muscle gene expression, notably that of the smooth muscle regulator Myocardin. Over-expression of Myocardin recapitulated many aspects of the miRNA-1/133 dKO phenotype, arresting the developing cardiomyocytes in an immature state. Interestingly, Myocardin activated the transcription of both miRNA-1/133a clusters suggesting that a negative feedback loop exists to restrict smooth muscle gene expression and to promote cardiomyocyte maturation51.
In mammals, cardiomyocyte contraction depends on two myosin heavy chain (MHC) proteins. The faster contracting isoform αMHC is expressed predominantly in adult mouse heart, whilst the embryonic heart expresses the slower contracting isoform βMHC52. In humans, βMHC expression continues into adulthood however hypertrophy induces the β form in humans and rodents alike53. Expression of α- and β-MHC isoforms is controlled by miRNA-208a, miRNA-208b and miRNA-49954–56. MiRNA-208a and miRNA-208b are encoded in an intron of the αMHC and βMHC gene respectively. Mice null for miRNA-208a are viable and show no changes in heart structure up to 20 weeks. However, a progressive decrease in heart contractility was observed from 2 months post-birth and this was concomitant with aberrant expression of fast skeletal muscle contractile proteins54. Despite the lack of gross structural changes in heart structure in miRNA-208a null mice there are significant electrical conduction defects; with a lack of P waves (atrial depolarization) preceding QRS complexes (right and left ventricle depolarization) and significantly prolonged PR intervals (the interval from where the P wave begins until the beginning of the QRS complex)55. Further underlying a role in the heart’s electrical conduction system overexpression of miRNA-208a induced arrhythmia55. MiRNA-208a null mice fail to show a hypertrophic response following such stimuli as transverse aortic banding and calcineurin54, 55. Moreover there was no increase in βMHC expression54, 55 indicating that miRNA-208a controls expression of this MHC isoform. Thyroid signaling is important in the switch of MHC expression following birth, activating expression of αMHC, whilst inhibiting that of βMHC57. MiRNA-208a was found to be important in this process by directly targeting Thyroid Hormone Receptor Associated Protein 1 (THRAP1)54.
MiRNA-208a not only controls βMHC expression but also that of the closely related βMHC isoform Myh7b56. Both βMHC and Myh7b are slow myosins; the genes for these proteins contain intronic miRNAs, miRNA-208b and miRNA-499 respectively. These miRNAs are important in the specification of the identity of muscle fibers; by stimulating slow myofiber gene programs at the expense of those that control fast myofiber gene expression56. Mice lacking the miRNA-499 gene have no obvious developmental defects56. However over-expression of miRNA-499 promotes hypertrophy58, 59.
The mammalian heart increases in size dramatically during embryonic development predominantly via an increase in cardiomyocyte numbers. Following birth mammalian cardiomyocytes exit the cell-cycle and this has a negative impact on cardiac regeneration following injury60, 61, 62. In contrast to mammals, lower vertebrates such as Zebrafish retain the ability to regenerate their hearts throughout life. In the case of the Zebrafish, this natural ability is particularly robust, as complete cardiac regeneration has been observed even when ~20% of the ventricular myocardium was removed63. Cardiomyocyte de-differentiation followed by reentry into the cell cycle underlies this process64. MiRNAs are critically important for cardiomyocyteproliferation. In a recent study, cardiac regeneration in the Zebrafish was found to be dependent upon a decrease in the levels of miRNA-99/100 and Let-7a/c65. Interestingly, this did not occur in the murine heart following injury. When the authors forcibly reduced miRNA-99/100 and Let-7a/c expression following MI in the mouse recovery was observed; indicating a species conserved miRNA program for cardiac regeneration62, 65. The miRNA-17-92 cluster has also been shown to be important for cardiomyocyte proliferation. Over-expression of this miRNA cluster induced cardiomyocyte proliferation in neonatal and adult hearts. Interestingly, transgenic over-expression of miRNA-17-92 in adult cardiomyocytes protected the heart from MI associated injury. The miRNA-17-92 cluster was found to reduce the expression of PTEN (phosphatase and tensin homolog); a proliferation repressor66. Studies have demonstrated that withdrawal of cardiomyocytes from the cell-cycle is dependent upon specific miRNAs. A microarray analysis for miRNAs, differentially regulated between P1 and P10, the point at which mouse cardiomyocytes exit the cell-cycle, identified miRNA-195 as the most highly upregulated miRNA. Overexpression of this miRNA in the embryonic heart caused premature cell-cycle exit, ventricular hypoplasia and ventricular septal defects. Checkpoint kinase 1 (Chek1) was identified as the miRNA-195 target. Knockdown of the miRNA-15 family, to which miRNA-195 belongs, was associated with increased cardiomyocyte proliferation67 and cardiac regeneration68, 69. MiRNA-29a also induces cell-cycle arrest in cultured cardiomyocytes by targeting cyclin-D170.MiRNAs are also important for promoting cardiomyocyte proliferation. Using a high-throughput functional screening method to identify miRNAs that could promote neonatal cardiomyocyte proliferation, Eulalio et al. identified a number of candidates. Two of these candidates, hsa-miRNA-590 and hsa-miRNA-199a, induced ex vivo cultured adult cardiomyocytes to re-enter the cell-cycle. Moreover these cells also showed signs of cytokinesis. Indeed, in vivo administration of these miRNAs markedly stimulated cardiac regeneration post-MI71. Taken together, the results of the above studies demonstrate the potential of activating or antagonizing specific miRNAs to induce cardiomyocyte proliferation for cardiac regenerative therapy.
miRNAs and the formation of iPS cells
iPS are an important source of cells for cardiac regeneration as upon differentiation they can form all of the cardiovascular cell-types72, 73. Indeed, intramyocardial delivery of iPS following myocardial infarction (MI) has been shown to restore contractile performance, cardiac tissue, and ventricular wall thickness74–76.
The first iPS cells were generated when Takahashi and Yamanaka over-expressed Oct4, Sox2, Klf4 and cMyc (OKSM) in fibroblasts77. The formation of iPS cells occurs via the dedifferentiation of somatic cells and this requires shifts in the patterns of expression of thousands of genes78, 79. A single miRNA can influence many hundreds of genes and as such there has been much interest in their possible role in forming iPS cells. Indeed a number of reports have shown that miRNAs can promote the formation of iPS cells either alone80, 81 or in combination with the OKSM factors82.
During the process of converting to iPS cells somatic cells adopt a pluripotent cell cycle phenotype that is characterized by rapid proliferation, a shortened S-phase, and very low expression of cell cycle inhibitors such as p21 and p5383, 84. Moreover, activation of the p53-p21 pathway suppresses iPS formation85. A number of miRNAs are involved in embryonic stem cell (ESC) proliferation86 and they have been exploited to generate iPS cells. MiRNA-302d, miRNA-291, miRNA-294, and miRNA-295 promote proliferation in ESCs. For example, when ectopically expressed in ESCs lacking the miRNA microprocessor subunit Dgcr8 (DiGeorge Syndrome Critical Region 8), which lack canonical miRNAs and proliferate slowly, miRNA-302d, miRNA-291, miRNA-294, and miRNA-295 reduced the number of cells in G1 to that typically found in wild-type ESCs86 by specifically targeting p2186 and retinoblastoma-like 2 protein (Rbl2)87, 88. MiRNA-291-3p, miRNA-294 and miRNA-295 are potentially downstream effectors of cMyc and act as a substitute for this transcription factor. The reprogramming efficiency of Oct4, Sox2 and Klf4 was increased by miRNA-291-3p, miRNA-294 and miRNA-295 or cMyc. However there was no additive effect when the miRNAs and cMyc were used together89. Similarly the human orthologs hsa-miRNA-372 and hsa-miRNA-302b promoted reprogramming in human foreskin and lung fibroblasts expressing Oct4, Sox2, Klf4, and cMyc82. Members of the let-7 miRNA family, which are highly expressed in somatic cells, oppose the effects of miRNAs involved in ESC proliferation90. Knockdown of let-7 miRNA by an antisense RNA inhibitor promoted the de-differentiation of MEFs to iPS when Oct4, Klf4, Sox2, and cMyc were over-expressed90. Inactivation of miRNA targets of the cell cycle inhibitor p53 also enhances reprogramming efficiency. MiRNA-199a-3p is up-regulated by p53 at the post-transcriptional level; induction of this miRNA significantly decreases reprogramming efficiency by arresting cells in G1. Moreover miRNA-199a-3p inhibition partially rescues iPS generation impaired by p5391. The expression of the miRNA-34 family is also p53 dependent92. MiRNA-34a, a member of the miRNA-34 family, restrains iPS reprogramming by acting in concert with p21 to suppress expression of Nanog, Sox2 and N-Myc93. The expression of p53 is controlled by miRNAs and this information has been used to augment reprogramming efficiency. For example, miRNA-138 enhances reprogramming to iPS by binding to the 3′ UTR of p53 and decreasing the expression of the protein94. Moreover, depletion of two MEF enriched miRNAs, miRNA-21 and miRNA-29a, enhances reprogramming efficiency in part through reduced p53 expression95.
MiRNAs also influence the mesenchymal-to-epithelial transition that occurs in the initiation stage of reprogramming by modulating the TGF-β signaling pathway. The TGF-β receptor-2 protein is a specific target of two miRNA clusters, miRNA-106b/25 and miRNA-302/367. Overexpression of the miRNA-302/367 cluster96, 97 or two components of miRNA-106b/25 cluster, miRNA-93 and miRNA-106b98, accelerates the mesenchymal-to-epithelial transition and increases the number of iPS cells derived from MEFs expressing OKSM or OKS. The miRNA-302/367 cluster (miRNA-302a/b/c/d and miRNA-367), in addition to its role in regulating TGF-β signaling, targets the BMP inhibitors TOB2, DAZAP2, and SLAIN199. The BMP and TGF-β signaling pathways converge on Smad proteins suggesting that miRNA-302/367 could potentially promote reprogramming via these proteins. Cross-talk between TGF-β and cell proliferation pathways also exists. One study, utilizing a library screen, found that the miRNA-130/301/721 family promoted generation of iPS cells by inhibiting expression of Mesenchyme Homeobox 2 (Meox2)100. TGF-β suppresses endothelial cell growth through activation of Meox2, which in turn triggers expression of p21101.
