

Abstract
Sickle cell disease (SCD) can be cured by allogeneic hematopoietic stem cell (HSC) transplant. However, this is only possible when a matched donor is available making the development of gene therapy using autologous HSCs a highly desired alternative. We used a culture model of human erythropoiesis to directly compare two insulated, self-inactivating, and erythroid-specific lentiviral vectors, encoding for γ-globin (V5m3-400) or a modified β-globin (βAS3-FB) for production of anti-sickling hemoglobin (Hb) and correction of red cell deformability after deoxygenation. Bone marrow CD34+ cells from three SCD patients were transduced using V5m3-400 or βAS3-FB and compared to mock transduced SCD or healthy donor CD34+ cells. Lentiviral transduction did not impair cell growth or differentiation, as gauged by proliferation and acquisition of erythroid markers. Vector copy number averaged ~1 copy per cell and corrective globin mRNA levels were increased more than 7-fold over mock-transduced controls. Erythroblasts derived from healthy donor and mock-transduced SCD cells produced a low level of HbF that was increased to 23.6 ± 4.1% per vector copy for cells transduced with V5m3-400. Equivalent levels of modified HbA of 17.6 ± 3.8% per vector copy were detected for SCD cells transduced with βAS3-FB. These levels of anti-sickling Hb production were sufficient to reduce sickling of terminal stage RBCs upon deoxygenation. We conclude that the achieved levels of HbF and modified HbA would likely prove therapeutic to SCD patients who lack matched donors.
Keywords: fetal hemoglobin, adult hemoglobin, lentivirus, sickle cell disease
Introduction
Individuals with sickle cell disease (SCD) have a mutation in β-globin that predisposes deoxygenated sickle hemoglobin (HbS; α2,βS2) to polymerize. HbS causes red blood cells (RBCs) to assume a rigid, sickle-shape leading to vaso-occlusion, painful crisis and organ damage (1). While hydroxyurea and other palliative therapies improve the quality/duration of life for many, treatment for SCD remains inadequate. Bone marrow (BM) transplantation can be curative but is only available to patients with matched donors (2). These considerations make gene therapy approaches a highly desired alternative to cure SCD.
SCD patients who continue to produce γ-globin containing fetal hemoglobin (HbF; α2,γ2) to levels >20% experience less severe disease (3-10). The protective activity of HbF is mediated by lowering HbS concentrations and inhibition of sickling due to incorporation of γ-globin into mixed hemoglobin tetramers (α2,γβS) that do not participate in polymer formation (1,11). The benefits of HbF, including its anti-sickling properties, may be achieved through ectopic expression of γ-globin such is possible with the V5m3-400 lentiviral vector. Alternatively, β-globin derivatives modified to include γglobin amino acid substitutions have increased affinity for α-globin, and these βglobin/α-globin tetramers inhibit HbS polymerization. The second lentiviral vector studied herein (βAS3-FB) encodes for modified β-globin sequences, and was shown to significantly inhibit HbS polymerization by conferring a competitive advantage over βS-globin for α-globin polypeptides with an oxygen affinity comparable with HbF (13).
Given the success of both approaches, gene therapy for SCD will likely make use of lentiviral vectors encoding for erythroid-specific expression of either γ- or modified βglobin coding sequences. However, no study has directly compared the biochemical and physiological potential of these treatments in late stage erythroblast derived from BM CD34+ cells obtained from SCD patients. Herein, we determined the activity of two candidate vectors for planned use in clinical trials for SCD.
Materials and Methods
CD34+ cells and Erythroid Culture
BM cells were obtained from patients diagnosed with homozygous SCD according to clinical protocols approved by the IRB of UCLA, CHLA and CHORC. CD34+ cells were enriched from BM samples by positive selection using the CD34 MicroBead Kit (MACS system, Miltenyi Biotec) following manufacturer’s instructions. Purity and viability of CD34+ cells was >75% for all the samples. CD34+ cells were transduced and cultured under conditions that promote differentiation into predominantly mature orthochromatic erythroid cells that were positive for glycophorin A (CD235) and negative for the nuclear dye (DRAQ5) as previously described (14).
