

Abstract
To properly understand the transcriptional network of animals, we must have a full, quantitative understanding of the spatial and temporal expression patterns of transcription factors and their targets. Visual inspection of embryos stained to reveal the patterns of genes shows levels of expression that change from cell to cell in complex manners. With our current wealth of understanding of the basic biology of animal genomes, their transcriptional regulatory networks and many of the components involved, combined with current technologies in optical microscopy, computing and image and vision analysis, we ought to be able to capture quantitative, three dimensional information about the transcriptional network, all the factors and targets, for an entire animal at cellular resolution. It should also be possible to assemble these data into a single computationally-analyzable database, an atlas, which could be the basis for uncovering new biology governing regulatory gene networks. Realizing these possibilities is the basis of this chapter. It will focus on one of the most studied model organisms, Drosophila melanogaster, describe a suite of high throughput methods that have been used to create the first quantitative three-dimensional description of gene expression and morphology at cellular resolution in a whole animal and present some of the new biology that has been revealed.
INTRODUCTION
This chapter describes the efforts of the Berkeley Drosophila Transcription Network Project (BDTNP) [http://bdtnp.lbl.gov] to produce a computationally-analyzable, cellular resolution, morphology and gene expression atlas, of a whole blastoderm embryo. This work has been a multidisciplinary collaboration involving expertise in biology, imaging, image analysis, mathematics, computer science, vision, visualization and database construction, and so it is not practical to create a how-to manual of all these areas in this chapter. However, the methods and references are described in detail in the BDTNP publications. The first part of the chapter is an essay that describes the thinking required to develop the strategy for this project and the second part briefly outlines the techniques that enabled the atlas to be produced and some of the biological discoveries made. The goal is to bring you this understanding and inspire you to think of the many possible endeavors that comprehensive morphology and gene expression data of an entire animal, like those described here, will enable.
THINKING ABOUT A STRATEGY
Looking at gene expression in Drosophila embryos
You have walked the Bungtown Road up to the sandbar and been fascinated by the prehistoric-looking horseshoe crabs found there on the beach and now, back in the lab, you find yourself peering down a microscope at an early-stage embryo of another arthropod, one of the most studied model organisms, Drosophila melanogaster. The goal is to better understand how the genome orchestrates transcriptional control during morphogenesis to produce such incredible biological structures. This has been a long standing problem that will continue to baffled and delighted us. The transcriptional regulatory network in each animal is comprised of thousands of different genetically controlled components, many of which directly and-or indirectly interact with one another with varying binding strengths. The output of such networks are intricate patterns of expression that vary in space and time and direct morphogenesis.
Drosophila embryos are only about 500μm long, an ideal size for high-resolution fluorescence imaging and this animal comes with over 100 years of research data, including extensive work on the genetic basis of morphogenesis, for which the Nobel Prize was awarded in 1995 [Nobel Prize in Physiology or Medicine 1995, Edward B. Lewis, Christiane Nusslein-Volhard, and Eric F. Wieschaus], and more recently the entire genetic sequence [Adams et al. 2000]. The embryo you are looking at, a blastoderm as it is known before gastrulation, has been fluorescently stained for one or two of the many genes that are involved in the animal’s development. The blastoderm, which exists for the first three hours of embryogenesis, is a multinucleated syncytium comprising a central yolk surrounded by a single layer of some 6,000 nuclei [Campos-Ortega et al. 1997]. The single layer of cells means that the gene expression patterning is on the embryo surface, which is why the patterning you are looking at is so evident. The transcriptional regulatory network is also relatively simple, comprising only 50 or so transcription factors which control about 1000 target genes [Lawrence 1992], and most of these have been well characterized. You make the following notes:
-
1)
The size of a Drosophila embryo makes it well suited for microscopy, and fits into a single field of view.
-
2)
The blastoderm is a syncytium where the expression patterning controlling the body plan is completely revealed on the embryo surface.
-
3)
Only about 50 transcription factors and 1000 genes are thought to be involved in the transcriptional network of the blastoderm
Switching to phase-microscopy you see that the embryo is in the process of cellularizing. Cell membranes are invaginating from the surface of the embryo, on the apical sides of the nuclei, and growing towards the basal side through the cytoplasm separating the newly forming cells. Closer observation of several embryo at different D/V orientations reveals that cellularization is more complete on the ventral side than the dorsal side. Cellularization occurs through stage five and does not quite complete before the embryo starts to gastrulate. You notice also that nuclei are more spherical at the beginning of cellularization and elongate during stage 5 with their long axis aligning with the normal of the embryo surface.
-
4)
Cellularization begins during stage 5, the period before the onset of gastrulation. Cell membranes grow from the embryo surface inwards and nuclei become elongated.
