

Abstract
The Kv7 (KCNQ) family of voltage-gated K+ channels regulates cellular excitability. The functional role of Kv7.2 has been hampered by the lack of a viable Kcnq2-null animal model. In this study, we generated homozygous Kcnq2-null sensory neurons using the Cre-Lox system; in these mice, Kv7.2 expression is absent in the peripheral sensory neurons, whereas the expression of other molecular components of nodes (including Kv7.3), paranodes, and juxtaparanodes is not altered. The conditional Kcnq2-null animals exhibit normal motor performance, but have increased thermal hyperalgesia and mechanical allodynia. Whole cell patch recording technique demonstrates that Kcnq2-null sensory neurons have increased excitability and reduced spike frequency adaptation. Taken together, our results suggest that the loss of Kv7.2 activity increases the excitability of primary sensory neurons.
Keywords: KCNQ, M-current, dorsal root ganglion, nociceptors, Kv7
INTRODUCTION
The Kv7 (KCNQ) family of K+ channels is made up of five members, Kv7.1 to Kv7.5 (Jentsch, 2000, Delmas and Brown, 2005, Brown and Passmore, 2009). Many peripheral nervous system (PNS) and central nervous system neurons express Kv7.2, Kv7.3, and K7.5, and Kv7.1 and Kv7.4 have recently been found in certain neuronal populations (Goldman et al., 2009, Heidenreich et al., 2011). In addition, Kv7.1 and Kv7.4 are prominently expressed in the cochlea, and Kv7.1 is expressed in the heart (Brown and Passmore, 2009). Kv7 channels contribute to the normal resting membrane potential, and also form the non-inactivating M-current, which is modulated by muscarinic agonists (Brown and Adams, 1980, Wang et al., 1998). Their importance in maintaining normal cellular excitability is demonstrated by the effects of dominant mutations of KCNQ1, KCNQ2, KCNQ3, and KCNQ4 – all decrease the Kv7 current and cause hereditary diseases in a cell autonomous manner (Singh et al., 1998) - in the heart (KCNQ1), brain (KCNQ2 and KCNQ3), and inner ear (KCNQ1 and KCNQ4) (Jentsch, 2000, Brown and Passmore, 2009). One KCNQ2 mutation causes neuromyotonia (Dedek et al., 2001), a form of peripheral nerve hyperexcitability, which is likely the result of diminished Kv7.2 and Kv7.3 current at nodes of Ranvier, where Kv7.2 and Kv7.3 are localized (Devaux et al., 2004). A conserved ankyrin-G binding motif located at the C-termini mediates the localization of Kv7.2 and Kv7.3 at nodes and axon initial segments (AIS) (Pan et al., 2006).
Previous investigations of the role of Kv7 in regulating neuronal excitability and nociceptive behaviors utilized pharmacological M-channel blockers and/or enhancers (Passmore et al., 2003, Yue and Yaari, 2004, Rivera-Arconada and Lopez-Garcia, 2006, Lang et al., 2008, Roza and Lopez-Garcia, 2008). Because homozygous Kcnq2-null mice die at birth due to pulmonary atelectasis, investigators studied heterozygous Kcnq2- knockout mice (Watanabe et al., 2000, Yang et al., 2003, Otto et al., 2006, Tzingounis and Nicoll, 2008), Kcnq3-null mice (Tzingounis and Nicoll, 2008), or mice expressing a dominate-negative human KCNQ2 mutation as a transgene (Peters et al., 2005). None of these studies, however, satisfy the need to develop an animal model with a complete absence of Kv7.2 expression. In addition, previous studies of heterozygous Kcnq2-knockout mice found that reduction of Kv7.2 expression have a decreased seizure threshold (Watanabe et al., 2000, Yang et al., 2003).
In the present study, we characterize mice in which Kcnq2 has been deleted in all somatic sensory neurons, using the Cre-Lox system (Sauer and Henderson, 1988, Nagy, 2000). These mice are viable and their myelinated sensory axons have a normal ultrastructure and normal molecular composition of nodes (including Kv7.3), paranodes, and juxtaparanodes. Kcnq2 mutant mice showed signs of thermal hyperalgesia and mechanical allodynia, and Kcnq2-null dorsal root ganglia (DRG) neurons showed increased excitability and reduced spike-frequency adaptation. Taken together, our results suggest that Kv7.2 regulates neuronal excitability, and a reduction of Kv7.2 expression could lead to altered nociception.
MATERIALS AND METHODS
All procedures involving rodents were approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania. Except when specified, all chemicals were from Sigma.
Generation of conditional Kcnq2-null mice
A floxed allele of Kcnq2 was designed (Figure 1A) to delete exons 3–5; these correspond to 463 bp (amino acid 130–285) of mouse Kcnq2 cDNA (GeneBank AF490773; (Wen and Levitan, 2002)); this deletion has been shown to result in a functional null allele (Watanabe et al., 2000). Two DNA fragments (EcoRV and Xhol) were cloned from R1 ES-cells genomic DNA, and used to generate a targeting vector in which a FRT-neomycin- FRT-loxP cassette was inserted into a unique Xhol site, with flanking 7kb and 3.4kb of homologous genomic DNA. An additional loxP site was inserted in an EcoRI site just upstream of the third exon. The targeting vector also includes a diphtheria toxin (DT) gene as a negative selection marker against ES clones which have randomly integrated the targeting vector. The linearized targeting vector was electroporated into R1 embryonic stem cells, selected with G418, and colonies were picked and analyzed by Southern blot. Positive ES cells were used to generate chimeras and then heterozygous mice (Gollan et al., 2003, Poliak et al., 2003). The presence of the targeted locus was confirmed by polymerase chain reaction (PCR) analysis of tail DNA. Heterozygous mice were first mated with mice carrying the FLP recombinase (129S4/SvJaeSor-Gt(ROSA)26Sortm1(FLP1)Dym/J, RRID:IMSR_JAX:003946; (Farley et al., 2000)), to remove the Neo cassette and to create a floxed Kcnq2 allele.
Figure 1. Targeting strategy to generate an inducible deletion allele of Kcnq2.

Panel A shows the genomic arrangement of the first seven exons of wild-type Kcnq2. The external probes a and b are indicated as horizontal green lines. The targeted allele contains two loxP sites (orange triangles facing down) before the third, and after the fifth exons. A cassette containing a neo gene under the herpes simplex Tk promoter (PtkNeo; yellow box), flanked by two FRT sites (purple triangles facing up) is inserted in a unique XhoI site in the intron between the fifth and the sixth exons. The targeting vector (not shown) is the same as the pictured targeted allele except that it includes a diphtheria toxin (DT) gene as a negative selection marker after the last EcoRV site. Mice carrying the floxed allele were obtained after the removal of the PtkNeo cassette by mating the targeted mice with a general FRT-deleter strain. The recombined allele lacks exons 3–5, is generated after Cre-mediated deletion. Panel B shows the expected sizes of the amplified DNA with the different primer pairs. Panel C shows the PCR results of DNA isolated from DRG, trigeminal ganglia, and liver of Kcnq2-null mice and their control littermates, along with tail DNA from control and heterozygous floxed Kcnq2 mice (Kcnq2 fl/+). Samples were subjected to PCR with the indicated primers, and the reaction products were separated by gel electrophoresis. A ~270 bp band, corresponding to the predicted size of wild-type Kcnq2 allele, was detected in both tail samples, as well as in all of the samples from the control littermates. A ~300 bp band, corresponding to the predicted size of the floxed Kcnq2 allele, was detected in the heterozygous tail, as well as in all of the Kcnq2-null samples. A ~380 bp band, corresponding to the predicted size of the recombined Kcnq2 allele, was detected in both the DRG and the trigeminal ganglia of the Kcnq2-null mice, but not in their liver.
We crossed mice expressing Pax3-Cre (RRID:IMSR_JAX:005549) with mice carrying the Rosa26 reporter gene (RRID:IMSR_JAX:003504). We examined X-gal expression in the brains, spinal cords, and lumbar DRG of 3 Pax3-Cre-positive//Rosa26 mice and 3 Pax3-Cre-negative//Rosa26 littermates, all 1-month-old, as previously described (Feltri et al., 1992, Arroyo et al., 1998). The mice were perfused with 0.5% glutaraldehyde in 0.1M phosphate buffer (PB; pH 7.4), their cerebra, cerebelli, and spinal cords were dissected and cut into slabs with a razor blade, and the resulting sections along with L4 and L5 DRG were fixed for 3 hours at 4°C, stained in X-Gal (Roche Diagnostic) at 37°C for 24–48 hours, rinsed in 0.1M PB, then re-fixed in 3% glutaraldehyde (in 0.1M PB) at 4°C overnight. The samples were photographed using a Nikon Coolpix 5000 camera mounted on a Leica MZ16 FA stereomicroscope. The DRG were osmicated, dehydrated, and embedded with the Embed 812 kit (EMS). Semi-thin sections (1 μm thick) were photographed using a cooled Hamamatsu camera mounted on Leica DMR light microscope.
Kcnq2 was deleted in sensory axons by crossing with mice that were heterozygous for both the floxed Kcnq2 allele and Pax3-Cre (Kcnq2f/+//Pax3-Cre). We chose this approach because it generates relatively more mice of the desired genotype, and homozygous Pax3-Cre mice fail to develop past E18.5 according to Jackson Laboratory. All offspring were genotyped by PCR. Tail DNA was digested with DirectPCR (Viagen) and proteinase K overnight at 55°C, heated to 85°C for 45 minutes to denature proteinase K, then PCR were then performed with REDTaq ReadyMix PCR reaction mix following manufacturer’s protocol in a Bio-Rad DNA Engine Peltier thermal cycler. Three primers were used together: KCNQ2A (GGGGCAGTTGTCTAACCCTC), KCNQ2C (TATGTGGTGCTCCCCAGAAG), and KCNQ2E (GGGAGGCTCTAGTGTCAGTG); see Figure 1B. After amplification, samples were separated in 1.5% agarose (GeneMate) gel in 1xTBS at 125V for 1 hour. To detect the presence of Pax3-Cre in the control littermates (Kcnq2+/+), three primers were used together: oIMR6977 (CTGCACTCAAGGGACTCCTC), oIMR6978 (GTGAAGGCGAGACGAAAAAG), and oIMR9074 (AGGCAAATTTTGGTGTACGG), following instruction provided by Jackson Labs.
