

Abstract
DNA replication errors are a major source of genome instability in all organisms. In the fission yeast Schizosaccharomyces pombe, the DNA damage response protein Brc1 binds phospho-histone H2A (γH2A)-marked chromatin during S-phase, but how Brc1 protects genome integrity remains unclear. Here we report that the non-homologous end-joining (NHEJ) protein Ku becomes critical for survival of replication stress in brc1∆ cells. Ku’s protective activity in brc1∆ cells does not involve its canonical NHEJ function or its roles in protecting telomeres or shielding DNA ends from Exo1 exonuclease. In brc1∆ pku80∆ cells, nuclear foci of Rad52 homologous recombination (HR) protein increase and Mus81-Eme1 Holliday junction resolvase becomes critical, indicating increased replication fork instability. Ku’s localization at a ribosomal DNA replication fork barrier associated with frequent replisome-transcriptosome collisions increases in brc1∆ cells and increased collisions correlate with an enhanced requirement for Brc1. These data indicate that Ku stabilizes replication forks in the absence of Brc1.
Introduction
Genome integrity is especially vulnerable during the DNA synthesis (S) phase of the cell cycle, when replisomes encounter DNA lesions, DNA-bound proteins and opposing transcriptosomes. Limiting supplies of deoxyribonucleotides and oncogene activation also cause replicative stress. Genome maintenance proteins insure accurate genome duplication during replicative stress, with the paramount factor being the master checkpoint protein kinase known as ATR in humans, Mec1 in the budding yeast Saccharomyces cerevisiae and Rad3 in the fission yeast Schizosaccharomyces pombe [1–4]. The carboxyl terminus of histone H2A in yeasts and H2AX in mammals is a key substrate of these checkpoint kinases [5]. Phospho-H2A/X (γH2A/X) is best known for its functions at double-strand breaks (DSBs) but it also marks a diverse array of genomic features during S-phase in fission yeast, including natural replication fork barriers, retrotransposons, heterochromatin in the centromeres and telomeres, and ribosomal RNA (rDNA) repeats [6]. Indeed, γH2A is required for full resistance to genotoxins that cause replicative stress [7]. A key role of γH2A in S-phase was revealed by the discovery that Brc1 genome protection protein forms nuclear foci by binding γH2A during replicative stress [8, 9]. X-ray crystallography of a Brc1-γH2A peptide complex showed that the C-terminal region of Brc1 consisting of tandem BRCT domains folds to form a highly sculpted docking pocket for the phospho-SQE motif at the carboxyl-tail of γH2A. Missense mutations in this docking pocket ablate Brc1 foci formation and confer sensitivity to replicative stress [8].
Despite these insights it remains unclear how Brc1 actually protects genome integrity during replicative stress. Brc1 was first identified as high copy suppressor of the hypomorphic smc6-74 mutation, which compromises the activity of the Smc5-Smc6 holocomplex that is essential for structural maintenance of chromosomes and has important but poorly understood roles in DNA repair [10–12]. Brc1 is not required for cellular viability but it is essential in strains with defective functions of the Smc5-Smc6 complex [11, 13, 14]. Brc1-null strains are sensitive to DNA damaging agents and drugs that cause replication fork arrest or collapse [9], and the appearance of Rad52 foci in untreated brc1∆ cells suggest DNA replication difficulties even in the absence of exogenous genotoxins [8, 15]. Brc1-coated chromatin might promote the bypass of DNA lesions by post-replication repair (PRR) or it might stabilize stalled replisomes [6, 15, 16]. Unstable replication forks are prone to collapse and breakage, necessitating engagement of the homologous recombination (HR) repair machinery to reestablish the replication fork [17, 18]. Indeed, the repair of a site-specific broken replication fork in fission yeast absolutely depends on key HR proteins such as Rad51 and Rad52 recombinases and Mus81-Eme1 Holliday junction resolvase, but not on other DSB repair factors such as Ku that are required for non-homologous end joining (NHEJ) [19]. The absence of an acute requirement for Rad51 or Mus81 in brc1∆ cells suggests that replication fork collapse does not increase in brc1∆ mutants [9], even though these cells have more Rad52 foci [8, 15].
Here, we report on our latest efforts to determine how Brc1 protects genome integrity during S-phase. One of our key findings is that elimination of Ku in brc1∆ cells reveals a critical requirement for Mus81, which indicates that Ku stabilizes replication forks in the absence of Brc1. Our studies shed new light on a non-canonical function of Ku and the role of Brc1 in protecting cells from replicative stress.
Materials and Methods
Strains and genetic methods
The strains used in this study are listed in S1 Table. Standard fission yeast methods were used as described previously [20]. Deletion mutations strains were constructed as described previously [21]. Successful deletion of these genes was verified by PCR. Tetrad analysis was performed to construct double mutants and verified by PCR.
The E-MAP screen methods and workflow were performed and normalized as previously described [22]. Genetic interaction score were determined with a simple growth phenotype that measures negative interactions, such as synthetic sick/lethal (SSL) interactions (E-MAP score < -2.5), as well as positive interactions (E-MAP score > 2) in which the double mutant is healthier than would be expected based on the growth of the two single mutants.
For synchronization of cells using cdc25-22 block and release, cells containing the temperature sensitive cdc25-22 allele were incubated at restrictive temperature (36°C) for 4 hours to arrest the cell cycle in G2-phase. Upon release to permissive temperature (25°C) the cells synchronously enter the cell cycle. Cell cycle progression was monitored microscopically by counting cells that contained septa, the appearance of which correlates with S-phase.
Survival assay
DNA damage sensitivity assays were performed by spotting 10-fold serial dilutions of exponentially growing cells onto yeast extract with glucose and supplements (YES) plates, and treated with indicated amounts of hydroxyurea (HU), camptothecin (CPT), and methyl methanesulfonate (MMS). For UV treatment, cells were serially diluted onto YES plates and irradiated using a Stratagene Stratalinker UV source. Cell survival was determined after 3–4 days at 30°C.
Telomere analysis
Genomic DNA isolated from each strain was digested overnight with EcoRI and resolved in 2% TAE agarose gel. DNA was transferred via capillary method to an Amersham Hybond-XL membrane (GE Healthcare Life Sciences) and probed with 32P-labeled TAS1 [23].