Chromatin remodeling influences the ability of miRNAs to reprogram somatic cells into iPS. Considerable epigenetic changes occur during reprogramming 102 and pharmacological inhibition of key chromatin modifiers such as HDACs (histone deacetylases) and DNMTs (DNA methyltransferases) increase the efficiency of reprogramming103, 104. HDAC2 suppression by valproic acid or genetic ablation allows the miRNA-302/367 cluster, a direct target of Oct4 and Sox2105, to reprogram MEFs into iPS cells without the need for transcription factors81. Similarly, human foreskin fibroblasts which naturally express low levels of HDAC2 were reprogrammed to iPS by expression of the miRNA-302/367 cluster in the absence of valproic acid81. Elevating the expression of a single miRNA, miRNA-302, to a level 1.3-fold above that found in human embryonic stem cells was sufficient to reprogram human hair follicle cells to iPS106. MiRNA-302 reprogramming to iPS required the repression of a number of epigenetic regulators, such as the lysine-specific demethylases AOF1 and AOF2 as well as the methyl-CpG-binding proteins MECP1-p66 and MECP2106. Apoptosis and senescence have also been proposed as possible mechanisms by which miRNAs regulate reprogramming to iPS however definitive roles have not been currently established107. Moreover, direct regulation of the OKSM factors is another putative mechanism. MiRNA-25 directly regulates Wwp2, an E3 ubiquitin ligase that targets Oct4 for ubiquitination, and Fbxw7, which is known to regulate c-Myc. By increasing levels of Oct4 and c-Myc miRNA-25 was found to promote the formation of iPS cells.108.
To summarize miRNAs are important tools for reprogramming somatic cells to iPS cells. However, the similarity between miRNA and transcription factor reprogrammed iPS has not been studied and differences may exist in the iPS generated by the two methods.
miRNAs, iPS, ESCs, and the acquisition of a cardiac phenotype
A number of microRNAs, including miRNA-1, miRNA-133, miRNA-208, and miRNA-499, have also been used to drive cardiac differentiation in ESCs and IPS cells109, 110.
In a 2D ES culture model levels of miRNA-1 and miRNA-133 were reduced following forced myocardial differentiation by trichostatin-A, a histone deacetylase inhibitor111. Moreover, overexpression of miRNA-1 or miRNA-133 by lentivirus reduced the expression of Nkx2.5, with miRNA-1 also inhibiting expression of αMHC. CDK9 was proposed to be involved in the pathway based on the finding that miRNA-1 reduced protein expression by targeting the 3′ UTR of the CDK9 mRNA111 as well as previous work by the authors which showed that CDK9 formed a transcriptional complex with p300/Gata4 to activate expression of Nkx2.5, ANF and β-MHC112. In support of these studies overexpression of the Drosophila miRNA-1 homolog (dmiR-1) in cardiac mesoderm resulted in fewer cardiac cells113.
However, the effects of miRNA-1 and miRNA-133 upon ES cardiac differentiation may be dependent upon the model employed as the findings with embyroid body (EB) based culture of ES cells were found to be markedly different109. Starting from the observation that both miRNA-1 and miRNA-133 were highly expressed in ES derived cardiomyocytes the authors found that expression of miRNA-1 or miRNA-133 in EB promoted mesoderm gene expression at the expense of ectoderm and endoderm differentiation. Moreover, whilst miRNA-1 promoted further differentiation towards a cardiac or skeletal muscle fate, miRNA-133 was inhibitory. The Notch ligand, Delta-like 1 (Dll1), was translationally repressed by miRNA-1 and indeed knockdown of Dll1 recapitulated the miRNA-1 overexpression phenotype in ESCs109. Further support for a role for miRNA-1 in the adoption of a cardiomyocyte phenotype has come from studies in Xenopus embryos42. Misexpression of miRNA-1 strongly inhibited myogenesis by targeting histone deacetylase 4 (HDAC4)42, which negatively regulates a protein critical for muscle differentiation, Mef2114. Moreover, overexpression of miRNA-1 in iPS cells led to the expression of cardiac transcription factors and sarcomeric proteins115. Similarly, in ESC-derived multipotent cardiovascular progenitors miRNA-1 promoted cardiomyocyte differentiation and suppressed endothelial cell commitment by modulating Wnt and FGF signaling pathways, with Frizzled Class Receptor 7 (FZD7) and Far1 Related Sequence (FRS1) being confirmed as miRNA-1 targets115.
MiRNA-1 may also be involved in the electrophysiological maturation of ES-derived cardiomyocytes116. Lentiviral mediated delivery of miRNA-1 into human ESC-derived cardiovascular progenitors had no effect on the yield of human ESC-derived ventricular cadiomyocytes. However, hallmarks of maturation were observed such as decreased action potential duration and hyperpolarized resting membrane potential/maximum diastolic potential. Ca2+ transient amplitude and kinetics were also augmented116.
Irrespective of the in vitro results over-expression of miRNA-1 has been shown to drive cardiac differentiation of ESCs in vivo117. When assessed two-weeks post-MI, injected ESCs expressing miRNA-1 demonstrated enhanced commitment to the cardiomyocyte lineage when compared to control ESCs and improved cardiac function was noted117. A paracrine mechanism may also be important in this model as the host myocardium displayed reduced apoptosis through Akt activation and caspase-3 inactivation117.
Levels of miRNA-499 also increase during the differentiation of ES cells into cardiomyocytes110. Overexpression of miRNA-499 in embryoid bodies upregulated αMHC and Mef2C expression110. Similarly increasing expression of miRNA-499 in human ESC- derived cardiovascular progenitors significantly augmented the yield of ventricular cardiomyocytes and contractile protein expression without affecting electrophysiological properties116.
MiRNA-363 is involved in ESC-derived cardiac subtype specification. Screening for miRNAs that potentially affected Hand-and-neural-crest-derivative-expressed (HAND) 1 and 2, genes involved in left and right ventricular development respectively, identified miRNA-363 as a candidate. Over-expression of miRNA-363 reduced HAND1 mRNA levels and suppression of this miRNA led to an enrichment of left ventricular ESC-derived cardiomyocytes118.
MiRNAs and direct cardiac reprogramming
Development of miRNA combo for the transdifferentiation of fibroblasts to cardiomyocytes
Based on their roles in cardiac development, our laboratory hypothesized that miRNAs would be capable of reprogramming fibroblasts directly into cardiomyocytes9. We selected six candidate miRNAs, miRNA-1, miRNA-126, miRNA-133a, miRNA-138, miRNA-206 and miRNA-208a, based on their function in cardiac muscle development and differentiation40, 47, 55, 119–121. Adopting a combinatorial approach, we identified that miRNA-1 and the combination of miRNA-1, miRNA-133a, and miRNA-208a induced the expression of early markers of commitment to the cardiomyocyte lineage9. Though miRNA-1 alone was found to be sufficient to drive cardiac gene expression, efficiency was higher in combination with miRNA-133a and miRNA-208a. Further studies showed that the addition of miRNA-499 further augmented the efficiency of cardiac reprogramming9. This combination of miRNAs, miRNA-1, miRNA-133a, miRNA-208a, and miRNA 499 we named miR combo. A single transient transfection of miR combo was found to be sufficient to induce fibroblasts to express cardiac markers such as Mef2C and αMHC 9, 10. The initial steps of reprogramming were relatively rapid in vitro. Mature cardiomyocyte markers such as αMHC and cardiac troponins were observed ~7 days after transfection. Full maturation of the reprogrammed cells was observed only after prolonged culture. Approximately 4 weeks following transfection, organized sarcomeres, contraction and spontaneous calcium transients were observed9.
miR combo regenerates the heart: reprogramming and the correlation between maturation and functional improvement
In a proof-of-principle experiment we used the Fsp1Cre:tdTomato model to validate reprogramming of fibroblasts in vivo. In this model the tdTomato protein permanently labels fibroblasts. Lentiviruses encoding for the individual miRNAs in the miR combo were injected into ischemic myocardium. One month following myocardial injury tdTomato+ cardiomyocytes were observed which provided evidence of direct reprogramming of fibroblasts in situ9. We found that 1 month post-MI tdTomato+ cardiomyocytes represented ~1% of the infarct border zone, two months post-MI, the number of tdTomato+ cardiomyocytes had risen to ~10% 9, 11. We observed that miR combo promoted a progressive improvement in cardiac function over a 3 month period associated with reduced fibrosis following MI11. The improvement of cardiac function by miR combo occurred in the absence of any effects on cardiomyocyte apoptosis and de novo vascularization. Thus the effect of miR combo in vivo on cardiac function was both progressive and time-delayed. This has also been observed with reprogramming strategies involving transcription factors. Based on our in vitro observation of time taken from reprogramming to cell maturation, we hypothesized that the delay in cardiac functional improvement in vivo is in part due to the time taken for reprogrammed fibroblasts to fully mature into cardiomyocytes and integrate into the myocardium. Indeed, at 2 months, the reprogrammed tdTomato+ cardiomyocytes expressed cardiomyocyte markers, sarcomeric organization, excitation-contraction coupling, and action potentials characteristic of maturing ventricular tdTomato- cardiomyocytes 11. Taken together, our findings suggest a correlation between maturation of reprogrammed cardiomyocytes and improved cardiac function (Figure 1A). Fully establishing that full maturation of reprogrammed fibroblasts into cardiomyocytes underlies the improvements in cardiac function will require a systematic study of the temporal effects of miR combo treatment at the cellular and functional level in conjunction with statistical modeling.
