

Abstract
Background
Here, we report application of high-throughput near-full-length genome (NFLG) and partial HIV-1 proviral genome deep sequencing to characterize HIV in recently infected blood donors at four major blood centers in Brazil.
Study Design and Methods
From 2007-2011, 341 HIV+ blood donors from 4 blood centers were recruited to participate in a case control study to identify HIV risk factors and motivations to donate. Forty-seven (17 from São Paulo [SP], 8 from Minas Gerais [MG], 11 from Pernambuco [PE] and 11 from Rio de Janeiro [RJ]) were classified as recently infected based on testing by less-sensitive enzyme immunoassays. Five overlapping amplicons spanning the HIV genome were PCR amplified from peripheral blood mononuclear cells (PBMCs). The amplicons were molecularly bar-coded, pooled, and sequenced by an Illumina paired-end protocol.
Results
Of the 47 recently infected donor samples studied, 39 (82.9%) NFLGs and 6 (12.7%) partial fragments were de novo assembled into contiguous sequences and successfully subtyped. Subtype B was the only non-recombinant virus identified in this study and accounted for 62.2% (28/45) of samples. The remaining 37.8% (17/45) of samples showed various patterns of subtype discordance in different regions of HIV-1 genomes, indicating 2- 4 circulating recombinant subtypes derived from clades B, F and C. Fourteen samples (31.1%) from this study harbored drug resistance mutations, indicating higher rate of drug resistance among Brazilian blood donors.
Conclusion
Our findings revealed a high proportion of HIV-1 recombinants among recently infected blood donors in Brazil which has implications for future blood screening, diagnosis, therapy and vaccine development.
Introduction
The immense genetic variability and constant evolution of human immunodeficiency virus type 1 (HIV-1) present major challenges for vaccine design as well as assuring sensitivity of blood donor screening and diagnostic assays and accuracy of viral load testing. The high rate of mutation coupled with rapid turnover of HIV-1 within each infected individual 1 allows for the generation and fixation of a variety of genetic changes, which are selected for by host immune responses in the context of relative fitness of variants within the evolving quasispecies. Recombination between infected strains or quasispecies variants also significantly contributes to the genetic diversification of HIV and may produce more virulent viruses, drug resistant viruses, or viruses with altered cell tropism that may compromise the effectiveness of antiretroviral therapy 2. Currently HIV-1 viruses are classified into four phylogenetic groups: M, O, N and P, which most likely reflect four independent events of cross-species transmission from chimpanzees 3-5. The M group, which dominates the current AIDS pandemic, is subdivided into subtypes (A–D, F–H, J and K), sub-subtypes (A1- A3, F1 and F2), circulating recombinant forms (CRFs) and unique recombinant forms (URFs)3,6. Recombination between the URFs and CRFs and between the existing CRFs (inter-CRF recombinants) results in emergence of novel second and third generation recombinant forms, which will continue to shape the future of HIV epidemic through the generation of other variants with improved fitness that may influence viral transmissibility 7. The existence of recombinant viruses is evidence of simultaneous infection of multiple viruses during a single transmission event (co-infection) or from the sequential infection of viruses during multiple transmission events (superinfection). It has been reported that recombinant viruses including the URFs and CRFs may account for at least 20% of all HIV infections 8.
As in European countries and North America, HIV-1 subtype B is the major genetic clade circulating in Brazil, but the overall prevalence of non-B strains has increased alarmingly, particularly URF BF1 variants9-11. Indeed, we have recently reported that 38% of 42 sampled children and adolescents are infected with HIV-1 BF1 recombinant variants and three of these patients had evidence of dual infections with the same or distinct HIV-1 subtypes12. These results further emphasized the importance of continuous monitoring of the circulating HIV-1 subtypes in Brazil's epidemic to assure sensitivity of screening, diagnostic and viral load assays and for directing development of future vaccines that are targeted to currently transmitted HIV strains. Recently, Alencar et al 13 performed a molecular epidemiological survey of HIV-1 in Brazil by analyzing partial pol gene sequences in 341 samples from seropositive blood donors collected between 2007 and 2011 at the four Brazilian blood centers participating in the REDS-II (Retrovirus Epidemiological Donor Study) International Program. Of these donors, forty-seven were classified as recently infected based on testing by less-sensitive or “detuned” enzyme immunoassay as previously described 14,15. Analysis of the 43 successfully sequenced pol subgenomic region revealed 97.8% were pure subtypes and only one sequence was recombinant of BF1 variants. In this study, we aimed to determine whether the classification of these strains from recently infected donors extends to the whole genome. An understanding of HIV- genetic variability in individuals recently acquired the virus provides a range of benefits that include a deeper understanding of the very recently transmitted HIV-1 strains, current dynamics of the epidemic, transmission networks, patterns of transmitted drug resistance and guide HIV prevention and intervention strategies. Our results demonstrate that analysis of HIV-1 NFLG and larger genomic fragments is needed for reliable evaluation of viral diversity and evolution of the current HIV epidemic in Brazil.
Materials and Methods
Study population
From 2007-2011, 341 HIV+ blood donors from 4 blood centers were recruited to participate in a case control study to identify risk factors for exposure to HIV and motivations to donate 16. Forty-seven of the 341 enrolled donors were classified as recently infected at the time of donation based on antibody levels consistent with recent seroconversion (17 from São Paulo [SP], 8 from Minas Gerais [MG], 11 from Pernambuco [PE] and 11 from Rio de Janeiro [RJ])13. HIV diagnosis was established using a fourth‐generation (HIV antigen/antibody combination detection assay) enzyme-linked immunosorbent assay (AxSYM; Abbott Laboratories, Wiesbaden, Germany). Reactive samples were not considered HIV-1-positive until confirmation by the HIV-1/2 Western blot immune assay (Gene Lab, Singapore). All study subjects provided written informed consent. The study was approved by the local ethical review committee of participating institutes as well as the REDS-II collaborating centers (Blood Systems Research Institute/University of California San Francisco) and Data Coordinating Center (Westat, Inc.) in the US.