Microscopy
Cells were photographed using a Nikon Eclipse E800 microscope equipped with a Photometrics Quantix charge-coupled device (CCD) camera and IPlab Spectrum software. All fusion proteins were expressed at their own genomic locus. Rad52-yellow fluorescence protein (YFP) and RPA(Rad11)-green fluorescence protein (GFP) expressing strains were grown in EMM media until mid-log phase for focus quantification assays. 500 or more nuclei were scored in three independent experiments.
Chromatin immunoprecipitation (ChIP) assay
Real time qPCR ChIP experiments were performed as described previously [6, 24] using anti-HA antibody (Roche Applied Science) conjugated to anti-mouse magnetic beads (Invitrogen) to precipitate Pku70-HA expressed at wild type levels from the endogenous locus under the control of the native pku70 + promoter. qPCR primers are from [6]. Percent of immunoprecipitation DNA (%IP) in the ChIP samples was calculated relative to the amount of DNA in the input samples. ChIP enrichment was calculated relative to act1. All error bars represent the standard error between technical triplicates.
Results
Requirement for Ku in brc1∆ mutant
To gain new functional insights into Brc1 we generated an epistatic miniarray profile (E-MAP) consisting of the quantitative analysis of genetic interactions between brc1∆ and a S. pombe gene deletion library of ~2,200 nonessential genes [25–27]. A full description of this E-MAP is in preparation; here, we focus on the synthetic sick/lethal (SSL) genetic interaction with Ku. The brc1∆ pku80∆ double mutant generated an E-MAP score of -5.4, which ranks 19th amongst the 2,027 E-MAP scores obtained in this screen in which an E-MAP score below -2.5 was judged to be significant. For comparison the previously analyzed E-MAP interaction with the deoxycytidylate deaminase dcd1∆ mutation, which causes a deoxyribonucleotide imbalance that creates replicative stress, were -7.8 and -7.6 for the two dcd1∆ mutants tested in the library [25].
Comparisons made with a panel of genotoxins that cause replicative stress showed that a brc1∆ pku80∆ double mutant was much more sensitive than either single mutant (Fig 1). This effect was observed with hydroxyurea (HU), which slows replication by inhibiting ribonucleotide reductase, and with agents such as UV light, camptothecin (CPT) and methyl methanesulfonate (MMS), which create several types of DNA lesions that cause replication forks to stall or collapse. Note that these serial dilution assays indicated only a weak SSL interaction in the brc1∆ pku80∆ double mutant grown in the absence of genotoxins, which attests to the sensitivity of the E-MAP assay (Fig 1).
Fig 1. DNA damaging agents enhance the requirement for Ku in brc1∆ cells.
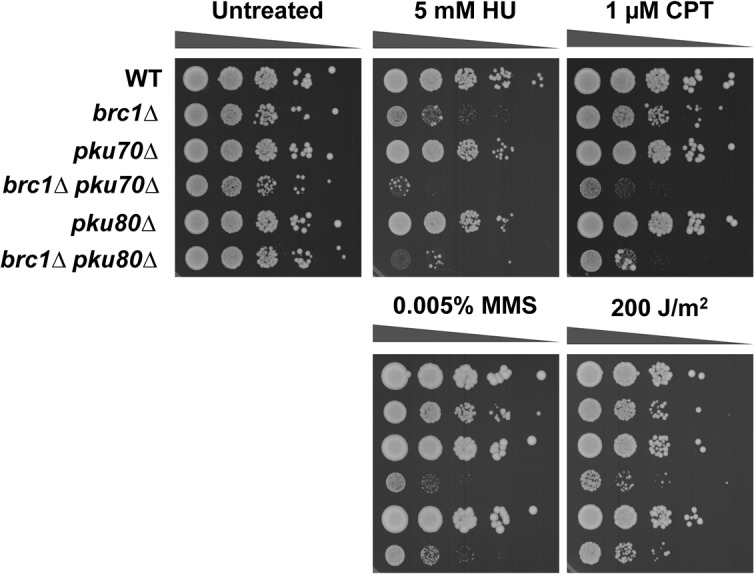
Tenfold serial dilutions of cells were exposed to the indicated DNA damaging agents. Plates were incubated at 30°C for 3 or 4 days.
The pku70∆ mutation was absent from the version of the haploid deletion library used in our screen. We therefore generated brc1∆ pku70∆ cells to specifically test whether the Ku heterodimer is important in the absence of Brc1. As predicted, these cells displayed an SSL interaction that was accentuated in the presence of the aforementioned genotoxins (Fig 1).
Non-canonical requirement for Ku in the absence of Brc1
Ku consists of the Ku70-Ku80 heterodimer that forms a ring that encircles duplex DNA by sliding onto the DNA end. Ku binds DSBs with very high affinity, whereupon it links DNA ends to initiate NHEJ repair [28]. NHEJ is not generally known to be involved in the repair of replication-associated DNA damage, thus its genetic interaction with Brc1 was unexpected. As Ku is absolutely essential for NHEJ, whereas Brc1 is one of many pathways that prevent replication-associated DNA damage or participate in its repair, we suspected that the Brc1-Ku SSL interaction indicated a role for Ku in recovery from replicative stress. Indeed, an SSL interaction between Ku and Rqh1 DNA helicase, which is involved in multiple DNA repair pathways and DNA replication but not NHEJ, suggested that Ku has an unappreciated role in genome protection during S-phase [29].
To formally examine whether the NHEJ defect of pku80∆ mutants is responsible for the SSL interaction with brc1∆, we tested whether DNA ligase IV, which is essential for the final ligation step of NHEJ, was required in brc1∆ cells. No SSL interaction was evident in brc1∆ lig4∆ cells (Fig 2B). Furthermore, we found that lig4∆ did not enhance genotoxin sensitivity in brc1∆ cells, unlike the effect of pku80∆ in brc1∆ cells. These findings establish that the requirement for Ku in brc1∆ cells does not involve NHEJ.
Fig 2. Mus81 Holliday junction resolvase is critical in brc1∆ pku80∆ cells.

Genetic interactions amongst Brc1, Mus81 and Pku80 (A) or Lig4 (B). Tenfold serial dilutions of cells were exposed to the indicated DNA damaging agents. Plates were incubated at 30°C for 3 to 4 days.
