

Abstract
MJC13, a novel FKBP52 targeting agent, has potential use for the treatment of castrate-resistant prostate cancer. The purpose of this work was to develop a solution formulation of MJC13, and obtain its efficacy profile in a human prostate cancer xenograft mouse model. Preformulation studies were conducted to evaluate the physicochemical properties. Co-solvent systems were evaluated for aqueous solubility and tolerance. A human prostate cancer xenograft mouse model was established by growing 22Rv1 prostate cancer cells in C.B-17 SCID mice. The optimal formulation was used to study the efficacy of MJC13 in this preclinical model of castrate-resistant prostate cancer. We found that MJC13 was stable (at least for 1 month), very lipophilic (logP = 6.49), poorly soluble in water (0.28 μg/mL), and highly plasma protein bound (> 98%). The optimal formulation consisting of PEG 400 and Tween 80 (1:1, v/v) allowed us to achieve a MJC13 concentration of 7.5 mg/mL, and tolerated an aqueous environment. After twice weekly intratumoral injection with 10 mg/kg MJC13 in this formulation for 4 consecutive weeks, tumor volumes were significantly reduced compared to vehicle-treated controls.
Keywords: MJC13, Preformulation, Formulation, Efficacy, Castration-resistant prostate cancer (CRPC), Androgen-independent
1. Introduction
Prostate cancer is the second leading cause of cancer deaths and the most commonly diagnosed cancer in American males. Death due to prostate cancer accounts for nearly 11% of cancer-related deaths in men (Siegel et al., 2013). Normal prostate growth and maintenance depend on androgens acting through the androgen receptor (AR). AR expression and activity is critical for prostate cancer development and progression. In early stage prostate cancer, which remains localized within the prostate, surgery, radiation therapy, and cryotherapy are often effective treatments (Goldman and Schafer, 2011; Thompson et al., 2007). In more advanced stages, when cancer has spread to other parts of the body, hormone therapy is frequently effective. All such treatment options either block androgen production or act as classic receptor antagonists by competing with androgens for binding in the AR hormone binding pocket (Mohler et al., 2009). However, hormone therapy eventually becomes ineffective due to mutation and/or deregulation of the AR. The term “castrate-resistant prostate cancer (CRPC)” is used to describe prostate cancer that has become resistant to hormone therapy (Seruga et al., 2011). CRPC is represents the most aggressive and lethal form of prostate cancer, and its clinical management remains a significant challenge. Thus, treatments that target novel surfaces on AR and/or novel AR regulatory mechanisms are needed to more effectively target AR activity in CRPC.
Recent efforts have focused on the development of molecules directed at alternative sites including the AR activation function 2 (AF2) (Gunther et al., 2009), AR hormone response elements (Chenoweth et al., 2009; Nickols and Dervan, 2007), a newly characterized binding surface on the AR termed binding function 3 (BF3) (Estebanez-Perpina et al., 2007), and AR inhibitors for which the binding sites are currently unknown (Jones et al., 2009; Joseph et al., 2009; Norris et al., 2009). This new class of AR inhibitors has been termed nuclear receptor alternate site modulators (NRAMS) (Moore et al., 2009). In addition to targeting alternative sites on the receptor, the targeting of regulatory proteins critical for receptor protein folding in the cytoplasm also represents an attractive therapeutic option. The folding of AR to a functional conformation that can bind hormone is a highly ordered process that involves a variety of chaperone and cochaperone proteins (Smith and Toft, 2008). Prior to hormone binding, functionally mature AR exists in the cytoplasm in complex with heat shock protein 90 (Hsp90), a 23 kDa cochaperone (p23), and the 52 kDa FK506-binding protein (FKBP52). FKBP52 has been shown to be a positive regulator of AR, glucocorticoid receptor (GR) and progesterone receptor (PR) in both cellular and whole animal models (Sivils et al., 2011; Storer et al., 2011). Given that FKBP52 is functionally specific for a small subset of Hsp90 client proteins (Riggs et al., 2004), targeting FKBP52 regulation of AR activity represents a promising therapeutic approach with the possibility of fewer side effects.
A series of small molecules that can specifically inhibit FKBP52-mediated potentiation of AR signaling through the putative targeting of the AR BF3 surface were recently identified and characterized (De Leon et al., 2011). The lead molecule, MJC13 (N-(2,3-dichlorophenyl)-cyclohexanecarboxamide, see Fig. 1 for structure), effectively blocks hormone-induced dissociation of the AR-chaperone complex leading to a loss of AR nuclear translocation, AR-dependent gene expression, and proliferation in prostate cancer cells at low micromolar concentrations. More importantly, these effects were observed in both hormone-dependent and hormone-independent cellular models of prostate cancer. Thus, MJC13 represents a potentially attractive therapeutic option for the treatment of CRPC.