DNA extraction and amplification of the NFLGs
Genomic DNA used for the current PCR analyses was from cryopreserved PBMCs extracted using the QIAamp blood kit (Qiagen GmbH, Hilden Germany) according to the manufacturer's instructions. NFLGs from five overlapping fragments were obtained by PCR using the Platinum Taq DNA Polymerase High Fidelity (5 U/μl) (Invitrogen, Life Technologies, Carlsbad, CA) as previously reported10,11. The amplified DNA fragments from the nested PCR products were separated by gel electrophoresis and purified using Freeze ‘N Squeeze DNA Gel Extraction Spin Columns (Bio-Rad, Hercules, CA, USA). Each purified amplicon was quantified using Quant-IT HS reagents (Invitrogen, Life Technologies, Carlsbad, CA), and all five amplicons from a single viral genome were pooled together at equimolar ratios.
Whole viral genome library preparation
Sequencing libraries were prepared as described previously17-19. Briefly, one ng of each sample amplicon pool was used in a fragmentation and tagmentation reaction mix using the Nextera XT DNA sample prep kit according to the manufacturer's protocol (Illumina, San Diego, CA). After neutralization of the fragmented DNA, a light 12-cycle PCR was performed with Illumina Ready Mix to add Illumina flowcell adaptors, indexes and common adapters for subsequent cluster generation and sequencing. Amplified DNA libraries were then purified using Agencourt AMPure XP beads (Beckman Coulter), which excluded very short library fragments. Finally, all libraries were pooled and loaded on an Illumina MiSeq for paired-end 250 sequencing.
Data analysis
Fastq files were generated by the Illumina MiSeq reporter for downstream analysis and validated to evaluate the distribution of quality scores and to ensure that quality scores did not drastically decline over each read. To take the sequencing error rate into account, we only considered variants detected at a frequency higher than 1% and Phred quality score of >30%, i.e., a base call accuracy of 99.9%. Validated fastq files from each viral genome were de novo assembled into contiguous sequences and annotated with CLC Genomics Workbench version 5.5 (CLC Bio, Aarhus, Denmark) with default parameters and were additionally assembled using Velvet implemented in the Sequencher program 5.2 (Gene Code Corp., Ann Arbor, MI). The contiguous genomic sequence from each virus strain was extracted from the assembly and used for further analysis. The full designation of samples, according to WHO-proposed nomenclature is YYBRCY_XXX, where YY stands for the year of study, BR for Brazil, CY for city of enrolment; XXX for sample number.
Screening for recombination events and Phylogenetic Analysis
The de novo assembled NFLGs and partial consensus sequences were aligned with reference sequences representing subtypes A–D, F–H, J and K obtained from the Los Alamos database (http://hiv-web.lanl.gov) using MAFFT 20. Aligned sequences were manually edited and trimmed to the minimal shared length in the BioEdit Sequence Alignment Editor Program. The gap-stripped aligned sequences were screened for the presence of recombination by the bootscan methods implemented in the SIMPLOT program v3.5.121,22, the jumping profile Hidden Markov Model 23 and the Neighbor-Net method implemented in SplitsTree4 version 4.3 24. Recombinant regions of the alignment as determined by the crossover points were analyzed separately by phylogenetic analysis.
Comparison of tree topologies or branching pattern between subgenomic regions was performed using the algorithm described by Nye et al 25. This algorithm provides a measure of distance between two trees from different genes or subgenomic sequences and match up branches that have similar topological characteristics.
Maximum likelihood trees were obtained by PhyML version 3.1 using the general time reversible gamma + proportion invariant (GTR+I+G) model 26. The approximate likelihood ratio test (aLRT) was used as a statistical test to calculate branch support. All trees were displayed using MEGA version 6.0 software.
Transmission clusters among recently infected blood donors was defined as those clades of subjects infected with HIV-1 strains whose phylogenetic analysis revealed a strong similarity, indicating a reduced or direct chain of viral transmission.
Transmitted drug resistant mutations
For provirus sequences generated in this study, the massively parallel sequencing (MPS) reads of partial pol gene associated with transmitted drug resistant mutations (TDRMs) in the protease and reverse transcriptase regions of the HIV-1 genome of each sample were aligned to their corresponding consensus sequence using the CLC Genomics Workbench version 5.5 (CLC Bio, Aarhus, Denmark). The minority HIV-1 resistant variants were identified using a threshold of >1.0% of the reads sequenced. Reads with <1% were discarded to account for potential errors due to the error rate of PCR. The TDRMs profile were inferred by using the last updated list for surveillance of transmitted drug-resistant strains in untreated patients available in the Calibrated Population Resistance (CPR) Tool Version 6.0 (http://cpr.stanford.edu/cpr.cgi). The CPR program estimates the prevalence of TDRMs and ignore natural polymorphisms and mutations that occur at a frequency higher than 0.5% in the absence of drug pressure 27.
GenBank accession numbers
All consensus genome assemblies generated in this study were submitted to NCBI's GenBank database (Accession numbers KJ849784 - KJ849827).
Results
Samples
Of the 341 recruited HIV positive blood donors, 47 were identified as having been recently infected with HIV-1 by the less-sensitive or detuned assay as reported in our previous study13. In that study, 43 of 47 were successfully subtyped based on pol subgenomic reverse transcriptase (RT) PCR of cell free viruses using a conventional bulk sequencing approach (Table 1). Of note, reanalysis of plasma bulk sequence data from the previous study revealed five samples (10BR_SP011, 10BR_MG017, 10BR_PE018, 10BR_SP046, and 10BR_SP057) that were erroneously classified to different subtypes (asterisked categorization in Table 1). The above sequences were thus corrected in this study and used for further analyses (Table 1). Here, the 47 samples were subjected to further amplification of the viral NFLGs followed by nucleotide MPS on the Illumina MiSeq instrument which has higher throughput and lower error rates than conventional Sanger sequencing 28.
Table 1. The near full-length genomic (NFLG) and partial fragments subtyping of HIV-1 from plasma and blood samples.