Figure 1. Schematic describing the hypothesis that the time-delayed improvements in cardiac function are dependent upon the maturation of reprogrammed cells.
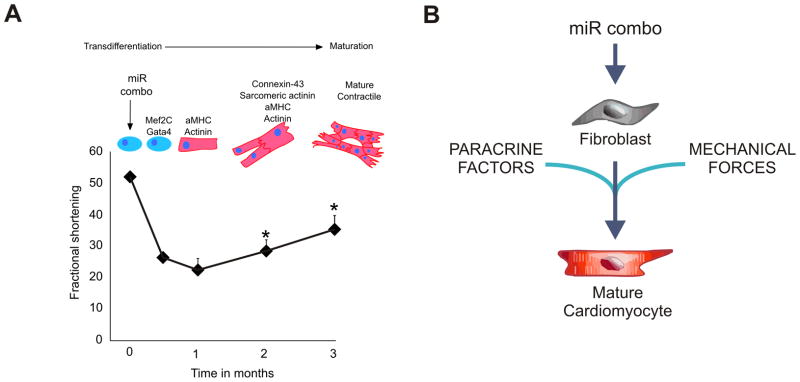
(A)The cardiac function data (fractional shortening) vs time are derived from Jayawardena et al. 11. The upper panel depicting the progression of transdifferentiation (with specific cardiac gene expression) to maturation is based partly on data11 and partly on hypothesis.
(B) The putative factors influencing cardiomyocyte maturation in vivo: paracrine factors released by other cardiac cells, the composition of the ECM, mechanical forces and/or cell:cell communication.
Maturation: why is there more in vivo?
Reprogramming with miR combo is more efficient in vivo than in vitro. Why this is the case is currently unknown. It is possible that other cell types in the heart, cardiomyocytes and cardiac progenitor cells, influence reprogramming either through the release of paracrine factors or by cell:cell communication through adherens junctions We anticipate that paracrine factors will play a particularly important role. In the past decade, substantial evidence has been provided to support the notion that stem cells exert their reparative and regenerative effects, in large part, through the release of biologically active molecules acting in a paracrine fashion on resident cells122. Pertinent to this review we have recently identified that a paracrine factor released by MSCs, Abi3bp, promotes cardiac progenitor cell differentiation123.
Tissue engineers have sought to recapitulate the native cardiac environment. They have had notable successes in generating cardiac tissue124 and their findings potentially shed light on the question of why maturation of miR combo reprogrammed fibroblasts is more efficient in vivo. What they have found is that 3D environments, decellularization of organs which leaves extracellular matrix intact, and stimulation, whether it be mechanical or electrical, promote the formation of cardiac tissue124. Thus, we hypothesize that components of the extracellular matrix, mechanical forces and the cell shape in the 3D environment is important (Figure 1B) for the maturation of miR combo reprogrammed cells. These mechanisms would promote maturation either by increasing the rate of the initial reprogramming event or by directly affecting the expression of mature cardiac structural and functional proteins.
Mechanisms for direct reprogramming of cardiac fibroblasts by miR combo
Knowledge of the mechanistic basis for the reprogramming of cardiac fibroblasts into cardiomyocytes by transcription factors or miRNAs is currently lacking. The epigenetic landscape of cardiac fibroblasts is likely to be very different to that of cardiomyocytes; such differences are believed to be important for the maintenance of a differentiated phenotype125. Differences between the epigenetic landscape of cardiac fibroblasts and cardiomyocytes would be expected to be a significant barrier to direct reprogramming125. The transcription factors currently employed, such as Mef2C, Gata4, and Tbx5, are known to be important for the development of heart. However, how these transcription factors access the silent promoters of cardiac genes in fibroblasts is currently unknown; presumably they act as “pioneer” transcription factors that recognize their target sites irrespective of the pre-existing chromatin state. Considering that the epigenetic landscape represents a “molecular roadblock” to reprogramming125 we conducted a genetic screen to identify epigenetic modifiers regulated by miR combo. We found that miRNA combo downregulates expression of the histone lysine N-methyltransferase Setdb2; a protein which specifically trimethylates lysine-9 of histone H3 (H3K9me3). Considering that H3K9me3 is associated with transcriptional repression our data indicates that miR combo induces reprogramming by alleviating the suppression of cardiac genes in fibroblasts (unpublished data). Future work is necessary to determine which constituent of miR combo downregulates Setdb2 expression as well as the possible role of other epigenetic modifications in mediating the effects of miR combo.
Enhancing direct reprogramming by combining miRNA and transcription factor strategies
Other researchers have used miRNA-1 and miRNA-133 in combination with transcription factors to increase the efficiency of mouse and human fibroblast reprogramming into cardiomyocytes in vitro. Nam et al. were able to drive expression of cardiac markers in neonatal and human fibroblasts using miRNA-1, miRNA-133, Gata4, Hand2, Tbx5, and myocardin. Four to eleven weeks following transfection sarcomeric like structures and calcium transients were observed, with a number of cells showing spontaneous contractility126. Gata4, Hand2, Tbx5, myocardin and Mef2C were originally identified as being able to reprogram fibroblasts into cardiomyocytes. Retroviral expression of miRNA-1 and miRNA-133 increased the number of cardiac troponin-T positive cells. Curiously removal of Mef2C enhanced reprogramming126. Underlying the importance of miRNA-133, genetic ablation of this miRNA reduced the number of cardiac troponin-T positive cells to almost background levels126. The authors speculated that miRNA-1 and miRNA-133 were important for the development of sarcomeres and suppressed myocardin activation of smooth muscle differentiation126.miRNA-133, in combination with Gata4, Mef2C, and Tbx5 (GMT), has also been shown to be important in the acquisition of a cardiac phenotype in murine embryonic fibroblasts (MEFs)127. Overexpression of miRNA-133, in combination with GMT, increased by greater than 5-fold the number of MEFs expressing α-sarcomeric actinin, αMHC, and/or cardiac troponin-T when compared to cells expressing GMT alone. Similar results were obtained with the number of beating cells and the time taken for maturation was found to be significantly shortened127. The authors showed that miRNA-133 enhanced GMT reprogramming by silencing fibroblast markers via suppression of the epithelial-to-mesenchymal transition regulator Snai1127.
miRNAs and adult cardiac stem cells
The heart contains resident cardiac progenitors which can potentially form all the major cell types of the heart, including cardiomyocytes, endothelial cells, and smooth muscle128. In two recent clinical trials, SCIPIO and CADUCEUS, cardiac progenitor cells have demonstrated their therapeutic potential129, 130. Strategies are needed to drive cardiac progenitor differentiation towards the cardiomyocyte lineage especially, as currently this is very inefficient. Despite the potential benefits of miRNAs to direct cardiac progenitor fate a relatively limited number of reports have been published regarding their use in this regard.
Transient transfection of miRNA-1 and miRNA-499 into cardiac progenitors derived from human fetuses reduced cell proliferation and enhanced differentiation to cardiomyocytes; for example reducing the time taken for spontaneously beating clusters to appear from 21 to 7 days131. The effects of miRNA-1 and miRNA-499 were likely due to the repression of histone deacetylase 4 or Sox6. Indeed siRNA mediated knockdown of Sox6 induced cardiomyocyte differentiation131. The results with miRNA-499 were confirmed by a separate study using c-Kit+ cardiac progenitors132. Here, the authors went one step further and injected cardiac progenitors expressing miRNA-499 into infarcted hearts. Increased cardiomyogenesis was observed, with an elevated cardiomyocyte mass132. The authors also identified a novel mechanism by which miRNA-499 can influence cardiac progenitor behavior. MiRNA-499 was found to translocate from C2C12 myoblasts to recipient cardiac progenitor cells via gap junctions between the two cell types. The function of the miRNA-499 was preserved following gap junction transfer to cardiac progenitors and this favored their differentiation into functionally competent cardiomyocytes132. MiRNA-1 is also important for the control of cardiac progenitor cell polarity during development in Drosophila133. Cell polarity is known to play multiple roles in cardiac differentiation and development134. MiRNAs are also involved in inhibiting differentiation. For example, miRNA-590 and miRNA-155 have opposing effects on cardiac progenitor differentiation. TGF-β1 promotes differentiation of cardiosphere-derived cells by the down-regulation of miRNA-590135. MiRNA-155 inhibits Sca-1+ cardiac progenitor cell differentiation by down-regulating β-arrestin-2136.
Differentiation of cardiac progenitors requires a halt in proliferation. Sirish et al. compared miRNA expression profiles in c-Kit+ cardiac progenitors derived from neonatal and adult hearts. When compared to adult cells neonatal c-Kit+ cardiac progenitors expressed higher levels of the proliferation marker Ki67 with a seven-fold higher doubling time. MiRNA-17 was also elevated in neonatal c-Kit+ cardiac progenitors and as a proof-of-principle expression of the miRNA-17-92 cluster in adult c-Kit+ progenitors increased their proliferation rate. The anti-proliferative cell cycle protein retinoblastoma-like 2 (Rbl2/p130) was proposed as a target for miRNA-17 on the basis of an interaction site within the 3′ UTR; though Rbl2/p130 mRNA levels were not significantly different, protein levels were significantly lower in neonatal c-Kit+ cardiac progenitor cells when compared to adult cells137. MiRNA-10a reduces cardiac progenitor cell proliferation by targeting Gata6138.
In summary, a number of microRNAs have been identified that regulate key aspects of cardiac progenitor biology. However, it is clear that more studies are necessary to fully characterize the full complement of microRNAs that influence cardiac progenitor differentiation and proliferation.