Ku participates in telomere maintenance in fission yeast [30]. Rearrangements of telomere-associated sequences (TAS) were reported to increase during meiosis and/or germination in pku80∆ cells [31]. To assess whether the SSL interaction between brc1∆ and pku80∆ might involve catastrophic loss of TAS sequences, we performed a cross of brc1∆ and pku80∆ and analyzed TAS by Southern blotting. Our data indicated that the brc1∆ pku80∆ double mutant retained TAS sequences in a pattern that was quite similar to the brc1∆ parent (Fig 3). Thus, the SSL interaction between brc1∆ and pku80∆ does not appear to involve defects in telomere maintenance.
Fig 3. A brc1∆ pku80∆ strain maintains telomeres.
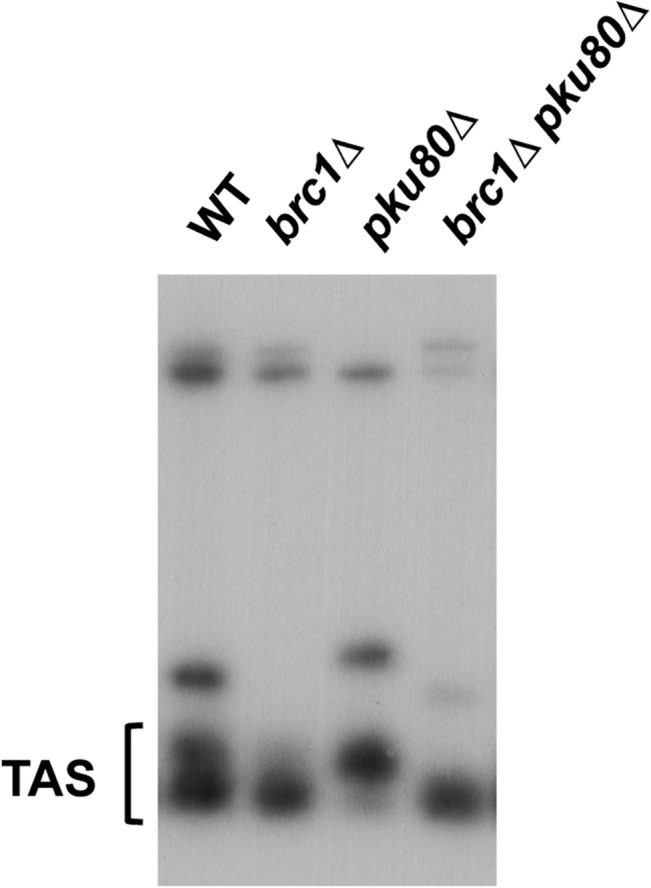
Southern blot analysis of EcoRI-digested genomic DNA from the indicated strains probed with telomere associated-1 (TAS1) DNA.
Critical requirement for Mus81-Eme1 in brc1∆ pku80∆ cells
The SSL phenotypes of brc1∆ pku80∆ cells suggested that they might suffer increased replication fork collapse or rearrangement in response to replication stress. Repair of these events typically requires resolution of Holliday junctions by Mus81-Eme1 endonuclease, which is the sole nuclear Holliday Junction resolvase in fission yeast. This requirement explains why mus81∆ mutants are acutely sensitive to genotoxins that cause replication fork collapse but are insensitive to ionizing radiation and other types of clastogens that create DSBs independently of DNA replication [32–34]. To test whether the SSL interaction involving Brc1 and Ku involves replication fork instability, we analyzed their genetic interactions with Mus81. In untreated conditions, brc1∆ mus81∆ and pku80∆ mus81∆ cells displayed nearly the same growth defect as mus81∆ cells, confirming that loss of neither Brc1 nor Ku leads to a large increase in replication fork collapse. In contrast, the triple mutant brc1∆ pku80∆ mus81∆ displayed a severe growth defect (Fig 2A, untreated). These data strongly suggested that SSL interaction involving brc1∆ and pku80∆ results in replication fork instability leading to the formation of HJs or related DNA structures that must be resolved by Mus81-Eme1. This interaction was apparent in the absence of genotoxins, indicating that endogenous conditions that impede replication are more likely to result in replication fork collapse or rearrangement in brc1∆ pku80∆ cells.
Interestingly, both brc1∆ mus81∆ and pku80∆ mus81∆ strains displayed enhanced sensitivity to HU and UV as compared to the relevant single mutants (Fig 2A). The genetic interaction between pku80∆ and mus81∆ is particularly interesting because it indicates that pku80∆ mutants suffer increased replication fork instability in the presence of these genotoxins. Note that potential synergistic effects in CPT and MMS could not be assessed in this experiment because of the acute sensitivity of the mus81∆ mutant to these genotoxins (Fig 2A).
In contrast to pku80∆, the lig4∆ mutation did not impair growth in the brc1∆ mus81∆ background, which further supports the conclusion that the requirement for Ku in brc1∆ does not involve its function in NHEJ (Fig 2B). The absence of genetic interactions involving lig4∆ was also apparent in cells treated with HU, although for unknown reasons lig4∆ did enhance UV sensitivity in the brc1∆ mus81∆ background (Fig 2B).
Increased Rad52 foci in brc1∆ pku80∆ cells
Rad52 (aka Rad22 in fission yeast) is essential for HR repair and many mutants with genome maintenance defects have increased numbers of Rad52 nuclear foci [35–37]. As an independent test of genome instability in brc1∆ pku80∆ cells, we monitored Rad52-YFP foci in this strain and its parents. The incidence of Rad52 foci was modestly increased in brc1∆ cells (9%) and pku80∆ (6.5%) cells compared to wild type (4%). There was a synergistic increase of Rad52 foci in brc1∆ pku80∆ cells, with 24% of these cells having at least one Rad52 focus (Fig 4A). This result supported the idea that the SSL interaction between brc1∆ and pku80∆ is caused by increased replication fork instability.
Fig 4. Increased Rad52 foci in brc1∆ pku80∆ and rqh1∆ pku80∆ cells.

Cells expressing Rad52-YFP (panel A) or Rad11(RPA)-GFP (panel B) were cultured in minimal medium at 25°C until mid-log phase. Foci were scored in three independent experiments. Error bars correspond to standard deviations of the means. Asterisk depicts statistically significant differences with wild type, + symbol with brc1∆, and ‡ with rqh1∆, as determined by two-tailed Student T-test, p-value ≤ 0.05.