Anti-sickling Lentiviral Vectors
The details regarding the lentiviral vectors encoding for γ-globin (V5m3-400) and modified β-globin (βAS3-FB) are reported elsewhere (14,15). Both vectors have self-inactivating (SIN) design, confer erythroid-specific expression of the respective globin gene and include insulator elements (16,17) in the deleted U3 portion of the 3′-long terminal repeat (LTR) to enhance safety.
Lentivirus Production, Titration and Transduction
Lentiviral particles were prepared using an established four-plasmid transient transfection system (18). Titers were determined by exposing 293T cells to varying volumes of viral supernatants in the presence of polybrene. After seven days of culture, cells were isolated of genomic DNA for Southern blot and quantitative PCR (qPCR) analysis (15,18).
The details regarding transduction of SCD BM CD34+ cells have been reported (15). After 18-24 hours of pre-stimulation, cells were exposed to vector particles to achieve a multiplicity of infection (m.o.i.) of 40. The next day, cells were collected and subjected to erythroid culture conditions (15).
Flow Cytometry Analysis
Live cells at terminal stages of differentiation were identified and tested for expression of CD235 and DRAQ5 using antibodies conjugated to either PE or APC on a LSRFortessa System using DIVA analysis software (both BD Biosciences).
Determination of Vector Copy Number
The average vector copy number (VCN) in transduced cell populations was determined by Southern blot and qPCR analysis using genomic DNA prepared from bulk populations of erythroid cells as described (14,15).
Globin mRNA Analysis
Total RNA was extracted from cells on culture day 14 and first-strand cDNA was synthesized using random primers. Quantitative PCR amplification of βAS3-, γ-, and αglobin transcripts was performed in a one-step RT-PCR with commercially available primer/probe sets for γ-globin: Hs00361131_g1 and α-globin: Hs00361191_g1(Life Technologies, and custom primers for βAS3-globin (HBBAS3 For: 5′-GGA GAA GTC TGC CGT TAC TG-3′; HBBAS3 Rev: 5′-CAC TAA AGG CAC CGA GCA CT-3′, HBBAS3 probe: 5′-FAM-ACA AGG TGA-ZEN-ACG TGG ATG CCG TTG-Iowa Black-3′) on a QX100 Droplet Digital PCR System (BioRad). Values of βAS3- and γglobin were calculated as fold-increase in mRNA levels relative to mock-transduced SCD controls, using α-globin as an internal control.
Hb Analysis and Quantification
Terminally differentiated cells were lysed (hemolysate reagent, Helena Laboratories) and supernatants were used to characterize Hb production by cellulose acetate electrophoresis using Hb standards (Helena Laboratories) or high performance liquid chromatography (HPLC) using calibrated samples for human Hbs (19). Peaks for Hb species were identified by retention time and the relative percentage of HbF or HbAS3 produced for each transduced sample calculated based on the sum total of areas under the curve for each of the primary hemoglobin peaks which included acetylated fetal hemoglobin, HbFAc; fetal hemoglobin, HbF; modified βAS3, HbAS3 or normal adult hemoglobin, HbA; and sickle hemoglobin, HbS.
SCD Phenotype
The detailed procedure for this assay has been described (15). Briefly, cells were collected after 21 days of erythroid culture, deoxygenated with 0.1 μg sodium metabisulfite (Sigma Aldrich), and incubated at 5% CO2, 37°C for 25–40 minutes. Cell images from consecutive fields at ×10 magnification were captured by inverse microscopy with a Nikon DS-Fi1 camera. Isolated cells within each field were analyzed for normal or sickle morphology in a randomized and unbiased fashion across treatment groups.