Future Perspectives
The studies described in this review highlight the fundamental role that miRNAs play in cardiac reprogramming, differentiation and development (Figure 2). Based on the understanding of above, we hypothesize that targeting specific miRNAs is a rational strategy for cardiac regenerative therapy.
Figure 2. miRNAs and reprogramming.

miRNAs promote the generation of cardiomyocytes via a number of mechanisms. Fibroblasts can be reprogrammed into cardiomyocytes by miRNAs directly or through an intermediate iPSC state. miRNAs also promote cardiac progenitor cell (CPC) and embryonic stem cell (ESC) cardiac differentiation. miRNAs can promote or inhibit cardiomycyte proliferation.
MiRNA based therapy can be used to promote cardiomyocyte proliferation, reprogram directly fibroblasts to cardiomyocytes or indirectly to iPSc as well as driving the differentiation of iPSCs, ESCs or CPCs to cardiomyocytes (Figure 2).
Key issues need to be addressed before miRNAs can be taken into the clinic. One such question is the combination of miRNAs that will induce cardiac regeneration. Species differences between mouse and human are likely to necessitate a different combination of miRNAs for efficient proliferation, direct or indirect reprogramming127. Another question is how these miRNAs will be delivered into the patient. There are a number of options such as viral delivery or chemical modification. Lenti-, retro-, and adenoviral associated-viruses are all suitable viral delivery systems each with their own specific advantages and disadvantages142, 143. The adenoviral associated virus (AAV) approach is potentially the most suitable as AAVs offer significant benefits over lenti- and retro-viruses such as non-integration into the host genome139, 140. A single virus containing all of the reprogramming factors is the obvious ideal approach; and this further highlights the benefits of miRNAs as a reprogramming strategy over transcription factors. Only a limited amount of DNA can be packaged into a virus and miRNAs are smaller DNA moieties than transcription factor genes. Currently, the in vivo use of transcription factors for reprogramming has relied on retroviruses containing a single transcription factor7. Another potential issue is the expression of multiple genes from a single DNA cassette; the genes do not necessarily express at similar levels141 and this could seriously affect reprogramming efficiency. Naturally occurring multicistronic miRNA constructs can be modified to express any miRNA combination of choice. For example, modification of the endogenous miRNA-17-92 cluster to express miRNAs that target the Hepatitis C viral genome prevents replication of the Hepatitis C virus142. This has been used to inhibit Hepatitis C virus replication for example, by modifying Packaging DNA into viral vectors due to their small size.
AntagomiRs, miRNAs modified with cholesterol and/or phosphorothioate moieties to increase their stability in vivo143, have been used to reduce the expression of miRNAs that mediate pathology following myocardial injury144. AntagomiRs may also prove to be a useful ally for cardiac reprogramming. It is well known that there are significant barriers to reprogramming125. Cell type-specific miRNAs prevent translation of lineage-inappropriate mRNAs125, 145. Targeting these cell type-specific miRNAs with antagomiRs is likely to enhance cardiac reprogramming.
Cardiomyocyte proliferation, differentiation and reprogramming involve the co-ordinated action of many proteins acting in multiple pathways. For this reason, of all the many strategies employed for reprogramming, miRNAs offer the most appropriate route as a single miRNA can influence multiple pathways at once. By virtue of their small size miRNAs are ideal for AAV based therapies; which have generated much interest due to their inherent safety. Finally, by adapting naturally occurring multicistronic miRNA constructs researchers have the ability to nuance the expression of reprogramming miRNAs, which is important for dosing concerns.
Supplementary Material
Acknowledgments
SOURCES OF FUNDING
This work was supported by National Heart, Lung, and Blood Institute grants RO1 HL81744, HL72010, and HL73219 (to V.J.D.); and the Edna and Fred L. Mandel Jr. Foundation (to V.J.D., and M.M.). M.M. was also supported by an American Heart Association National Scientist Development Award (10SDG4280011).
NON-STANDARD ABBREVIATIONS AND ACRONYMS
- dKO
double knockout
- Fsp1
fibroblast-specific protein 1
- Hand
Heart- and neural crest derivatives-expressed protein
- MHC
myosin heavy chain
- MI
myocardial infarction
- miRNA(s)/miR(s)
microRNA(s)
- RISC
RNA induced silencing complex
- tdTomato
tandem dimer Tomato (fluorescent protein)
- UTR
untranslated region
Footnotes
DISCLOSURES
None of the authors have any real or apparent conflict(s) of interest to disclose.
References
- 1.Fan D, Takawale A, Lee J, Kassiri Z. Cardiac fibroblasts, fibrosis and extracellular matrix remodeling in heart disease. Fibrogenesis & tissue repair. 2012;5:15. doi: 10.1186/1755-1536-5-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Dal-Pra S, Mirotsou M. Reprogramming approaches in cardiovascular regeneration. Current treatment options in cardiovascular medicine. 2014;16:327. doi: 10.1007/s11936-014-0327-0. [DOI] [PubMed] [Google Scholar]
- 3.Buckingham M, Meilhac S, Zaffran S. Building the mammalian heart from two sources of myocardial cells. Nature reviews Genetics. 2005;6:826–35. doi: 10.1038/nrg1710. [DOI] [PubMed] [Google Scholar]
- 4.Olson EN. Gene regulatory networks in the evolution and development of the heart. Science. 2006;313:1922–7. doi: 10.1126/science.1132292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Srivastava D. Making or breaking the heart: from lineage determination to morphogenesis. Cell. 2006;126:1037–48. doi: 10.1016/j.cell.2006.09.003. [DOI] [PubMed] [Google Scholar]
- 6.Song K, Nam YJ, Luo X, Qi X, Tan W, Huang GN, Acharya A, Smith CL, Tallquist MD, Neilson EG, Hill JA, Bassel-Duby R, Olson EN. Heart repair by reprogramming non-myocytes with cardiac transcription factors. Nature. 2012;485:599–604. doi: 10.1038/nature11139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Qian L, Huang Y, Spencer CI, Foley A, Vedantham V, Liu L, Conway SJ, Fu JD, Srivastava D. In vivo reprogramming of murine cardiac fibroblasts into induced cardiomyocytes. Nature. 2012;485:593–8. doi: 10.1038/nature11044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Protze S, Khattak S, Poulet C, Lindemann D, Tanaka EM, Ravens U. A new approach to transcription factor screening for reprogramming of fibroblasts to cardiomyocyte-like cells. Journal of molecular and cellular cardiology. 2012;53:323–32. doi: 10.1016/j.yjmcc.2012.04.010. [DOI] [PubMed] [Google Scholar]
- 9.Jayawardena TM, Egemnazarov B, Finch EA, Zhang L, Payne JA, Pandya K, Zhang Z, Rosenberg P, Mirotsou M, Dzau VJ. MicroRNA-mediated in vitro and in vivo direct reprogramming of cardiac fibroblasts to cardiomyocytes. Circulation research. 2012;110:1465–73. doi: 10.1161/CIRCRESAHA.112.269035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Jayawardena T, Mirotsou M, Dzau VJ. Direct reprogramming of cardiac fibroblasts to cardiomyocytes using microRNAs. Methods in molecular biology. 2014;1150:263–72. doi: 10.1007/978-1-4939-0512-6_18. [DOI] [PubMed] [Google Scholar]
- 11.Jayawardena TM, Finch EA, Zhang L, Zhang H, Hodgkinson C, Pratt RE, Rosenberg PB, Mirotsou M, Dzau VJ. MicroRNA Induced Cardiac Reprogramming In Vivo: Evidence for Mature Cardiac Myocytes and Improved Cardiac Function. Circulation research. 2015;116:418–424. doi: 10.1161/CIRCRESAHA.116.304510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ghildiyal M, Zamore PD. Small silencing RNAs: an expanding universe. Nature reviews Genetics. 2009;10:94–108. doi: 10.1038/nrg2504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Schraivogel D, Meister G. Import routes and nuclear functions of Argonaute and other small RNA-silencing proteins. Trends in biochemical sciences. 2014;39:420–431. doi: 10.1016/j.tibs.2014.07.004. [DOI] [PubMed] [Google Scholar]
- 14.Kozomara A, Griffiths-Jones S. miRBase: annotating high confidence microRNAs using deep sequencing data. Nucleic acids research. 2014;42:D68–73. doi: 10.1093/nar/gkt1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Friedman RC, Farh KK, Burge CB, Bartel DP. Most mammalian mRNAs are conserved targets of microRNAs. Genome research. 2009;19:92–105. doi: 10.1101/gr.082701.108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Ha M, Kim VN. Regulation of microRNA biogenesis. Nature reviews Molecular cell biology. 2014;15:509–24. doi: 10.1038/nrm3838. [DOI] [PubMed] [Google Scholar]
- 17.Coolen M, Bally-Cuif L. MicroRNAs in brain development and physiology. Current opinion in neurobiology. 2009;19:461–70. doi: 10.1016/j.conb.2009.09.006. [DOI] [PubMed] [Google Scholar]