Fig. 1.
Molecular structure of MCJ13.
In this study, we sought to determine whether MJC13 is also effective in an animal xenograft model of CRPC. For this purpose, we developed a formulation able to efficiently deliver MJC13 in vivo. Due to a lack of hydrophilic functional groups, the aqueous solubility of MJC13 was predicted to be low, making the development of a suitable formulation challenging. Given the lack of previous formulation data, preformulation studies were needed to determine the basic physicochemical properties of MJC13 to allow further formulation development. Therefore, we performed a preformulation evaluation of MJC13; we developed a solution formulation suitable for in vivo administration; and we evaluated the formulated MJC13 in a CRPC xenograft mouse model.
2. Materials and methods
2.1. Materials
MJC13 was custom synthesized (purity ≥ 99%) by Chembridge (San Diego, CA). Water, ethanol, propylene glycol, polyethylene glycol 400 (PEG 400), polysorbate 80 (Tween 80), olive oil, peanut oil, soybean oil, dimethyl sulfoxide (DMSO), 1-octanol, 0.9% sodium chloride solution (normal saline), fetal bovine serum (FBS), and 0.25% (w/v) Trypsin-0.53mM EDTA solution were purchased from Sigma-Aldrich (St. Louis, MO). Captex® 200 was from ABITEC (Columbus, OH). Labrafac™ Lipophile WL1349 was from Gattefosse (Lyon, France). All chemicals and reagents were used as received. The 22Rv1 human prostate carcinoma epithelial cell line and its base medium RPMI-1640 were purchased from ATCC (Manassas, VA). C.B-17 SCID mice (male, 15–20 g) were purchased from Charles River Laboratories (Wilmington, MA).
2.2. Assay
The assay used in this work to determine the concentration of MJC13 in various fluids utilized a published LC-MS/MS method that was developed and validated previously (Liang et al., 2014).
2.3. Preformulation
2.3.1. Solid-state stability
Dry samples of MJC13 were stored at three different temperatures (−20 °C, 4 °C and room temperature) and analyzed via LC-MS/MS at various time intervals for up to 1 month to determine the amount of MJC13 remaining. Experiments were conducted in triplicate.
2.3.2. Solubility
The solubility of MJC13 in water, ethanol, propylene glycol, polyethylene glycol 400, Tween 80, olive oil, peanut oil, soybean oil, Captex 200 and Labrafac Lipophile WL1349 was determined by shake-flask method. Briefly, an excess amount of MJC13 was added to each capped glass bottle containing selected vehicle and mixed for 48 h at room temperature using a reciprocating shaker. Samples were centrifuged at 3000 rpm for 10 min and subsequently filtered through a 0.22 μm ultrafiltration unit. The filtrates were analyzed by LC-MS/MS to determine the concentration of drug dissolved. Experiments were conducted in triplicate.
2.3.3. Lipophilicity
The lipophilicity of MJC13 was determined as the logarithm of partition coefficient (logP) of the solute between water and 1-octanol. Equal volumes of two-phase solution (water and 1-octanol) was mixed well in a capped glass bottle and then placed in a shaker for 24 h to make sure that saturation equilibrium had been achieved. 1 mL of MJC13 dissolved in the two-phase solution was added the bottle. Then, MJC13 was partitioned between aqueous and 1-octanol for 72 h at room temperature using the shake-flask method. The mixture from the bottle was centrifuged at 3000 rpm for 10 min and subsequently filtered through a 0.22 μm ultrafiltration unit, and then moved to a separatory funnel for phase separation. The aqueous and 1-octanol phases were analyzed by LC-MS/MS to determine the concentration of drug dissolved. Experiments were conducted in triplicate. The logP was calculated according to Equation 1:
| (1) |
where Co and Cw represent the concentrations of MJC13 in 1-octanol and aqueous phase, respectively.