Statistical Analysis
Microsoft Excel was used to determine descriptive statistics mean, standard deviation, or standard error mean and significant differences between mean values using Student t-test and pairwise comparisons.
Results and Discussion
Efficient anti-sickling globin lentiviral vector gene transfer into bone marrow CD34+ cells from SCD patients
Autologous stem cell gene therapy is an attractive option for treating SCD due to limited pharmacological options and restricted numbers of HLA-matched donors. Given that high-level, erythroid-specific expression of γ- or modified β-globin coding sequences following lentivirus gene delivery could be curative, we directly compared V5m3-400 and βAS3-FB vectors (Fig. 1A) in CD34+ cells from three SCD patients by monitoring erythroid differentiation, gene transfer efficiency (vector copy number), globin gene expression, Hb production, and RBC sickling following deoxygenation (phenotypic correction) (Fig. 1B). Lentivirus particles pseudotyped with VSV-G were produced by transient transfection and concentrated by ultracentrifugation. Analysis of genomic DNA isolated from transduced HEK293T cells demonstrated that unconcentrated titer (transducing units (TU)/mL) was similar for both vectors by Southern blot (V5m3-400: 5.0 × 106 TU/mL; βAS3-FB: 2.5 × 106 TU/mL) and qPCR (V5m3-400: 2.2 × 106 TU/mL; βAS3-FB: 2.6 × 106 TU/mL). Despite subtle differences in vector design both anti-sickling globin vectors yielded nearly equivalent infectious titer using an established transient transfection protocol.
Figure 1. Anti-sickling globin lentiviral vectors demonstrate efficient gene transfer into human sickle cell hematopoietic stem cells (HSCs).
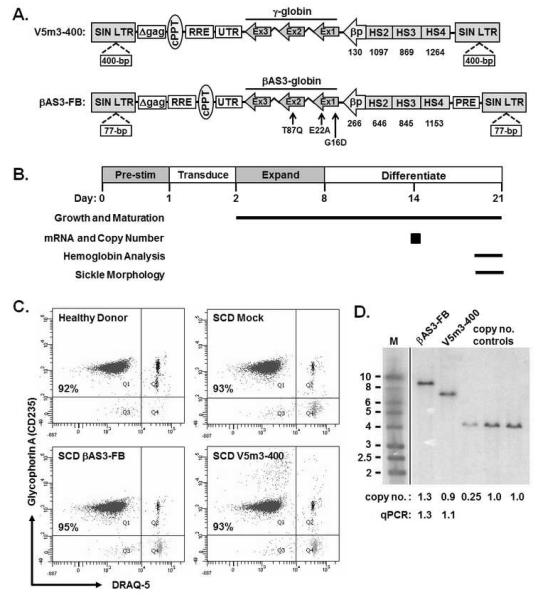
(A) Schematic representation of the integrated γ-globin (V5m3-400, top) and modified β-globin (βAS3-FB, bottom) lentiviral vector forms. V5m3-400 encodes for γ-globin genomic sequences and the 3′-untranslated region (UTR) of β-globin controlled by 3.1-kb of regulatory sequences from the β-globin locus control region (LCR) and a 130-bp beta-globin promoter. βAS3-FB encodes for β-globin genomic sequences mutated such that aspartic acid replaces glycine at position 16 (G16D), glutamic acid replaces alanine at position 22 (E22A) and glutamine replaces threonine at position 87 (T87Q) and β-globin 3′-UTR driven by 3.4-kb of regulatory sequences and a 266-bp β-globin promoter. Both vectors are self-inactivating (SIN) and include insulator elements in the deleted U3 portion of the 3′-LTR (400-bp core of the chicken HS4 for V5m3-400 or 77-bp FB element for βAS3-FB). cPPT, central polypurine tract; RRE, Rev-responsive element; βp, human β-globin promoter; HS, DNase1 hypersensitive sites; PRE, woodchuck post-transcriptional regulatory element; LTR, long terminal repeat. (B) Experimental schema with time course of pre-stimulation, transduction, expansion and differentiation phases as indicated. Time points or intervals are indicated for the specific determinations shown at left. (C) Dot plots of terminal stage orthochromatic erythroblasts derived from CD34+ cells of a healthy donor (HD) or sickle cell disease (SCD) patient and transduced using mock conditions or with the indicated lentiviral vectors and reacted with antibodies to glycophorin A (CD235) or the nuclear dye DRAQ5 and analyzed by flow cytometry (D) Southern blot analysis of genomic DNA, digested with BglII to release a nearly full-length pro-viral fragment, from SCD cell populations transduced with the indicated lentiviral vectors. A vertical line was inserted to represent repositioned lanes on the gel image. Average vector copy number determined by densitometry relative to a K562 clone that contains a single copy of an integrated GFP-encoding lentiviral vector or quantitative PCR (qPCR) is provided below each lane. M, molecular size marker (in kilobases).