- 18.Malizia AP, Wang DZ. MicroRNAs in cardiomyocyte development. Wiley interdisciplinary reviews Systems biology and medicine. 2011;3:183–90. doi: 10.1002/wsbm.111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Ambros V. MicroRNAs and developmental timing. Current opinion in genetics & development. 2011;21:511–7. doi: 10.1016/j.gde.2011.04.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Vidigal JA, Ventura A. Embryonic stem cell miRNAs and their roles in development and disease. Seminars in cancer biology. 2012;22:428–36. doi: 10.1016/j.semcancer.2012.04.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sokol NS. Small temporal RNAs in animal development. Current opinion in genetics & development. 2012;22:368–73. doi: 10.1016/j.gde.2012.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kuppusamy KT, Sperber H, Ruohola-Baker H. MicroRNA regulation and role in stem cell maintenance, cardiac differentiation and hypertrophy. Current molecular medicine. 2013;13:757–64. doi: 10.2174/1566524011313050007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wang XH. MicroRNA in myogenesis and muscle atrophy. Current opinion in clinical nutrition and metabolic care. 2013;16:258–66. doi: 10.1097/MCO.0b013e32835f81b9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Sun AX, Crabtree GR, Yoo AS. MicroRNAs: regulators of neuronal fate. Current opinion in cell biology. 2013;25:215–21. doi: 10.1016/j.ceb.2012.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Luo W, Nie Q, Zhang X. MicroRNAs involved in skeletal muscle differentiation. Journal of genetics and genomics = Yi chuan xue bao. 2013;40:107–16. doi: 10.1016/j.jgg.2013.02.002. [DOI] [PubMed] [Google Scholar]
- 26.Cremisi F. MicroRNAs and cell fate in cortical and retinal development. Frontiers in cellular neuroscience. 2013;7:141. doi: 10.3389/fncel.2013.00141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Turner ML, Schnorfeil FM, Brocker T. MicroRNAs regulate dendritic cell differentiation and function. Journal of immunology. 2011;187:3911–7. doi: 10.4049/jimmunol.1101137. [DOI] [PubMed] [Google Scholar]
- 28.Rao PK, Kumar RM, Farkhondeh M, Baskerville S, Lodish HF. Myogenic factors that regulate expression of muscle-specific microRNAs. Proceedings of the National Academy of Sciences of the United States of America. 2006;103:8721–6. doi: 10.1073/pnas.0602831103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Takaya T, Nishi H, Horie T, Ono K, Hasegawa K. Roles of microRNAs and myocardial cell differentiation. Progress in molecular biology and translational science. 2012;111:139–52. doi: 10.1016/B978-0-12-398459-3.00006-X. [DOI] [PubMed] [Google Scholar]
- 30.Joladarashi D, Thandavarayan RA, Babu SS, Krishnamurthy P. Small Engine, Big Power: MicroRNAs as Regulators of Cardiac Diseases and Regeneration. International journal of molecular sciences. 2014;15:15891–15911. doi: 10.3390/ijms150915891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cai X, Hagedorn CH, Cullen BR. Human microRNAs are processed from capped, polyadenylated transcripts that can also function as mRNAs. Rna. 2004;10:1957–66. doi: 10.1261/rna.7135204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Gommans WM, Berezikov E. Controlling miRNA regulation in disease. Methods in molecular biology. 2012;822:1–18. doi: 10.1007/978-1-61779-427-8_1. [DOI] [PubMed] [Google Scholar]
- 33.Ogata H, Audic S, Renesto-Audiffren P, Fournier PE, Barbe V, Samson D, Roux V, Cossart P, Weissenbach J, Claverie JM, Raoult D. Mechanisms of evolution in Rickettsia conorii and R. prowazekii. Science. 2001;293:2093–8. doi: 10.1126/science.1061471. [DOI] [PubMed] [Google Scholar]
- 34.Azuma-Mukai A, Oguri H, Mituyama T, Qian ZR, Asai K, Siomi H, Siomi MC. Characterization of endogenous human Argonautes and their miRNA partners in RNA silencing. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:7964–9. doi: 10.1073/pnas.0800334105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Iwasaki S, Kobayashi M, Yoda M, Sakaguchi Y, Katsuma S, Suzuki T, Tomari Y. Hsc70/Hsp90 chaperone machinery mediates ATP-dependent RISC loading of small RNA duplexes. Molecular cell. 2010;39:292–9. doi: 10.1016/j.molcel.2010.05.015. [DOI] [PubMed] [Google Scholar]
- 36.Kawamata T, Seitz H, Tomari Y. Structural determinants of miRNAs for RISC loading and slicer-independent unwinding. Nature structural & molecular biology. 2009;16:953–60. doi: 10.1038/nsmb.1630. [DOI] [PubMed] [Google Scholar]
- 37.Chen JF, Murchison EP, Tang R, Callis TE, Tatsuguchi M, Deng Z, Rojas M, Hammond SM, Schneider MD, Selzman CH, Meissner G, Patterson C, Hannon GJ, Wang DZ. Targeted deletion of Dicer in the heart leads to dilated cardiomyopathy and heart failure. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:2111–6. doi: 10.1073/pnas.0710228105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.da Costa Martins PA, Bourajjaj M, Gladka M, Kortland M, van Oort RJ, Pinto YM, Molkentin JD, De Windt LJ. Conditional dicer gene deletion in the postnatal myocardium provokes spontaneous cardiac remodeling. Circulation. 2008;118:1567–76. doi: 10.1161/CIRCULATIONAHA.108.769984. [DOI] [PubMed] [Google Scholar]
- 39.Saxena A, Tabin CJ. miRNA-processing enzyme Dicer is necessary for cardiac outflow tract alignment and chamber septation. Proceedings of the National Academy of Sciences of the United States of America. 2010;107:87–91. doi: 10.1073/pnas.0912870107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Zhao Y, Ransom JF, Li A, Vedantham V, von Drehle M, Muth AN, Tsuchihashi T, McManus MT, Schwartz RJ, Srivastava D. Dysregulation of cardiogenesis, cardiac conduction, and cell cycle in mice lacking miRNA-1-2. Cell. 2007;129:303–17. doi: 10.1016/j.cell.2007.03.030. [DOI] [PubMed] [Google Scholar]
- 41.Zhao Y, Samal E, Srivastava D. Serum response factor regulates a muscle-specific microRNA that targets Hand2 during cardiogenesis. Nature. 2005;436:214–20. doi: 10.1038/nature03817. [DOI] [PubMed] [Google Scholar]
- 42.Chen JF, Mandel EM, Thomson JM, Wu Q, Callis TE, Hammond SM, Conlon FL, Wang DZ. The role of microRNA-1 and microRNA-133 in skeletal muscle proliferation and differentiation. Nature genetics. 2006;38:228–33. doi: 10.1038/ng1725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Liu N, Williams AH, Kim Y, McAnally J, Bezprozvannaya S, Sutherland LB, Richardson JA, Bassel-Duby R, Olson EN. An intragenic MEF2-dependent enhancer directs muscle-specific expression of microRNAs 1 and 133. Proceedings of the National Academy of Sciences of the United States of America. 2007;104:20844–9. doi: 10.1073/pnas.0710558105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Heidersbach A, Saxby C, Carver-Moore K, Huang Y, Ang YS, de Jong PJ, Ivey KN, Srivastava D. microRNA-1 regulates sarcomere formation and suppresses smooth muscle gene expression in the mammalian heart. eLife. 2013;2:e01323. doi: 10.7554/eLife.01323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wei Y, Peng S, Wu M, Sachidanandam R, Tu Z, Zhang S, Falce C, Sobie EA, Lebeche D, Zhao Y. Multifaceted roles of miR-1s in repressing the fetal gene program in the heart. Cell research. 2014;24:278–92. doi: 10.1038/cr.2014.12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Yin VP, Lepilina A, Smith A, Poss KD. Regulation of zebrafish heart regeneration by miR-133. Developmental biology. 2012;365:319–27. doi: 10.1016/j.ydbio.2012.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Liu N, Bezprozvannaya S, Williams AH, Qi X, Richardson JA, Bassel-Duby R, Olson EN. microRNA-133a regulates cardiomyocyte proliferation and suppresses smooth muscle gene expression in the heart. Genes & development. 2008;22:3242–54. doi: 10.1101/gad.1738708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Care A, Catalucci D, Felicetti F, Bonci D, Addario A, Gallo P, Bang ML, Segnalini P, Gu Y, Dalton ND, Elia L, Latronico MV, Hoydal M, Autore C, Russo MA, Dorn GW, 2nd, Ellingsen O, Ruiz-Lozano P, Peterson KL, Croce CM, Peschle C, Condorelli G. MicroRNA-133 controls cardiac hypertrophy. Nature medicine. 2007;13:613–8. doi: 10.1038/nm1582. [DOI] [PubMed] [Google Scholar]
- 49.Li Q, Lin X, Yang X, Chang J. NFATc4 is negatively regulated in miR-133a-mediated cardiomyocyte hypertrophic repression. American journal of physiology Heart and circulatory physiology. 2010;298:H1340–7. doi: 10.1152/ajpheart.00592.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Dong DL, Chen C, Huo R, Wang N, Li Z, Tu YJ, Hu JT, Chu X, Huang W, Yang BF. Reciprocal repression between microRNA-133 and calcineurin regulates cardiac hypertrophy: a novel mechanism for progressive cardiac hypertrophy. Hypertension. 2010;55:946–52. doi: 10.1161/HYPERTENSIONAHA.109.139519. [DOI] [PubMed] [Google Scholar]
- 51.Wystub K, Besser J, Bachmann A, Boettger T, Braun T. miR-1/133a clusters cooperatively specify the cardiomyogenic lineage by adjustment of myocardin levels during embryonic heart development. PLoS genetics. 2013;9:e1003793. doi: 10.1371/journal.pgen.1003793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Lompre AM, Nadal-Ginard B, Mahdavi V. Expression of the cardiac ventricular alpha- and beta-myosin heavy chain genes is developmentally and hormonally regulated. The Journal of biological chemistry. 1984;259:6437–46. [PubMed] [Google Scholar]