Perhaps the pattern you are viewing under fluorescence is one of the maternally expressed genes, like bicoid, or maybe it is one of the later more intricate patterns expressed by the developing embryo along either its anterior/posterior (A/P) axis, or around its dorsal/ventral (D/V) axis. What is striking is how clever the underlying transcriptional regulatory information encoded in animal genomes must be, for such exquisitely varying patterns to be created. Because the patterning is on the surface, a simple adjust of the focus is all that is needed to clearly see the rise and fall of expression levels along the A/P direction. It becomes clear that this complexity can only occur if expression levels vary in precisely determined, quantitative, ways from one cell to another. Further, while these cell to cell expression variations are clearly observed along the A/P axis, they are difficult to appreciate around the D/V axis. This is due to a combination of the geometry of the embryo, the limited depth of focus of high numerical aperture lenses, and the difficulty of perceiving patterns in the third z-dimension. Never-the-less, there is no reason to think that expression level variations around the D/V axis are any less complex.
-
5)
Complex patterning requires expression level variations between individual cells. These variations are precisely controlled over the entire animal,
-
6)
Variations of expression are clearly seen along the A/P axis. It is difficult to see around the D/V axis, but patterning in this direction is probably just as complex.
You then lower the magnification by switching objectives, or view the embryo under a binocular dissecting fluorescence microscope. The longer depth of focus created by the lower numerical aperture of the lens, a simple geometric “pin-hole” effect, allows the three dimensional nature of the pattern, enveloping the embryo, to be revealed. And even though the numerical aperture and resolving power at this magnification are lower, the cellular-level dependence of the patterning is still appreciated. To overcome the limited perception around the D/V axis, you imagine inventing a rotisserie system for the embryo, or a potato peeler whereby the embryo could be skinned and the skin laid flat for better observation. You could squash the embryo and analyze one side, but that would perturb the delicate system being studied and make it difficult to determine the D/V orientation. Either way you wish for a better way to quantitatively examine this beautifully patterned developmental “canvas” of life - than skinning or squashing.
Pattern Dynamics: looking at fixed versus live embryos
Gene expression pattern formation is dynamic. Broadly expressed maternal proteins initiated patterning of increasingly complex target gene expression. The earliest patterns in the system can be divided into those having dominantly A/P or D/V dependence. These early A/P and D/V patterns act largely independently at first but at some stage their interaction is needed to create later patterns of greater complexity that have both A/P and D/V dependence. How do these regulators interact and what is the shape of the network?
Pattern dynamics result from individual nuclei changing their expression level with time. Are these nuclei uniformly distributed on the embryo surface or are they, like the pattern, dynamic as well? Nuclear spatial dynamics would certainly impact the underlying mechanism of pattern formation. The nuclei were long assumed to be uniformly distributed at the periphery of the embryo during stage 5 - but they undergo great dynamics during the earlier stages when the nuclei migrated to the embryo surface - and as soon as the embryo gastrulates, the nuclei are neither stationary nor distributed uniformly. So there is really no evidence to suggest that nuclei should be uniformly distributed during stage 5. To understand embryonic pattern formation it is likely important to map both gene expression levels and nuclear position through time. In fact, it would not be surprising if there were significant rearrangements of nuclear positions that correlated spatially with the gastrulation events that were immanent.
-
7)
To track expression pattern dynamics a precise temporal staging system will be required.
-
8)
How do the early A/P and D/V expression patterns interact?
-
9)
What is the role of nuclear position on the regulatory “canvas”. Are the nuclei distributed uniformly through stage 5? Do the nuclei reorganize in preparation for gastrulation?
Then you have another thought, why are we looking at fixed embryo when certainly, for studying dynamics, live animals would be better? A few calculations quickly reveal that live animals bring other complexities. Firstly, the yolk is opaque, because it is granulated and optically similar to a bag of glass marbles in air. The fixed embryos have been carefully optically-cleared by a mounting medium that has made the central yolk transparent, in much the same way as thoes marbles would become clear by mounting then in molten glass. The mountant also has a refractive index that matches that of the cover glass and this removes the possibility of spherical aberration - caused by refractive index mismatch at the optical interface of the cover glass and biology - which increasingly obscures image formation with acquisition depth. Having a single refractive index throughout the object allows you to see clearly all the way through the embryo. Secondly, you realize that stage 5 only lasts for a little less than an hour. To properly capture the pattern dynamics you will need to acquire each image within a minute or so. This is a problem because the acquisition time for a high resolution 3D image of an entire blastoderm takes almost an order of magnitude longer on the laser scanning microscope you are using. Finally, you realize that it is far easier to create probes that can be hybridized to any gene product for the 1000 genes needed than it is to create live animals that express fluorescent proteins for a 1000 genes. You decide to focus, at least initially, on fixed embryos and start to develop an image-based strategy to capture the morphology and gene expression patterns of what you are seeing.
-
10)
Live animal would be ideal but complicate the imaging. Initially, let’s focus on fixed embryos.
High throughput imaging and analysis to create a computationally-analyzable gene expression atlas
High throughput biology has become common place in many areas including proteomics, genomics and drug discovery, where the research drives screening and analysis of repetitious biological experiments. Fluorescence microscopy is a well established tool in biology but going fast are the days of “point-and-click” where a single representative image is all that is required to reveal the underlying biology being sort. Current technologies in microscopy and computing are allowing new kinds of experiments to be envisioned and performed, which require automated image acquisition, large repositories for data management and storage, fast automated image-analysis and quantitative visualization techniques.