Potentially therapeutic levels of anti-sickling Hb in erythroid cells derived from CD34+ bone marrow cells of SCD patients
To evaluate vector performance, we used CD34+ cells isolated from BM of healthy donors (HD) or SCD patients. Cells were grown under erythroid culture conditions with those from HD serving as controls and those from SCD patients transduced using mock conditions or with V5m3-400 or βAS3-FB lentivirus. As previously observed, (14,15,19) transduction had negligible effects on cell growth and differentiation with all cell populations achieving a >50-fold expansion and similar numbers of enucleated RBC (CD235+/DRAQ5−) after differentiation (Fig. 1C; Table 1). Southern blot confirmed transmission of intact viral genomes (Fig. 1D) that was one copy per cell by qPCR analysis (V5m3 0.9 ± 0.2; βAS3-FB 1.1 ± 0.1). For this level of gene transfer, γ-globin and βAS3 mRNA levels were increased 7-fold compared to mock transduced controls (Table 1). These results indicated a high level of gene transfer and expression for both vectors at a level (~1 copy per cell) which limits risk of insertional genotoxicity (20).
Table 1.
High levels of anti-sickling hemoglobin in erythroblasts derived from BM CD34+ cells from SCD patients transduced with V5m3-400 or βAS3-FB lentiviral vectors.
| Condition |
aCell Growth |
b(%) CD235+/Draq5− |
cVector Copy No. |
dmRNA Levels |
e(%) HbA/AS3 |
e(%) HbF |
f(%) Correction |
|---|---|---|---|---|---|---|---|
| HD CD34 | 82 ± 18 | 85 ± 4 | ND | ND | 83.6 ± 1.8 | 5.8 ± 0.1 | ND |
| SCD mock | 76 ± 21 | 86 ± 5 | ND | 0 | 4.9 ± 1.2 | 6.9 ± 0.4 | 0 |
| βAS3-FB | 83 ± 19 | 86 ± 5 | 1.1 ± 0.1 | 7.6 ± 1.9 | 18.9 ± 3.1 | 8.1 ± 1.3 | 21.5 ± 5.0 |
| V5m3-400 | 70 ± 13 | 88 ± 2 | 0.9 ± 0.2 | 7.3 ± 2.2 | 7.0 ± 2.2 | 20.8 ± 1.6 | 22.1 ± 7.7 |
Mean ± s.e.m (n=3 independent donors with matched controls) are reported for each characteristic except for % Corrected (mean ± s.d., n=2 independent donors with matched controls)
Fold-increase in viable cells from day of transduction to culture day 11 or 12
Erythroid differentiation was determined by flow cytometry analysis of bulk cell populations for enucleated RBC (CD235+/ Draq5−) on culture day 21
Genomic DNA was isolated from the bulk cell population on day 8 of culture and copy number determined by quantitative PCR and/or Southern blot analysis
Relative levels of βAS3- and γ-globin transcripts determined by droplet digital quantitative RT-PCR with normalization to α-globin before subtraction of results for SCD mock
% of adult hemoglobin (HbA) or HbAS3 and fetal hemoglobin (HbF) quantified by densitometry analysis of cellulose acetate Hb electrophoresis gel results or HPLC
% red blood cells that did not sickle when deoxygenated with sodium metabisulfite and normalized per vector copy using the formula: % correction = (% sickle RBC in SCD mock - % sickle RBC in vector treated sample)/vector copy number.