- 53.Chien KR. Genomic circuits and the integrative biology of cardiac diseases. Nature. 2000;407:227–32. doi: 10.1038/35025196. [DOI] [PubMed] [Google Scholar]
- 54.van Rooij E, Sutherland LB, Qi X, Richardson JA, Hill J, Olson EN. Control of stress-dependent cardiac growth and gene expression by a microRNA. Science. 2007;316:575–9. doi: 10.1126/science.1139089. [DOI] [PubMed] [Google Scholar]
- 55.Callis TE, Pandya K, Seok HY, Tang RH, Tatsuguchi M, Huang ZP, Chen JF, Deng Z, Gunn B, Shumate J, Willis MS, Selzman CH, Wang DZ. MicroRNA-208a is a regulator of cardiac hypertrophy and conduction in mice. The Journal of clinical investigation. 2009;119:2772–86. doi: 10.1172/JCI36154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.van Rooij E, Quiat D, Johnson BA, Sutherland LB, Qi X, Richardson JA, Kelm RJ, Jr, Olson EN. A family of microRNAs encoded by myosin genes governs myosin expression and muscle performance. Developmental cell. 2009;17:662–73. doi: 10.1016/j.devcel.2009.10.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Morkin E. Control of cardiac myosin heavy chain gene expression. Microscopy research and technique. 2000;50:522–31. doi: 10.1002/1097-0029(20000915)50:6<522::AID-JEMT9>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 58.Shieh JT, Huang Y, Gilmore J, Srivastava D. Elevated miR-499 levels blunt the cardiac stress response. PloS one. 2011;6:e19481. doi: 10.1371/journal.pone.0019481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Matkovich SJ, Hu Y, Eschenbacher WH, Dorn LE, Dorn GW., 2nd Direct and indirect involvement of microRNA-499 in clinical and experimental cardiomyopathy. Circulation research. 2012;111:521–31. doi: 10.1161/CIRCRESAHA.112.265736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Laflamme MA, Murry CE. Heart regeneration. Nature. 2011;473:326–35. doi: 10.1038/nature10147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Soonpaa MH, Field LJ. Assessment of cardiomyocyte DNA synthesis in normal and injured adult mouse hearts. The American journal of physiology. 1997;272:H220–6. doi: 10.1152/ajpheart.1997.272.1.H220. [DOI] [PubMed] [Google Scholar]
- 62.Hodgkinson CPaDVJ. Conserved microRNA program as key to mammalian cardiac regeneration: insights from Zebrafish. Circulation research. 2015 doi: 10.1161/CIRCRESAHA.115.305852. In Press. [DOI] [PubMed] [Google Scholar]
- 63.Poss KD, Wilson LG, Keating MT. Heart regeneration in zebrafish. Science. 2002;298:2188–90. doi: 10.1126/science.1077857. [DOI] [PubMed] [Google Scholar]
- 64.Jopling C, Boue S, Izpisua Belmonte JC. Dedifferentiation, transdifferentiation and reprogramming: three routes to regeneration. Nature reviews Molecular cell biology. 2011;12:79–89. doi: 10.1038/nrm3043. [DOI] [PubMed] [Google Scholar]
- 65.Aguirre A, Montserrat N, Zacchigna S, Nivet E, Hishida T, Krause MN, Kurian L, Ocampo A, Vazquez-Ferrer E, Rodriguez-Esteban C, Kumar S, Moresco JJ, Yates JR, 3rd, Campistol JM, Sancho-Martinez I, Giacca M, Izpisua Belmonte JC. In vivo activation of a conserved microRNA program induces mammalian heart regeneration. Cell stem cell. 2014;15:589–604. doi: 10.1016/j.stem.2014.10.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Chen J, Huang ZP, Seok HY, Ding J, Kataoka M, Zhang Z, Hu X, Wang G, Lin Z, Wang S, Pu WT, Liao R, Wang DZ. mir-17-92 cluster is required for and sufficient to induce cardiomyocyte proliferation in postnatal and adult hearts. Circulation research. 2013;112:1557–66. doi: 10.1161/CIRCRESAHA.112.300658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Porrello ER, Johnson BA, Aurora AB, Simpson E, Nam YJ, Matkovich SJ, Dorn GW, 2nd, van Rooij E, Olson EN. MiR-15 family regulates postnatal mitotic arrest of cardiomyocytes. Circulation research. 2011;109:670–9. doi: 10.1161/CIRCRESAHA.111.248880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Porrello ER, Mahmoud AI, Simpson E, Johnson BA, Grinsfelder D, Canseco D, Mammen PP, Rothermel BA, Olson EN, Sadek HA. Regulation of neonatal and adult mammalian heart regeneration by the miR-15 family. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:187–92. doi: 10.1073/pnas.1208863110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Hullinger TG, Montgomery RL, Seto AG, Dickinson BA, Semus HM, Lynch JM, Dalby CM, Robinson K, Stack C, Latimer PA, Hare JM, Olson EN, van Rooij E. Inhibition of miR-15 protects against cardiac ischemic injury. Circulation research. 2012;110:71–81. doi: 10.1161/CIRCRESAHA.111.244442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cao X, Wang J, Wang Z, Du J, Yuan X, Huang W, Meng J, Gu H, Nie Y, Ji B, Hu S, Zheng Z. MicroRNA profiling during rat ventricular maturation: A role for miR-29a in regulating cardiomyocyte cell cycle re-entry. FEBS letters. 2013;587:1548–55. doi: 10.1016/j.febslet.2013.01.075. [DOI] [PubMed] [Google Scholar]
- 71.Eulalio A, Mano M, Dal Ferro M, Zentilin L, Sinagra G, Zacchigna S, Giacca M. Functional screening identifies miRNAs inducing cardiac regeneration. Nature. 2012;492:376–81. doi: 10.1038/nature11739. [DOI] [PubMed] [Google Scholar]
- 72.Martinez-Fernandez A, Nelson TJ, Yamada S, Reyes S, Alekseev AE, Perez-Terzic C, Ikeda Y, Terzic A. iPS programmed without c-MYC yield proficient cardiogenesis for functional heart chimerism. Circulation research. 2009;105:648–56. doi: 10.1161/CIRCRESAHA.109.203109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Mauritz C, Schwanke K, Reppel M, Neef S, Katsirntaki K, Maier LS, Nguemo F, Menke S, Haustein M, Hescheler J, Hasenfuss G, Martin U. Generation of functional murine cardiac myocytes from induced pluripotent stem cells. Circulation. 2008;118:507–17. doi: 10.1161/CIRCULATIONAHA.108.778795. [DOI] [PubMed] [Google Scholar]
- 74.Nelson TJ, Martinez-Fernandez A, Yamada S, Perez-Terzic C, Ikeda Y, Terzic A. Repair of acute myocardial infarction by human stemness factors induced pluripotent stem cells. Circulation. 2009;120:408–16. doi: 10.1161/CIRCULATIONAHA.109.865154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Priori SG, Napolitano C, Di Pasquale E, Condorelli G. Induced pluripotent stem cell-derived cardiomyocytes in studies of inherited arrhythmias. The Journal of clinical investigation. 2013;123:84–91. doi: 10.1172/JCI62838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Yoshida Y, Yamanaka S. iPS cells: a source of cardiac regeneration. J Mol Cell Cardiol. 2011;50:327–32. doi: 10.1016/j.yjmcc.2010.10.026. [DOI] [PubMed] [Google Scholar]
- 77.Takahashi K, Yamanaka S. Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 2006;126:663–76. doi: 10.1016/j.cell.2006.07.024. [DOI] [PubMed] [Google Scholar]
- 78.Hochedlinger K, Plath K. Epigenetic reprogramming and induced pluripotency. Development. 2009;136:509–23. doi: 10.1242/dev.020867. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Polo JM, Anderssen E, Walsh RM, Schwarz BA, Nefzger CM, Lim SM, Borkent M, Apostolou E, Alaei S, Cloutier J, Bar-Nur O, Cheloufi S, Stadtfeld M, Figueroa ME, Robinton D, Natesan S, Melnick A, Zhu J, Ramaswamy S, Hochedlinger K. A molecular roadmap of reprogramming somatic cells into iPS cells. Cell. 2012;151:1617–32. doi: 10.1016/j.cell.2012.11.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Miyoshi N, Ishii H, Nagano H, Haraguchi N, Dewi DL, Kano Y, Nishikawa S, Tanemura M, Mimori K, Tanaka F, Saito T, Nishimura J, Takemasa I, Mizushima T, Ikeda M, Yamamoto H, Sekimoto M, Doki Y, Mori M. Reprogramming of mouse and human cells to pluripotency using mature microRNAs. Cell stem cell. 2011;8:633–8. doi: 10.1016/j.stem.2011.05.001. [DOI] [PubMed] [Google Scholar]
- 81.Anokye-Danso F, Trivedi CM, Juhr D, Gupta M, Cui Z, Tian Y, Zhang Y, Yang W, Gruber PJ, Epstein JA, Morrisey EE. Highly efficient miRNA-mediated reprogramming of mouse and human somatic cells to pluripotency. Cell stem cell. 2011;8:376–88. doi: 10.1016/j.stem.2011.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Subramanyam D, Lamouille S, Judson RL, Liu JY, Bucay N, Derynck R, Blelloch R. Multiple targets of miR-302 and miR-372 promote reprogramming of human fibroblasts to induced pluripotent stem cells. Nature biotechnology. 2011;29:443–8. doi: 10.1038/nbt.1862. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Stead E, White J, Faast R, Conn S, Goldstone S, Rathjen J, Dhingra U, Rathjen P, Walker D, Dalton S. Pluripotent cell division cycles are driven by ectopic Cdk2, cyclin A/E and E2F activities. Oncogene. 2002;21:8320–33. doi: 10.1038/sj.onc.1206015. [DOI] [PubMed] [Google Scholar]