Anatomical studies
One-month-old Kcnq2-mutant mice of either sex (Kcnq2fl/fl//Pax3-Cre; n=3) and their control littermates (Kcnq2+/+//Pax3-Cre; n=3) were anesthetized with ketamine/xylazine mix and killed by decapitation. Sciatic nerves, DRG (from L4–L6 spinal levels) with the ventral and dorsal roots attached, and femoral nerves (motor and sensory branches separately) were dissected, and quickly embedded in OCT mounting media cooled in an acetone/dry ice slurry. The sciatic and femoral nerve fibers were teased apart with fine needles, mounted on SuperFrost Plus glass slides (Fisher Scientific, Pittsburgh, PA), dried overnight, and stored at −20°C. Ten μm thick cryostat sections were thaw-mounted onto SuperFrost slides, and stored at −20°C. Teased fibers and OCT sections were immersed in −20°C acetone for 10 minutes, rinsed in Tris-buffered saline (TBS; pH 7.4), blocked at room temperature for one hour in TBS containing 5% fish skin gelatin and 0.5% Triton X-100, and incubated overnight at 4°C with various combination of primary antibodies diluted in blocking solution. The slides were washed with TBS, incubated with the appropriate FITC- and TRITC-conjugated donkey cross-affinity-purified secondary antibodies (Jackson ImmunoResearch, 1:200) at room temperature for one hour, washed with TBS, counterstained with 4′,6-diamidino-2-phenylindole dihydrochloride (DAPI; Invitrogen), mounted with Vectashield (Vector Laboratories), then examined with a Leica DMR light microscope with a cooled Hamamatsu camera under the control of Openlab software (PerkinElmer, http://www.perkinelmer.com/pages/020/cellularimaging/products/openlab.xhtml, RRID:rid_000096).
To analyze the structure of myelinated axons, one-month-old Kcnq2 mutant mice of either sex (Kcnq2fl/fl//Pax3-Cre; n=3) and littermates controls (Kcnq2+/+//Pax3-Cre; n=3) were perfused with 0.9% NaCl, followed by fixative (2% glutaraldehyde/2% paraformaldehyde in 0.1M PB) for 10 minutes, then the femoral sensory and motor branches as well as the L4 and L5 DRG were dissected, fixed for 4 hours at 4°C, osmicated, dehydrated, and embedded with the Embed 812 kit. Semi-thin (1 μm) sections were stained with toluidine blue and examined as above; thin (80 nm thick) sections were stained with lead citrate and examined with a JEM-1010 transmission electron microscope (JEOL USA) outfitted with a digital camera.
Antibody Characterization
Please see Table 1 for a summary of all primary antibodies used. The KCNQ2N antiserum (Dr. Edward Cooper, Cat# KCNQ2N, RRID:AB_2312342) stained nodes and AISs of teased nerve fibers in an identical pattern as previously shown (Devaux et al., 2004; Pan et al., 2006), and is directed against residues 13–37 (GEKKLKVGFVGLDPGAPDSTRDC) from the intracellular terminal region of human KCNQ2 (Cooper et al. 2001). The KCNQ3C antiserum (Dr. Edward Cooper, Cat# KCNQ3C, RRID:AB_2312343), which detects residues 578–604 (STPKHKKSQKGSAFTFPSQQSPRNEPYc) of human KCNQ3 (Pan et al. 2006), also stained nodes in an identical pattern as previously described (Pan et al., 2006). To ascertain the specificity of the Kv7.2 antisera, Hela cells were transiently transfected with the cDNA encoding human Kv7.2 or Kv7.3, (kindly provided by Dr. Edward Cooper) using the Lipofectamine 2000 (Invitrogen) method as previously described (Rasmussen et al., 2007), and immunostained 1 day post-transfection with the Kv7.2 and Kv7.3 antisera. The KCNQ2N antiserum positively stained Hela cells that were transiently transfected with the Kv7.2 cDNA but did not stain Hela cells transfected with Kv7.3 cDNA; the KCNQ3C positively stained transfected Hela cells expressing Kv7.3, but did not label transfected cells expressing Kv7.2 (data not shown).
Table 1.
List of Primary Antibodies Used
| Name | Manufacturer | Dilution | Species | Type | Immunogen |
|---|---|---|---|---|---|
| KCNQ2N | A gift from Dr. Edward Cooper | 1:200 | Rabbit | Polyclonal | Human KCNQ2 N-terminus residues 13–37 (GEKKLKVGFVGLDPGAPDSTRDC) |
| KCNQ3C | A gift from Dr. Edward Cooper | 1:200 | Rabbit | Polyclonal | Human KCNQ3 C-terminus residues 578–604 (STPKHKKSQKGSAFTFPSQQSPRNEPYc) |
| KCNQ5 | Chemicon; AB5599 | 1:1000 | Rabbit | Polyclonal | KCNQ5 N-terminus (M1–R88:GAAGLWVRSGAAAAAGAGGGRPGSGMKDVESGRGRVLLNSAAARGDGLLLLGTRAAALGGGGGGLRESRRGKQGARMSLLGK) |
| panNav | Sigma; clone K58/35 (S8809) | 1:250 | Mouse | Monoclonal | CTEEQKKYYNAMKKLGSKK from the intracellular IIIIV loop of Na+ channels |
| Kv1.1 | NeuroMab; clone K20/78 | 1:250 | Mouse | Monoclonal | Rat Kv1.1 amino acids 458–476 (CEEDMNNSIAHYRQANIRTG) |
| Caspr | A gift from Dr. Elior Peles; Clone 275 | 1:100 | Mouse | Monoclonal | Contained within extracellular domain of human Caspr (amino acids 1–1282) |
The sequences of immunogen targeted by the KCNQ5 antiserum (Millipore (CHEMICON/Upstate/Linco), Cat# AB5599, RRID:AB_210806) is a stretch of 88 amino acids from the N-terminal sequence of KCNQ5 (M1–R88:GAAGLWVRSGAAAAAGAGGGRPGSGMKDVESGRGRVLLNSAAARGDGLLLLGTRAAALGGGGGGLRESRRGKQGARMSLLGK), and were ascertained from both the manufacturer and from Caminos et al. (2007). The antiserum was able to positively label Hela cells transfected by the Kv7.5 cDNA (kindly provided by Dr. Thomas Jentsch) but did not label Hela cells transfected by the Kv7.2 cDNA (data not shown).
The mouse panNav monoclonal antiserum (Sigma-Aldrich, Cat# S8809, RRID:AB_477552) targets multiple voltage-gated Na+ channels, and detects immunogen (CTEEQKKYYNAMKKLGSKK) from the intracellular III–IV loop of Na+ channels (Rasband et al., 1999). Its specificity was confirmed by Western blotting of rat brain extracts recognizing a 260 kDa protein (manufacturer’s technical information), and has previously been shown to specifically stain the AISs and nodes of a wide range of nervous tissues (Rasband et al., 1999; Devaux et al., 2004; Pan et al., 2006); in our stainings it recognized the nodes of both rat and mouse sciatic nerves, as expected.
The mouse monoclocal Kv1.1 antiserum (NeuroMab, Cat# K20/78, RRID:AB_2312366) was prepared against a synthetic peptide representing amino acids 458–476 (CEEDMNNSIAHYRQANIRTG) of rat Kv1.1 (Rasband et al., 1998). Our Western blot analysis showed a strong band at around 85 kDa, and a weaker band at around 65 kDa, identical with the information provided by the manufactuer (data not shown). In our staining it labeled the juxtaparanodal component of myelinated axons, as had been demonstrated previously (Rasband et al., 2001).
The mouse monoclonal contactin-associated protein (Caspr) antiserum (Peles et al., 1997, Cat# contactin-associated protein (Caspr), RRID:AB_2311776) was generated by Dr. Poliak by immunizing mice with a fusion protein composed of the extracellular domain of human Caspr (amino acids 1–1282; Peles et al., 1997) fused to the Fc region of human immunoglobulin G, and stained a single band of 180kD molecular weight on Western blot (Poliak et al., 1999); in our staining it recognized the paranodes of mouse sciatic nerve, identical to previous descriptions (Poliak et al., 1999, Poliak et al., 2003, Ogawa et al., 2010).
Behavior Testing
Three-month-old Kcnq2-mutant (Kcnq2fl/fl//Pax3-Cre; 5 males and 4 females) and their littermates (Kcnq2+/+//Pax3-Cre; 5 males and 4 females), derived from 3 litters were studied. Mechanical allodynia was measured using Chaplan’s up-down threshold method (Chaplan et al., 1994, Hubbard and Winkelstein, 2005, Lee et al., 2008, Quinn et al., 2010). Mice were confined in plexiglass enclosure placed on a wire-mesh platform and allowed to acclimate for at least 30 minutes before each test. Three rounds of testing were performed over three consecutive days, with each round comprised of five stimulations of either the right or the left mid-plantar hindpaw in a random order, with a series of ascending von Frey filament strength (0.4 g, 0.6 g, 1.0 g, 1.4 g, 2.0 g; Stoelting Co.) held perpendicular for 6–8 seconds against the skin with enough force to cause slightly buckling. A positive response is recorded for sharp paw withdrawal or if immediate flinching was observed upon removal of filament. Ambulation was considered an ambiguous response and in such case the stimulus was repeated. If two consecutive filament strengths elicited a withdrawal response, the lower of the two filament strengths was recorded as the threshold. Any mouse that failed to display a response with the highest filament strength was recorded as having a threshold of 2.0 g. Testing in the opposite direction (descending filament strength) was also performed during each round to confirm the withdrawal threshold. The average threshold of the three rounds was recorded for each mouse.
The thermal nociceptive response was assessed using a paw thermal stimulator system (UARDG, University of California San Diego; otherwise referred to as Hargreaves’ apparatus) as previously described (Hargreaves et al., 1988, Dirig et al., 1997). Four rounds were performed over four consecutive days. Briefly, the mice were allowed to acclimate on the glass plate of the apparatus (maintained at 30°C) for at least 30 minutes, then either the left or the right mid-plantar hindpaw was randomly heated with a thermocouple set at 5 amperes. A timer is automatically started with the thermal source, and response latency is defined as the time required for the hindpaw to show an abrupt withdrawal (maximum 20 seconds). Paw withdrawal is automatically detected by an array of photodiode motion sensors mounted on the stimulus tower that stops the timer and terminates the stimulus. Stimulus current is monitored continuously. Six trials were performed during each round, with a minimum of 5 minutes between each trial to allow the hindpaws to return to normothermic baseline (Dirig et al., 1997). The average threshold of the four rounds was recorded for each mouse.
Motor function was measured with a rotarod apparatus (Ugo Basile, Stoelting Co.) as previously described (Wood et al., 2005, Oliveira et al., 2006). Briefly, the rotarod has a 3-cm diameter rotating rod raised 16 cm above a platform and divided into five sections for testing multiple mice simultaneously. Mice were acclimated on the first day by allowing them to run on the rotarod with the slowest rotation speed. Three rounds of testing were performed during the three subsequent days, with three trials during each round. For each trial, mice were placed on the rotarod, and the rotation speed was gradually increased from 4 to 40 rpm over the course of 5 minutes. Each trial ended when mice fell off (maximum 300 seconds), and the latency to fall was recorded for each trial. Test was considered valid if mice ran forward on the rotarod for at least 10 seconds. Mice were given 1-hour rest between each trial. The average time to fall of each mouse was used as the outcome.
Whole cell patch recording
DRG neurons were dissected and cultured from three-month-old Kcnq2 mutant mice of either sex (Kcnq2fl/fl//Pax3-Cre; n=5) and their littermate controls (Kcnq2+/+//Pax3-Cre; n=5) from 2 litters, as previously described (Malin et al., 2007). Lumbar DRGs were rapidly removed, transferred to ice-cold Ca2+/Mg2+-free HBSS (GIBCO), then incubated first in papain solution (60U papain; Worthington), 3μl saturated NaHCO3, 1 mg L-cysteine free base, 1.5 ml Ca2+/Mg2+-free HBSS (GIBCO) for 10 minutes at 37°C, then in collagenase/dispase solution (0.1 U/ml collagenase 0.8 U/ml dispase (Roche), 3 ml Ca2+/Mg2+-free HBSS) for 1 hour at 37°C. Neurons were dissociated by trituration using fire-polished glass Pasteur pipettes, suspended in F12 medium (GIBCO) containing 10% FCS (Invitrogen) and 1% penicillin/streptomycin (Invitrogen), then plated onto laminin/poly-D-lysine (Beckton Dickson) coated coverslips (Fisher). DRG neurons adhered to coverslips and were maintained in culture for 12–48 hours after plating at 37°C prior to recording.