2.3.4. Plasma protein binding
In vitro rat plasma samples of MJC13 were prepared by diluting the stock solution with acetonitrile and spiking in rat plasma at five different concentrations: 100, 500, 1000, 2000 and 5000 ng/mL to approximate the plasma concentrations of MJC13 in rats after 2 mg/kg intravenous bolus dosing. The plasma was incubated at 37 °C for 1 h before being transferred to Amicon Ultra-0.5 mL Centrifugal Filters (EMD Millipore Corporation, Billerica, MA) for ultrafiltration at 3000 rpm for 10 min. Filtrate and nonfiltrate concentrations were determined by LC-MS/MS. The fraction unbound, fu, was determined as Equation 2:
| (2) |
where Cu is the unbound concentration and Ct is the total concentration. And the plasma protein binding was determined by (1 – fu).
2.4. Formulation
2.4.1. Co-solvency
Co-solvent systems with various compositions and ratios of DMSO, ethanol, PEG 400 and Tween 80 were prepared with a range MJC13 concentrations (5-10 mg/mL). Each system was diluted with normal saline at the ratios of 1:1, 1:3, 1:7 and 1:15 (v/v) to evaluate whether MJC13 precipitation would occur in 4 h. No precipitation occurring in the diluted formulation after 4 h indicated that the formulation had a good capacity to dissolve MJC13 in an aqueous environment, and that an intravenous dose of this formulation will not cause precipitation at the site of administration. A system with high MJC13 solubility and greatest aqueous tolerance was selected as the optimal co-solvent formulation for further studies. And the MJC13 concentration of the optimal co-solvent formulation was confirmed with the LC-MS/MS method.
2.4.2. Formulation stability
The optimal co-solvent formulation of MJC13 was stored at three different temperatures (−20 °C, 4 °C and room temperature) and analyzed via LC-MS/MS at various time intervals for up to 1 month to determine the amount of MJC13 present. Experiments were conducted in triplicate.
2.5. Xenograft model
2.5.1. Preparation of tumor cells
22Rv1 cells were grown in complete medium (10% v/v FBS in RPMI-1640 medium). When cells were 70–80% confluent, 3–4 h before harvesting, medium was replaced with fresh medium to remove dead and detached cells. Then fresh medium was removed, and cells were washed with PBS. After adding a minimum amount of trypsin-EDTA, cells were dispersed by adding complete medium (5:1), and then centrifuged immediately at 1000 rpm for 3 min. After resuspending with PBS, cells were counted using a hemocytometer.
2.5.2. Tumor inoculation
The work area was prepared by disinfecting all hood surfaces with 70% ethanol. The inoculation area of each mouse was cleaned and sterilized with an alcohol pad. A freshly prepared cell suspension was agitated to prevent the cells from settling, and then mixed with matrigel. One mL of the mixture (containing 1 × 107 cells) was injected subcutaneously into the lower flank of 10 C.B-17 SCID mice. Tumor diameters were measured with digital calipers, and the tumor volume was calculated using Equation 3:
| (3) |
where V is the tumor volume, W is the tumor width, and L is the tumor length.
2.6. Preclinical Efficacy
Therapy was started 2 weeks after inoculation, when the tumors reached an average volume of about 25 mm3. Mice were randomized into two groups with 5 mice in each group. The work area was prepared by disinfecting all hood surfaces with 70% ethanol. The tumor site of each mouse was cleaned and sterilized with an alcohol pad. The test group was administered 10 mg/kg MJC13 by intratumoral administration in the optimal co-solvent formulation twice weekly for 4 consecutive weeks. The control group was administered the equivalent amount of co-solvent vehicle without API intratumorally following the same schedule. Tumor volumes were recorded prior to each treatment.
2.7. Statistical analysis
All values are expressed as the mean ± standard deviation (SD). Tumor volumes were analyzed using Student's t-test to determine the significance between test and control groups at each time point. Statistical significance was reported if p-value was <0.05. Statistical tests were performed with SYSTAT 12 (Systat Software, Chicago, IL).
3. Results
3.1. Preformulation
To investigate the effect of storage temperature on compound stability, dry samples of MJC13 were stored at −20 °C, 4 °C, and room temperature, and then sampled and analyzed at 3-day, 1-week, 2-week, and 1-month intervals. The results are summarized in Table 1. After 1 month, the samples stored in three different conditions displayed 97.3 ± 2.5%, 99.7 ± 3.5%, and 95.3 ± 2.8% average recoveries, respectively. These data indicate that MJC13 powder was stable at room temperature at least for 1 month.
Table 1.