We also used this assay to assess the SSL interaction between rqh1∆ and pku80∆ [29]. We found that the rqh1∆ mutation caused a ~3-fold increase in cells with Rad52 foci (15%), indicating that replication fork instability increases in rqh1∆ cells (Fig 4A). There was a further increase in Rad52 foci in rqh1∆ pku80∆ cells to 25%. A comparable value was obtained in brc1∆ rqh1∆ cells, which comports with the observation that these cells and rqh1∆ pku80∆ cells reveal strong SSL interactions.
The same assay was also performed with RPA, which is the major single-stranded DNA binding factor in fission yeast (Fig 4B). All of the single mutants had an increased percentage of cells with RPA foci (16–18%) as compared to wild type (7.5%), but there was little or no increase in the double mutant combinations (14–23%). All of strains increased RPA foci when treated with CPT. Taken together these data strongly suggest that Ku has an important role in protecting genome integrity in the absence of Brc1 or Rqh1.
Exo1 contributes to cell survival in brc1∆ pku80∆ cells
Ku and the Mre11-Rad50-Nbs1 (MRN) protein complex rapidly associate with DSBs in vivo [38, 39]. In the presence of the MRN cofactor Ctp1, which is orthologous to CtIP in mammals and Sae2 in budding yeast, the MRN endonuclease complex initiates 5’-to-3’ resection of the DSB and this process dislodges Ku [24, 40]. Exo1 exonuclease subsequently binds the 3’ single-strand tail and catalyzes further resection. In the absence of Ctp1 or even the entire MRN complex, Ku blocks the resection activity of Exo1 exonuclease. This DNA end blocking activity of Ku is independent of Ligase IV and other NHEJ factors [39]. We wondered whether this anti-Exo1 activity might explain the requirement for Ku in brc1∆ cells. In support of this possibility, studies in S. cerevisiae showed that elimination of Exo1 stabilizes replication forks and reduces genotoxin sensitivity in cells lacking the checkpoint kinase Rad53, which is orthologous to human Chk2 and fission yeast Cds1. Other studies have also suggested that Exo1 might process reversed replication forks, which depending on the circumstances might enhance or impair cell survival [41, 42].
To explore whether Ku has an important Exo1-blocking activity in brc1∆ cells we analyzed the genetic interactions of brc1, pku80 and exo1. Deletion of Exo1 did not impair growth although it did appear to modestly increase sensitivity to HU and MMS, suggesting it plays a positive role in survival of replication stress (Fig 5). Deletion of Exo1 did not appear to impair or enhance growth in untreated brc1∆ or pku80∆ cells, although it did modestly increase MMS sensitivity in brc1∆ cells. Most strikingly, elimination of Exo1 in brc1∆ pku80∆ cells clearly impaired growth in the absence of genotoxins (Fig 5, untreated). These data argue against the anti-Exo1 activity of Ku playing a positive role in brc1∆ cells; indeed, Exo1 contributes to cell survival in the brc1∆ pku80∆ background.
Fig 5. Elimination of Exo1 enhances genotoxin sensitivity in brc1∆ pku80∆ cells.
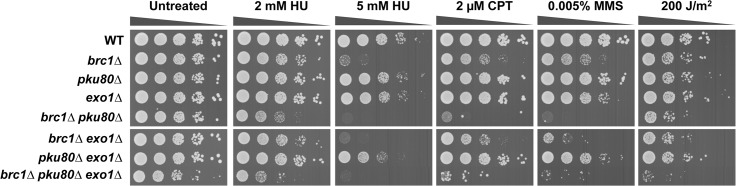
Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days.
Ku localizes near the RFP4 replication fork barrier in the rDNA
The synergistic growth defect of brc1∆ pku80∆ cells, their hypersensitivity to genotoxins that disturb DNA replication, and the critical requirement for Mus81 in these cells, all strongly suggested that Ku plays an important role in stabilizing replication forks in brc1∆ cells. We therefore investigated whether Ku co-localizes with stalled forks. For these studies we focused on the replication fork barriers (RFBs) in the ribosomal DNA (rDNA) loci because we had previously found that the majority of spontaneous Brc1 foci co-localize with the nucleolus, which contains the rDNA occurring as tandem repeats in the subtelomeric arms of chromosome 3 [8, 43, 44]. A diagram of a single rDNA repeat is shown in Fig 6A. Each rDNA repeat consists of the 35S rDNA genes, a replication origin (ars3001), and four distinct replication fork barriers (RFB1-3 and RFP4). The rDNA genes are particularly vulnerable to recombination triggered by replication fork stalling or collapse because the repetitive sequences provide good substrates for homology-directed repair [45].
Fig 6. Enrichment of Pku70 at the rDNA RFP4 replication fork barrier in brc1∆ and ctp1∆ mutants.

(A) Diagram of a single rDNA repeat (not to scale) shows the location of the four replication fork barriers (red vertical bars) relative to the 35S rDNA genes, the direction of replication (upper black arrow) from the ars3001 replication origin, the direction of 35S rDNA transcription (lower black arrow) and qPCR primer locations, below graph. (B) Cells were synchronized in G2-phase using the cdc25-22 allele and S-phase progression was monitored using septation index. (C) Pku70 ChIP at the rDNA was performed in untagged, wild type, brc1∆, and ctp1∆ strains synchronized by cdc25-22 block and analyzed by qPCR with the indicated primers. The G2-phase and S-phase samples correspond to 0 and 60 minutes respectively in this experiment.
We carried out chromatin immunoprecipitation (ChIP) to measure Pku70 enrichment throughout the rDNA locus using the indicated primers (Fig 6A). We performed these ChIP studies on cells enriched in S-phase by using the cdc25-22 arrest and release protocol [6]. We did not detect enrichment of Pku70 at any of the rDNA sites in the wild type brc1 + strain. However, ChIP revealed ~2-fold enrichment of Pku70 in brc1∆ cells specifically using the RFP4 and rDNA primer sets (Fig 6C). These primers flank the RFP4 barrier. Fork pausing at RFP4 is caused by collisions between the transcription and replication machineries, whereas replication pausing at RFB1-3 is programmed and depends on the Swi1-Swi3 complex [44]. Unexpectedly, Pku70 enrichment near RFP4 was approximately equal in the G2 and S phase samples, suggesting delayed proteolytic removal of Ku topologically trapped on double-stranded DNA either by HR repair or formation of extrachromosomal ribosomal DNA circles [46].