- 84.Hanna J, Saha K, Pando B, van Zon J, Lengner CJ, Creyghton MP, van Oudenaarden A, Jaenisch R. Direct cell reprogramming is a stochastic process amenable to acceleration. Nature. 2009;462:595–601. doi: 10.1038/nature08592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Hong H, Takahashi K, Ichisaka T, Aoi T, Kanagawa O, Nakagawa M, Okita K, Yamanaka S. Suppression of induced pluripotent stem cell generation by the p53-p21 pathway. Nature. 2009;460:1132–5. doi: 10.1038/nature08235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Wang Y, Baskerville S, Shenoy A, Babiarz JE, Baehner L, Blelloch R. Embryonic stem cell-specific microRNAs regulate the G1-S transition and promote rapid proliferation. Nature genetics. 2008;40:1478–83. doi: 10.1038/ng.250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tian Y, Zhang Y, Hurd L, Hannenhalli S, Liu F, Lu MM, Morrisey EE. Regulation of lung endoderm progenitor cell behavior by miR302/367. Development. 2011;138:1235–45. doi: 10.1242/dev.061762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Benetti R, Gonzalo S, Jaco I, Munoz P, Gonzalez S, Schoeftner S, Murchison E, Andl T, Chen T, Klatt P, Li E, Serrano M, Millar S, Hannon G, Blasco MA. A mammalian microRNA cluster controls DNA methylation and telomere recombination via Rbl2-dependent regulation of DNA methyltransferases. Nature structural & molecular biology. 2008;15:268–79. doi: 10.1038/nsmb.1399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Judson RL, Babiarz JE, Venere M, Blelloch R. Embryonic stem cell-specific microRNAs promote induced pluripotency. Nature biotechnology. 2009;27:459–61. doi: 10.1038/nbt.1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Melton C, Judson RL, Blelloch R. Opposing microRNA families regulate self-renewal in mouse embryonic stem cells. Nature. 2010;463:621–6. doi: 10.1038/nature08725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Wang J, He Q, Han C, Gu H, Jin L, Li Q, Mei Y, Wu M. p53-facilitated miR-199a-3p regulates somatic cell reprogramming. Stem cells. 2012;30:1405–13. doi: 10.1002/stem.1121. [DOI] [PubMed] [Google Scholar]
- 92.He L, He X, Lim LP, de Stanchina E, Xuan Z, Liang Y, Xue W, Zender L, Magnus J, Ridzon D, Jackson AL, Linsley PS, Chen C, Lowe SW, Cleary MA, Hannon GJ. A microRNA component of the p53 tumour suppressor network. Nature. 2007;447:1130–4. doi: 10.1038/nature05939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Choi YJ, Lin CP, Ho JJ, He X, Okada N, Bu P, Zhong Y, Kim SY, Bennett MJ, Chen C, Ozturk A, Hicks GG, Hannon GJ, He L. miR-34 miRNAs provide a barrier for somatic cell reprogramming. Nature cell biology. 2011;13:1353–60. doi: 10.1038/ncb2366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Ye D, Wang G, Liu Y, Huang W, Wu M, Zhu S, Jia W, Deng AM, Liu H, Kang J. MiR-138 promotes induced pluripotent stem cell generation through the regulation of the p53 signaling. Stem cells. 2012;30:1645–54. doi: 10.1002/stem.1149. [DOI] [PubMed] [Google Scholar]
- 95.Yang CS, Li Z, Rana TM. microRNAs modulate iPS cell generation. Rna. 2011;17:1451–60. doi: 10.1261/rna.2664111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Rosa A, Spagnoli FM, Brivanlou AH. The miR-430/427/302 family controls mesendodermal fate specification via species-specific target selection. Developmental cell. 2009;16:517–27. doi: 10.1016/j.devcel.2009.02.007. [DOI] [PubMed] [Google Scholar]
- 97.Liao B, Bao X, Liu L, Feng S, Zovoilis A, Liu W, Xue Y, Cai J, Guo X, Qin B, Zhang R, Wu J, Lai L, Teng M, Niu L, Zhang B, Esteban MA, Pei D. MicroRNA cluster 302-367 enhances somatic cell reprogramming by accelerating a mesenchymal-to-epithelial transition. The Journal of biological chemistry. 2011;286:17359–64. doi: 10.1074/jbc.C111.235960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Li Z, Yang CS, Nakashima K, Rana TM. Small RNA-mediated regulation of iPS cell generation. The EMBO journal. 2011;30:823–34. doi: 10.1038/emboj.2011.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Lipchina I, Elkabetz Y, Hafner M, Sheridan R, Mihailovic A, Tuschl T, Sander C, Studer L, Betel D. Genome-wide identification of microRNA targets in human ES cells reveals a role for miR-302 in modulating BMP response. Genes & development. 2011;25:2173–86. doi: 10.1101/gad.17221311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Pfaff N, Fiedler J, Holzmann A, Schambach A, Moritz T, Cantz T, Thum T. miRNA screening reveals a new miRNA family stimulating iPS cell generation via regulation of Meox2. EMBO reports. 2011;12:1153–9. doi: 10.1038/embor.2011.176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Valcourt U, Thuault S, Pardali K, Heldin CH, Moustakas A. Functional role of Meox2 during the epithelial cytostatic response to TGF-beta. Molecular oncology. 2007;1:55–71. doi: 10.1016/j.molonc.2007.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Koche RP, Smith ZD, Adli M, Gu H, Ku M, Gnirke A, Bernstein BE, Meissner A. Reprogramming factor expression initiates widespread targeted chromatin remodeling. Cell stem cell. 2011;8:96–105. doi: 10.1016/j.stem.2010.12.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Huangfu D, Maehr R, Guo W, Eijkelenboom A, Snitow M, Chen AE, Melton DA. Induction of pluripotent stem cells by defined factors is greatly improved by small-molecule compounds. Nature biotechnology. 2008;26:795–7. doi: 10.1038/nbt1418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Mikkelsen TS, Hanna J, Zhang X, Ku M, Wernig M, Schorderet P, Bernstein BE, Jaenisch R, Lander ES, Meissner A. Dissecting direct reprogramming through integrative genomic analysis. Nature. 2008;454:49–55. doi: 10.1038/nature07056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Card DA, Hebbar PB, Li L, Trotter KW, Komatsu Y, Mishina Y, Archer TK. Oct4/Sox2-regulated miR-302 targets cyclin D1 in human embryonic stem cells. Molecular and cellular biology. 2008;28:6426–38. doi: 10.1128/MCB.00359-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Lin SL, Chang DC, Lin CH, Ying SY, Leu D, Wu DT. Regulation of somatic cell reprogramming through inducible mir-302 expression. Nucleic acids research. 2011;39:1054–65. doi: 10.1093/nar/gkq850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Anokye-Danso F, Snitow M, Morrisey EE. How microRNAs facilitate reprogramming to pluripotency. Journal of cell science. 2012;125:4179–87. doi: 10.1242/jcs.095968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lu D, Davis MP, Abreu-Goodger C, Wang W, Campos LS, Siede J, Vigorito E, Skarnes WC, Dunham I, Enright AJ, Liu P. MiR-25 regulates Wwp2 and Fbxw7 and promotes reprogramming of mouse fibroblast cells to iPSCs. PloS one. 2012;7:e40938. doi: 10.1371/journal.pone.0040938. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Ivey KN, Muth A, Arnold J, King FW, Yeh RF, Fish JE, Hsiao EC, Schwartz RJ, Conklin BR, Bernstein HS, Srivastava D. MicroRNA regulation of cell lineages in mouse and human embryonic stem cells. Cell stem cell. 2008;2:219–29. doi: 10.1016/j.stem.2008.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wilson KD, Hu S, Venkatasubrahmanyam S, Fu JD, Sun N, Abilez OJ, Baugh JJ, Jia F, Ghosh Z, Li RA, Butte AJ, Wu JC. Dynamic microRNA expression programs during cardiac differentiation of human embryonic stem cells: role for miR-499. Circulation Cardiovascular genetics. 2010;3:426–35. doi: 10.1161/CIRCGENETICS.109.934281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Takaya T, Ono K, Kawamura T, Takanabe R, Kaichi S, Morimoto T, Wada H, Kita T, Shimatsu A, Hasegawa K. MicroRNA-1 and MicroRNA-133 in spontaneous myocardial differentiation of mouse embryonic stem cells. Circulation journal : official journal of the Japanese Circulation Society. 2009;73:1492–7. doi: 10.1253/circj.cj-08-1032. [DOI] [PubMed] [Google Scholar]
- 112.Sunagawa Y, Morimoto T, Takaya T, Kaichi S, Wada H, Kawamura T, Fujita M, Shimatsu A, Kita T, Hasegawa K. Cyclin-dependent kinase-9 is a component of the p300/GATA4 complex required for phenylephrine-induced hypertrophy in cardiomyocytes. The Journal of biological chemistry. 2010;285:9556–68. doi: 10.1074/jbc.M109.070458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Kwon C, Han Z, Olson EN, Srivastava D. MicroRNA1 influences cardiac differentiation in Drosophila and regulates Notch signaling. Proceedings of the National Academy of Sciences of the United States of America. 2005;102:18986–91. doi: 10.1073/pnas.0509535102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Lu J, McKinsey TA, Zhang CL, Olson EN. Regulation of skeletal myogenesis by association of the MEF2 transcription factor with class II histone deacetylases. Molecular cell. 2000;6:233–44. doi: 10.1016/s1097-2765(00)00025-3. [DOI] [PubMed] [Google Scholar]