For the Drosophila blastoderm atlas the goal is to produce a computationally-analyzable record of the pattern of protein expression some 50 transcription factors and the mRNA expression of 1,000 genes, the primary patterning regulators and their targets. While it is relatively straight forward to produce probes that are specific for any given gene, current staining and imaging technology does not allow 1,000 different genes to be stained and imaged in a single embryo. Instead several thousand embryo images will be required if we are to capture all of the patterns of interest in a single atlas. To enable such a large project, automation must be developed for acquiring and analyzing the images and a database will be needed to track the associated metadata. Embryos should be imaged at as high a numerical aperture as possible, but the lens must have sufficient working distance to allow whole embryo to be imaged. Each image will have to be analyzed to remove the inherent image acquisition artifacts, and to quantify and tabulate the gene expression and morphology at cellular resolution to produce computationally-analyzable data sets. This analysis will condense the information from a large 3D multichannel image into a much smaller PointCloud, a table of the nuclear coordinates and the relative expression per nucleus for the genes imaged [Luengo Hendriks et al. 2006].
To create the atlas, the PointClouds of multiple embryos will need to be registered together, onto an average morphological framework. This framework will allow new patterns to be added, and the atlas to be built in a sequential manner. Image registration is a field of research in computer science and vision. As microprocessor power increases new computational techniques are being developed to automatically find objects of interest in an image and track them over multiple images. One of the goals of such work is the identification of unique fiducial points in two images that correspond to the same object. Here we will apply image registration techniques to the PointCloud data, rather than trying to register the large raw image files. Embryo PointCloud registration is complicated by the simplicity of the blastoderm morphology which limits the availability of fiducial markers needed to coordinate reciprocal positions between different embryos. PointClouds can be coarsely registered by aligning the embryo’s A/P and D/V axes, as well as registering their surfaces, but this coarse registration is only a first step and does not provide the final accuracy needed. Different embryos at the same stage differ in size, shape, and in the relative location of expression patterns [Luengo Hendriks et al. 2006, Keränen et al. 2006]. At late stage 5, for example, the maxima of pair-rule stripes is only two or three cells wide and the relative position of these stripes varies slightly between different embryos. Coarsely registered embryos would average these biological variations, by incorrectly blurring the pair-rule stripe patterns over too many nuclei, and produce representations of patterns that were incorrect. To circumvent this, expression patterns need to be registered on a nucleus by nucleus basis. Even so, it would be difficult to uniquely identify nuclei based on morphology alone. However, if each embryo was stained for a common gene which had a complex pattern of expression, then many nuclei could be uniquely identified based on their relative position to the A/P and D/V axes and their relative expression level. These fiducial marks could then be used to establish one-to-one correspondences between all the nuclei of two embryo to produce pair-wise averages that do not blur the expression patterns [Fowlkes et al. 2008].
The imaging and registration strategy now becomes clear. To allow nuclear segmentation, each embryo must be stained and imaged for fluorescent markers of total DNA. One common marker gene is needed, to aid registration, and other expression patterns imaged can be registered to the atlas. Embryo PointClouds are first coarsely registered to the framework based on the A/P and D/V axis, and then finely registered based on a set of fiducial marks defined by the coordinates of individual nuclei. For practical reasons, based on the number of acquisition channels of the microscope and the complexity of hybridizing probes to an embryo, 3-channel imaging provides a simple solution. This means that one fluorescent channel will be used to capture new expression patterns that will be added to the atlas. Finally, the atlas will be complex and three dimensional, and we will need a sophisticated visualization tool to allow the data to be quantitatively explored. This strategy then defines an imaging and acquisition pipeline involving the following steps.
Staining, Mounting and Imaging
Temporal Staging
Converting Images to PointClouds
Registering PointClouds into a Virtual Embryo
PointCloudXplore: A Visualization Tool
EXPERIMENTAL PROTOCOL: THE PIPELINE
Staining, Mounting and Imaging
One of the crucial steps in acquiring images is proper staining and mounting. Poor images result from a lack of communication between the disciplines of biology and imaging - the staining has not worked well, the auto fluorescence is overwhelming and-or the mounting is not optimal. It is not uncommon that many experiments are needed to figure out the proper staining and mounting for an application. For high resolution imaging, attention to detail is required down to the scale of one half micron, the emission wavelength, the diffraction limited resolution of the system.
For the BDTNP pipeline, RNA probes for specific genes were visualized using tyramide signal amplification reactions using many fluorescent dyes, like Cy3 and coumarin, and total DNA was stained with Sytox-Green [Luengo Hendriks et al. 2006]. These dyes have well separated emission spectrum and are all excited by 2-photon absorption of a single wavelength, which allowed three-channel images to be acquired simultaneously. The stained embryos were mounted in a xylene-based plastic, DePex (Electron Microscopy Sciences, Hatfield, PA, USA). This mountant has the advantages of creating permanent solid slides that protect the fluorophors from oxygen, making the samples resistant to photo bleaching. The mountant also optically clears the embryo yolk, and has a refractive index similar to that of the mounting cover-glass. Accurate determination of the refractive index of mountant was critical because it sets the length scale along the optical z-direction. The refractive index can be determined from geometric measurements made from the images of embryo morphology, which is assumed to be independent of the orientation of the embryo when it is imaged.