MJC13 solid-state stability.
| Storage condition | 3-day recovery (%) | 1-week recovery (%) | 2-week recovery (%) | 1-month recovery (%) |
|---|---|---|---|---|
| −20 °C | 99.3 ± 3.5 | 97.7 ± 1.5 | 97.0 ± 1.0 | 97.3 ± 2.5 |
| 4 °C | 99.3 ± 1.5 | 99.3 ± 0.6 | 100.7 ± 1.5 | 99.7 ± 3.5 |
| Room temperature | 100.3 ± 3.2 | 100.0 ± 3.6 | 101.3 ± 3.8 | 95.3 ± 2.8 |
The solubility of MJC13 in water, ethanol, propylene glycol, PEG 400, Tween 80, olive oil, peanut oil, soybean oil, Captex 200 and Labrafac Lipophile WL1349 was determined. The results are summarized in Table 2. According to the classification in The Pharmacopoeia of the United States of America (USP), MJC13 is poorly soluble in water (3571 parts of water is needed for 1 part of MJC13).
Table 2.
Solubility of MJC13 in various solvents.
| Solvent | Solubility (mg/mL) |
|---|---|
| Water | (2.8 ± 0.2) × 10-4 |
| DMSO | 100∼200 |
| Ethanol | 35.6 ± 1.8 |
| Propylene glycol | 5.7 ± 0.2 |
| PEG 400 | 23.0 ± 0.7 |
| Polysorbate 80 | 28.2 ± 0.7 |
| Olive oil | 16.2 ± 0.3 |
| Peanut oil | 15.6 ± 0.4 |
| Soybean oil | 17.3 ± 0.5 |
| Captex 200 | 31.9 ± 1.6 |
| Labrafac Lipophile WL1349 | 24.0 ± 1.1 |
The experimental logP of MJC13 between water and 1-octanol tested by the shake-flask method was 6.49 ± 0.37.
The plasma protein binding of MJC13 in rat plasma at concentrations of 100, 500, 1000, 2000, and 5000 ng/mL were found to be 98.1 ± 1.5%, 97.6 ± 2.7%, 99.6 ± 1.7%, 97.2 ± 2.8%, and 97.9 ± 2.4%, respectively. These data indicate that MJC13 is highly protein bound, and the binding is concentration independent.
3.2. Formulation
The co-solvent solubility and aqueous tolerance of MJC13 in various compositions and ratios of solvents are summarized in Table 3. Other 26 formulations without or lower Tween 80 presence have been prepared and evaluated, they all showed MJC13 precipitation even with 1:1 (v/v) dilution with normal saline (data didn't show). The optimal formulation consisted of PEG 400 and Tween 80 (1:1, v/v) and yielded a MJC13 concentration of 7.5 mg/mL, which was confirmed with LC-MS/MS analysis.
Table 3.
MJC13 co-solvent systems: composition, ratio and precipitation upon dilution with normal saline at the ratio of 1:1, 1:3, 1:7 and 1:15 (v/v) in 4 hr*.
| Label | Composition and ratio of solvent (v/v) | MJC13 Concentration (mg/mL) | Precipitation upon dilution with normal saline (v/v) | ||||||
|---|---|---|---|---|---|---|---|---|---|
| DMSO | Ethanol | PEG 400 | Tween 80 | 1:1 | 1:3 | 1:7 | 1:15 | ||
| PT2 | - | - | 7 | 3 | 5 | N | N | Y | Y |
| PT3 | - | - | 5 | 5 | 5 | N | N | N | N |
| PT4** | - | - | 5 | 5 | 7.5 | N | N | N | N |
| PT5 | - | - | 5 | 5 | 10 | N | Y | Y | Y |
| ET2 | - | 7 | - | 3 | 5 | N | N | Y | Y |
| ET3 | - | 5 | - | 5 | 5 | N | N | N | N |
| ET4 | - | 5 | - | 5 | 7.5 | N | N | N | Y |
| DPT2 | 1 | - | 6 | 3 | 5 | N | N | N | Y |
| DPT3 | 1 | - | 4 | 5 | 5 | N | N | N | N |
| DPT4 | 1 | - | 4 | 5 | 7.5 | N | N | N | N |
| DPT5 | 1 | - | 4 | 5 | 10 | N | N | Y | Y |
| DPT7 | 3 | - | 4 | 3 | 10 | N | N | Y | Y |
| DPT8 | 3 | - | 2 | 5 | 10 | N | N | N | Y |
Other 26 formulations with no or lower Tween 80 presence have been prepared and evaluated, they all showed MJC13 precipitation even with 1:1 (v/v) dilution with normal saline. Data didn't show;
Formulation PT4 consisting of PEG 400 and Tween 80 (1:1, v/v) with a MJC13 concentration of 7.5 mg/mL was selected as the optimal co-solvent formulation.