As mentioned above, we had previously found that the MRN protein complex and Ctp1 are required to displace Ku from DSBs through endonuclease processing of the DNA end [24]. To investigate whether this process applies in the RFP4 region, we used ChIP to monitor Pku70 localization in a ctp1∆ strain. Interestingly, in both the G2 and S phase samples we again found Pku70 enrichment using primers that flanked RFP4 (Fig 6C). The overall pattern was similar to that observed in brc1∆ cells except there was greater enrichment in ctp1∆ cells. These findings suggest that Ku localizes to DNA ends formed regressed or broken replication forks at RFP4, and the MRN protein complex and Ctp1 displace Ku from these DNA ends by the same mechanism that occurs at DSBs formed by clastogens or DNA endonucleases.
Increased requirement for Brc1 in swi1∆ and swi3∆ cells
The enrichment of Ku specifically near the RFP4 pause site in brc1∆ cells suggests that Brc1 may be important for stabilizing forks in regions of replisome-transcriptosome collisions. Previous studies revealed that elimination of RFB1-3 fork pause sites in swi1∆ and swi3∆ mutants dramatically increases fork pausing at RFP4, likely resulting from increased replisome-transcriptosome collisions [44]. This effect correlates with increased Rad52 foci and a critical requirement for Mus81 and other HR enzymes in swi1∆ and swi3∆ mutants [35, 47]. If Brc1 is important for stabilizing replication forks at sites of replisome-transcriptosome collisions, as suggested by our results and recent studies [16, 48], we would expect a significant SSL interaction between brc1∆ and swi1∆ or swi3∆. Indeed, we detected an interaction with swi3∆ in our brc1∆ E-MAP screen, with the brc1∆ swi3∆ mutant ranking 26th with an E-MAP score of -4.6. This interaction was confirmed by construction of a brc1∆ swi3∆ strain by tetrad analysis (Fig 7). This negative genetic interaction was enhanced in the presence of genotoxins. Construction of a brc1∆ swi1∆ yielded an obvious SSL interaction only in the presence of genotoxins (Fig 7). The stronger SSL interaction with swi3∆ is consistent with earlier studies showing that swi3∆ mutants are more sensitive to replicative stress [35, 47].
Fig 7. Requirement for Brc1 is enhanced in swi1∆ and swi3∆ mutants.
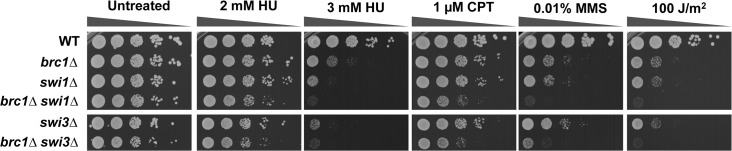
Tenfold serial dilutions of cells were exposed to the indicated DNA-damaging agents, and plates were incubated at 30°C for 3 to 4 days.
Discussion
Here, we have described how Ku contributes to cell survival in the absence of Brc1. This genetic interaction suggested that brc1∆ cells suffer increased DSBs that require NHEJ for repair, but we found that Ligase IV was not required in brc1∆ cells. As Ligase IV is essential for NHEJ, we concluded that the requirement for Ku in brc1∆ cells does not involve NHEJ. Instead, we propose that Ku protects DNA ends that arise at stalled replication forks in brc1∆ cells.
Our data indicates that replicative stress from endogenous sources is toxic in brc1∆ pku80∆ cells. Rad3 (ATR) creates γH2A at key genomic features during S-phase, including natural replication fork barriers, retrotransposons, heterochromatin domains and rDNA repeats [6]. The rDNA repeats have multiple fork barriers and a region of frequent collisions between DNA and RNA polymerases; indeed, we found that most spontaneous Brc1 nuclear foci localize in the nucleolus with the rDNA repeats [8]. Brc1 is also enriched in pericentromeric heterochromatin, which is another chromosomal domain of frequent polymerase collisions [16, 49].
The brc1∆ pku80∆ SSL interaction is enhanced when cells are treated with genotoxins that cause replicative stress. These genetic interactions are impressive because pku80∆ cells are largely insensitive to replication stress. Indeed, NHEJ should be useless as a repair mechanism for collapsed replication forks. This supposition is consistent with the absence SSL interactions between brc1∆ and lig4∆ mutations. Indeed, NHEJ defective strains are not generally sensitive to clastogens because G1 phase is very brief except during nutrient limitation [20, 50]. These considerations make the SSL interaction between brc1∆ and pku80∆ all the more striking.
The significantly higher percentage of cells with Rad52 foci in brc1∆ pku80∆ cells suggests that DNA replication-associated lesions are increased or inefficiently repaired. The relatively modest increase of Rad52 foci in brc1∆ cells might involve activation of dormant origins, which could explain the epistatic genetic interactions involving Brc1 and Mus81 [9, 15]. As Rad52 is essential for all HR repair, the increased Rad52 foci in brc1∆ pku80∆ cells might indicate increased replication fork breakage. However, Rad52 might associate with ssDNA at stalled, regressed, rearranged, terminated or collapsed replication forks. These structures might be irreparable or repaired without HR. In either case, Ku’s DNA end-binding specificity suggests that broken or regressed forks are connected to the large increase in Rad52 foci in brc1∆ pku80∆ cells.
Mus81-Eme1 is the only Holliday junction resolvase in fission yeast and is therefore essential for repairing broken replication forks [19]. The poor growth of mus81∆ mutants is not exacerbated by brc1∆ or pku80∆ mutations, indicating that neither mutation increases fork collapse or breakage. However, the very poor growth of mus81∆ brc1∆ pku80∆ cells indicates that Ku prevents replication fork collapse, rearrangement or breakage in the absence of Brc1. This result indicates that many of the Rad52 foci in brc1∆ pku80∆ cells indicate HR repair of replication forks.