| Sample ID | Plasma (pol) bulk | Provirus Sequence Fragment | Provirus deep sequencing | ||||||
|---|---|---|---|---|---|---|---|---|---|
|
| |||||||||
| Subtype | A (546-2598) | B1 (2157-3791) | B2 (3236-5220) | C (4890-7808) | D (7719-9537) | subtype | Number of Reads | Av. coverage | |
| 10BR_PE001 | B | + | + | + | + | + | B | 219420 | 5.521 |
| 10BR_MG003j | F | + | + | + | + | + | BF1 | 765498 | 11.566 |
| 10BR_SP003 | B | + | + | + | + | + | B | 173299 | 4.084 |
| 10BR_MG007 | B | + | + | + | + | + | B | 6983 | 117 |
| 10BR_SP008 | B | - | - | - | + | + | B | 83849 | 2.948 |
| 10BR_SP009 | B | - | - | + | + | + | B | 65287 | 2.080 |
| 10BR_MG010j | F | + | + | + | + | + | BCF1 | 822016 | 17.667 |
| 10BR_PE010 | B | + | + | + | + | + | B | 138685 | 6.081 |
| 10BR_SP011* | A1K | + | + | + | + | + | CRF45_cpx | 15018 | 357 |
| 10BR_SP014 | B | + | + | + | + | + | B | 92894 | 5.880 |
| 10BR_RJ0153 | F | + | + | + | + | + | BF1 | 207553 | 22.907 |
| 10BR_SP017¶ | B | + | + | + | + | + | BF1 | 12793 | 788 |
| 10BR_MG017* | BF1 | + | - | + | + | + | BF1 | 42175 | 621 |
| 10BR_PE018* | BC | + | + | + | + | + | BC | 5314 | 59 |
| 10BR_SP021 | B | + | + | + | + | + | B | 231493 | 7.677 |
| 10BR_RJ023¶ | F | + | + | + | + | + | BF1 | 103542 | 9.575 |
| 10BR_RJ026j | B | + | + | + | + | + | BF1 | 254997 | 6.677 |
| 10BR_MG029 | B | + | + | + | + | + | B | 20077 | 294 |
| 10BR_PE029¶ | B | + | + | + | + | + | BCF1 | 43370 | 16.704 |
| 10BR_RJ032 | B | + | + | + | + | + | B | 17313 | 16.000 |
| 10BR_MG034j | B | + | + | + | + | + | BF1 | 117626 | 1.211 |
| 10BR_PE034 | B | + | + | + | + | + | B | 288411 | 7.027 |
| 10BR_MG035 | B | + | + | + | + | + | B | 337914 | 18.735 |
| 10BR_SP038 | B | + | + | + | + | + | B | 137747 | 14.088 |
| 10BR_RJ041¶ | B | + | + | + | + | + | BF1 | 87138 | 1.989 |
| 10BR_SP042 | B | + | + | + | + | + | B | 128310 | 2.504 |
| 10BR_SP043 | B | + | + | + | + | + | B | 147789 | 4.138 |
| 10BR_SP045 | B | + | + | + | + | + | B | 20832 | 220 |
| 10BR_RJ046j | F | + | + | + | + | + | CRF70_BF1 | 409028 | 11.101 |
| 10BR_SP046* | CF1 | + | + | + | + | + | CF1 | 161346 | 8.061 |
| 10BR_SP048j | F | + | + | + | + | + | BF1 | 49706 | 1.240 |
| 10BR_SP049 | B | + | + | + | + | + | B | 109344 | 1.891 |
| 10BR_RJ050 | B | + | + | + | + | + | B | 401040 | 9.266 |
| 10BR_SP050 | B | + | + | + | + | + | B | 360395 | 10.949 |
| 10BR_RJ051 | - | - | + | + | + | B | 270325 | 6.991 | |
| 10BR_PE053¶ | F | + | + | + | + | + | B | 259906 | 7.520 |
| 10BR_RJ053 | BF1 | + | + | + | + | + | BF1 | 209533 | 14.057 |
| 10BR_RJ054 | B | + | + | + | + | + | B | 470004 | 10.042 |
| 10BR_SP055 | B | + | + | + | + | + | B | 147158 | 10.808 |
| 10BR_PE056 | + | + | + | + | - | B | 49981 | 588 | |
| 10BR_SP057* | BF1 | + | + | + | + | + | BF1 | 162546 | 3.206 |
| 10BR_PE091 | B | + | + | + | + | + | B | 52565 | 2.734 |
| 10BR_PE097 | B | + | + | + | + | + | B | 110231 | 7.077 |
| 10BR_PE098 | B | + | + | + | + | - | B | 92541 | 12.856 |
| 10BR_PE100 | B | + | + | + | + | + | B | 243863 | 15.560 |
Reanalysis of misclassified plasma bulk sequences from the previous study
Samples diplayed dual infections
Sequences of partial pol from plasma bulk sequencing turned out to be recombinant sequences when re-analyzed by NFLG of their cell-associated viruses
Analysis of the proviral NFLGs and partial consensus sequences revealed that all except one variant retained intact reading frames for a majority of their genes and no gross deletions or rearrangements were observed. A deletion of 64 bp (Nucleotide position from start of HXB2 gp120; 2507–2571) was observed in sample 10BR_RJ041 (characterized as subclade BF1).
HIV variants and sequences
Sequences were obtained for all five overlapping fragments that cover the NFLGs for 39 participants. Partial sequences were obtained from at least one fragment derived from 6 samples as shown in Table 1. Two samples did not amplify for any fragment. This might be a result of technical difficulties in recovering the provirus, but it is also possible that cells other than PBMCs are infected during the early stages of HIV infection. These 2 samples were not considered further, and the analyses includes only the 45 samples whose subtypes were successfully determined (43 of these had subtype classifications based on the previous bulk sequencing of plasma virus13).
Characterization and distribution of non-recombinant HIV-1 NFLG subtypes
Phylogenetic analysis of the 39 HIV-1 incident samples with NFLG sequences revealed 23 non-recombinant subtype B infections (58.9%) and 17 recombinants subtypes (43.6%) over a mean length of 8894 bp (range: 8382–9341). Figure 1 shows the ML tree of 119 NFLG sequences, including the 23 subtype B variants sequenced in this study, 57 non-redundant B sequences from Brazil and 39 reference strains (GenBank and Los Alamos database) representing subtypes A-D, F-H, J and K. The 23 sequences fell into the clade B reference group (100% aLRT), with an overall mean genetic diversity of 9.9%. Close inspection of the tree indicated small transmissions clusters (an excess of 80% aLRT support) of only two groups composed of 2 to 3 infections per cluster sampled from Pernambuco, but in general there was no clustering of subtype B NFLG viruses based on their geographical region. Similar levels of genetic diversity were observed when sequences were compared from PE to those from RJ, SP, and MG.
Figure 1.