To investigate the effect of storage temperature on formulation stability, the optimal formulation of MJC13 was stored at −20 °C, 4 °C, and room temperature, sampled, and analyzed at 3-day, 1-week, 2-week, and 1-month intervals. The results are summarized in Table 4. Storage for 1 month in the three different conditions yielded 98.0 ± 6.1%, 97.3 ± 1.5%, and 96.7 ± 1.5% average recoveries, respectively. These data indicated that the optimal MJC13 co-solvent formulation was stable at room temperature for, at least, 1 month.
Table 4.
MJC13 optimal co-solvent formulation stability.
| Storage condition | 3-day recovery (%) | 1-week recovery (%) | 2-week recovery (%) | 1-month recovery (%) |
|---|---|---|---|---|
| −20 °C | 101.0 ± 2.0 | 99.0 ± 1.0 | 99.0 ± 2.6 | 98.0 ± 6.1 |
| 4 °C | 97.7 ± 1.2 | 96.3 ± 2.1 | 100.3 ± 2.5 | 97.3 ± 1.5 |
| Room temperature | 98.0 ± 2.0 | 98.3 ± 2.1 | 98.3 ± 3.5 | 96.7 ± 1.5 |
3.3. Preclinical Efficacy
Fig. 2 shows the tumor volume vs. time for control (vehicle-treated) and test (MJC13-treated) groups. The average tumor volumes for each group at each time point are summarized in Table 5. Our data showed that 10 mg/kg MJC13 administered twice weekly via intratumoral injection significantly reduced the rate of tumor growth, but failed to cause tumor regression.
Fig. 2.
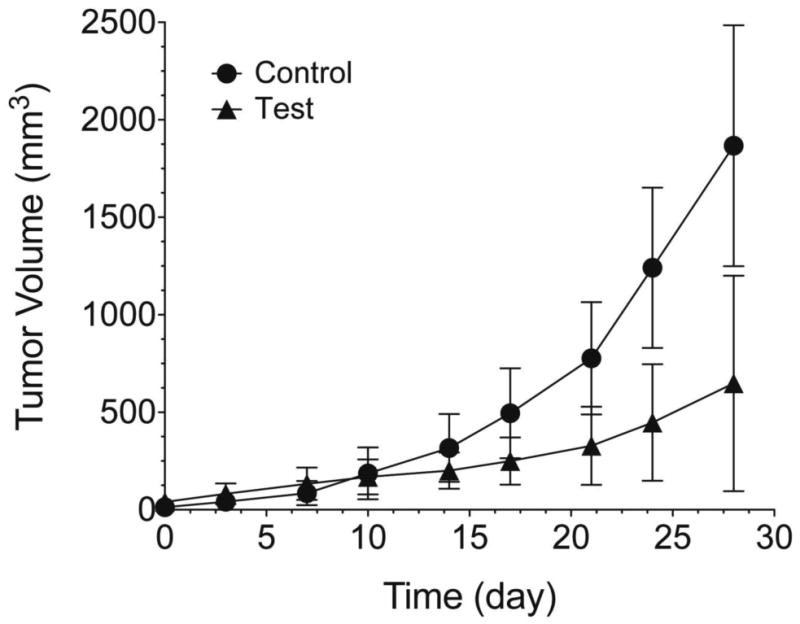
Tumor volume (Mean ± SD) vs. time curves for (•) control (n = 5) and (▴) test (n = 5) group. Vehicle or MJC13 was administered by intratumoral injection twice weekly for 4 consecutive weeks. Tumor growth rate between groups over the 4-week period was significantly different (p < 0.05).
Table 5.
Tumor volumes (Mean ± SD) of control and test group at each time point.
| Time (day) | Control tumor volume (mm3, n = 5) | Test tumor volume (mm3, n = 5) | P-value |
|---|---|---|---|
| 0 | 12.99 ± 10.56 | 39.08 ± 30.08 | 0.127 |
| 3 | 41.56 ± 31.83 | 81.07 ± 52.87 | 0.198 |
| 7 | 85.21 ± 62.00 | 132.50 ± 83.03 | 0.34 |
| 10 | 186.6 ± 132.89 | 167.23 ± 89.89 | 0.795 |
| 14 | 316.75 ± 173.69 | 199.99 ± 92.59 | 0.232 |
| 17 | 495.37 ± 230.41 | 248.68 ± 121.21 | 0.078 |
| 21 | 776.87 ± 287.80 | 327.16 ± 200.27 | 0.024* |
| 24 | 1241.04 ± 411.48 | 446.58 ± 298.52 | 0.009* |
| 28 | 1866.95 ± 617.68 | 646.94 ± 552.30 | 0.011* |
Statistical significance between control and test group.