Three channel images were then acquired using 2-photon excitation at 750nm (Figure 1A–C) [Luengo Hendriks et al. 2006]. Simultaneous multichannel acquisition of conventional dyes results in channel cross-talk, due to the long wavelength emission tails of the dyes. For quantitative analysis, it is critical to unmix the channels so relative fluorescence levels can be measured correctly. For the large numbers of images, an automated channel unmixing algorithem was developed and used [Luengo Hendriks et al. 2007]. One bottleneck for imaging is the wait-time while the microscope is acquiring. Most manufacturers allow the user to develop macros to automate microscope control. Manufacturers will soon realize, hopefully, that high throughput biology has arrived, and will develop and provide these tools. In the mean time, to automate image acquisition a macro was developed that lets the user record the state of the microscope for each embryo to be imaged. The macro then sequentially reset the microscope to the configuration for each embryo and acquired and saved the embryo images along with the microscope configuration data. This approach minimized the necessary user-microscope interaction and allowd the microscope to sequentially image multiple embryos without user intervention. Because all the acquisition parameters were recorded, a slide could be put back on the microscope and any or all of the embryos that have been previously imaged, automatically revisited and/or re-imaged.
Figure 1. Segmentation Analysis: Creating a PointCloud.
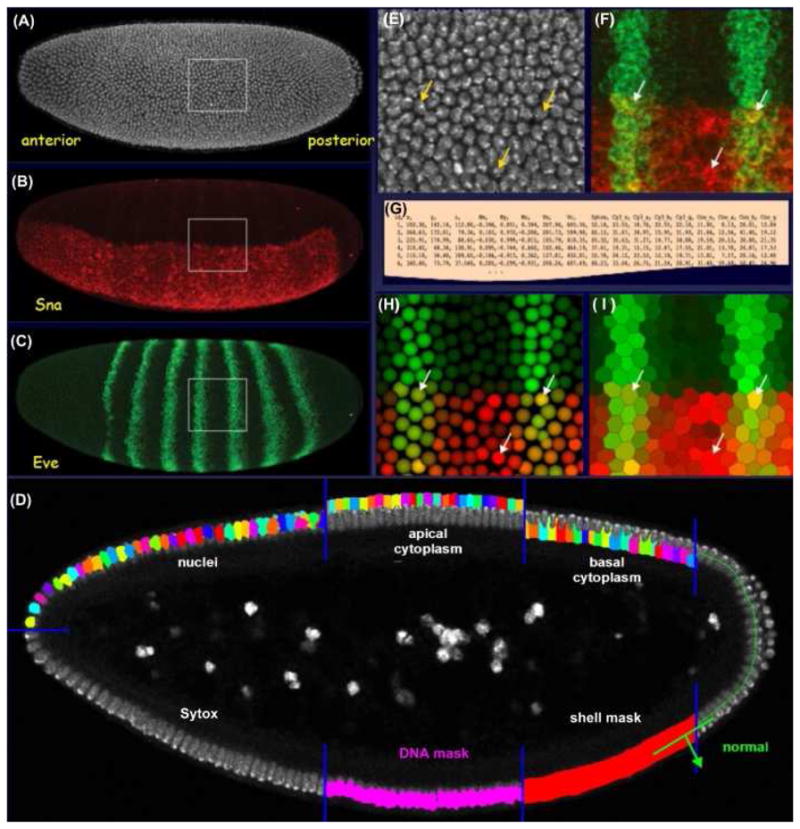
(A – C) The three channels of a three-dimensional embryo image; (A) the nuclear stain (white); (B) a snail mRNA stain (red); and (C) an eve mRNA stain (green). The position and extent of the nuclei on the surface of the blastoderm embryo were defined by a model-based three-dimensional segmentation analysis (D). The analysis defines the embryo surface. It locates individual nuclei based on the DNA stain, using a gray-weighted watershed algorithm to grow nuclear seeds along the embryo surface normals. Apical and basal cytoplasmic regions were estimated about each nucleus by Voronoi tessellation. Magnifications of the raw image are shown of the nuclear stain (E) and the mRNA stains for eve and sna (F). The resulting segmentation mask was used to quantify expression levels and the resulting data were simply tabulated to create a PointCloud file (G). Two different renderings of the PointCloud are shown for this region of interest, as derived from this image made using the visualization tool PointCloudXplore (H, I). The arrows indicate the locations of the same three cells in these panels.
Temporal Staging
To study pattern dynamics, a precise temporal staging system was required. Embryo can be staged by visual inspection under phase microscopy and broadly classified via the stages previously described [Campos-Ortega et al. 1997]. But a finer temporal staging system was needed to fully capture the rapid changes in morphology and gene expression. During stage 5, the percentage of membrane invagination was used as a temporal marker. Membrane invagination is clearly seen under phase contract microscopy, but only accurately measured in the X-Y plane. Further, because embryo were mounted in random D/V orientations and membrane invagination is more advanced on the ventral side than the dorsal side, the final membrane invagination percentage was extrapolated from the value measured on the ventral-most surface, and the D/V orientation of the embryo, which was accurately determined after the PointCloud was made.