Mre11 complex and Sae2/Ctp1 are required to remove Ku from DNA ends [38, 39]. Elimination of Ku improves HR repair in mutants lacking Mre11 endonuclease activity or Sae2/Ctp1, but this suppression requires Exo1, suggesting that Ku blocks Exo1-mediated resection of DSBs. However, the Exo1-blocking activity of Ku does not explain the brc1∆ pku80∆ SSL interaction because we found that that exo1∆ exacerbates the brc1∆ pku80∆ growth defect. The brc1∆ exo1∆ strain did not have an obvious growth defect, suggesting that elimination of Exo1 does not increase replicative stress. Instead, we suspect that the DNA end resection activity of Exo1 facilitates repair of broken forks in brc1∆ pku80∆ cells.
Taken together, these data suggest that Ku prevents replication fork collapse or rearrangement in the absence of Brc1. The increased association of Ku at RFB4 but not RFB1 in brc1∆ cells suggests that fork pausing caused by transcriptosome-replisome collisions creates a requirement for Ku in brc1∆ cells. This model is consistent with SSL interactions of brc1∆ with swi1∆ and swi3∆ mutations, which increase transcriptosome-replisome collisions at RFB4.
Our findings contribute to a growing body of evidence that Ku has an under appreciated role in responding to replication stress. Of special note are the studies of Ishikawa and colleagues, who described a critical non-NHEJ requirement for Ku in rqh1∆ cells exposed to replication stress [29]. Extending their findings, we found that eliminating Ku increases Rad52 foci in rqh1∆ cells in the absence of replication stress genotoxins. This non-canonical role of Ku in S-phase might be conserved because Ku improves replication stress survival in budding yeast mutants lacking Sgs1, which is the ortholog of Rqh1 [51, 52]. Furthermore, studies with mammalian cells have implicated NHEJ factors such as DNA-PKcs and Artemis in recovery from replication stress [53].
In Fig 8 we propose a model that we believe most economically reconciles our findings with the known biochemical properties of the involved proteins, particularly Ku’s high affinity for double-stranded DNA ends. In this model Brc1 stabilizes replication forks to prevent their regression. Increased fork regression in the absence of Brc1 generates a chicken foot structure containing a DNA end that is bound by Ku. By binding nascent chicken feet, Ku stabilizes the replication fork and reduces the probability that it will break or undergo homologous recombination to form Holliday junctions that require resolution by Mus81-Eme1. Our future experiments will be aimed at testing this model.
Fig 8. Model proposed to explain the requirement for Ku in brc1∆ cells.
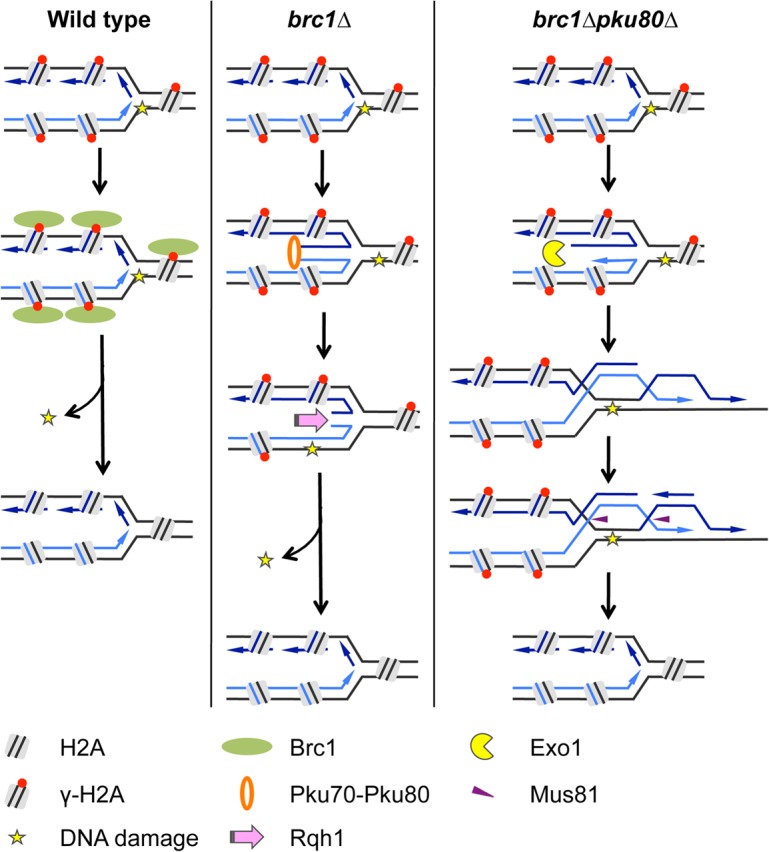
In wild type cells, replication fork stalling leads to phosphorylation of histone H2A followed by recruitment of Brc1, which stabilizes the replication fork. In brc1∆ cells, stalled replication forks are prone to fork reversal. Ku binds the exposed DNA end of the chicken foot structure to prevent fork collapse or rearrangement. This activity of Ku favors resetting of replication fork which increases the likelihood of successful completion of DNA replication. In brc1∆ pku80∆ cells, stalled replication forks undergo homologous recombination without DSB formation as shown or collapse (possibly through a Mus81-dependent mechanism) and reform through homologous recombination (not shown). In either case Mus81-Eme1 is required to resolve Holliday junction-like structures and Exo1 contributes to resection necessary for HR.
Supporting Information
(DOCX)
Acknowledgments
We thank Oliver Limbo for expert advice and assistance.
Data Availability
All relevant data are within the paper and its Supporting Information files.