4. Discussion
We first evaluated the solid-state stability of MJC13 and we found that MJC13 powder was quite stable even stored at room temperature at least for 1 month, so stability was not an issue need to be considered in the formulation development followed. To develop a solution formulation of MJC13 suitable for in vivo administration, the solubility of MJC13 is the most significant property needed to be determined. Since there is no ionizable function group on the molecule, the aqueous solubility of MJC13 was expected to be low and barely affect by pH. Our studies showed that MJC13 was highly lipophilic (logP = 6.49) and very poorly soluble in water (only 0.28 μg/mL). Moreover, we found that MJC13 was highly plasma protein bound, and the binding is concentration-independent. Based on the free drug hypothesis, only the free unbound fraction of the drug is available for therapeutic activity. Therefore, whether we can successfully develop an appropriate formulation that can deliver a sufficient amount of drug to the site of action was critical for preclinical efficacy studies.
The solubility of a non-water-soluble molecule can be increased by the addition of water-miscible solvent in which the molecule has good solubility. The solvents used in combination to increase the solubility of the solute are known as co-solvents. Commonly, water-miscible co-solvents are utilized in intravenous formulations of non-water-soluble drugs. However, drug precipitation often occurs upon addition of intravenous fluids or blood to the co-solvent system if the system does not have decent aqueous tolerance. Co-solvent systems are commonly used in cancer drug formulations. For example, the clinical formulation of the well-established chemotherapy drug Taxotere consists of ethanol and Tween 80 (1:1, v/v) to dissolve the lipophilic active pharmaceutical ingredient (Engels et al., 2007). In this work, co-solvent systems were evaluated to develop the optimal solution formulation of MJC13. The optimal formulation was selected based on consideration of four factors: MJC13 solubility, MJC13 precipitation upon aqueous dilution, solvent toxicity, and formulation stability. Our preformulation studies found that MJC13 had good solubility in DMSO (greater than 100 mg/mL), but the use of DMSO in co-solvent systems cannot prevent MJC13 precipitation upon dilution and can often cause undesired toxic effects. We also found that MJC13 has relatively good solubility in ethanol and PEG 400, but ethanol or PEG 400 alone without the presence of surfactant cannot prevent MJC13 precipitation upon dilution, unless Tween 80 is present in the co-solvent systems. Either ethanol or PEG 400 combined with a high percentage of Tween 80 can achieve a relatively satisfactory solubility of MJC13 and prevent MJC13 precipitation upon dilution. Considering ethanol is a volatile solvent, which may cause the formulation to be unstable during storage, the formulation of PEG 400 and Tween 80 was preferred. The final optimal MJC13 co-solvent formulation consisted of PEG 400 and Tween 80 (1:1, v/v), which yielded an achievable MJC13 concentration of 7.5 mg/mL. This formulation can be diluted with normal saline to the desired concentration of MJC13 before various routes of administration, such as oral administration, intravenous (bolus/infusion) injection, intramuscular injection, and intratumoral injection. Meanwhile, this optimal formulation was stable even at room temperature for at least 1 month, therefore having great potential in further preclinical and clinical applications.
This study was the first to evaluate MJC13 in an animal tumor model. Absent any previous knowledge on pharmacokinetic profile, therapeutic window and toxicity of the agent, we chose to deliver the drug by direct intratumoral injection, thus bypassing the major obstacles to systemic delivery while taking advantage of solid tumor barriers to prevent rapid drug clearance and promote local drug retention (French et al., 2010; Holback and Yeo, 2011). According to our previous study, intratumoral injection of therapeutic agents can achieve higher retention in tumor and lower concentration in other healthy tissues (Xie et al., 2012). MJC13, as a hydrophobic drug, is predicted to have a poor pharmacokinetic profile (maintain a low central compartmental level and distribute widely within the body). Intratumoral administration of MJC13 avoids these problem that insufficient of drug reaches the site of action. Meanwhile, relatively higher amount of MJC13 could be dosed without significant systematic side effects, since it would cause more drug localized on the tumor site and less drug in the system circulation. Our data showed that 10 mg/kg MJC13 twice a week intratumoral dose of the co-solvent formulation significantly slowed the rate of tumor growth. Given the aggressive nature and androgen independence of the 22Rv1 tumor model (He et al., 2013), these findings are encouraging. Our data warrant further preclinical dose ranging and schedule evaluation studies of MJC13 in CRPC tumor models.