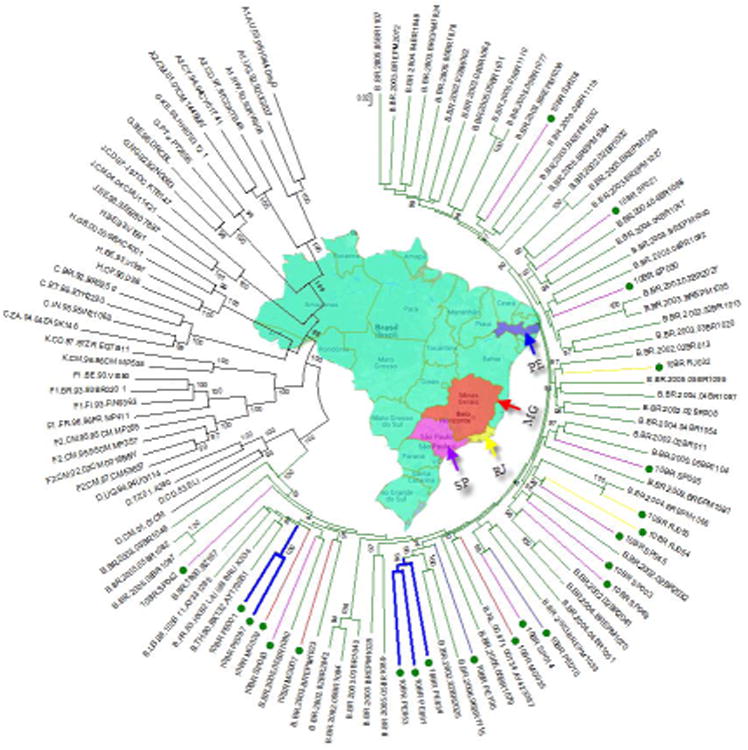
Phylogenetic tree constructed using a maximum-likelihood method from the NFLG sequences of 23 blood donor samples, 57 non recombinant subtype B NFLG published sequences from Brazil and 37 HIV-1 reference sequences from the Los Alamos HIV-1 database representing 11 genetic subtypes. Sequences identified in this study are indicated by green circles and the geographic origin of each sequence is color-coded. The 23 genuine subtype B infected donors were identified in the four HemoCenter regions; 7 in PE, 3 in MG, 9 in SP, and 3 in RJ. Monophyletic clusters from the same geographic origin are indicated by thick branches. For clarity purposes, the tree was midpoint rooted. The approximate likelihood ratio test (aLRT) values of ≥80% are indicated at nodes. The scale bar represents 0.02 nucleotide substitutions per site.
The stability and order of the 23 sequences were further investigated with ML trees independently made with the gag-pol and env regions alignment. For convenience, only the alignment of gag-pol and env from the 23 subtype B sequences obtained in this study are represented in Figures 2A and B. The phylogenetic trees from both regions received an overall topological score of 53.3% as scored by the Nye et al. algorithm 25. Examination of both subgenomic trees revealed a shifting of topological position across numerous sequences in a manner suggestive of intra-subtype recombination 29. For instance, isolates 10BR_PE10 and 10BR_SP14 changed their topological positions over the gag-pol and env regions of their genomes. The computed topological score of the sequences in both regions was 33.3%, with a branch length match of 100%. Similarly, isolate 10BR_SP045 placed the gag-pol region with 10BR_RJ054, while env strongly grouped with isolate 10BR_SP014.
Figure 2.

Comparison of alternative phylogenies of sequences identified in this study inferred from full-length gag-pol and env nucleotide sequences. Thick branches marked with arrows indicate those receiving a low topological score. The yellow markers indicate a match between branches.
Characterization and distribution of the NFLG recombinant sequences
Among the 17 NFLG variants with strong evidence of recombination, 10 were mosaic sequences consisting of subtype BF1, and the rest belonged to seven different HIV-1 recombinant subtypes (Table 1). One BF1 sequence from RJ (10BR_RJ046) displayed recombinant structure identical to the newly described CRF70_BF1 circulating in PE described by our group (Pessoa et al., submitted). None of the other putative BF1 recombinants had a recombination profile identical to the other BF mosaics described in Brazil or elsewhere. Moreover, the jumping profile Hidden Markov Model analysis identified one sequence (10BR_SP011) with largely identical structures to CRF45 cpx originating from Central Africa30. The blast of this sequence against the NCBI nucleotide database revealed a significant number of similar full genome hits with a Central African isolate 97CD.MBFE185 (GenBank: FN392874).
Characterization and distribution of HIV-1 subtypes by partial sequencing
Partial sequences were obtained from at least one fragment derived from six blood samples as shown in Table 1. All but one of the six strains analyzed in partial genomes were subtype B; the exception was a unique BF1 recombinant detected in a patient from MG (10BR_MG017).
Comparison of HIV-1 subtypes in plasma and PBMC
The relationships of the proviral DNA sequences from patients' PBMCs to the RNA sequences derived from plasma viruses within the same regions (n = 41) were examined for each patient to assess the viral diversity in both compartments (Table 1). Surprisingly, the intra-individual plasma and proviral sequence variation in five patients (10BR_SP017, 10BR_RJ023, 10BR_PE029, 10BR_RJ041, and 10BR_PE053) in the partial pol regions depicted were remarkably high, indicating that the plasma viruses were derived from a population significantly distinct from those of the cellular sources, results consistent with dual infection with different subtypes. Except for those with dual infections, all the other sequences from both compartments were located close to one another on the same branch and had plasma RNA and proviral DNA variation ranging between 0.0–1.6%. Dual infection with two distinct viruses of subclade F1 and subtype B was observed in patient 010BR_PE053 in plasma and PBMCs, respectively; subclade F1 seen in the plasma was completely absent in the PBMCs.
Overall, 29 viruses that were classified as B (n = 23), BF1 (n = 3), A1K (n = 1), BC (n = 1) and CF1 (n = 1) on partial pol plasma bulk analysis were confirmed by NFLG analysis (Table 1). The NFLG and large fragment analysis of seven sequence strains that were subtype F (n = 5) and B (n = 2) when sequenced in pol were found to be recombinants (Table 1). Sequences with evidence of double infections were observed in 5 donors. Based on our analysis of the 45 sequences generated in this study, it is evident that non-recombinant viruses account for 62.2% of HIV-1 subtype's circulating among recently infected blood donors while 37.8% were caused by recombinant viruses.
Geographical distribution of HIV-1 genetic forms
Figure 3 shows the regional distribution of HIV-1 subtypes in the four regions. Although sample sizes from these regions were small, the genetic diversity in the proviral NFLGs and larger fragment sequences for cases not yielding NFLGs was significant in all four regions. Multiple HIV-1 subtypes were cocirculating in infected blood donors, with at least 4 subtypes in SP and 3 in PE, RJ and MG. Subtype B was detected among donors in all four regions: PE (9 of 11), MG (3 of 7), and RJ (4 of 10), and SP (12 of 17). The HIV BF1 recombinant, which is the second-most prevalent subtype after B in Brazil, was found in MG (3 of 7), RJ (6 of 10), and SP (3 of 17). In contrast, the BF1 recombinant variants were not detected among blood donors in PE probably indicating that the HIV-1 epidemic among these donors is largely influenced by locally circulating variants.