Whole cell patch-clamp techniques (Hamill et al., 1981, Lancaster et al., 2001) were employed with an Axopatch 200B amplifier and Axon Instruments PCLAMP 9 software (pClamp, http://www.moleculardevices.com/products/software/pclamp.html, RRID:rid_000085). Patch pipettes (1–4 MΩ) were fabricated from glass capillaries (MTW150F-4, World Precision Instruments). Pipettes were filled with a variant of a solution described previously (Ikeda et al., 1986), with a composition of (in mM) 140 KCl, 2 MgATP, 10 N-[2-hydroxyethyl] piperazine-N9-[2-ethanesulfonic acid] (HEPES), 11 ethylene glycolbis(b-aminoethyl ether)-N,N,N9,N9-tetraacetic acid (EGTA), and 2 CaCl2; titrated to pH 7.3 with KOH, and to 314 mOsm with sucrose. Pipette voltage offset was neutralized prior to the formation of a giga-seal. Membrane input resistance (Rin), series resistance (Rs), and capacitance (Cm) were determined from current transients elicited by 5 mV depolarizing steps from a holding potential of −60 mV, delivered using the Membrane Test application of PCLAMP9. Criteria for cell inclusion in the study were as follows: Rs ≤ 10 MΩ, Rin ≥ 100 MΩ, and stable recording during the entire experiment. Cover slips were superfused (2–4 ml/min) continuously during recording at 34–36°C extracellular solution (composition in mM: 10 glucose, 140 NaCl, 3 KCl, 0.6 MgCl2, 2.5 CaCl2, 10 HEPES, titrated to pH 7.4 with TRIS base, to 325 mOsm with sucrose if needed). Tetraethylammonium (TEA) was fully dissolved in the extracellular solution prior to application; 3 mM TEA was applied for at least 2 minutes before determining its effects on membrane properties. To prevent bias in cell selection or analysis, the electrophysiologist was blinded to genotypes of the cells (control versus Kcnq2-null) during the experiments and measurements of cell properties. A total of 25 neurons from mutant mice, and 25 neurons from control littermates, were recorded and analyzed.
Statistical Tests
Other than the following exceptions, all comparisons between two samples were explored using unpaired two-samples Student’s t-test, with equal variance first established with the equality of variance test. All statistical tests were performed with SAS 9.2 (Statistical Analysis System, http://www.sas.com/en_us/software/sas9.html, RRID:nif-0000-31484), with all data given as mean ± standard error of mean (SEM). Comparison of Kv7.2- and Kv7.3-positive nodes in different peripheral nerves was tested with two-way ANOVA; motor function results over the course of nine sessions were analyzed with repeated measures ANOVA; two-way repeated measures ANOVA was used to compare mutant and control neurons before and after 3 mM TEA across multiple rheobases, followed by the Tukey test to identify specific significant differences. The minimum a priori criteria for statistical significance is p < 0.05.
RESULTS
Generation of conditional Kcnq2-null mice
We generated sensory neurons lacking Kcnq2 expression using the Cre-Lox system (Sauer and Henderson, 1988, Nagy, 2000). Mice containing a floxed allele of Kcnq2 (Fig. 1A) were crossed with mice expressing a Pax3-Cre transgene, which is expressed by cells derived from the neural crest (Li et al., 2000). To determine whether Pax3-Cre deleted exons 3–5 of Kcnq2, we examined three Kcnq2fl/fl//Pax3-Cre mice by amplifying genomic DNA with PCR and appropriate primers (Fig. 1A–B). As shown in Figure 1C, while both the floxed and the recombined Kcnq2 alleles were detected in the DRG and trigeminal ganglia, only the floxed allele was detected in the liver, indicating that the Kcnq2 allele was being selectively deleted in neural-crest-derived tissues.
We examined the expression of the Pax3-Cre by crossing the mice with Rosa26 lacZ reporter mice, in which Cre removes a floxed stop cassette so that lacZ is expressed (Soriano, 1999). Using litters that contained mice of the informative genotypes, we stained the brain, spinal cord, and DRG for beta-galactoside activity with X-gal as the chromogen. None of the samples taken from the control animals showed X-gal staining, whereas in Pax3-Cre//Rosa26 mice, there was robust staining in much of the cerebellum, regions of the cerebrum (including the cortex and the thalamus), the dorsal part of the spinal cord (including the dorsal roots), and the DRG (data not shown). The ventral part of the spinal cord and the ventral roots were unstained, suggesting that motor neurons do not express Pax3-Cre. Semi-thin sections demonstrated that virtually all DRG neurons from the Pax3-Cre-positive mice showed X-gal staining (Fig. 2), indicating that DRG neurons and/or their embryonic precursors express Pax3-Cre.
Figure 2. Sensory neurons expressed Pax3-Cre.
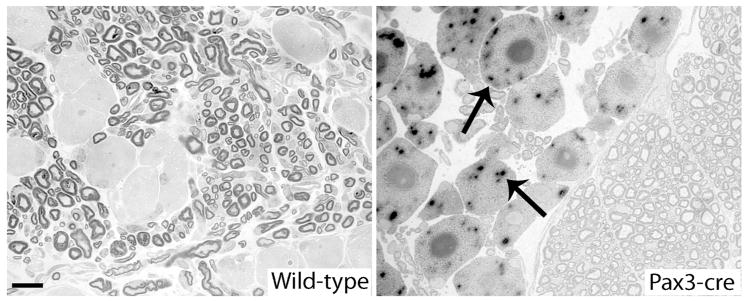
These are digital images of 1 μm thick epoxy sections of DRG neurons from Pax3-Crepositive (n=3) and -negative (n=3) mice that also expressed the Rosa26 reporter gene. The DRGs had been labeled with X-gal. Almost all of the Pax3-Cre positive neurons showed X-gal staining (two are indicated with arrows), whereas none of the neurons from control mice showed X-gal staining. Scale bar: 20 μm.
Primary sensory neurons in conditional Kcnq2-null mice lacked Kv7.2
To determine whether Pax3-Cre resulted in the loss of Kv7.2 expression in sensory neurons, we examined three mutants (Kcnq2fl/fl//Pax3-Cre) and three control littermates (Kcnq2+/+//Pax3-Cre). We immunostained unfixed teased nerve fibers from femoral sensory and motor branches, and from dorsal and ventral roots, as well as sections of the lumbar DRG, for Kv7.2 or Kv7.3, combined with a mouse monoclonal antibody against voltage-gated Na+ channels (panNav). As shown in Figure 3, in the control mice, all nodes of Ranvier of both the motor and sensory branches of the femoral nerve were Kv7.2- and panNav-positive, agreeing with our previously published results (Devaux et al., 2004, Pan et al., 2006). In mutant animals, all nodes were panNav-positive, but none of the nodes of the femoral sensory branch, and only some nodes of the femoral motor branch, were Kv7.2-positive. The lack of nodal staining of the Kcnq2-null sensory axons supports for the specificity of the Kv7.2 antiserum, which also selectively labeled HeLa cells transiently transfected to express human Kv7.2, but did not label transfected cells expressing Kv7.3 (data not shown). Similarly, none of the nodes in the dorsal roots (which are purely sensory), and all of the nodes in the ventral roots (which are purely motor), were Kv7.2-positive (Fig 4). Another Kv7.2 antiserum (provided by Dr. Jérôme Devaux) gave similar results (data not shown).
Figure 3. Selective loss of Kcnq2 from the nodes of sensory axons.

These are digital images of unfixed teased fibers, double-labeled for Kv7.2 (red) and voltage-gated sodium channels (panNav; green). In the femoral sensory branch (columns A and B), Kv7.2 is found at all nodes of Ranvier (arrows) in control animals, but is not detected at any nodes (arrowheads) in Kcnq2-null animals. In the femoral motor branch (columns C and D), Kv7.2 is found at all nodes of Ranvier (arrows) in control animals, but is not detected at some nodes (arrowheads) in Kcnq2-null animals. Scale bar: 10 μm.
Figure 4. Selective loss of Kcnq2 from the nodes of sensory axons.
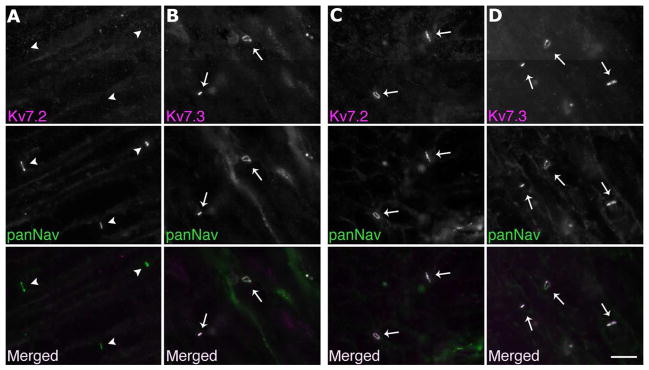
These are digital images of unfixed mouse teased nerves, double-labeled for either Kv7.2 or Kv7.3 (red) and panNav (green). In the dorsal roots of mutant animals (columns A and B), Kv7.2 is not detected at any node (arrowheads), whereas Kv7.3 is found at every node (arrows). In the ventral roots of mutant mice (columns C and D), note that all nodes are Kv7.2- and Kv7.3-positive (arrows). Scale bar: 10 μm.
We also immunostained sections of lumbar DRG and trigeminal ganglia with the Kv7.2 antiserum. As shown in Figure 5, in control animals, the Kv7.2 antiserum predominately labeled larger diameter DRG neurons (most of which were panNav-negative), while the panNav antibody primarily labeled smaller diameter neurons. In contrast, in the mutants, we did not detect robust Kv7.2 staining in any neurons, while the panNav antibody retained its staining pattern. Notably, similar to the teased fibers, nodes in the Kcnq2-null DRG were Kv7.2-negative. We also found similar results in the trigeminal ganglia (data not shown), with a loss of Kv7.2-immunostaining in the neurons and nodes of mutant animals. Thus, we conclude that Pax3-Cre deleted Kcnq2 from virtually all primary sensory neurons.
Figure 5. Primary sensory neurons of Kcnq2-null mice do not express Kv7.2.

These are images of unfixed sections of lumbar DRG, double-labeled with a rabbit antiserum against Kv7.2 (red) and a panNav monoclonal antibody (green). In control DRG (column A), the Kv7.2 antiserum labels large diameter neurons, one of which is indicated (arrow); these are relatively unlabeled by the panNav antibody. In contrast, in Kcnq2-null DRG (column B), the Kv7.2 antiserum does not label large neurons (arrow). In both control and Kcnq2-null DRG, the panNav monoclonal labels small neurons, two of which are indicated (arrowhead). In addition, all nodes of control neurons are doublelabeled by both Kv7.2 and panNav (some enclosed within a circle), while none of the nodes of mutant neurons are Kv7.2-positive. Scale bar: 20 μm.