5. Conclusion
We successfully developed and optimized a co-solvent formulation allowing for in vivo administration of MJC13 that is based on the compound's physicochemical properties. The optimal co-solvent formulation is composed of PEG 400 and Tween 80 (1:1, v/v) with a MJC13 concentration of 7.5 mg/mL. It is stable and suitable for further preclinical and clinical evaluations of safety and efficacy. This formulation was successfully applied in a preliminary preclinical efficacy study in an androgen-independent (but AR-dependent) prostate cancer xenograft mouse model and showed significant inhibition of tumor growth, supporting further preclinical development of MJC13.
Acknowledgments
HX has research grant support from an NIH/RTRN grant (5U54RR022762-05), NIH/NIGMS grant (1SC3GM102018-01) and from NIH/NIMHD/RCMI grant (5G12RR003045-21). MBC has research grant support from NIH/NCRR/RCMI grant (5G12RR008124), NIH/NIMHD/RCMI grant (G12MD007592), NIH/NIGMS grant (SC1GM084863) and State of Texas CPRIT grant (RP110444-P2).
Footnotes
Conflict of Interest: The authors report no declarations of interest.
References
- Chenoweth DM, Harki DA, Phillips JW, Dose C, Dervan PB. Cyclic pyrrole-imidazole polyamides targeted to the androgen response element. J Am Chem Soc. 2009;131:7182–7188. doi: 10.1021/ja901309z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Leon JT, Iwai A, Feau C, Garcia Y, Balsiger HA, Storer CL, Suro RM, Garza KM, Lee S, Kim YS, Chen Y, Ning YM, Riggs DL, Fletterick RJ, Guy RK, Trepel JB, Neckers LM, Cox MB. Targeting the regulation of androgen receptor signaling by the heat shock protein 90 cochaperone FKBP52 in prostate cancer cells. Proceedings of the National Academy of Sciences of the United States of America. 2011;108:11878–11883. doi: 10.1073/pnas.1105160108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engels FK, Mathot RA, Verweij J. Alternative drug formulations of docetaxel: a review. Anti-cancer drugs. 2007;18:95–103. doi: 10.1097/CAD.0b013e3280113338. [DOI] [PubMed] [Google Scholar]
- Estebanez-Perpina E, Arnold LA, Nguyen P, Rodrigues ED, Mar E, Bateman R, Pallai P, Shokat KM, Baxter JD, Guy RK, Webb P, Fletterick RJ. A surface on the androgen receptor that allosterically regulates coactivator binding. Proc Natl Acad Sci U S A. 2007;104:16074–16079. doi: 10.1073/pnas.0708036104. Epub 12007 Oct 16072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- French JT, Goins B, Saenz M, Li S, Garcia-Rojas X, Phillips WT, Otto RA, Bao A. Interventional therapy of head and neck cancer with lipid nanoparticle-carried rhenium 186 radionuclide. Journal of vascular and interventional radiology : JVIR. 2010;21:1271–1279. doi: 10.1016/j.jvir.2010.02.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldman L, Schafer AI. Cecil medicine: expert consult premium edition. Elsevier Health Sciences; 2011. [Google Scholar]
- Gunther JR, Parent AA, Katzenellenbogen JA. Alternative inhibition of androgen receptor signaling: peptidomimetic pyrimidines as direct androgen receptor/coactivator disruptors. ACS Chem Biol. 2009;4:435–440. doi: 10.1021/cb900043e. [DOI] [PMC free article] [PubMed] [Google Scholar]
- He S, Zhang C, Shafi AA, Sequeira M, Acquaviva J, Friedland JC, Sang J, Smith DL, Weigel NL, Wada Y, Proia DA. Potent activity of the Hsp90 inhibitor ganetespib in prostate cancer cells irrespective of androgen receptor status or variant receptor expression. International journal of oncology. 2013;42:35–43. doi: 10.3892/ijo.2012.1698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Holback H, Yeo Y. Intratumoral drug delivery with nanoparticulate carriers. Pharmaceutical research. 