Converting Images to PointClouds
To quantify the relative expression levels from images, the image positions of all the nuclei were required. The total-DNA Sytox image (Figure 1A) was used to determine the position and extent all embryonic nuclei to produce a segmentation mask. Many basic algorithms have been developed in the field of image analysis to enable image segmentation, but segmentation remains a problem that is not trivial. Model-based approaches are usually required where the segmentation algorithm relies on a priori knowledge of the objects to be segmented and high image quality is essential.
For speed, the segmentation analysis was restricted to the embryo surface by creating a three-dimensional binary mask, the ‘shell mask’ (Figure 1D). The mask was produced by taking an adaptive threshold of the Sytox image which isolates the bright DNA fluorescence and adapts for signal attenuation with image depth. The shell mask was then used to direct spectral unmixing and allowed attenuation correction of the Sytox channel required for the segmentation. The shell mask was refined to produce a ‘DNA mask’ (Figure 1D) that identified the regions in the image belonging to the nuclei. The Sytox image within this mask was then convolved with a narrow Gaussian and the position of individual nuclei, termed ‘seeds’, are defined by local maxima in this image. To eliminate multiple seeds per nucleus, the embryo surface normal for each seed was computed and neighboring seeds that lied along this normal were removed. Each seed was then grown to fill the nuclei, using a region-growing technique that combined a watershed algorithm and a gray-weighted distance transform [Luengo Hendriks et al. 2006]. The combination of these two algorithms created nuclear boundaries that match actual boundaries when visible, yet divided distances between seeds equally when boundaries where not distinguishable.
To capture the labeled mRNA expression levels, the cellular extent surrounding each nucleus was estimated. This was achieved by growing the nuclear segmentation mask along the apical and basal directions, into the cytoplasm by tessellation (Figure 1D). The distances grown were established by determining the cytoplasmic extent which can be estimated by its autofluorescence. The resulting cytoplasmic mask was then used in combination with the nuclear mask to divide each cell into three regions: apical, nuclear and basal (Figure 1D). Expression levels were estimated on a per cell basis as the average value within each of these region for all the channels (Figure 1E, F). These values, together with the center of mass of the nuclei, the volumes of the various cellular regions, and the neighborhood relationships between cells were then written to a text “PointCloud” file (Figure 1G). The PointCloud file captured the information from the image and allowed the data to be visualized (Figure 1H, I). For subsequent analysis, expression values from the PointClouds were corrected for signal attenuation by dividing the results from the Cy3 and coumarin channels by those from the Sytox channel. This approach assumed that the average Sytox intensity was constant from nucleus to nucleus, and that it was representative of the attenuation of the other channels.
Registering PointClouds into a Virtual Embryo
The atlas was constructed by registering individual PointClouds onto a common framework, created as an average over an ensemble of PointClouds. The registration takes into account the significant variation between embryo sizes, number of cells, and the relative locations of gene expression patterns. The key was identifying corresponding cells, fiducial marks, between images of multiple embryos, and using these correspondences to combine measurements of expression and morphology [Fowlkes et al. 2008] (Figure 2A). Fiducial marks were established by using the pair-rule patterns of Even-skipped (eve) and Fushi tarazu (ftz) as common expression markers. These patterns are used, along with the A/P and D/V coordinates, to define a sets of cells along pattern boundaries which act as the correspondence points between pairs of embryos. Once this was established it was straight forward to find the correspondence for the remaining cells in the embryo, so that an average Virtual Embryo could be built. Multiple frameworks were required because both embryo morphology and the relative position of nuclei change during stage 5. Thus to properly understand the dynamics, a temporal correspondence was required between different frameworks for the temporal cohorts (Figure 2A) [Fowlkes et al. 2008].
Figure 2. Construction of a VirtualEmbryo.

Images from hundreds of embryos were grouped into temporal cohorts and each cohort registered based on a common pair-rule marker gene, shown in red. Expression patterns of many other genes stained in different embryos were thus brought into a common frame. Temporal correspondence between registered cohorts produces the VirtualEmbryo atlas of multiple genes over multiple times (A). The accuracy of the VirtualEmbryo was demonstrated by plotting the co-expression of gt and eve for a set of cells along the anterior boarder of eve stripe 2, which are known to be repressed by gt (B).
A plot of the co-expression of gt and eve in embryos co-stained for eve and gt shows a clear anti-correlation (C). A plot of the co-expression of gt and eve for these cells in embryos co-stained for eve-ftz and gt-ftz and registered using ftz, show the same anti-correlation (D). However, a plot of co-expression of gt and eve in embryos co-stained for eve-ftz and gt-ftz which were only coarsly registered is much less accurate and does not reveal the anti-correlation (E).