In vitro studies have shown that Kv7.2 and Kv7.3 can form heteromeric channels, and that Kv7.2 increases cell membrane expression of Kv7.3 (Wang et al., 1998, Selyanko et al., 2001, Shah et al., 2002, Maljevic et al., 2003, Gomez-Posada et al., 2010) Because Kv7.2 and Kv7.3 are co-localized at nodes in vivo (Pan et al., 2006, Schwarz et al., 2006), we investigated whether nodal expression of Kv7.3 requires Kv7.2. As shown in Figures 4 and 6, all nodes of mutant mice showed robust Kv7.3 immunostaining – the femoral motor and sensory branches, as well as the dorsal and ventral roots. Thus, nodal expression of Kv7.3 does not require Kv7.2.
Figure 6. Kv7.3 is found at all nodes in Kcnq2-null mice.

These are digital images of unfixed mouse teased nerves, double-labeled for Kv7.3 (red) and panNav (green). In the femoral sensory (columns A and B) and motor branches (columns C and D) of both mutant and control mice, all panNav positive nodes are also Kv7.3-positive (arrows). Scale bar: 10 μm.
To quantify these results, we calculated the proportion of Kv7.2- or Kv7.3-positive nodes in each of the four nerve branches (femoral sensory, femoral motor, ventral root, and dorsal root) out of all panNav-positive nodes. As shown in Figure 7, the proportion of Kv7.2-positive nodes in the dorsal root (1.4 ± 0.06%), femoral sensory branch (0.48 ± 0.48%), and femoral motor branch (47 ± 1.6%) of the mutant animals were significantly less than in control littermates (99 ± 0.55%, 98 ± 0.53%, 96 ± 0.95% respectively). As expected, the proportion of Kv7.2-positive nodes in the ventral root is not different between the mutants (99 ± 0.60%) and the controls (97 ± 0.41%), and the proportions of Kv7.3-positive nodes in all four nerve branches was found to be nearly 100% in both mutant and control mice.
Figure 7. Selective loss of Kcnq2, but not Kcnq3, from the nodes of sensory axons.

The bars represent average proportion of panNav-positive nodes that are Kv7.2- (panel A) or Kv7.3- (panel B) positive from the indicated source (n=3 for all samples). Note that ~0% of nodes in the dorsal root and femoral sensory branch, and 50% in the femoral motor branch, were Kv7.2-positive. Error bars represent SEM; * p<0.05; ** p<0.01 (twoway ANOVA).
We examined the structure of Kcnq2-null myelinated axons. Teased fibers from mutant sciatic nerves (in which approximately 20% of myelinated axons are motor and the remaining are sensory; (Schmalbruch, 1986), were immunostained for Kv7.2 and either panNav, Caspr, or Kv1.1. As shown in Figure 8, the localization of selected nodal (panNav), paranodal (Caspr) and juxtaparanodal (Kv1.1) components were the same for myelinated axons from either Kv7.2-positive (motor axons) or Kv7.2-negative (sensory axons) nodes. The motor and sensory axons of the femoral nerves and dorsal roots of the mutant animals appeared normal in semi-thin cross-sections (data not shown). Longitudinal thin sections of femoral sensory nerves were visualized by electron microscopy. The paranodal loops, nodal microvilli, and nodal axolemma all appeared normal (data not shown).
Figure 8. The molecular components of nodes, paranodes and juxtaparanodes are maintained in Kcnq2-null sensory axons.

These are digital images of teased fibers from unfixed mutant sciatic nerves, doublelabeled for Kv7.2 (red) and either panNav (column A), Caspr (column B), or Kv1.1 (column C); all green. The selected nodal (panNav), paranodal (Caspr) and juxtaparanodal (Kv1.1) components are the same for myelinated axons with Kv7.2- positive (motor axons) or Kv7.2-negative (sensory axons) nodes. Scale bar: 10 μm.
Finally, because we had previously demonstrated that Remak fibers, but not nodes of Ranvier, express Kv7.5 (King and Scherer, 2012), we wished to determine whether this pattern of expression is retained in Kcnq2-mutant mice. As shown in figure 9, in both the femoral sensory and femoral motor branches, Remak fibers of both control and mutant mice showed robust Kv7.5 immunostaining. Nodes were Kv7.5-negative, indicating that Kv7.3 forms homomeric channels in the Kcnq2-null axons.
Figure 9. Remak fiber expression of Kv7.5 is maintained in Kcnq2 mutant mice.

These are digital images of unfixed mouse teased nerves, double-labeled for Kv7.5 (red) and panNav (green). In the femoral sensory (columns A and B) and motor (columns C and D) branches of both mutant and wild-type mice, all panNav-positive Remak fibers are Kv7.5-positive (chevrons), while all panNav-positive nodes are Kv7.5-negative (arrowheads). Scale bar: 10 μm.
Behavioral testing of mutant and control mice
We tested 9 mutants (Kcnq2fl/fl//Pax3-Cre) and 9 control littermates (Kcnq2+/+//Pax3-Cre) from three litters in a battery of tests. Mutant and control mice performed the rotarod test equally well (Fig. 10A), and showed similar performance improvement over the nine testing sessions across three consecutive days (data not shown). The average time to fall for all sessions was not statistically different between the mutants (163 ± 13 seconds) and controls (170. ± 6.1 seconds) groups (P = 0.62). To determine whether mutant mice have altered thermal hyperalgesia, we measured the withdrawal latency from noxious thermal stimuli using the Hargreaves’ test over four consecutive days. As shown in Figure 10B, the mutant mice on average had significantly shorter withdrawal latency (3.7 ± 0.16 seconds) than did controls (4.3 ± 0.16 seconds; p < 0.05). To determine whether mutant mice have altered mechanical allodynia, we measured pain withdrawal threshold using von Frey filaments over three consecutive days. As shown in Figure 10C, the mutants had a significantly lower average withdrawal threshold (0.65 ± 0.07 g) compared to the controls (0.93 ± 0.09 g; p < 0.05). Taken together, our behavior tests suggest that the loss of Kv7.2 expression leads to increased thermal hyperalgesia and mechanical allodynia.
Figure 10. Behavior testing of Kcnq2-null mice and control mice.
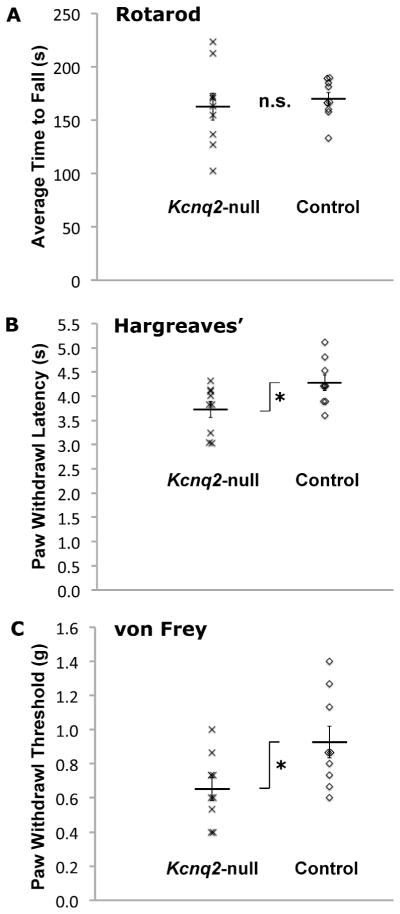
Panel A: Mutant mice (n=9) and their control littermates (n=9) were subjected to three consecutive days of testing on a rotarod (three tests per day), with motor performance measured by the time it took the mice to fall off the rotarod. There was no statistical difference in the overall average time to fall between the mutant and control animals.
Panel B: Both mutant mice (n=9) and their control littermates (n=9) were subjected to four consecutive days (six trials per day) of thermal hyperalgesia measured by the withdrawal latency after thermal stimulation (on a Hargreaves’ chamber). The overall average of all tests showed mutant animals have a statistically significant increase in thermal hyperalgesia.
Panel C: Both mutant mice (n=9) and control littermates (n=9) were subjected to three consecutive days of testing with a series of von Frey hair filaments (0.4 g, 0.6 g, 1.0 g, 1.4 g, 2.0 g; one up-and-down session per day). Mechanical allodynia determined by the lowest strength of hair filament capable of inducing a positive withdrawal response. The overall average of all tests showed mutant animals have a statistically significant increase in mechanical allodynia.
For all three behavior tests, error bars represent SEM; n.s. = no significant difference; * p<0.05 (unpaired two-sample Student’s t-test with equal variance).
Electrophysiological studies of DRG neurons
We examined the electrophysiological characteristics of acutely isolated DRG neurons from the lumbar DRG from 5 mutants (Kcnq2fl/fl//Pax3-Cre) and 5 controls (Kcnq2+/+//Pax3-Cre) from two litters, using whole cell-patch clamp recordings (Table 2). From each genotype, we recorded from 25 neurons of various sizes; the average cell size recorded from each genotype (as estimated from membrane capacitance) was not statistically different between the two groups (39 ± 3.3 pF versus 45 ± 4.1 pF, respectively). The initial resting membrane potential was also not statistically different between the two groups (−58 ± 2.5 mV versus −56 ± 1.6 mV, respectively).
Table 2.
Passive and active membrane properties of control and Kcnq2-null DRG neurons.
| control | Kcnq2-null | |
|---|---|---|
| Passive properties | ||
| Cm (pF) | 39 ± 3.3 | 45 ± 4.1 |
| Initial RMP (mV) | −58 ± 2.5 | −56 ± 1.6 |
| Pre-drug firing properties | ||
| Fast AHP peak (mV) | −83 ± 1.3 | −86 ± 0.97* |
| AP rheobase (nA) | 0.77 ± 0.23 | 0.81 ± 0.16 |
| 1 times rheobase (#APs) | 2.1 ± 0.38 | 2.9 ± 0.75 |
| 2 times rheobase (#APs) | 5.8 ± 1.3 | 11 ± 2.8 |
| 3 times rheobase (#APs) | 8.0 ± 1.3 | 17. ± 4.4* |
| Absolute stimulus (#APs) | 16 ± 1.6 | 25 ± 5.2* |
| Slow AHP peak (mV) | −74 ± 1.9 | −68 ± 1.2** |
| Post-drug firing properties | ||
| Fast AHP peak (mV) | −82 ± 0.97 | −84 ± 1.21 |
| AP rheobase (nA) | 0.39 ± 0.07 | 0.88 ± 0.19* |
| 1 times rheobase (#APs) | 4.0 ± 1.1 | 2.5 ± 0.66 |
| 2 times rheobase (#APs) | 11 ± 2.0 | 9.2 ± 2.2 |
| 3 times rheobase (#APs) | 13 ± 2.2 | 16 ± 2.9 |
| Absolute stimulus (#APs) | 21 ± 2.2 | 23 ± 3.9 |
| Slow AHP peak (mV) | −66 ± 1.2 | −67 ± 1.6 |
Twenty-five DRG neurons from five animals were recorded in the control and Kcnq2-null groups. Values are means ± SEM. Cm = membrane capacitance; RMP = resting membrane potential; AHP = afterhyperpolarization potential; fast AHP peak = AHP undershoot peak magnitude after single 3-ms stimulus; AP rheobase = either the minimum amount of current required to evoke a single AP, or 0.1 nA (whichever one was smallest); 1, 2, and 3 times rheobase = the number of APs fired by a DRG neuron during a 500-ms depolarizing current step of a magnitude 1, 2, or 3 times its rheobase, respectively; absolute = the maximum number of APs fired in response to a single 500-ms depolarizing current step of 0.1–0.9 nA magnitude (in 0.1-nA increments); slow AHP peak = AHP undershoot magnitude after 500-ms stimulus at 0.4nA. Values of Cm of the control DRG neurons were compared to mutant neurons using unpaired 2-sample Student’s t-test with equal variance; AHP and AP rheobase were compared with 2-way ANOVA with Tukey test; numbers of APs were compared with two-way repeated measures ANOVA.
p<0.05;
p<0.01.