Funding Statement
This research was funded by NIH grants GM059447, CA077325 and CA117638 awarded to PR. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
References
- 1. Zeman MK, Cimprich KA. Causes and consequences of replication stress. Nature cell biology. 2014;16(1):2–9. 10.1038/ncb2897 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Lambert S, Carr AM. Impediments to replication fork movement: stabilisation, reactivation and genome instability. Chromosoma. 2013;122(1–2):33–45. 10.1007/s00412-013-0398-9 . [DOI] [PubMed] [Google Scholar]
- 3. Hills SA, Diffley JF. DNA replication and oncogene-induced replicative stress. Current biology: CB. 2014;24(10):R435–44. 10.1016/j.cub.2014.04.012 . [DOI] [PubMed] [Google Scholar]
- 4. Branzei D, Foiani M. Maintaining genome stability at the replication fork. Nat Rev Mol Cell Biol. 2010;11(3):208–19. 10.1038/nrm2852 . [DOI] [PubMed] [Google Scholar]
- 5. Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, et al. GammaH2AX and cancer. Nature reviews Cancer. 2008;8(12):957–67. 10.1038/nrc2523 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Rozenzhak S, Mejia-Ramirez E, Williams JS, Schaffer L, Hammond JA, Head SR, et al. Rad3 decorates critical chromosomal domains with gammaH2A to protect genome integrity during S-Phase in fission yeast. PLoS Genet. 2010;6(7):e1001032 10.1371/journal.pgen.1001032 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Nakamura TM, Du LL, Redon C, Russell P. Histone H2A phosphorylation controls Crb2 recruitment at DNA breaks, maintains checkpoint arrest, and influences DNA repair in fission yeast. Mol Cell Biol. 2004;24(14):6215–30. 10.1128/MCB.24.14.6215-6230.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Williams JS, Williams RS, Dovey CL, Guenther G, Tainer JA, Russell P. gammaH2A binds Brc1 to maintain genome integrity during S-phase. EMBO J. 2010;29(6):1136–48. Epub 2010/01/23. 10.1038/emboj.2009.413 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Sheedy DM, Dimitrova D, Rankin JK, Bass KL, Lee KM, Tapia-Alveal C, et al. Brc1-mediated DNA repair and damage tolerance. Genetics. 2005;171(2):457–68. Epub 2005/06/24. 10.1534/genetics.105.044966 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Wehrkamp-Richter S, Hyppa RW, Prudden J, Smith GR, Boddy MN. Meiotic DNA joint molecule resolution depends on Nse5-Nse6 of the Smc5-Smc6 holocomplex. Nucleic acids research. 2012;40(19):9633–46. 10.1093/nar/gks713 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Verkade HM, Bugg SJ, Lindsay HD, Carr AM, O'Connell MJ. Rad18 is required for DNA repair and checkpoint responses in fission yeast. Mol Biol Cell. 1999;10(9):2905–18. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Pebernard S, Wohlschlegel J, McDonald WH, Yates JR 3rd, Boddy MN. The Nse5-Nse6 dimer mediates DNA repair roles of the Smc5-Smc6 complex. Mol Cell Biol. 2006;26(5):1617–30. 10.1128/MCB.26.5.1617-1630.2006 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Pebernard S, McDonald WH, Pavlova Y, Yates JR 3rd, Boddy MN. Nse1, Nse2, and a novel subunit of the Smc5-Smc6 complex, Nse3, play a crucial role in meiosis. Mol Biol Cell. 2004;15(11):4866–76. 10.1091/mbc.E04-05-0436 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Morikawa H, Morishita T, Kawane S, Iwasaki H, Carr AM, Shinagawa H. Rad62 protein functionally and physically associates with the smc5/smc6 protein complex and is required for chromosome integrity and recombination repair in fission yeast. Mol Cell Biol. 2004;24(21):9401–13. 10.1128/MCB.24.21.9401-9413.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bass KL, Murray JM, O'Connell MJ. Brc1-dependent recovery from replication stress. J Cell Sci. 2012;125(Pt 11):2753–64. 10.1242/jcs.103119 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Lee SY, Rozenzhak S, Russell P. gammaH2A-binding protein Brc1 affects centromere function in fission yeast. Mol Cell Biol. 2013;33(7):1410–6. 10.1128/MCB.01654-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. McGlynn P, Lloyd RG. Recombinational repair and restart of damaged replication forks. Nat Rev Mol Cell Biol. 2002;3(11):859–70. 10.1038/nrm951 . [DOI] [PubMed] [Google Scholar]
- 18. Costes A, Lambert SA. Homologous recombination as a replication fork escort: fork-protection and recovery. Biomolecules. 2012;3(1):39–71. 10.3390/biom3010039 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Roseaulin L, Yamada Y, Tsutsui Y, Russell P, Iwasaki H, Arcangioli B. Mus81 is essential for sister chromatid recombination at broken replication forks. EMBO J. 2008;27(9):1378–87. 10.1038/emboj.2008.65 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Forsburg SL, Rhind N. Basic methods for fission yeast. Yeast. 2006;23(3):173–83. 10.1002/yea.1347 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Bahler J, Wu JQ, Longtine MS, Shah NG, McKenzie A 3rd, Steever AB, et al. Heterologous modules for efficient and versatile PCR-based gene targeting in Schizosaccharomyces pombe. Yeast. 1998;14(10):943–51. 10.1002/(SICI)1097-0061(199807)14:10<943::AID-YEA292>3.0.CO;2-Y . [DOI] [PubMed] [Google Scholar]
- 22. Roguev A, Bandyopadhyay S, Zofall M, Zhang K, Fischer T, Collins SR, et al. Conservation and rewiring of functional modules revealed by an epistasis map in fission yeast. Science. 2008;322(5900):405–10. 10.1126/science.1162609 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Nakamura TM, Moser BA, Russell P. Telomere binding of checkpoint sensor and DNA repair proteins contributes to maintenance of functional fission yeast telomeres. Genetics. 2002;161(4):1437–52. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Langerak P, Mejia-Ramirez E, Limbo O, Russell P. Release of Ku and MRN from DNA ends by Mre11 nuclease activity and Ctp1 is required for homologous recombination repair of double-strand breaks. PLoS Genet. 2011;7(9):e1002271 Epub 2011/09/21. 10.1371/journal.pgen.1002271 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Sanchez A, Sharma S, Rozenzhak S, Roguev A, Krogan NJ, Chabes A, et al. Replication fork collapse and genome instability in a deoxycytidylate deaminase mutant. Mol Cell Biol. 2012;32(21):4445–54. 10.1128/MCB.01062-12 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Roguev A, Wiren M, Weissman JS, Krogan NJ. High-throughput genetic interaction mapping in the fission yeast Schizosaccharomyces pombe. Nat Methods. 2007;4(10):861–6. 10.1038/nmeth1098 . [DOI] [PubMed] [Google Scholar]