The accuracy of the resulting registration was tested by comparing average expression levels recorded in the VirtualEmbryo to those measured directly in embryos co-stained for the expression of two genes. For this experiment, a set of cells along the anterior edge of eve stripe 2 were selected, where the transcription factor giant (gt) is known to repress eve expression (Figure 2B). gt versus eve expression was then plotted for these cells from a set of embryos co-stained for eve and gt (Figure 2C) and from a set of eve-ftz and gt-ftz embryos, registered using the ftz expression (Figure 2D). Figure 2D shows that the “virtual” measurement of eve and gt replicates the relation derived from the co-stained embryos (Figure 2C). Inferences about regulatory relationships made using the virtual embryo would thus be similar to those made from individual embryos. As a comparison, the co-expression of eve and gt is plotted for a set of coarsely registered embryo (Figure 2E). This experiment demonstrates the superiority of the fine registration technique and shows that building cellular resolution expression atlases from multiple embryos is not only possible but biologically accurate and provides useful data for understanding transcriptional regulation.
PointCloudXplore: A Visualization Tool
To date the BDTNP has registered PointCloud data of some 3,000 embryos containing around 100 different mRNA and protein expression patterns into a VirtualEmbryo representation of the blastoderm. One of the challenges of such a large multidimensional dataset is making it accessible for visual observation and quantitative exploration. The BDTNP has developed a suite of visualization tools (PointCloudXplore) which show the data in multiple views [Rubel et al. 2007, Weber et al. 2009]. PointCloudXplore is a freely available executable which can downloaded, along with the latest blastoderm atlas from the BDTNP public web site [http://bdtnp.lbl.gov/Fly-Net/bioimaging.jsp?w=pcx]. Expression patterns like eve and sna can be displayed on an average embryo morphology for multiple temporal cohorts (Figure 3A) or these patterns can be unrolled and displayed as a surface projection (Figure 3B) Other relationships can be seen for multiple genes (Figure 3C), and height maps of the expression patterns can be added (Figure 3D). More complex analyses such as scatter plots and parallel coordinates are also possible within PointCloudXplore.
Figure 3. PointCloudXplore.
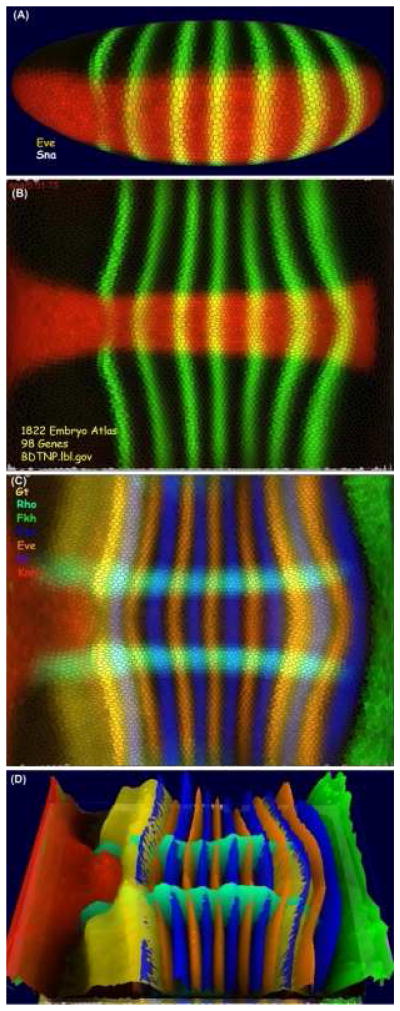
The visualization tools of the blastoderm project provide multiple physical views. This figure shows expression patterns of eve and sna (A, B) and gt, rho, fkh, ftz, kr and kni (C, D) from the blastoderm atlas. The patterns can be visualized in 3D on a virtual embryo, which can be rotated to any angle, (A) or the embryo surface can be unrolled to reveal the entire pattern in a 2D view (B, C). This view can also be rotated and height maps of the relative expression for each gene added (D).
NEW BIOLOGY REVEALED
The PointCloud and VirtualEmbryo data can be used to reveal new features of the blastoderm system. Calculations of the local spatial density of nuclei has revealed intricate morphology of nuclear density patterns that changes during stage 5 (Figure 4A–F) [Keränen et al. 2006]. An overlay of these density maps onto the expression patterns of eve and sna, for example(Figure 4G), shows that the pattern of lower nuclear densities corresponds to the embryo locations that are about to gastrulate and form the cephalic and the ventral furrows. The location of pair-rule expression stripes has also been shown to move relative to the field of nuclei [Keränen et al. 2006]. These two results had critical impact on the choice of method for temporal registration of the PointCloud data. The later result indicates that a reference gene pattern cannot be used to register data between temporal cohorts and the former result shows that temporal correspondences cannot be obtained by using common spatial coordinates. Thus, to register average frameworks of different temporal cohorts a model of the nuclear position dynamics was required [Fowlkes et al. 2008].
Figure 4. Complex Motions of Nuclear Density and patterns.