Figure 3.

Summary of the subtype distribution of viruses from the 45 blood donors analyzed. The number of viruses that belong to each subtype is indicated in the relevant pie section. The regions of origin are color-coded and indicated by arrows.
Transmitted Drug Resistance Mutation Analysis
To test whether the MPS of proviral DNA would provide additional information compared to plasma RNA testing, the profiles of TDRMs between both compartments were compared. A summary of these results are provided in Table 2. The overall prevalence of TDRMs was 31.1% (14/45; 95% CI 17.58% to 44.62%). Discordant data between the cell free viruses and the PBMC-viruses were found in 10 participants and five of which had detectable TDRMs at prevalence between 1-20% of the sequenced population. All of the discordant mutations were detected in the proviral MPS data and absent in corresponding plasma specimens. For the coding region of the protease gene, M46I mutations were only detected in the proviral DNA of five blood donors. In four patients, reverse transcriptase mutations (3 M184I and 1 K103N) were identified only in the proviral compartment but not in the viral RNA sequences. In one patient (10BR_RJ026), we were able to detect a mutation at codon 46 (M46I) at a prevalence higher than 20% of the MPS provirus variants that had not been identified using plasma bulk sequencing. Overall, we found drug resistance in 14 out of 45 (14/45; 95% CI 17.58% to 44.62%) blood donors' proviral DNA (about 22.1%), however this resistance was at least partially evident in four blood donors with a conventional plasma sequencing approach. These results indicate that MPS approach permits characterization of considerable heterogeneity in the diversity and frequency variations in the proviral DNA 31.
Table 2. Transmitted drug resistance mutations detected with bulk sequencing (plasma) and massively parallel sequencing (PBMCs).
| Plasma | PBMCs | Plasma | PBMCs | Plasma | PBMCs | |
|---|---|---|---|---|---|---|
| 10BR_MG003 | L90M | L90M | ||||
| 10BR_SP003 | M46I* | |||||
| 10BR_SP011 | M184I | |||||
| 10BR_SP017 | K103N | |||||
| 10BR_RJ026 | M46I | M184I* | ||||
| 10BR_MG034 | I85V | I85V | ||||
| 10BR_SP038 | V75M | V75M | ||||
| 10BR_RJ041 | M46I* | |||||
| 10BR_SP042 | K103N, P225H | K103N, P225H | ||||
| 10BR_SP049 | Y188L | Y188L | ||||
| 10BR_SP055 | M46I* | M41L, T215DE | M41L, T215DE | K103N | K103N | |
| 10BR_PE056 | M184I | |||||
| 10BR_SP057 | M46I* | |||||
| 10BR_PE100 | D67G, M184V, T215S, K219Q | D67G, M184V, T215S, K219Q | K101E | K101E |
Transmitted drug resistance mutations at prevalence < 20% of the sequenced population
Discussion
This study describes the genetic diversity of HIV-1 proviral NFLGs and larger PCR fragments from cases not yielding NFLGs from 45 recently infected first-time blood donors at four blood centers in Brazil who had previously been diagnosed as infected with different subtype based on pol subgenomic RT-PCR fragment from cell free viruses using conventional bulk sequencing. A higher level genetic diversity was documented with six subtypes including recombinants found to be cocirculating. Little regional differences were observed in subtype distribution, but in each region at least 3 to 4 different clades cocirculated. Overall, subtype B was predominant in PE and SP, with decreasing proportions in MG and RJ. These results must be interpreted with caution given that the numbers of strains analysed from these regions were relatively small. However, the findings are consistent with previous studies that show a predominance of subtype B in other population groups within Brazil11,32-34. Notably, analysis of the 43 pol subgenomic region sequences from the same population group reported in our previous study 13 revealed that 97.8% were pure subtypes and only one sequence was a recombinant of BF1 variants. However, when the NFLGs and larger fragments were used in this study for subtype determination a significantly higher proportion (37.8%) of a variety of circulating and unique recombinant strains, particularly BF1, was observed. This difference is not surprising, because small fragments of the HIV-1 genome were characterized in the previous study, indicating that misclassifications can very easily arise. For example, the previous report identified six donors infected with subclade F1 all of which turned out to be recombinants when their NFLGs were sequenced. These results are in accordance with the conclusions that most, if not all, HIV-1 subclade F1 strains circulating in Brazil may contain recombinant genomes35,36 that are unrecognized by partial sequencing. The lack of prototypic subclade F1 reported here lends further support to the reduced rate of transmission of this subclade estimated in a previous study 37. It is possible that this decline is linked to increased transmission of BF1 recombinant variants in the Brazilian epidemic. We believe that the main evolutionary driving force behind the decline and probably replacement of subclade F1 and emergence of BF1 recombinants might be due to stochastic events facilitated by the lower population size of the prototypic subclade F1, rather than an increased viral fitness or selective pressure. Also, the complete lack of subtype C and lower prevalence of its recombinants in our group agree with other results from the Northeast, Central-West and Southeast regions of Brazil11,32,33,38. In contrast, results from previous studies showed that most infections in the frontier municipalities in the southern region of Brazil are caused by subtype C and BC recombinants, including CRF31 BC 10,39,40. Thus, the uneven distribution of subtype C across Brazilian regions may largely be influenced by limited founder viruses with subsequent locally circulating variants.
The approach used in this study also permitted the description of mixed infections with distinct subtypes. In five samples with dual infections, one subtype seen in the plasma was completely absent in the PBMCs. Discordances in the HIV subtypes in both compartments may suggest differential sources of infecting viruses. It is also conceivable that the discordances in the HIV subtypes in PBMCs and plasma are due to low-level minority strains present that are not detected with bulk plasma sequencing or that the replicating viruses shed in the plasma were more fit. The evidence of dual infections in this study adds support to previous studies12,41-44, demonstrating that this event is far more common where multiple subtypes co-circulate which is now the case in Brazil. It is unclear from this study whether the occurrence of multiple distinct HIV strains was the result of superinfection with a second variant at a later time, or whether simultaneous infection with multiple viral strains occurred during a single transmission event. However, the circulation of multiple subtypes in Brazil fortifies the possibility of both scenarios. The overall results indicate that the rate of HIV-1 mixed infections within this Brazilian group is higher than 12%. These results demonstrate that the use of PBMC DNA in addition to plasma RNA provides the highest sensitivity to detect mixed infection. Whether dual infections and/or recombination had an impact on the clinical outcomes of the blood donors in this study is unknown, since the available clinical data was limited to one assessment that sought to understand risk exposures and motivations to donate blood16.