To investigate the fast and slow afterhyperpolarization (AHP) properties, we elicited single action potential (AP) with brief (3 ms) depolarizing current steps, and measured the peak undershoot (most negative potential during the AHP), as shown schematically in Figure 11A. Prior to applying TEA, the average fast AHP peak undershoot of mutant DRG neurons was 3 mV more negative than control neurons (−86 ± 0.97 mV versus −83 ± 1.3 mV, respectively; p < 0.05). After applying 3 mM TEA, the fast AHP peak magnitudes were not significantly different between the mutant and control neurons (−84 ± 1.2 mV versus −82 ± 0.97 mV, respectively; p = 0.24) (Figure 11B). In contrast, prior to 3 mM TEA, the average slow AHP peak undershoot of mutant neurons was 6 mV more positive than that of control neurons (−68 ± 1.2 mV versus −74 ± 1.9 mV, respectively; p<0.01). After applying 3 mM TEA, the slow AHP peak undershoot of control neurons became 8 mV more positive (−66 ± 1.2 mV; p<0.01), while the slow AHP peak undershoot of Kcnq2-null neurons did not change significantly (−67 ± 1.6 mV; Fig. 11C). Because 3 mM TEA should selectively block primarily the Kv7.2 homomers and Kv7.2/Kv7.3 heteromers and not Kv7.3 homomers (Wang et al., 1998, Hadley et al., 2000, Lerche et al., 2000, Hadley et al., 2003), these results indicate that inhibiting Kv7.2 contributes to the decreased slow AHP peak undershoot observed in the mutant DRG neurons.
Figure 11. Kcnq2-null DRG neurons have a diminished slow afterhyperpolization (AHP) that is not affected by 3 mM TEA.

Panel A shows representative responses from a control DRG neuron given a brief (3 ms) depolarizing current step that generated a single action potential (AP), and a prolonged (500 ms) depolarizing current step that generated multiple APs, from which the magnitude of the fast and slow AHP peak undershoots, respectively, were measured.
Panel B shows that before 3 mM TEA, the average fast AHP peak undershoot of mutant neurons (n=25) was slightly more negative than that of control neurons (n=25). After applying 3 mM TEA, the fast AHP peak undershoot of both control and Kcnq2-null neurons did not change by a statistically significant amount.
Panel C shows that before 3 mM TEA, the average slow AHP peak undershoot of mutant neurons (n=25) was 6 mV more positive than that of control neurons (n=25). After applying 3 mM TEA for 2 minutes, the slow AHP peak undershoot of control neurons became 8 mV more positive, while the slow AHP peak undershoot of Kcnq2-null neurons did not change by a statistically significant amount. * p<0.05; ** p<0.01 (2-way ANOVA).
We examined the AP firing patterns and spike-frequency adaptation by injecting 500 ms depolarizing current steps into the DRG neurons. First, the threshold current (rheobase) for each neuron was determined using incremental (100 pA) 500 ms current steps (starting from 0.1 nA). Then we recorded the number of APs evoked by 500 ms depolarizing current steps of 1x, 2x, and 3x rheobase. To determine the responsiveness of DRG neurons to absolute (as opposed to relative threshold as described above) depolarizing stimuli, we also injected a series of 500 ms current steps from 0.4 nA to 3.6 nA (in increments of 0.4 nA). As shown in Figure 12, the Kcnq2-null neurons produced significantly more APs and exhibited less spike-frequency adaptation than did control neurons, and 3 mM TEA significantly increased the number of APs and decreased spike-frequency adaptation in control neurons, but not in Kcnq2-null neurons (numbers of AP from each group - control neurons: 1x rheobase 2.1, 2x rheobase 5.8, 3x rheobase 8.04, absolute 15.6; Kcnq2-null neurons: 1x rheobase 2.9, 2x rheobase 11.2, 3x rheobase 17.44, absolute 24.4; two-way repeated ANOVA ). In addition, 3 mM TEA decreased the rheobase of control neurons by 49% (Table 1; 0.39 ± 0.07 nA from 0.77 ± 0.23 nA; p < 0.05), which is also statistically different than mutant neurons after TEA (0.88 ± 0.19 nA; p < 0.05). In contrast, the rheobase of the mutant neurons was not affected by TEA (0.88 ± 0.19 nA from 0.81 ± 0.16 nA; p = 0.78). In summary, mutant DRG neurons displayed increased excitability and decreased spike-frequency adaptation, and this hyperexcitability could be replicated in control neurons by 3 mM TEA.
Figure 12. Kcnq2-null DRG neurons have decreased spike-frequency adaption that is not affected by 3 mM TEA.
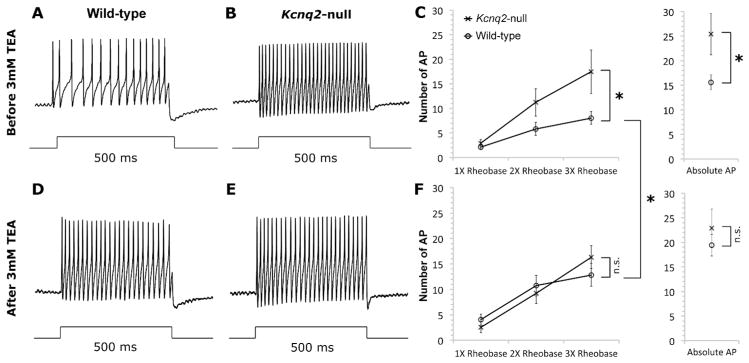
Single DRG neurons were injected with 500 ms depolarizing current steps of 1x, 2x, or 3x rheobase (A–F) from a holding level of between −50 to −60 mV before and after application of 3 mM TEA. Panels A–D show representative recordings of control (A&D) and mutant (B&E) neurons, before and after application of 3 mM TEA for 2 minutes. Panels C and F are summary diagrams showing the number of APs in mutant and control neurons, before and after applying TEA, respectively (* p<0.05; two-ways repeated measures ANOVA). Notably, after TEA application, control DRG neurons also showed a decrease in spike frequency adaption, and generated statistically similar numbers of APs at each rheobase as compared to mutant (n.s. = no significant difference; two-ways repeated measures ANOVA). In contrast, TEA application did not reduce spike-frequency adaptation of the mutant DRG neurons. In addition, we also injected 500 ms incremental current at set current step from 0.4nA to 3.6nA (in increments of 0.4nA) in order to determine the responsiveness of DRG neurons to absolute (as opposed to relative threshold as described above) depolarizing stimuli, and the spike frequency adaption was also significantly reduced in mutant DRG neurons in comparison to control neurons before TEA application; after TEA application the control neurons generated statistically similar number of APs in response to absolute stimuli as compared to mutant neurons.
DISCUSSION
Deleting Kcnq2 enabled us to investigate directly the role of Kv7.2 in sensory neurons. As expected, deleting Kv7.2 had no discernible effect on the structure of myelinated axons, or on the motor performance of the mutant animals. There were modest effects on both acute thermal and mechanical nociceptive behaviors, and on the electrophysiological properties of sensory neurons. Without a direct experimental test, we cannot exclude the possibility that the lack of KCNQ2 in developing neurons contributes to the observed phenotype or that compensation may have diminished the effects of deleting Kcnq2.
Kv7.3 nodal expression does not depend on Kv7.2
Our data confirm that Kv7.2 and Kv7.3 are found at nodes (Devaux et al., 2004, Pan et al., 2006, Schwarz et al., 2006), although we document an even greater extent of their colocalization. This finding suggests that Kv7.2/Kv7.3 heteromers are the main Kv7 channels at PNS nodes. The localization of Kv7.3 at Kcnq2-null nodes demonstrates that Kv7.3 surface expression does not require Kv7.2, in contrast to previous in vitro studies (Schwake et al., 2000, Gomez-Posada et al., 2010). Because both Kv7.2 and Kv7.3 contain an ankyrin-G binding domain (Pan et al., 2006), it is reasonable to expect that Kv7.3 could be selectively retained at nodes and AIS. Indeed, the deletion of the ankyrin-G binding motif in Kv7.2 alone does not alter the AIS localization of Kv7.2/Kv7.3 heteromers (Rasmussen et al., 2007). In addition, the normal molecular components of nodes (panNav), paranodes (Caspr), and juxtaparanodes (Kv1.1), as well as ultrastructure of Kcnq2-null sensory myelinated axons, shows that these features do not depend on Kv7.2. Finally, we found that Kv7.5 expression in Remak bundles is maintained in Kcnq2-null nerves. While Kv7.5 and Kv7.3 can form heteromeric channels in vitro (Schroeder et al., 2000), we did not detect Kv7.5-immunoreactivity at Kcnq2-null nodes, suggesting that in our mutant animals, the nodal Kv7 channels are comprised of Kv7.3 homomers.
Kv7.2 contributes to the regulation of neuronal excitability
Kv7 channels activate at subthreshold potentials and do not become inactivated, thereby contributing to the regulation of neuronal excitability (Brown and Passmore, 2009). Kv7 blockers (linopridine or XE991), dominant-negative Kv7.2 mutants, or decreased Kv7.2 expression have all been shown to increase excitability (decreased spike-frequency adaptation and/or increased number of action potentials) of hippocampal neurons (Aiken et al., 1995, Yue and Yaari, 2004, Gu et al., 2005, Peters et al., 2005, Shah et al., 2008) and of somatic and visceral sensory neurons (Passmore et al., 2003, Rivera-Arconada and Lopez-Garcia, 2005, Wladyka and Kunze, 2006, Wladyka et al., 2008, Zheng, 2013). A Kv7 enhancer (retigabine) produces the opposite effects (Lerche et al., 2000, Brueggemann et al., 2007). In isolated rat peripheral nerves, retigabine slows axonal conduction, and these effects can be reversed by application of linopridine or TEA (Devaux et al., 2004). Schwarz et al. (2006) showed that XE991 both abolishes the slow accommodation to the depolarization and the post-depolarization undershoot of action potential at nodes, as well as increases repetitive firing and decreases spike-frequency adaptation in rat motor axons. Taken together, these results indicate that Kv7 channels regulate both neuronal and axonal activity.