- 27. Kim DU, Hayles J, Kim D, Wood V, Park HO, Won M, et al. Analysis of a genome-wide set of gene deletions in the fission yeast Schizosaccharomyces pombe. Nat Biotechnol. 2010;28(6):617–23. 10.1038/nbt.1628 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annual review of biochemistry. 2010;79:181–211. 10.1146/annurev.biochem.052308.093131 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Miyoshi T, Kanoh J, Ishikawa F. Fission yeast Ku protein is required for recovery from DNA replication stress. Genes Cells. 2009;14(9):1091–103. 10.1111/j.1365-2443.2009.01337.x . [DOI] [PubMed] [Google Scholar]
- 30. Baumann P, Cech TR. Protection of telomeres by the Ku protein in fission yeast. Mol Biol Cell. 2000;11(10):3265–75. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Kibe T, Tomita K, Matsuura A, Izawa D, Kodaira T, Ushimaru T, et al. Fission yeast Rhp51 is required for the maintenance of telomere structure in the absence of the Ku heterodimer. Nucleic acids research. 2003;31(17):5054–63. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Doe CL, Ahn JS, Dixon J, Whitby MC. Mus81-Eme1 and Rqh1 involvement in processing stalled and collapsed replication forks. The Journal of biological chemistry. 2002;277(36):32753–9. 10.1074/jbc.M202120200 . [DOI] [PubMed] [Google Scholar]
- 33. Boddy MN, Lopez-Girona A, Shanahan P, Interthal H, Heyer WD, Russell P. Damage tolerance protein Mus81 associates with the FHA1 domain of checkpoint kinase Cds1. Mol Cell Biol. 2000;20(23):8758–66. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Boddy MN, Gaillard PH, McDonald WH, Shanahan P, Yates JR 3rd, Russell P. Mus81-Eme1 are essential components of a Holliday junction resolvase. Cell. 2001;107(4):537–48. doi: S0092-8674(01)00536-0 [pii]. . [DOI] [PubMed] [Google Scholar]
- 35. Noguchi E, Noguchi C, Du LL, Russell P. Swi1 prevents replication fork collapse and controls checkpoint kinase Cds1. Mol Cell Biol. 2003;23(21):7861–74. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Meister P, Poidevin M, Francesconi S, Tratner I, Zarzov P, Baldacci G. Nuclear factories for signalling and repairing DNA double strand breaks in living fission yeast. Nucleic acids research. 2003;31(17):5064–73. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Du LL, Nakamura TM, Moser BA, Russell P. Retention but not recruitment of Crb2 at double-strand breaks requires Rad1 and Rad3 complexes. Mol Cell Biol. 2003;23(17):6150–8. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Mimitou EP, Symington LS. DNA end resection—unraveling the tail. DNA repair. 2011;10(3):344–8. 10.1016/j.dnarep.2010.12.004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philosophical transactions of the Royal Society of London Series B, Biological sciences. 2011;366(1584):3562–71. 10.1098/rstb.2011.0070 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell. 2007;28(1):134–46. doi: S1097-2765(07)00621-1 [pii] 10.1016/j.molcel.2007.09.009 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Segurado M, Diffley JF. Separate roles for the DNA damage checkpoint protein kinases in stabilizing DNA replication forks. Genes Dev. 2008;22(13):1816–27. doi: 22/13/1816 [pii] 10.1101/gad.477208 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Cotta-Ramusino C, Fachinetti D, Lucca C, Doksani Y, Lopes M, Sogo J, et al. Exo1 processes stalled replication forks and counteracts fork reversal in checkpoint-defective cells. Mol Cell. 2005;17(1):153–9. 10.1016/j.molcel.2004.11.032 . [DOI] [PubMed] [Google Scholar]
- 43. Sanchez-Gorostiaga A, Lopez-Estrano C, Krimer DB, Schvartzman JB, Hernandez P. Transcription termination factor reb1p causes two replication fork barriers at its cognate sites in fission yeast ribosomal DNA in vivo. Mol Cell Biol. 2004;24(1):398–406. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Krings G, Bastia D. swi1- and swi3-dependent and independent replication fork arrest at the ribosomal DNA of Schizosaccharomyces pombe. Proc Natl Acad Sci U S A. 2004;101(39):14085–90. 10.1073/pnas.0406037101 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Tsang E, Carr AM. Replication fork arrest, recombination and the maintenance of ribosomal DNA stability. DNA repair. 2008;7(10):1613–23. 10.1016/j.dnarep.2008.06.010 . [DOI] [PubMed] [Google Scholar]
- 46. Postow L. Destroying the ring: Freeing DNA from Ku with ubiquitin. FEBS letters. 2011;585(18):2876–82. 10.1016/j.febslet.2011.05.046 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Noguchi E, Noguchi C, McDonald WH, Yates JR 3rd, Russell P. Swi1 and Swi3 are components of a replication fork protection complex in fission yeast. Mol Cell Biol. 2004;24(19):8342–55. 10.1128/MCB.24.19.8342-8355.2004 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Lee SY, Russell P. Brc1 links replication stress response and centromere function. Cell Cycle. 2013;12(11):1665–71. 10.4161/cc.24900 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Zaratiegui M, Castel SE, Irvine DV, Kloc A, Ren J, Li F, et al. RNAi promotes heterochromatic silencing through replication-coupled release of RNA Pol II. Nature. 2011;479(7371):135–8. 10.1038/nature10501 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Ferreira MG, Cooper JP. Two modes of DNA double-strand break repair are reciprocally regulated through the fission yeast cell cycle. Genes Dev. 2004;18(18):2249–54. 10.1101/gad.315804 . [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Ui A, Seki M, Ogiwara H, Onodera R, Fukushige S, Onoda F, et al. The ability of Sgs1 to interact with DNA topoisomerase III is essential for damage-induced recombination. DNA repair. 2005;4(2):191–201. 10.1016/j.dnarep.2004.09.002 . [DOI] [PubMed] [Google Scholar]
- 52. Yamana Y, Maeda T, Ohba H, Usui T, Ogawa HI, Kusano K. Regulation of homologous integration in yeast by the DNA repair proteins Ku70 and RecQ. Mol Genet Genomics. 2005;273(2):167–76. 10.1007/s00438-005-1108-y . [DOI] [PubMed] [Google Scholar]
- 53. Allen C, Ashley AK, Hromas R, Nickoloff JA. More forks on the road to replication stress recovery. J Mol Cell Biol. 2011;3(1):4–12. 10.1093/jmcb/mjq049 . [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
(DOCX)
Data Availability Statement
All relevant data are within the paper and its Supporting Information files.