Other important observation of this study is the underestimation of transmitted resistance obtained by routine plasma analysis that is revealed by the examination of the MPS populations of the archived proviruses in PBMCs. In this study, comparing both sources would have detected 10 DNA provirus disclosed TDRMs by MPS previously missed by plasma bulk analysis. Five of these strains had detectable TDRMs at prevalence between 1-20% of the MPS population. It is well known that conventional Sanger sequencing of bulk PCR products are limited to the detection of high-frequency variants that present in greater than 20% of the total sequenced viral population 45,46. It is possible that these minority variants occur in a very small percent of PBMCs and thus not be detectable by standard bulk sequencing. The relatively higher proportion of recently infected donors carried TDRMs leads us to conclude that the rate of the current transmitted DRMs is underestimated. These results justify the inclusion of proviral DNA from PMBCs as a valuable source for resistance analysis, which is in agreement with previous reports 47,48. Despite the small number of genotyped samples, this study revealed high prevalence of HIV-1 TDRMs among treatment-naive recently HIV-infected blood donors compared to those reported in other drug naive populations11,49,50 but in line with other study in Brazil51. It is possible that a wide use of antiretroviral drugs in Brazil could result in an increase in the prevalence of resistant variants. These results support the demand of resistance investigation before initiation of therapy.
The cross-sectional design, relatively low sample size, restriction of samples from recently infected blood donors, and lack of MPS analysis of HIV NFLGs from plasma may have biased the findings from this study. Despite these limitations, the results of this analysis indicate that HIV-1 recombination, dual infections and DRMs are much more frequent than thought previously among recently infected blood donors in Brazil. More studies with larger sample sizes from recently infected persons with broad demographic and geographic are required to unravel the mechanisms underlying the emergence of these recombinants and their implications for HIV clinical outcomes and control. Continued efforts to monitor the genetic makeup of HIV strains, preferably based on analysis of NFLG sequences, are critically important to assess the ongoing nature of the HIV epidemic in different regions as shown by this study.
Acknowledgments
This work was supported by the Retrovirus Epidemiology Donor Study-II (HHSN268200417175C) and the Recipient Epidemiology and Donor Outcomes Study-III (HHSN268201100007I) contracts from NHLBI, and grants 2011/11090-5 and 2011/12297-2 from the Fundação de Amparo à Pesquisa do Estado de São Paulo.
The Recipient Epidemiology and Donor Outcomes Study-III is the responsibility of the following persons:
Fundação Pró-Sangue/Hemocentro São Paulo (São Paulo): C. Almeida-Neto, A. Mendrone Jr.
Hemominas (Belo Horizonte, Minas Gerais): Anna Bárbara de F. Carneiro-Proietti, Fundação Hemope (Recife, Pernambuco): D.A. Sampaio, P. Loureiro
Fundação Hemorio (Rio de Janeiro, RJ): C. Lobo, M. E. Lopes
University of São Paulo: E.C. Sabino, L. Capuani, J. E. Ferreira, P.L Takecian
University of Sao Joao de Rey C L Oliveira
Blood Systems Research Institute and University of California San Francisco: M.P. Busch, B. Custer, T. Gonçalez
RTI international: D Brambilla, C McClure
National Heart, Lung, and Blood Institute, NIH: S.A. Glynn.
Abbreviations
- HIV-1
Human Immunodeficiency Virus Type 1
- NFLG
Near Full-Length Genome
- SP
São Paulo
- MG
Minas Gerais
- PE
Pernambuco
- RJ
Rio de Janeiro
- PBMCs
Peripheral Blood Mononuclear Cells
- CRFs
Circulating Recombinant Forms
- URFs
Unique Recombinant Forms
- REDS
Retrovirus Epidemiological Donor Study
- ML
Maximum likelihood
- MPS
Massively parallel sequencing Introduction
Footnotes
The authors report no conflicts of interest
References
- 1.Perelson AS, Neumann AU, Markowitz M, et al. HIV-1 dynamics in vivo: virion clearance rate, infected cell life-span, and viral generation time. Science. 1996;271(5255):1582–6. doi: 10.1126/science.271.5255.1582. [DOI] [PubMed] [Google Scholar]
- 2.Cohen OJ, Fauci AS. Transmission of multidrug-resistant human immunodeficiency virus--the wake-up call. N Engl J Med. 1998;339(5):341–3. doi: 10.1056/NEJM199807303390511. [DOI] [PubMed] [Google Scholar]
- 3.Robertson DL, Anderson JP, Bradac JA, et al. HIV-1 nomenclature proposal. Science. 2000;288(5463):55–6. doi: 10.1126/science.288.5463.55d. [DOI] [PubMed] [Google Scholar]
- 4.Gao F, Bailes E, Robertson DL, et al. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature. 1999;397(6718):436–41. doi: 10.1038/17130. [DOI] [PubMed] [Google Scholar]
- 5.Plantier JC, Leoz M, Dickerson JE, et al. A new human immunodeficiency virus derived from gorillas. Nat Med. 2009;15(8):871–2. doi: 10.1038/nm.2016. [DOI] [PubMed] [Google Scholar]
- 6.Tebit DM, Nankya I, Arts EJ, Gao Y. HIV diversity, recombination and disease progression: how does fitness “fit” into the puzzle? AIDS Rev. 2007;9(2):75–87. [PubMed] [Google Scholar]
- 7.Thomson MM, Perez-Alvarez L, Najera R. Molecular epidemiology of HIV-1 genetic forms and its significance for vaccine development and therapy. Lancet Infect Dis. 2002;2(8):461–71. doi: 10.1016/s1473-3099(02)00343-2. [DOI] [PubMed] [Google Scholar]