Our analysis of Kcnq2-null DRG neurons confirmed and extended these prior works. By using TEA at a Kv7.2-specific concentration (Wang et al., 1998, Hadley et al., 2000, Lerche et al., 2000, Hadley et al., 2003), we showed that Kv7.2 contributes to the spike-frequency adaptation of sensory neurons. Because TEA did not further decrease spike-frequency adaptation in Kcnq2-null neurons, decreased Kv7.2 activity is the most parsimonious explanation for the increased excitability of the Kcnq2-null neurons.
Classically, AHP can be subdivided into three phases: fast (1–5 ms), medium (50–200 ms), and slow (500 ms to several seconds) AHP (Madison and Nicoll, 1984, Storm, 1990, Gu et al., 2005). Because the slow activation speed of the M channels (tens of milliseconds), they do not contribute materially to the fast AHP (Brown and Passmore, 2009), which is instead considered to be mediated by the Big Potassium (BK) family of potassium channels (Storm, 1990). Indeed, our Kcnq2-null neurons exhibited only slightly more negative fast AHP amplitude compared to control neurons, and the application of TEA did not statistically change fast AHP amplitudes in both mutant and control neurons. The negative TEA results indicate that Kv7.2 does not play an appreciable role in the fast AHP. While we cannot explain why the fast AHP amplitude of our Kcnq2-null neurons was slightly more negative compared to the control neurons, the fact that Kv7.2-specific concentration of TEA did not change the fast AHP amplitude of the control neurons suggest that this difference may not be due to the lack of Kv7.2 activity.
On the other hand, the identity of the channel(s) that mediates the slow AHP is still uncertain, but decreases in Kv7.2 or Kv7.5 activity has been shown to reduce slow AHP in mouse hippocampal neurons (Tzingounis and Nicoll, 2008, Tzingounis et al., 2010). Our Kcnq2-null DRG neurons also displayed a decrease in slow AHP amplitude compared to control neurons, and 3 mM TEA was able to reduce the slow AHP amplitude of control neurons, while having no effect on mutant neurons. While a previous study have shown that 10 μM XE991 application in hippocampal had actually enhanced slow AHP, other experiments showed that muscarine, which suppresses Kv7 channels (Brown and Adams, 1980), was able to inhibit slow AHP in the supraoptic neurons (Ghamari-Langroudi and Bourque, 2004, Hu and Mooney, 2005), which has also been shown to express Kv7 (Zhang et al., 2009). Therefore, while the exact contribution of Kv7 channels to slow AHP remained uncertain, our results raise the possibility that Kv7.2 activity does contribute, at least in part, to slow AHP in DRG neurons.
Increased mechanical allodynia and thermal hyperalgesia in Kcnq2-null mice
The results regarding the role of Kv7 channels on mechanical allodynia and thermal hyperalgesia are inconsistent. In one study, retigabine increases the tail withdrawal threshold to noxious thermal stimuli at a dose-dependent manner (Dost et al., 2004), but because this effect was not reversed by the co-administration of linopridine, it may be due to non-Kv7-specific effect of retigabine. This finding also conflicts with another study using different methodology, in which retigabine did not affect the withdrawal response from noxious thermal stimuli (Blackburn-Munro and Jensen, 2003). Similarly, intraplantar injections of XE991 into the rat hindpaws did not induce thermal hyperalgesia or mechanical allodynia (Linley et al., 2008). The above studies shared the important limitation that these enhancers and blockers likely act on most or all Kv7 subunits, and potentially on other channels (for example GABAA (Otto et al., 2002)). Unmyelinated axons express Kv7.5 (King and Scherer, 2012), which could also be the site of these pharmacological agents.
In our current study we sought to minimize these confounding factors by analyzing a type of myelinated sensory axons that lacks Kv7.2 - the A-delta fibers. While technical limitations do not allow us to completely differentiate A-delta fiber mediated nociception from C-fiber mediated nociception, previous studies suggest that withdrawal reflex behavior from both acute noxious thermal stimuli (Price and Dubner, 1977, Dubner and Bennett, 1983, Yeomans and Proudfit, 1996, Hargreaves et al., 1998, Cuellar et al., 2010) and punctate mechanical stimuli (Dubner and Bennett, 1983, Koltzenburg et al., 1993, Ziegler et al., 1999) are both primarily mediated by A-delta fibers. Specifically, Yeomans and Proudfit (1996) found that radiant heating of rat hindpaw at a high rate of 6.5°C/second (°C/s) for 6 seconds primarily evokes A-delta fiber response, while a low rate of 0.9°C/s for 20 seconds primarily activates C-fibers response; notably, the C-fiber response at either heating rate did not begin until 5–8 seconds after onset of heating. Because the thermocouple of our Hargreaves apparatus had been shown to raise rat hindpaw temperature from 30°C to 49°C in 5 seconds (3.8°C/s) (Dirig et al., 1997), and because the average withdrawal latency of both mutant and control animals in our Hargreaves’ test were less than 5 seconds (3.7 s and 4.3 s, respectively), our results suggest that the increased thermal hyperalgesia behavior of our Kcnq2-null mutant animal was, at least in part, due to increased A-delta fiber activity. Furthermore, Ji et al. (2007) reported that rats treated with spinal nerve ligation displayed decreased mechanical threshold of A-delta fibers, but not of C fibers, and simultaneously exhibited increased mechanical allodynia (as tested with von Frey filaments), suggesting that the increased mechanical allodynia of our Kcnq2-null animals may also be due to increase Adelta fiber activity. Lastly, Passmore et al. (2012) recently showed that M-current inhibition by XE991 (at concentration that is relatively selective against Kv7.2) enhanced the response of A-delta fibers, but not C fibers, to noxious heat stimulation. Taken together, the increased thermal hyperalgesia and mechanical allodynia exhibited by our Kcnq2-null mice suggest that the lack of Kv7.2 expression in the A-delta fibers may produce increased acute thermal and mechanical nociception. Because our rotarod test provided no evidence of different motor performance between our mutant and control animals, the increased nociceptive responses observed in the Kcnq2-null mice is unlikely to be a consequence of altered motor behavior. However, because our X-gal stainings indicate that Pax3-Cre expression may also be present in regions of the brain, the possibility exists that the loss of Kv7.2 expression within the central nervous system may also have played a role in the increased thermal hyperalgesia and mechanical allodynia exhibited by our Kcnq2 mutant mice.
Taken together, our work raises the possibility that decreasing Kv7.2 activity can increase sensory neuronal excitability, and lead to increased perception of mechanical and acute thermal pain.
Acknowledgments
Support/Grant Information: This work was supported by the NIH R01NS43174, NIH/NINDS NS50220, and the Binational Science Foundation. Elior Peles is the Incumbent of the Hanna Hertz Professorial Chair for Multiple Sclerosis and Neuroscience.
We like to thank Drs. Edward Cooper, Jérôme Devaux, and Virginia Lee for cDNAs and antisera, Dr. Gordon Barr for loaning us the Hargreaves’ Apparatus, Dr. Ted Abel for loaning us the rotarod, Dr. Beth Winkelstein for advice regarding the von Frey hair filament test, Dr. Marc Dichter for the use of their electrophysiology rig, and Dr. Jian Li for help with the histology.
Footnotes
Conflict of interest: The authors declare that they do not have any conflicts of interest.
Role of authors: All authors had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Study concept and design: CHK, EP, and SSS. Acquisition of data: CHK, EL, and DS. Analysis and interpretation of data: CHK, EL, DS, EP, and SSS. Drafting of the manuscript: CHK and SSS. Critical revision of the manuscript for important intellectual content: CHK, EL, DS, EP, and SSS. Statistical analysis: CHK. Obtained funding: EP and SSS. Administrative, technical, and material support: CHK, EL, DS, EP, and SSS. Study supervision: EP and SSS.
References
- Aiken SP, Lampe BJ, Murphy PA, Brown BS. Reduction of spike frequency adaption and blockade of M-crrent in rat CA1 pyramidal neurones by linopirdine (DuP 996), a neurotransmitter release enhancer. Br J Pharmacol. 1995;115:1163–1168. doi: 10.1111/j.1476-5381.1995.tb15019.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arroyo EJ, Bermingham JRJ, Rosenfeld MG, Scherer SS. Promyelinating Schwann cells express Tst-1/SCIP/Oct-6. J Neurosci. 1998;18:7891–7902. doi: 10.1523/JNEUROSCI.18-19-07891.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blackburn-Munro G, Jensen BS. The anticonvulsant retigabine attenuates nociceptive behaviours in rat models of persistent and neuropathic pain. European Journal of Pharmacology. 2003;460:109–116. doi: 10.1016/s0014-2999(02)02924-2. [DOI] [PubMed] [Google Scholar]
- Brown DA, Adams PR. Muscarinic suppression of a novel voltage sensitve K+ current in a vertebrate neuron. Nature. 1980;283:673–676. doi: 10.1038/283673a0. [DOI] [PubMed] [Google Scholar]
- Brown DA, Passmore GM. Neural KCNQ (Kv7) channels. Br J Pharmacol. 2009;156:1185–1195. doi: 10.1111/j.1476-5381.2009.00111.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brueggemann LI, Moran CJ, Barakat JA, Yeh JZ, Cribbs LL, Byron KL. Vasopressin stimulates action potential firing by protein kinase C-dependent inhibition of KCNQ5 in A7r5 rat aortic smooth muscle cells. Am J Physiol Heart Circ Physiol. 2007;292:H1352–1363. doi: 10.1152/ajpheart.00065.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caminos E, Garcia-Pino E, Martinez-Galan JR, Juiz JM. The potassium channel KCNQ5/Kv7.5 is localized in synaptic endings of auditory brainstem nuclei of the rat. J Comp Neurol. 2007;503:363–378. doi: 10.1002/cne.21497. [DOI] [PubMed] [Google Scholar]
- Chaplan SR, Bach FW, Pogrel JW, Chung JM, Yaksh TL. Quantitative assessment of tactile allodynia in the rat paw. J Neurosci Methods. 1994;53:55–63. doi: 10.1016/0165-0270(94)90144-9. [DOI] [PubMed] [Google Scholar]