- 115.Lu TY, Lin B, Li Y, Arora A, Han L, Cui C, Coronnello C, Sheng Y, Benos PV, Yang L. Overexpression of microRNA-1 promotes cardiomyocyte commitment from human cardiovascular progenitors via suppressing WNT and FGF signaling pathways. Journal of molecular and cellular cardiology. 2013;63:146–54. doi: 10.1016/j.yjmcc.2013.07.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fu JD, Rushing SN, Lieu DK, Chan CW, Kong CW, Geng L, Wilson KD, Chiamvimonvat N, Boheler KR, Wu JC, Keller G, Hajjar RJ, Li RA. Distinct roles of microRNA-1 and -499 in ventricular specification and functional maturation of human embryonic stem cell-derived cardiomyocytes. PloS one. 2011;6:e27417. doi: 10.1371/journal.pone.0027417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Glass C, Singla DK. MicroRNA-1 transfected embryonic stem cells enhance cardiac myocyte differentiation and inhibit apoptosis by modulating the PTEN/Akt pathway in the infarcted heart. American journal of physiology Heart and circulatory physiology. 2011;301:H2038–49. doi: 10.1152/ajpheart.00271.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Wagh V, Pomorski A, Wilschut KJ, Piombo S, Bernstein HS. MicroRNA-363 negatively regulates the left ventricular determining transcription factor HAND1 in human embryonic stem cell-derived cardiomyocytes. Stem cell research & therapy. 2014;5:75. doi: 10.1186/scrt464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Fish JE, Santoro MM, Morton SU, Yu S, Yeh RF, Wythe JD, Ivey KN, Bruneau BG, Stainier DY, Srivastava D. miR-126 regulates angiogenic signaling and vascular integrity. Developmental cell. 2008;15:272–84. doi: 10.1016/j.devcel.2008.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Morton SU, Scherz PJ, Cordes KR, Ivey KN, Stainier DY, Srivastava D. microRNA-138 modulates cardiac patterning during embryonic development. Proceedings of the National Academy of Sciences of the United States of America. 2008;105:17830–5. doi: 10.1073/pnas.0804673105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Townley-Tilson WH, Callis TE, Wang D. MicroRNAs 1, 133, and 206: critical factors of skeletal and cardiac muscle development, function, and disease. The international journal of biochemistry & cell biology. 2010;42:1252–5. doi: 10.1016/j.biocel.2009.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Gnecchi M, Zhang Z, Ni A, Dzau VJ. Paracrine mechanisms in adult stem cell signaling and therapy. Circulation research. 2008;103:1204–19. doi: 10.1161/CIRCRESAHA.108.176826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Hodgkinson CP, Gomez JA, Payne AJ, Zhang L, Wang X, Dal-Pra S, Pratt RE, Dzau VJ. Abi3bp regulates cardiac progenitor cell proliferation and differentiation. Circulation research. 2014;115:1007–16. doi: 10.1161/CIRCRESAHA.115.304216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Georgiadis V, Knight RA, Jayasinghe SN, Stephanou A. Cardiac tissue engineering: renewing the arsenal for the battle against heart disease. Integrative biology : quantitative biosciences from nano to macro. 2014;6:111–26. doi: 10.1039/c3ib40097b. [DOI] [PubMed] [Google Scholar]
- 125.Vierbuchen T, Wernig M. Molecular roadblocks for cellular reprogramming. Molecular cell. 2012;47:827–38. doi: 10.1016/j.molcel.2012.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nam YJ, Song K, Luo X, Daniel E, Lambeth K, West K, Hill JA, DiMaio JM, Baker LA, Bassel-Duby R, Olson EN. Reprogramming of human fibroblasts toward a cardiac fate. Proceedings of the National Academy of Sciences of the United States of America. 2013;110:5588–93. doi: 10.1073/pnas.1301019110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Muraoka N, Yamakawa H, Miyamoto K, Sadahiro T, Umei T, Isomi M, Nakashima H, Akiyama M, Wada R, Inagawa K, Nishiyama T, Kaneda R, Fukuda T, Takeda S, Tohyama S, Hashimoto H, Kawamura Y, Goshima N, Aeba R, Yamagishi H, Fukuda K, Ieda M. MiR-133 promotes cardiac reprogramming by directly repressing Snai1 and silencing fibroblast signatures. The EMBO journal. 2014;33:1565–81. doi: 10.15252/embj.201387605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Leri A, Kajstura J, Anversa P. Role of cardiac stem cells in cardiac pathophysiology: a paradigm shift in human myocardial biology. Circulation research. 2011;109:941–61. doi: 10.1161/CIRCRESAHA.111.243154. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 129.Bolli R, Chugh AR, D’Amario D, Loughran JH, Stoddard MF, Ikram S, Beache GM, Wagner SG, Leri A, Hosoda T, Sanada F, Elmore JB, Goichberg P, Cappetta D, Solankhi NK, Fahsah I, Rokosh DG, Slaughter MS, Kajstura J, Anversa P. Cardiac stem cells in patients with ischaemic cardiomyopathy (SCIPIO): initial results of a randomised phase 1 trial. Lancet. 2011;378:1847–57. doi: 10.1016/S0140-6736(11)61590-0. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 130.Makkar RR, Smith RR, Cheng K, Malliaras K, Thomson LE, Berman D, Czer LS, Marban L, Mendizabal A, Johnston PV, Russell SD, Schuleri KH, Lardo AC, Gerstenblith G, Marban E. Intracoronary cardiosphere-derived cells for heart regeneration after myocardial infarction (CADUCEUS): a prospective, randomised phase 1 trial. Lancet. 2012;379:895–904. doi: 10.1016/S0140-6736(12)60195-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Sluijter JP, van Mil A, van Vliet P, Metz CH, Liu J, Doevendans PA, Goumans MJ. MicroRNA-1 and -499 regulate differentiation and proliferation in human-derived cardiomyocyte progenitor cells. Arteriosclerosis, thrombosis, and vascular biology. 2010;30:859–68. doi: 10.1161/ATVBAHA.109.197434. [DOI] [PubMed] [Google Scholar]
- 132.Hosoda T, Zheng H, Cabral-da-Silva M, Sanada F, Ide-Iwata N, Ogorek B, Ferreira-Martins J, Arranto C, D’Amario D, del Monte F, Urbanek K, D’Alessandro DA, Michler RE, Anversa P, Rota M, Kajstura J, Leri A. Human cardiac stem cell differentiation is regulated by a mircrine mechanism. Circulation. 2011;123:1287–96. doi: 10.1161/CIRCULATIONAHA.110.982918. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 133.King IN, Qian L, Liang J, Huang Y, Shieh JT, Kwon C, Srivastava D. A genome-wide screen reveals a role for microRNA-1 in modulating cardiac cell polarity. Developmental cell. 2011;20:497–510. doi: 10.1016/j.devcel.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Qian L, Liu J, Bodmer R. Slit and Robo control cardiac cell polarity and morphogenesis. Current biology : CB. 2005;15:2271–8. doi: 10.1016/j.cub.2005.10.037. [DOI] [PubMed] [Google Scholar]
- 135.Tousi SE, Soltani BM, Sadeghizadeh M, Mowla SJ, Parsi S, Soleimani M. Inhibitory effect of hsa-miR-590-5p on cardiosphere-derived stem cells differentiation through downregulation of TGFB signaling. Journal of cellular biochemistry. 2014 doi: 10.1002/jcb.24957. [DOI] [PubMed] [Google Scholar]
- 136.Zhao J, Feng Y, Yan H, Chen Y, Wang J, Chua B, Stuart C, Yin D. beta-arrestin2/miR-155/GSK3beta regulates transition of 5′-azacytizine-induced Sca-1-positive cells to cardiomyocytes. Journal of cellular and molecular medicine. 2014;18:1562–70. doi: 10.1111/jcmm.12339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Sirish P, Lopez JE, Li N, Wong A, Timofeyev V, Young JN, Majdi M, Li RA, Chen HS, Chiamvimonvat N. MicroRNA profiling predicts a variance in the proliferative potential of cardiac progenitor cells derived from neonatal and adult murine hearts. Journal of molecular and cellular cardiology. 2012;52:264–72. doi: 10.1016/j.yjmcc.2011.10.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liang D, Zhen L, Yuan T, Huang J, Deng F, Wuyahan, Zhang H, Pan L, Liu Y, The E, Yu Z, Zhu W, Zhang Y, Li L, Peng L, Li J, Chen YH. miR-10a regulates proliferation of human cardiomyocyte progenitor cells by targeting GATA6. PloS one. 2014;9:e103097. doi: 10.1371/journal.pone.0103097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kotterman MA, Schaffer DV. Engineering adeno-associated viruses for clinical gene therapy. Nature reviews Genetics. 2014;15:445–51. doi: 10.1038/nrg3742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tilemann L, Ishikawa K, Weber T, Hajjar RJ. Gene therapy for heart failure. Circulation research. 2012;110:777–93. doi: 10.1161/CIRCRESAHA.111.252981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Mizuguchi H, Xu Z, Ishii-Watabe A, Uchida E, Hayakawa T. IRES-dependent second gene expression is significantly lower than cap-dependent first gene expression in a bicistronic vector. Molecular therapy : the journal of the American Society of Gene Therapy. 2000;1:376–82. doi: 10.1006/mthe.2000.0050. [DOI] [PubMed] [Google Scholar]
- 142.Yang X, Marcucci K, Anguela X, Couto LB. Preclinical evaluation of an anti-HCV miRNA cluster for treatment of HCV infection. Molecular therapy : the journal of the American Society of Gene Therapy. 2013;21:588–601. doi: 10.1038/mt.2012.247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.van Rooij E, Olson EN. MicroRNA therapeutics for cardiovascular disease: opportunities and obstacles. Nature reviews Drug discovery. 2012;11:860–72. doi: 10.1038/nrd3864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Thum T, Gross C, Fiedler J, Fischer T, Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, Castoldi M, Soutschek J, Koteliansky V, Rosenwald A, Basson MA, Licht JD, Pena JT, Rouhanifard SH, Muckenthaler MU, Tuschl T, Martin GR, Bauersachs J, Engelhardt S. MicroRNA-21 contributes to myocardial disease by stimulating MAP kinase signalling in fibroblasts. Nature. 2008;456:980–4. doi: 10.1038/nature07511. [DOI] [PubMed] [Google Scholar]
- 145.Hornstein E, Shomron N. Canalization of development by microRNAs. Nature genetics. 2006;38 (Suppl):S20–4. doi: 10.1038/ng1803. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.