Stage 5 blastoderm embryos show a complex dynamic pattern of nuclear packing densities. Spatial nuclear densities were calculated for several temporal cohorts of embryos through stage 5 as the number of nuclear centers per μm2. The resulting density maps were computationally unrolled into a 2D projection (A–F). Nuclear densities are represented as a color heat-map with lowest densities (0.025 nuclei/μm2) in blue and the highest densities in red (0.05nuclei/μm2). Isodensity contours were overlayed with expression patterns of sna and eve (G). Expression of pair-rule stripes of ftz and eve vary significantly around the D/V axis (H and I). The stripes show marked differences in expression profiles and have unique modes of expression variation along the D/V axis. The error bars give the 95% confidence intervals for the means. The data were derived from 155 embryos.
The atlas makes it possible to quantitatively describe expression patterns along both the A/P and D/V axes. Expression pattern gradients were used to define the pattern boundaries, and their dynamics and relative expression levels plotted [Keränen et al. 2006]. For example, it is now easy to plot the relative expression of pair-rule stripes around the D/V axis (Figure 4H, I). These plots show that there are unsuspected quantitative changes along the D/V axis of pair-rule stripes [Luengo Hendriks et al. 2006]. Interestingly, the D/V dependencies of pair-rule expression stripes are almost lost in dorsalizing mutants, suggesting that D/V acting factors regulate A/P expression patterns [Keränen et al. 2006]. Although the atlas is an average over 1000s of embryos, the biological variability is not lost and can be studied from the individual embryo PointClouds. For example, plotting the number of nuclei per embryo versus A/P egg-length [Fowlkes et al. 2008], shows that embryo size correlates with number of nuclei, suggesting that egg size depends on mitosis rate.
The chief motivation for developing the blastoderm expression atlas was to understand the regulation of target genes by transcription factors. Using the atlas, it has been shown that the pattern of many transcription factors correlate or anti correlate with the patterns of other genes [Fowlkes et al. 2008], in the same way that the expression of gt and eve stripe 2 anti correlate (Figure 2B – E). From these correlations, it has been possible to infer potential regulatory relationships within the network, a number of which meet the expectations of previous molecular genetic data [Fowlkes et al. 2008]. While such inferences cannot be used to imply that a transcription factor directly binds and regulates a target gene, in combination with other classes of data such as genome-wide in vivo binding data and in vitro DNA specificity data [Li et al 2008], they will provide a significant constraint on possible models for the structure of the regulatory network.
CONCLUSION
Current technologies in optical microscopy and computing are allowing high-throughput image-based investigations of multicellular biological systems. The output are multidimensional computationally-analyzable maps providing unprecedented fidelity about the biology of a system at the macromolecular level, on a per cell basis. Realizing this potential has resulted in the first quantitative, 3D atlas of gene expression and morphology of early embryonic development in Drosophila. These technologies are profound and have a broad and exciting future.
Acknowledgments
I am privileged in collaborating with an outstanding group of scientists from multiple disciplines who as part of the BDTNP brought their expertise to bear on this problem. Special thanks to Mark D. Biggin. Many biologists are overwhelmed by today’s image-based technologies and what they can provide. Mark quickly understood the possibilities and provided leadership and drive, without which this project would not have been realized. Thanks to Soile V.E. Keränen and Mark for helpful suggestions with this chapter.
References