- 8.Arien KK, Vanham G, Arts EJ. Is HIV-1 evolving to a less virulent form in humans? Nat Rev Microbiol. 2007;5(2):141–51. doi: 10.1038/nrmicro1594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sanabani S, Kleine Neto W, Kalmar EM, et al. Analysis of the near full length genomes of HIV-1 subtypes B, F and BF recombinant from a cohort of 14 patients in Sao Paulo, Brazil. Infect Genet Evol. 2006;6(5):368–77. doi: 10.1016/j.meegid.2006.01.003. [DOI] [PubMed] [Google Scholar]
- 10.Sanabani S, Neto WK, de Sa Filho DJ, et al. Full-length genome analysis of human immunodeficiency virus type 1 subtype C in Brazil. AIDS Res Hum Retroviruses. 2006;22(2):171–6. doi: 10.1089/aid.2006.22.171. [DOI] [PubMed] [Google Scholar]
- 11.Sanabani SS, Pastena ER, da Costa AC, et al. Characterization of partial and near full-length genomes of HIV-1 strains sampled from recently infected individuals in Sao Paulo, Brazil. PLoS One. 2011;6(10):e25869. doi: 10.1371/journal.pone.0025869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sanabani SS, Pessoa R, Soares de Oliveira AC, et al. Variability of HIV-1 genomes among children and adolescents from Sao Paulo, Brazil. PLoS One. 2013;8(5):e62552. doi: 10.1371/journal.pone.0062552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Alencar CS, Sabino EC, Carvalho SM, et al. HIV genotypes and primary drug resistance among HIV-seropositive blood donors in Brazil: role of infected blood donors as sentinel populations for molecular surveillance of HIV. J Acquir Immune Defic Syndr. 2013;63(3):387–92. doi: 10.1097/QAI.0b013e31828ff979. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Rawal BD, Degula A, Lebedeva L, et al. Development of a new less-sensitive enzyme immunoassay for detection of early HIV-1 infection. J Acquir Immune Defic Syndr. 2003;33(3):349–55. doi: 10.1097/00126334-200307010-00009. [DOI] [PubMed] [Google Scholar]
- 15.Sabino EC, Goncalez TT, Carneiro-Proietti AB, et al. Human immunodeficiency virus prevalence, incidence, and residual risk of transmission by transfusions at Retrovirus Epidemiology Donor Study-II blood centers in Brazil. Transfusion. 2012;52(4):870–9. doi: 10.1111/j.1537-2995.2011.03344.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.de Almeida-Neto C, Goncalez TT, Birch RJ, et al. Risk factors for human immunodeficiency virus infection among Brazilian blood donors: a multicentre case-control study using audio computer-assisted structured interviews. Vox Sang. 2013;105(2):91–9. doi: 10.1111/vox.12028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Pessoa R, Watanabe JT, Nukui Y, et al. Molecular characterization of human T-cell lymphotropic virus type 1 full and partial genomes by illumina massively parallel sequencing technology. PLoS One. 2014;9(3):e93374. doi: 10.1371/journal.pone.0093374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Pessoa R, Carneiro Proietti AB, Busch MP, Sanabani SS. Identification of a Novel HIV-1 Circulating Recombinant Form (CRF72_BF1) in Deep Sequencing Data from Blood Donors in Southeastern Brazil. Genome Announc. 2014;2(3) doi: 10.1128/genomeA.00386-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pessoa R, Lopes ME, Sanabani SS. Genetic Characterization of HIV-1 Subtype D Near-Full-Length Proviral Genomes by Illumina Massively Parallel Sequencing Technology. Genome Announc. 2014;2(3) doi: 10.1128/genomeA.00586-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Katoh K, Misawa K, Kuma K, Miyata T. MAFFT: a novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002;30(14):3059–66. doi: 10.1093/nar/gkf436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Salminen MO, Carr JK, Burke DS, McCutchan FE. Identification of breakpoints in intergenotypic recombinants of HIV type 1 by bootscanning. AIDS Res Hum Retroviruses. 1995;11(11):1423–5. doi: 10.1089/aid.1995.11.1423. [DOI] [PubMed] [Google Scholar]
- 22.Lole KS, Bollinger RC, Paranjape RS, et al. Full-length human immunodeficiency virus type 1 genomes from subtype C-infected seroconverters in India, with evidence of intersubtype recombination. J Virol. 1999;73(1):152–60. doi: 10.1128/jvi.73.1.152-160.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Schultz AK, Zhang M, Leitner T, et al. A jumping profile Hidden Markov Model and applications to recombination sites in HIV and HCV genomes. BMC Bioinformatics. 2006;7:265. doi: 10.1186/1471-2105-7-265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Huson DH, Bryant D. Application of phylogenetic networks in evolutionary studies. Mol Biol Evol. 2006;23(2):254–67. doi: 10.1093/molbev/msj030. [DOI] [PubMed] [Google Scholar]
- 25.Nye TM, Lio P, Gilks WR. A novel algorithm and web-based tool for comparing two alternative phylogenetic trees. Bioinformatics. 2006;22(1):117–9. doi: 10.1093/bioinformatics/bti720. [DOI] [PubMed] [Google Scholar]
- 26.Guindon S, Dufayard JF, Lefort V, et al. New algorithms and methods to estimate maximum-likelihood phylogenies: assessing the performance of PhyML 3.0. Syst Biol. 2010;59(3):307–21. doi: 10.1093/sysbio/syq010. [DOI] [PubMed] [Google Scholar]
- 27.Shafer RW, Rhee SY, Pillay D, et al. HIV-1 protease and reverse transcriptase mutations for drug resistance surveillance. AIDS. 2007;21(2):215–23. doi: 10.1097/QAD.0b013e328011e691. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Quail MA, Smith M, Coupland P, et al. A tale of three next generation sequencing platforms: comparison of Ion Torrent, Pacific Biosciences and Illumina MiSeq sequencers. BMC Genomics. 2012;13:341. doi: 10.1186/1471-2164-13-341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Worobey M, Holmes EC. Homologous recombination in GB virus C/hepatitis G virus. Mol Biol Evol. 2001;18(2):254–61. doi: 10.1093/oxfordjournals.molbev.a003799. [DOI] [PubMed] [Google Scholar]