Abbreviaions: HD, healthy donor; SCD, sickle cell disease; ND, not determined.
Erythroid cells derived from HD or transduced SCD progenitors were evaluated for anti-sickling Hb production by cellulose acetate electrophoresis and levels quantified by HPLC (Figure 2A and 2B; Table 1). When corrected for vector copy number, anti-sickling Hb levels were 17.6 ± 3.8% (βAS3) and 23.6 ± 4.1% (V5m3-400), a difference that was not statistically significant (P = 0.34). The functional effect of βAS3- or γ-globin expression on RBC sickling was assessed by an in vitro deoxygenation assay (15). Differentiated RBCs for two donors were harvested at the end of erythroid culture and deoxygenated with sodium metabisulfite to induce HbS polymer formation. RBC morphology was analyzed for 1000 to 2000 cells per treatment and the percentage of sickle RBC determined for the combined donors (Figure 2C) or percentage “corrected” sickle RBC calculated per vector copy to normalize for transduction efficiency (Table 1). As previously observed, sickling was minimal for HD RBC, high for SCD mock transduced RBC, and reduced for cells with anti-sickling globin expression (15). We conclude that the achieved levels of HbF and HbAS3 would likely provide therapeutic benefit to SCD patients who lack matched donors. However, curative SCD therapy may require HbF levels of 30% (11). Such therapies could be achieved through combined expression of a structural anti-sickling globin gene with induction of endogenous HbF, via knockdown of BCL11A (21); or via inhibition of βS-globin (22). Successful design and production of these combination vectors could prove effective treatments for all SCD patients.
Figure 2. Therapeutic production of anti-sickling hemoglobins in erythroblast derived from CD34+ BM cells of SCD patients transduced with V5m3-400 or βAS3-FB lentiviral vectors.
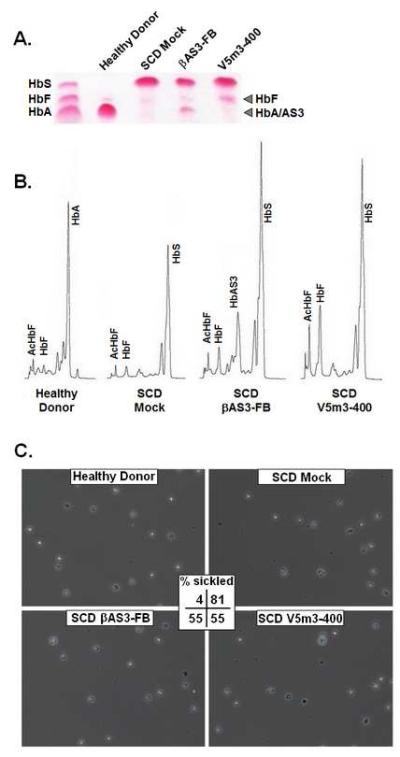
Bone marrow CD34+ cells of SCD patients were transduced using mock conditions or with V5m3-400 or βAS3-FB vectors and grown under erythroid culture conditions. CD34+ cells from healthy donors (HD) served as controls. (A) Cellulose acetate Hb electrophoresis of lysates derived from CD34+ BM cells of a HD or SCD patient and transduced either under mock conditions or with the indicated vectors. (B) Representative Hb HPLC traces from terminal stage erythroblasts derived from the indicated samples. (C) Terminal stage erythroid cultures for two independent SCD donors and matched with healthy controls were treated with sodium metabisulfite and cell morphology assessed using phase contrast microscopy. Representative photomicrographs are shown for the indicated samples, original magnification x 10. Reported for each is the average percentage of sickled red blood cells (% sickle) following deoxygenation calculated by the formula: (% sickle = total number of sickled cells / total number of sickled and nonsickled cells). Actual numbers of sickled (S) and nonsickled (NS) cells for each sample set: Healthy Donor (38S; 1024NS), SCD Mock (1675S; 385NS), SCD βAS3-FB (844S; 689NS), SCD V5m3-400 (865S; 696NS). High resolution versions of these photomicrographs for use with the Virtual Microscope are available as eSlides: VM00499, VM00500, VM00501, VM00502
Highlights.