2011;28:1819–1830. doi: 10.1007/s11095-010-0360-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jones JO, Bolton EC, Huang Y, Feau C, Guy RK, Yamamoto KR, Hann B, Diamond MI. Non-competitive androgen receptor inhibition in vitro and in vivo. Proc Natl Acad Sci U S A. 2009;106:7233–7238. doi: 10.1073/pnas.0807282106. Epub 2009 Apr 7210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Joseph JD, Wittmann BM, Dwyer MA, Cui H, Dye DA, McDonnell DP, Norris JD. Inhibition of prostate cancer cell growth by second-site androgen receptor antagonists. Proc Natl Acad Sci U S A. 2009;106:12178–12183. doi: 10.1073/pnas.0900185106. Epub 12009 Jul 12172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang S, Bian X, Sivils J, Neckers LM, Cox MB, Xie H. Quantification of a New Anti-Cancer Molecule MJC13 Using a Rapid, Sensitive, and Reliable Liquid Chromatography-Tandem Mass Spectrometry Method. American Journal of Modern Chromatography. 2014;1:1–11. [PMC free article] [PubMed] [Google Scholar]
- Mohler ML, Bohl CE, Jones A, Coss CC, Narayanan R, He Y, Hwang DJ, Dalton JT, Miller DD. Nonsteroidal selective androgen receptor modulators (SARMs): dissociating the anabolic and androgenic activities of the androgen receptor for therapeutic benefit. J Med Chem. 2009;52:3597–3617. doi: 10.1021/jm900280m. [DOI] [PubMed] [Google Scholar]
- Moore TW, Mayne CG, Katzenellenbogen JA. Minireview: Not picking pockets: nuclear receptor alternate-site modulators (NRAMs) Mol Endocrinol. 2009;24:683–695. doi: 10.1210/me.2009-0362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nickols NG, Dervan PB. Suppression of androgen receptor-mediated gene expression by a sequence-specific DNA-binding polyamide. Proc Natl Acad Sci U S A. 2007;104:10418–10423. doi: 10.1073/pnas.0704217104. Epub 12007 Jun 10412. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Norris JD, Joseph JD, Sherk AB, Juzumiene D, Turnbull PS, Rafferty SW, Cui H, Anderson E, Fan D, Dye DA, Deng X, Kazmin D, Chang CY, Willson TM, McDonnell DP. Differential presentation of protein interaction surfaces on the androgen receptor defines the pharmacological actions of bound ligands. Chem Biol. 2009;16:452–460. doi: 10.1016/j.chembiol.2009.01.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riggs DL, Cox MB, Cheung-Flynn J, Prapapanich V, Carrigan PE, Smith DF. Functional specificity of co-chaperone interactions with Hsp90 client proteins. Crit Rev Biochem Mol Biol. 2004;39:279–295. doi: 10.1080/10409230490892513. [DOI] [PubMed] [Google Scholar]
- Seruga B, Ocana A, Tannock IF. Drug resistance in metastatic castration-resistant prostate cancer. Nature reviews. Clinical oncology. 2011;8:12–23. doi: 10.1038/nrclinonc.2010.136. [DOI] [PubMed] [Google Scholar]
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2013. CA: a cancer journal for clinicians. 2013;63:11–30. doi: 10.3322/caac.21166. [DOI] [PubMed] [Google Scholar]
- Sivils JC, Storer CL, Galigniana MD, Cox MB. Regulation of steroid hormone receptor function by the 52-kDa FK506-binding protein (FKBP52) Curr Opin Pharmacol. 2011;11:314–319. doi: 10.1016/j.coph.2011.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith DF, Toft DO. Minireview: the intersection of steroid receptors with molecular chaperones: observations and questions. Mol Endocrinol. 2008;22:2229–2240. doi: 10.1210/me.2008-0089. Epub 2008 May 2221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Storer CL, Dickey CA, Galigniana MD, Rein T, Cox MB. FKBP51 and FKBP52 in signaling and disease. Trends Endocrinol Metab. 2011;22:481–490. doi: 10.1016/j.tem.2011.08.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson I, Thrasher JB, Aus G, Burnett AL, Canby-Hagino ED, Cookson MS, D'Amico AV, Dmochowski RR, Eton DT, Forman JD. Guideline for the management of clinically localized prostate cancer: 2007 update. The Journal of urology. 2007;177:2106–2131. doi: 10.1016/j.juro.2007.03.003. [DOI] [PubMed] [Google Scholar]
- Xie H, Goins B, Bao A, Wang ZJ, Phillips WT. Effect of intratumoral administration on biodistribution of 64Cu-labeled nanoshells. International journal of nanomedicine. 2012;7:2227–2238. doi: 10.2147/IJN.S30699. [DOI] [PMC free article] [PubMed] [Google Scholar]