- 30.Niama FR, Vidal N, Bazepeo SE, et al. CRF45_AKU, a circulating recombinant from Central Africa, is probably the common ancestor of HIV type 1 MAL and HIV type 1 NOGIL. AIDS Res Hum Retroviruses. 2009;25(12):1345–53. doi: 10.1089/aid.2009.0169. [DOI] [PubMed] [Google Scholar]
- 31.Bansode V, McCormack GP, Crampin AC, et al. Characterizing the emergence and persistence of drug resistant mutations in HIV-1 subtype C infections using 454 ultra deep pyrosequencing. BMC Infect Dis. 2013;13:52. doi: 10.1186/1471-2334-13-52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.de Castro CA, Grinsztejn B, Veloso VG, et al. Prevalence, estimated HIV-1 incidence and viral diversity among people seeking voluntary counseling and testing services in Rio de Janeiro, Brazil. BMC Infect Dis. 2010;10:224. doi: 10.1186/1471-2334-10-224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Brennan CA, Brites C, Bodelle P, et al. HIV-1 strains identified in Brazilian blood donors: significant prevalence of B/F1 recombinants. AIDS Res Hum Retroviruses. 2007;23(11):1434–41. doi: 10.1089/aid.2007.0121. [DOI] [PubMed] [Google Scholar]
- 34.Cavalcanti AM, Lacerda HR, Brito AM, et al. Antiretroviral resistance in individuals presenting therapeutic failure and subtypes of the human immunodeficiency virus type 1 in the Northeast Region of Brazil. Mem Inst Oswaldo Cruz. 2007;102(7):785–92. doi: 10.1590/s0074-02762007005000109. [DOI] [PubMed] [Google Scholar]
- 35.Sanabani SS, Pastena ER, Kleine Neto W, et al. Near full-length genome analysis of low prevalent human immunodeficiency virus type 1 subclade F1 in Sao Paulo, Brazil. Virol J. 2009;6:78. doi: 10.1186/1743-422X-6-78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Carr JK, Avila M, Gomez Carrillo M, et al. Diverse BF recombinants have spread widely since the introduction of HIV-1 into South America. AIDS. 2001;15(15):F41–7. doi: 10.1097/00002030-200110190-00002. [DOI] [PubMed] [Google Scholar]
- 37.Bello G, Eyer-Silva WA, Couto-Fernandez JC, et al. Demographic history of HIV-1 subtypes B and F in Brazil. Infect Genet Evol. 2007;7(2):263–70. doi: 10.1016/j.meegid.2006.11.002. [DOI] [PubMed] [Google Scholar]
- 38.Stefani MM, Pereira GA, Lins JA, et al. Molecular screening shows extensive HIV-1 genetic diversity in Central West Brazil. J Clin Virol. 2007;39(3):205–9. doi: 10.1016/j.jcv.2007.04.012. [DOI] [PubMed] [Google Scholar]
- 39.Soares EA, Martinez AM, Souza TM, et al. HIV-1 subtype C dissemination in southern Brazil. AIDS. 2005;19(Suppl 4):S81–6. doi: 10.1097/01.aids.0000191497.00928.e4. [DOI] [PubMed] [Google Scholar]
- 40.Soares EA, Santos RP, Pellegrini JA, et al. Epidemiologic and molecular characterization of human immunodeficiency virus type 1 in southern Brazil. J Acquir Immune Defic Syndr. 2003;34(5):520–6. doi: 10.1097/00126334-200312150-00012. [DOI] [PubMed] [Google Scholar]
- 41.Soares de Oliveira AC, Pessoa de Farias R, da Costa AC, et al. Frequency of subtype B and F1 dual infection in HIV-1 positive, Brazilian men who have sex with men. Virol J. 2012;9:223. doi: 10.1186/1743-422X-9-223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Ramos A, Tanuri A, Schechter M, et al. Dual and recombinant infections: an integral part of the HIV-1 epidemic in Brazil. Emerg Infect Dis. 1999;5(1):65–74. doi: 10.3201/eid0501.990108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Diaz RS, Sabino EC, Mayer A, et al. Dual human immunodeficiency virus type 1 infection and recombination in a dually exposed transfusion recipient. The Transfusion Safety Study Group. J Virol. 1995;69(6):3273–81. doi: 10.1128/jvi.69.6.3273-3281.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Blackard JT, Mayer KH. HIV superinfection in the era of increased sexual risk-taking. Sex Transm Dis. 2004;31(4):201–4. doi: 10.1097/01.olq.0000118082.45312.1f. [DOI] [PubMed] [Google Scholar]
- 45.Van Laethem K, Van Vaerenbergh K, Schmit JC, et al. Phenotypic assays and sequencing are less sensitive than point mutation assays for detection of resistance in mixed HIV-1 genotypic populations. J Acquir Immune Defic Syndr. 1999;22(2):107–18. doi: 10.1097/00126334-199910010-00001. [DOI] [PubMed] [Google Scholar]
- 46.Gunthard HF, Wong JK, Ignacio CC, et al. Comparative performance of high-density oligonucleotide sequencing and dideoxynucleotide sequencing of HIV type 1 pol from clinical samples. AIDS Res Hum Retroviruses. 1998;14(10):869–76. doi: 10.1089/aid.1998.14.869. [DOI] [PubMed] [Google Scholar]
- 47.Jakobsen MR, Tolstrup M, Sogaard OS, et al. Transmission of HIV-1 drug-resistant variants: prevalence and effect on treatment outcome. Clin Infect Dis. 2010;50(4):566–73. doi: 10.1086/650001. [DOI] [PubMed] [Google Scholar]
- 48.Bon I, Alessandrini F, Borderi M, et al. Analysis of HIV-1 drug-resistant variants in plasma and peripheral blood mononuclear cells from untreated individuals: implications for clinical management. New Microbiol. 2007;30(3):313–7. [PubMed] [Google Scholar]
- 49.Ferreira AS, Cardoso LP, Stefani MM. Moderate prevalence of transmitted drug resistance and high HIV-1 genetic diversity in patients from Mato Grosso State, Central Western Brazil. J Med Virol. 2011;83(8):1301–7. doi: 10.1002/jmv.22128. [DOI] [PubMed] [Google Scholar]
- 50.Pilotto JH, Grinsztejn B, Veloso VG, et al. Moderate prevalence of transmitted drug resistance mutations among antiretroviral-naive HIV-infected pregnant women in Rio de Janeiro, Brazil. AIDS Res Hum Retroviruses. 2013;29(4):681–6. doi: 10.1089/AID.2011.0333. [DOI] [PubMed] [Google Scholar]
- 51.Sucupira MC, Caseiro MM, Alves K, et al. High levels of primary antiretroviral resistance genotypic mutations and B/F recombinants in Santos, Brazil. AIDS Patient Care STDS. 2007;21(2):116–28. doi: 10.1089/apc.2006.0079. [DOI] [PubMed] [Google Scholar]