Therapeutic levels of anti-sickling globins were achieved in differentiated RBCs.
Anti-sickling globin expression improved red cell deformability after deoxygenation.
βAS3- or γ-globin vectors may be applied to clinical gene therapy of SCD.
Acknowledgments
This work was supported by the Doris Duke Charitable Foundation (2011054: DAP, DBK, CBC and AW; 2009092 and 2013158: ZR, JW and DBK) and National Heart, Lung and Blood Institute (PO1HL053749: DAP, PWH, CBC and AW; PO1HL074104: DBK, SG, MLK, and RPH) and the California Institute for Regenerative Medicine (DR1-01452: FU and DBK). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final citable form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Bunn HF. Pathogenesis and treatment of sickle cell disease. N. Engl. J. Med. 1997;337:762–769. doi: 10.1056/NEJM199709113371107. [DOI] [PubMed] [Google Scholar]
- 2.Walters MC. Stem cell therapy for sickle cell disease: transplantation and gene therapy. Hematology. Am. Soc. Hematol. Educ. Program. 2005:66–73. doi: 10.1182/asheducation-2005.1.66. [DOI] [PubMed] [Google Scholar]
- 3.Stamatoyannopoulos G, Wood WG, Papayannopoulou T, Nute PE. A new form of hereditary persistence of fetal hemoglobin in blacks and its association with sickle cell trait. Blood. 1975;46:683–692. [PubMed] [Google Scholar]
- 4.Serjeant GR, Serjeant BE, Mason K. Heterocellular hereditary persistence of fetal haemoglobin and homozygous sickle-cell disease. Lancet. 1977;1:795–796. doi: 10.1016/s0140-6736(77)92976-2. [DOI] [PubMed] [Google Scholar]
- 5.Labie D, Pagnier J, Lapoumeroulie C, et al. Common haplotype dependency of high G gamma-globin gene expression and high HbF levels in beta-thalassemia and sickle cell anemia patients. Proc. Natl. Acad. Sci. U.S.A. 1985;82:2111–2114. doi: 10.1073/pnas.82.7.2111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Weatherall DJ. The Thalassemias. In: Stamatoyannopoulos G, Majerus P, Perlmutter R, Varmus HE, editors. The Molecular Basis for Blood Disorders. W.B. Saunders Company; Philadelphia: 2001. pp. 183–226. [Google Scholar]
- 7.Powars DR, Weiss JN, Chan LS, Schroeder WA. Is there a threshold level of fetal hemoglobin that ameliorates morbidity in sickle cell anemia? Blood. 1984;63:921–926. [PubMed] [Google Scholar]
- 8.Bunn HF. Induction of fetal hemoglobin in sickle cell disease. Blood. 1999;93:1787–1789. [PubMed] [Google Scholar]
- 9.Stevens MC, Hayes RJ, Vaidya S, Serjeant GR. Fetal hemoglobin and clinical severity of homozygous sickle cell disease in early childhood. J. Pediatr. 1981;98:37–41. doi: 10.1016/s0022-3476(81)80529-x. [DOI] [PubMed] [Google Scholar]
- 10.Ngo DA, Aygun B, Akinsheye I, et al. Fetal haemoglobin levels and haematological characteristics of compound heterozygotes for haemoglobin S and deletional hereditary persistence of fetal haemoglobin. Br J Haematol. 2012;156(2):259–264. doi: 10.1111/j.1365-2141.2011.08916.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Bunn HF. Subunit assembly of hemoglobin: an important determinant of hematologic phenotype. Blood. 1987;69:1–6. [PubMed] [Google Scholar]