- Adams MD, Celniker SE, Holt RA, Evans CA, Gocayne JD, Amanatides PG, Scherer SE, Li PW, Hoskins RA, Galle RF, George RA, Lewis SE, Richards S, Ashburner M, Henderson SN, Sutton GG, Wortman JR, Yandell MD, Zhang Q, Chen LX, Brandon RC, Rogers YH, Blazej RG, Champe M, Pfeiffer BD, Wan KH, Doyle C, Baxter EG, Helt G, Nelson CR, Gabor GL, Abril JF, Agbayani A, An HJ, Andrews-Pfannkoch C, Baldwin D, Ballew RM, Basu A, Baxendale J, Bayraktaroglu L, Beasley EM, Beeson KY, Benos PV, Berman BP, Bhandari D, Bolshakov S, Borkova D, Botchan MR, Bouck J, Brokstein P, Brottier P, Burtis KC, Busam DA, Butler H, Cadieu E, Center A, Chandra I, Cherry JM, Cawley S, Dahlke C, Davenport LB, Davies P, de Pablos B, Delcher A, Deng Z, Mays AD, Dew I, Dietz SM, Dodson K, Doup LE, Downes M, Dugan-Rocha S, Dunkov BC, Dunn P, Durbin KJ, Evangelista CC, Ferraz C, Ferriera S, Fleischmann W, Fosler C, Gabrielian AE, Garg NS, Gelbart WM, Glasser K, Glodek A, Gong F, Gorrell JH, Gu Z, Guan P, Harris M, Harris NL, Harvey D, Heiman TJ, Hernandez JR, Houck J, Hostin D, Houston KA, Howland TJ, Wei MH, Ibegwam C, Jalali M, Kalush F, Karpen GH, Ke Z, Kennison JA, Ketchum KA, Kimmel BE, Kodira CD, Kraft C, Kravitz S, Kulp D, Lai Z, Lasko P, Lei Y, Levitsky AA, Li J, Li Z, Liang Y, Lin X, Liu X, Mattei B, McIntosh TC, McLeod MP, McPherson D, Merkulov G, Milshina NV, Mobarry C, Morris J, Moshrefi A, Mount SM, Moy M, Murphy B, Murphy L, Muzny DM, Nelson DL, Nelson DR, Nelson KA, Nixon K, Nusskern DR, Pacleb JM, Palazzolo M, Pittman GS, Pan S, Pollard J, Puri V, Reese MG, Reinert K, Remington K, Saunders RD, Scheeler F, Shen H, Shue BC, Sidén-Kiamos I, Simpson M, Skupski MP, Smith T, Spier E, Spradling AC, Stapleton M, Strong R, Sun E, Svirskas R, Tector C, Turner R, Venter E, Wang AH, Wang X, Wang ZY, Wassarman DA, Weinstock GM, Weissenbach J, Williams SM, Woodage T, Worley KC, Wu D, Yang S, Yao QA, Ye J, Yeh RF, Zaveri JS, Zhan M, Zhang G, Zhao Q, Zheng L, Zheng XH, Zhong FN, Zhong W, Zhou X, Zhu S, Zhu X, Smith HO, Gibbs RA, Myers EW, Rubin GM, Venter JC. The genome sequence of Drosophila melanogaster. Science. 2000;287:2185–2195. doi: 10.1126/science.287.5461.2185. [DOI] [PubMed] [Google Scholar]
- Campos-Ortega JA, Hartenstein V. The Embryonic Development of Drosophila melanogaster. 2. Springer; Berlin: 1997. [Google Scholar]
- Fowlkes CC, Luengo Hendriks CL, Keränen SVE, Weber GH, Rübel O, Huang MY, Chatoor S, DePace AH, Simirenko L, Henriquez C, Beaton A, Weiszmann R, Celniker S, Hamann B, Knowles DW, Biggin MD, Eisen MB, Malik J. A quantitative spatiotemporal atlas of gene expression in the Drosophila blastoderm. Cell. 2008;133:364–374. doi: 10.1016/j.cell.2008.01.053. [DOI] [PubMed] [Google Scholar]
- Keränen SVE, Fowlkes CC, Luengo Hendriks CL, Sudar D, Knowles DW, Malik J, Biggin MD. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution II: dynamics. Genome Biol. 2006;7:R124. doi: 10.1186/gb-2006-7-12-r124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lawrence PA. The Making of a Fly: The Genetics of Animal Design. Blackwell-Scientific; Oxford: 1992. [Google Scholar]
- Li X, MacArthur S, Bourgon R, Nix D, Pollard DA, Iyer VN, Hechmer A, Simirenko L, Stapleton M, Luengo Hendriks CL, Chu HC, Ogawa N, Inwood W, Sementchenko V, Beaton A, Weiszmann R, Celniker SE, Knowles DW, Gingeras T, Speed TP, Eisen MB, Biggin MD. Transcription Factors Bind Thousands of Active and Inactive Regions in the Drosophila Blastoderm. PLoS Biol. 2008;6:e27. doi: 10.1371/journal.pbio.0060027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo Hendriks CL, Keränen SVE, Fowlkes CC, Simirenko L, Weber GH, DePace AH, Henriquez C, Kaszuba DW, Hamann B, Eisen MB, Malik J, Sudar D, Biggin MD, Knowles DW. Three-dimensional morphology and gene expression in the Drosophila blastoderm at cellular resolution I: data acquisition pipeline. Genome Biol. 2006;7:R123. doi: 10.1186/gb-2006-7-12-r123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo Hendriks CL, Keränen SVE, Mark D, Biggin MD, Knowles DW. Automatic channel unmixing for high-throughput quantitative analysis of fluorescence images. Optics Express. 2007;15:12306–12317. doi: 10.1364/oe.15.012306. [DOI] [PubMed] [Google Scholar]
- Rübel O, Weber GH, Keränen SVE, Fowlkes CC, Luengo Hendriks CL, Simirenko L, Shah NY, Eisen MB, Biggin MD, Hagen H, Sudar D, Malik J, Knowles DW, Hamann B. In: Santos BC, Ertl T, Joy K, editors. PointCloudXplore: Visual analysis of 3D gene expression data using physical views and parallel coordinates; Data Visualization (Proceedings of the Eurographics/IEEE-VGTC Symposium on Visualization); Lisbon, Portugal. 2006.2006. [Google Scholar]
- Weber GH, Rübel O, Huang M, DePace AH, Fowlkes CC, Keränen SVE, Luengo Hendriks CL, Hagen H, Knowles DW, Malik J, Biggin MD, Hamann B. Visual Exploration of Three-dimensional Gene Expression Using Physical Views and Linked Abstract Views. Transactions on Computational Biology and Bioinformatics. IEEE/ACM. 2009;6:296–309. doi: 10.1109/TCBB.2007.70249. [DOI] [PMC free article] [PubMed] [Google Scholar]