- Cooper EC, Harrington E, Jan YN, Jan LY. M channel KCNQ2 subunits are localized to key sites for control of neuronal network oscillations and synchronization in mouse brain. J Neurosci. 2001;21:9529–9540. doi: 10.1523/JNEUROSCI.21-24-09529.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuellar JM, Manering NA, Klukinov M, Nemenov MI, Yeomans DC. Thermal nociceptive properties of trigeminal afferent neurons in rats. Mol Pain. 2010;6:39. doi: 10.1186/1744-8069-6-39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dedek K, Kunath B, Kananura C, Reuner U, Jentsch TJ, Steinlein OK. Myokymia and neonatal epilepsy caused by a mutation in the voltage sensor of the KCNQ2 K+ channel. Proc Natl Acad Sci U S A. 2001;98:12272–12277. doi: 10.1073/pnas.211431298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Delmas P, Brown DA. Pathways modulating neural KCNQ/M (Kv7) potassium channels. Nat Rev Neurosci. 2005;6:850–862. doi: 10.1038/nrn1785. [DOI] [PubMed] [Google Scholar]
- Devaux JJ, Kleopa KA, Cooper EC, Scherer SS. KCNQ2 is a nodal K+ channel. J Neurosci. 2004;24:1236–1244. doi: 10.1523/JNEUROSCI.4512-03.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dirig DM, Salami A, Rathbun ML, Ozaki GT, Yaksh TL. Characterization of variables defining hindpaw withdrawl latency evoked by radiant thermal stimuli. J Neurosci Methods. 1997;76:183–191. doi: 10.1016/s0165-0270(97)00097-6. [DOI] [PubMed] [Google Scholar]
- Dost R, Rostock A, Rundfeldt C. The anti-hyperalgesic activity of retigabine is mediated by KCNQ potassium channel activation. Naunyn-Schmiedeberg’s archives of pharmacology. 2004;369:382–390. doi: 10.1007/s00210-004-0881-1. [DOI] [PubMed] [Google Scholar]
- Dubner R, Bennett GJ. Spinal and trigeminal mechanisms of nociception. Annu Rev Neurosci. 1983;6:381–418. doi: 10.1146/annurev.ne.06.030183.002121. [DOI] [PubMed] [Google Scholar]
- Farley FW, Soriano P, Steffen LS, Dymecki SM. Widespread recombinase expression using FLPeR (Flipper) mice. Genesis. 2000;28:106–110. [PubMed] [Google Scholar]
- Feltri ML, Scherer SS, Wrabetz L, Kamholz J, Shy ME. Mitogen-expanded Schwann cells retain the capacity to myelinate regenerating axons after transplantation into rat sciatic nerve. Proc Natl Acad Sci U S A. 1992;89:8827–8831. doi: 10.1073/pnas.89.18.8827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghamari-Langroudi M, Bourque CW. Muscarinic receptor modulation of slow afterhyperpolarization and phasic firing in rat supraoptic nucleus neurons. J Neurosci. 2004;24:7718–7726. doi: 10.1523/JNEUROSCI.1240-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman AM, Glasscock E, Yoo J, Chen TT, Klassen TL, Noebels JL. Arrhythmia in heart and brain: KCNQ1 mutations link epilepsy and sudden unexplained death. Science translational medicine. 2009;1:2ra6. doi: 10.1126/scitranslmed.3000289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gollan L, Salomon D, Salzer JL, Peles E. Caspr regulates the processing of contactin and inhibits its binding to neurofascin. J Cell Biol. 2003;163:1213–1218. doi: 10.1083/jcb.200309147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomez-Posada JC, Etxeberria A, Roura-Ferrer M, Areso P, Masin M, Murrell-Lagnado RD, Villarroel A. A pore residue of the KCNQ3 potassium M-channel subunit controls surface expression. J Neurosci. 2010;30:9316–9323. doi: 10.1523/JNEUROSCI.0851-10.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gu N, Vervaeke K, Hu H, Storm JF. Kv7/KCNQ/M and HCN/h, but not KCa2/SK channels, contribute to the somatic medium after-hyperpolarization and excitability control in CA1 hippocampal pyramidal cells. J Physiol. 2005;566:689–715. doi: 10.1113/jphysiol.2005.086835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Noda M, Selyanko AA, Wood IC, Abogadie FC, Brown DA. Differential tetraethylammonium sensitivity of KCNQ1-4 potassium channels. Br J Pharmacol. 2000;129:413–415. doi: 10.1038/sj.bjp.0703086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hadley JK, Passmore GM, Tatulian L, Al-Qatari M, Ye F, Wickenden AD, Brown DA. Stoichiometry of expressed KCNQ2/KCNQ3 potassium channels and subunit composition of native ganglionic M channels deduced from block by tetraethylammonium. J Neurosci. 2003;23:5012–5019. doi: 10.1523/JNEUROSCI.23-12-05012.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, Sigworth FJ. Improved patch-clamp techniques for high-resolution current recording from cells and cell free membrane patches. Pflugers Arch. 1981;391:85–100. doi: 10.1007/BF00656997. [DOI] [PubMed] [Google Scholar]
- Hargreaves K, Dubner R, Brown F, Flores C, Joris J. A new and sensitive method for measuring thermal nociception in cutaneous hyperalgesia. Pain. 1988;32:77–88. doi: 10.1016/0304-3959(88)90026-7. [DOI] [PubMed] [Google Scholar]
- Hargreaves KM, Swift JQ, Roszkowski MT, Bowles W, Garry MG, Jackson DL. Pharmacology of peripheral neuropeptide and inflammatory mediator release. Oral Surg Oral Med Oral Pathol. 1998;78:503–510. doi: 10.1016/0030-4220(94)90045-0. [DOI] [PubMed] [Google Scholar]
- Heidenreich M, Lechner SG, Vardanyan V, Wetzel C, Cremers CW, De Leenheer EM, Aránguez G, Moreno-Pelayo MÁ, Jentsch TJ, Lewin GR. KCNQ4 K+ channels tune mechanoreceptors for normal touch sensation in mouse and man. Nature Neuroscience. 2011;15:138–145. doi: 10.1038/nn.2985. [DOI] [PubMed] [Google Scholar]
- Hu B, Mooney DM. Burst firing induces a slow after hyperpolarization in rat auditory thalamus. Neurosci Lett. 2005;375:162–164. doi: 10.1016/j.neulet.2004.11.005. [DOI] [PubMed] [Google Scholar]
- Hubbard RD, Winkelstein BA. Transient cervical nerve root compression in the rat induces bilateral forepaw allodynia and spinal glial activation: mechanical factors in painful neck injuries. Spine (Phila Pa 1976) 2005;30:1924–1932. doi: 10.1097/01.brs.0000176239.72928.00. [DOI] [PubMed] [Google Scholar]
- Ikeda SR, Schofield GG, Weight FF. Na+ and Ca2+ currents of acutely isolated adult rat nodose ganglion cells. J Neurophysiol. 1986;55:527–539. doi: 10.1152/jn.1986.55.3.527. [DOI] [PubMed] [Google Scholar]
- Jentsch TJ. Neuronal KCNQ potassium channels: physiology and role in disease. Nat Rev Neurosci. 2000;1:21–30. doi: 10.1038/35036198. [DOI] [PubMed] [Google Scholar]
- Ji G, Zhou S, Kochukov MY, Westlund KN, Carlton SM. Plasticity in intact A delta- and C-fibers contributes to cold hypersensitivity in neuropathic rats. Neuroscience. 2007;150:182–193. doi: 10.1016/j.neuroscience.2007.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- King CH, Scherer SS. Kv7.5 is the primary Kv7 subunit expressed in C-fibers. J Comp Neurol. 2012;520:1940–1950. doi: 10.1002/cne.23019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Koltzenburg M, Lundberg LE, Torebjork HE. Dynamic and static components of mechanical hyperalgesia in human hairy skin. Pain. 1993;51:207–219. doi: 10.1016/0304-3959(92)90262-A. [DOI] [PubMed] [Google Scholar]
- Lancaster E, Oh EJ, Weinreich D. Vagotomy decreases excitability in primary vagal afferent somata. J Neurophysiol. 2001;85:247–253. doi: 10.1152/jn.2001.85.1.247. [DOI] [PubMed] [Google Scholar]
- Lang PM, Fleckenstein J, Passmore GM, Brown DA, Grafe P. Retigabine reduces the excitability of unmyelinated peripheral human axons. Neuropharmacology. 2008;54:1271–1278. doi: 10.1016/j.neuropharm.2008.04.006. [DOI] [PubMed] [Google Scholar]
- Lee KE, Davis MB, Winkelstein BA. Capsular ligament involvement in the development of mechanical hyperalgesia after facet joint loading: behavioral and inflammatory outcomes in a rodent model of pain. J Neurotrauma. 2008;25:1383–1393. doi: 10.1089/neu.2008.0700. [DOI] [PubMed] [Google Scholar]
- Lerche C, Scherer CR, Seebohm G, Derst C, Wei AD, Busch AE, Steinmeyer K. Molecular cloning and functional expression of KCNQ5, a potassium channel subunit that may contribute to neuronal M-current diversity. J Biol Chem. 2000;275:22395–22400. doi: 10.1074/jbc.M002378200. [DOI] [PubMed] [Google Scholar]
- Li J, Chen F, Epstein JA. Neural crest expression of Cre recombinase directed by the proximal Pax3 promoter in transgenic mice. Genesis. 2000;26:162–164. doi: 10.1002/(sici)1526-968x(200002)26:2<162::aid-gene21>3.0.co;2-r. [DOI] [PubMed] [Google Scholar]
- Linley JE, Rose K, Patil M, Robertson B, Akopian AN, Gamper N. Inhibition of M current in sensory neurons by exogenous proteases: a signaling pathway mediating inflammatory nociception. J Neurosci. 2008;28:11240–11249. doi: 10.1523/JNEUROSCI.2297-08.2008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madison DV, Nicoll RA. Control of the repetitive discharge of rat CA1 pyramidal neurons in vitro. J Physiol. 1984;354:319–331. doi: 10.1113/jphysiol.1984.sp015378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Malin SA, Davis BM, Molliver DC. Production of dissociated sensory neuron cultures and considerations for their use in studying neuronal function and plasticity. Nat Protoc. 2007;2:152–160. doi: 10.1038/nprot.2006.461. [DOI] [PubMed] [Google Scholar]
- Maljevic S, Lerche C, Seebohm G, Alekov AK, Busch AE, Lerche H. C-terminal interaction of KCNQ2 and KCNQ3 K+ channels. J Physiol. 2003;548:353–360. doi: 10.1113/jphysiol.2003.040980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nagy A. Cre recombinase: the universal reagent for genome tailoring. Genesis. 2000;26:99–109. [PubMed] [Google Scholar]
- Ogawa Y, Oses-Prieto J, Kim MY, Horresh I, Peles E, Burlingame AL, Trimmer JS, Meijer D, Rasband MN. ADAM22, a Kv1 channel-interacting protein, recruits membrane-associated guanylate kinases to juxtaparanodes of myelinated axons. J Neurosci. 2010;30:1038–1048. doi: 10.1523/JNEUROSCI.4661-09.2010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oliveira AM, Abel T, Brindle PK, Wood MA. Differential role for CBP and p300 CREB-binding domain in motor skill learning. Behav Neurosci. 2006;120:724–729. doi: 10.1037/0735-7044.120.3.724. [DOI] [PubMed] [Google Scholar]
- Otto JF, Kimball MM, Wilcox KS. Effects of the anticonvulsant retigabine on cultured cortical neurons: changes in electroresponsive properties and synaptic transmission. Mol Pharmacol. 2002;61:921–927. doi: 10.1124/mol.61.4.921. [DOI] [PubMed] [Google Scholar]