- 12.Cavazzana-Calvo M, Payen E, Negre O, et al. Transfusion independence and HMGA2 activation after gene therapy of human β-thalassemia. Science. 2010;467:318–323. doi: 10.1038/nature09328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Levasseur DN, Ryan TM, Reilly MP, McCune SL, Asakura T, Townes TM. A recombinant human hemoglobin with anti-sickling properties greater than fetal hemoglobin. J. Biol. Chem. 2004;279:27518–27524. doi: 10.1074/jbc.M402578200. [DOI] [PubMed] [Google Scholar]
- 14.Romero Z, Urbinati F, Geiger S, et al. β-globin gene transfer to human bone marrow for sickle cell disease. J Clin. Invest. 2013;123(8):3317–3330. doi: 10.1172/JCI67930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Wilber A, Tschulena U, Hargrove PW, et al. A zinc-finger transcriptional activator designed to interact with the gamma-globin gene promoters enhances fetal hemoglobin production in primary adult erythroblasts. Blood. 2010;115(15):3033–3041. doi: 10.1182/blood-2009-08-240556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Emery DW, Yannaki E, Tubb J, Nishino T, Li Q, Stamatoyannopoulos G. Development of virus vectors for gene therapy of beta chain hemoglobinopathies: flanking with a chromatin insulator reduces gamma-globin silencing in vivo. Blood. 2002;100(6):2012–2019. doi: 10.1182/blood-2002-01-0219. [DOI] [PubMed] [Google Scholar]
- 17.Ramezani A, Hawley TS, Hawley RG. Combinatorial incorporation of enhancer-blocking components of the chicken β-globin 5′HS4 and human T-cell receptor α/δ BEAD-1 insulators in self-inactivating retroviral vectors reduces their genotoxic potential. Stem Cells. 2008;26:3257–3266. doi: 10.1634/stemcells.2008-0258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hanawa H, Kelly PF, Nathwani AC, et al. Comparison of various envelope proteins for their ability to pseudotype lentiviral vectors and transducer primitive hematopoietic cells from human blood. Mol. Ther. 2002;5:242–251. doi: 10.1006/mthe.2002.0549. [DOI] [PubMed] [Google Scholar]
- 19.Wilber A, Hargrove PW, Kim YS, et al. Therapeutic Levels of fetal hemoglobin in erythroid progeny of β-thalassemic CD34+ cells following lentiviral vector-mediated gene transfer. Blood. 2011;117:2817–2826. doi: 10.1182/blood-2010-08-300723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Modlich U, Bohne J, Schmidt M, von Kalle C, Knöss S, Schambach A, Baum C. Cell-culture assays reveal the importance of retroviral vector design for insertional genotoxicity. Blood. 2006;108(8):2545–2553. doi: 10.1182/blood-2005-08-024976. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sankaran VG, Menne TF, Xu J, et al. Human fetal hemoglobin expression is regulated by the developmental stage-specific repressor BCL11A. Science. 2008;322(5909):1839–1842. doi: 10.1126/science.1165409. [DOI] [PubMed] [Google Scholar]
- 22.Samakoglu S, Lisowski L, Budak-Alpdogan T, et al. A genetic strategy to treat sickle cell anemia by coregulating globin transgene expression and RNA interference. Nat. Biotechnol. 2006;24(1):89–94. doi: 10.1038/nbt1176. [DOI] [PubMed] [Google Scholar]