- Otto JF, Yang Y, Frankel WN, White HS, Wilcox KS. A spontaneous mutation involving Kcnq2 (Kv7.2) reduces M-current density and spike frequency adaptation in mouse CA1 neurons. J Neurosci. 2006;26:2053–2059. doi: 10.1523/JNEUROSCI.1575-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan Z, Kao T, Horvath Z, Lemos J, Sul JY, Cranstoun SD, Bennett V, Scherer SS, Cooper EC. A common ankyrin-G-based mechanism retains KCNQ and NaV channels at electrically active domains of the axon. J Neurosci. 2006;26:2599–2613. doi: 10.1523/JNEUROSCI.4314-05.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Passmore GM, Selyanko AA, Mistry M, Al-Qatari M, Marsh SJ, Matthews EA, Dickenson AH, Brown TA, Burbidge SA, Main M, Brown DA. KCNQ/M currents in sensory neurons: significance for pain therapy. J Neurosci. 2003;23:7227–7236. doi: 10.1523/JNEUROSCI.23-18-07227.2003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peles E, Nativ M, Lustig M, Grumet M, Schilling J, Martinez R, Plowman GD, Schlessinger J. Identification of a novel contactin-associated transmembrane receptor with multiple domains implicated in protein-protein interactions. EMBO J. 1997;16:978–988. doi: 10.1093/emboj/16.5.978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters HC, Hu H, Pongs O, Storm JF, Isbrandt D. Conditional transgenic suppression of M channels in mouse brain reveals functions in neuronal excitability, resonance and behavior. Nature Neuroscience. 2005;8:51–60. doi: 10.1038/nn1375. [DOI] [PubMed] [Google Scholar]
- Poliak S, Gollan L, Martinez R, Custer A, Einheber S, Salzer JL, Trimmer JS, Shrager P, Peles E. Caspr2, a new member of the neurexin superfamily, is localized at the juxtaparanodes of myelinated axons and associated with K+ channels. Neuron. 1999;24:1037–1047. doi: 10.1016/s0896-6273(00)81049-1. [DOI] [PubMed] [Google Scholar]
- Poliak S, Salomon D, Elhanany H, Sabanay H, Kiernan B, Pevny L, Stewart CL, Xu X, Chiu SY, Shrager P, Furley AJ, Peles E. Juxtaparanodal clustering of Shaker-like K+ channels in myelinated axons depends on Caspr2 and TAG-1. J Cell Biol. 2003;162:1149–1160. doi: 10.1083/jcb.200305018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Price DD, Dubner R. Mechanisms of first and second pain in the peripheral and central nervous systems. J Invest Dermatol. 1977;69:167–171. doi: 10.1111/1523-1747.ep12497942. [DOI] [PubMed] [Google Scholar]
- Quinn KP, Dong L, Golder FJ, Winkelstein BA. Neuronal hyperexcitability in the dorsal horn after painful facet joint injury. Pain. 2010;151:414–421. doi: 10.1016/j.pain.2010.07.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Park EW, Vanderah TW, Lai J, Porreca F, Trimmer JS. Distinct potassium channels on pain-sensing neurons. Proc Natl Acad Sci U S A. 2001;98:13373–13378. doi: 10.1073/pnas.231376298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Peles E, Trimmer JS, Levinson SR, Lux SE, Shrager P. Dependence of nodal sodium channel clustering on paranodal axoglial contact in the developing CNS. J Neurosci. 1999;19:756–7528. doi: 10.1523/JNEUROSCI.19-17-07516.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasband MN, Trimmer JS, Schwarz TL, Levinson SR, Ellisman MH, Schachner M, Shrager P. Potassium Channel Distribution, Clustering, and Function in Remyelinating Rat Axons. J Neurosci. 1998;18:36–47. doi: 10.1523/JNEUROSCI.18-01-00036.1998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rasmussen HB, Frokjaer-Jensen C, Jensen CS, Jensen HS, Jorgensen NK, Misonou H, Trimmer JS, Olesen SP, Schmitt N. Requirement of subunit co-assembly and ankyrin-G for M-channel localization at the axon initial segment. J Cell Sci. 2007;120:953–963. doi: 10.1242/jcs.03396. [DOI] [PubMed] [Google Scholar]
- Rivera-Arconada I, Lopez-Garcia JA. Effects of M-current modulators on the excitability of immature rat spinal sensory and motor neurones. The European journal of neuroscience. 2005;22:3091–3098. doi: 10.1111/j.1460-9568.2005.04507.x. [DOI] [PubMed] [Google Scholar]
- Rivera-Arconada I, Lopez-Garcia JA. Retigabine-induced population primary afferent hyperpolarisation in vitro. Neuropharmacology. 2006;51:756–763. doi: 10.1016/j.neuropharm.2006.05.015. [DOI] [PubMed] [Google Scholar]
- Roza C, Lopez-Garcia JA. Retigabine, the specific KCNQ channel opener, blocks ectopic discharges in axotomized sensory fibres. Pain. 2008;138:537–545. doi: 10.1016/j.pain.2008.01.031. [DOI] [PubMed] [Google Scholar]
- Sauer B, Henderson N. Site-specific DNA recombination in mammalian cells by the Cre recombinase of bacteriophage P1. Proc Natl Acad Sci U S A. 1988;85:5166–5170. doi: 10.1073/pnas.85.14.5166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schmalbruch H. Fiber composition of the rat sciatic nerve. Anat Rec. 1986;215:71–81. doi: 10.1002/ar.1092150111. [DOI] [PubMed] [Google Scholar]
- Schroeder BC, Hechenberger M, Weinreich F, Kubisch C, Jentsch TJ. KCNQ5, a novel potassium channel broadly expressed in brain, mediates M-type currents. J Biol Chem. 2000;275:24089–24095. doi: 10.1074/jbc.M003245200. [DOI] [PubMed] [Google Scholar]
- Schwake M, Pusch M, Kharkovets T, Jentsch TJ. Surface expression and single channel properties of KCNQ2/KCNQ3, M-type K+ channels involved in epilepsy. J Biol Chem. 2000;275:13343–13348. doi: 10.1074/jbc.275.18.13343. [DOI] [PubMed] [Google Scholar]
- Schwarz JR, Glassmeier G, Cooper EC, Kao TC, Nodera H, Tabuena D, Kaji R, Bostock H. KCNQ channels mediate IKs, a slow K+ current regulating excitability in the rat node of Ranvier. J Physiol. 2006;573:17–34. doi: 10.1113/jphysiol.2006.106815. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Selyanko A, Hadley JK, Brown DA. Properties of single M-type KCNQ2/KCNQ3 potassium channels expressed in mammalian cells. J Physiol. 2001;534:15–24. doi: 10.1111/j.1469-7793.2001.00015.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Migliore M, Valencia I, Cooper EC, Brown DA. Functional significance of axonal Kv7 channels in hippocampal pyramidal neurons. Proc Natl Acad Sci U S A. 2008;105:7869–7874. doi: 10.1073/pnas.0802805105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah MM, Mistry M, Marsh SJ, Brown DA, Delmas P. Molecular correlates of the M-current in cultured rat hippocampal neurons. J Physiol. 2002;544:29–37. doi: 10.1113/jphysiol.2002.028571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh NA, Charlier C, Stauffer D, Dupont BR, Leach RJ, Melis R, Ronen GM, Bjerre I, Quattlebaum T, Murphy JV. A novel potassium channel gene, KCNQ2, is mutated in an inherited epilepsy of newborns. Nature Genetics. 1998;18:25–29. doi: 10.1038/ng0198-25. [DOI] [PubMed] [Google Scholar]
- Soriano P. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nature Genetics. 1999;21:70–71. doi: 10.1038/5007. [DOI] [PubMed] [Google Scholar]
- Storm JF. Potassium currents in hippocampal pyramidal cells. Prog Brain Res. 1990;83:161–187. doi: 10.1016/s0079-6123(08)61248-0. [DOI] [PubMed] [Google Scholar]
- Tzingounis AV, Heidenreich M, Kharkovets T, Spitzmaul G, Jensen HS, Nicoll RA, Jentsch TJ. The KCNQ5 potassium channel mediates a component of the afterhyperpolarization current in mouse hippocampus. Proc Natl Acad Sci U S A. 2010;107:10232–10237. doi: 10.1073/pnas.1004644107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tzingounis AV, Nicoll RA. Contribution of KCNQ2 and KCNQ3 to the medium and slow afterhyperpolarization currents. Proc Natl Acad Sci U S A. 2008;105:19974–19979. doi: 10.1073/pnas.0810535105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang HS, Pan Z, Shi W, Brown BS, Wymore RS, Cohen IS, Dixon JE, MckKinnon D. KCNQ2 and KCNQ3 Potassium Channel Subunits: Molecular Correlates of the M-Channel. Science. 1998;282:1890–1893. doi: 10.1126/science.282.5395.1890. [DOI] [PubMed] [Google Scholar]
- Watanabe H, Nagata E, Kosakai A, Nakamura M, Yokoyama M, Tanaka K, Sasai H. Disruption of the epilepsy KCNQ2 gene results in neural hyperexcitability. J Neurochem. 2000;75:28–33. doi: 10.1046/j.1471-4159.2000.0750028.x. [DOI] [PubMed] [Google Scholar]
- Wen H, Levitan IB. Calmodulin is an auxiliary subunit of KCNQ2/3 potassium channels. J Neurosci. 2002;22:7991–8001. doi: 10.1523/JNEUROSCI.22-18-07991.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Feng B, Glazebrook PA, Schild JH, Kunze DL. The KCNQ/Mcurrent modulates arterial baroreceptor function at the sensory terminal in rats. J Physiol. 2008;586:795–802. doi: 10.1113/jphysiol.2007.145284. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wladyka CL, Kunze DL. KCNQ/M-currents contribute to the resting membrane potential in rat visceral sensory neurons. J Physiol. 2006;575:175–189. doi: 10.1113/jphysiol.2006.113308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wood MA, Kaplan MP, Park A, Blanchard EJ, Oliveira AM, Lombardi TL, Abel T. Transgenic mice expressing a truncated form of CREB-binding protein (CBP) exhibit deficits in hippocampal synaptic plasticity and memory storage. Learn Mem. 2005;12:111–119. doi: 10.1101/lm.86605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Y, Beyer BJ, Otto JF, O’Brien TP, Letts VA, White HS, Frankel WN. Spontaneous deletion of epilepsy gene orthologs in a mutant mouse with a low electroconvulsive threshold. Human Molecular Genetics. 2003;12:975–984. doi: 10.1093/hmg/ddg118. [DOI] [PubMed] [Google Scholar]
- Yeomans DC, Proudfit HK. Nociceptive responses to high and low rates of noxious cutaneous heating are mediated by different nociceptors in the rat: electrophysiological evidence. Pain. 1996;68:141–150. doi: 10.1016/S0304-3959(96)03177-6. [DOI] [PubMed] [Google Scholar]
- Yue C, Yaari Y. KCNQ/M channels control spike afterdepolarization and burst generation in hippocampal neurons. J Neurosci. 2004;24:4614–4624. doi: 10.1523/JNEUROSCI.0765-04.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng QF, Liu D, Cai M, Wan J, Han Y, Xing J-S, G-G Suppression of KCNQ/M (Kv7) potassium channels in dorsal root ganglion neurons contributes to the development of bone cancer pain in a rat model. Pain. 2013;154:434–448. doi: 10.1016/j.pain.2012.12.005. [DOI] [PubMed] [Google Scholar]
- Ziegler EA, Mageri W, Meyer RA, Treede RD. Secondary hyperalgesia to punctate mechanical stimuli: central sensitization to A-fibre nociceptor input. Brain. 1999;122:2245–2257. doi: 10.1093/brain/122.12.2245. [DOI] [PubMed] [Google Scholar]